

Abstract
Molecular markers of sorafenib efficacy in metastatic renal cell carcinoma (mRCC) patients are not available. The purpose of this study was to discover genetic markers of survival in mRCC patients treated with sorafenib. Germline variants from 56 genes were genotyped in 295 mRCC patients. Variant-overall survival (OS) associations were tested in multivariate regression models. Mechanistic studies were conducted to validate clinical associations. VEGFA rs1885657, ITGAV rs3816375, and WWOX rs8047917 (sorafenib arm), and FLT4 rs307826 and VEGFA rs3024987 (sorafenib and placebo arms combined) were associated with shorter OS. FLT4 rs307826 increased VEGFR-3 phosphorylation, membrane trafficking and receptor activation. VEGFA rs1885657 and rs58159269 increased transcriptional activity of the constructs containing these variants in endothelial and RCC cell lines, and VEGFA rs58159269 increased endothelial cell proliferation and tube formation. FLT4 rs307826 and VEGFA rs58159269 led to reduced sorafenib cytotoxicity. Genetic variation in VEGFA and FLT4 could affect survival in sorafenib-treated mRCC patients. These markers should be examined in additional malignancies treated with sorafenib, and in other angiogenesis inhibitors used in mRCC.
Keywords: sorafenib, single nucleotide polymorphism, overall survival
Introduction
Sorafenib tosylate (sorafenib, Nexavar®) is a vascular endothelial growth factor (VEGF)-pathway inhibitor approved by the U.S. Food and Drug Administration (FDA) for the treatment of advanced and metastatic renal cell carcinoma (mRCC), unresectable hepatocellular carcinoma (HCC), radioactive iodine-refractory differentiated thyroid carcinoma, and is becoming increasingly utilized off-label for FLT3-mutated acute myeloid leukemia (AML). Sorafenib is a potent inhibitor of VEGFR-2 and VEGFR-3, PDGFR-β, FLT-3, c-KIT, RAF-1, and BRAF (1).
The phase III Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) was a double-blind, randomized, placebo-controlled, multicenter study of 903 mRCC patients who had failed previous cytokine therapy (2). Based upon results from TARGET and a phase II study that preceded TARGET, the U.S. FDA approved sorafenib for the treatment of mRCC (2,3). Currently, sorafenib is primarily used in mRCC after progression on first-line therapy (4). Recently, immunotherapy with checkpoint inhibitors nivolumab and ipilimumab has proven to be efficacious in patients with mRCC (5, 6).
Clear cell RCC is a malignancy characterized by high vascularization, resulting from molecular mechanisms that abrogate the activity of the VHL tumor suppressor gene (5,6). Sorafenib affects tumor vascular endothelium, the tumor microenvironment, but importantly the host vascular endothelium and pericytes (7,8). Because RCC is dependent on angiogenesis, the VEGF pathway has been shown to be a viable target for drug therapy (9), and sorafenib has been shown to inhibit angiogenic targets in multiple RCC models (9,10). As angiogenesis is primarily a host-mediated process (11), germline variants that regulate angiogenic processes are likely to be associated with both disease progression and sorafenib efficacy.
Clinically, response to sorafenib is highly variable (2,3,12), and molecular mechanisms underlying the interpatient variability in sorafenib efficacy have yet to be elucidated. While the armamentarium of treatment for mRCC has been expanded over the past decade, there is still uncertainty concerning selection and sequencing of these agents, particularly after patients progress on first-line therapy. Moreover, there is a dearth of validated molecular biomarkers to help inform clinicians about disease progression and drug efficacy.
Clinical studies using genetic analyses are ideal to select novel molecular candidates for testing in experimental models. Through this reverse translational approach (i.e., bedside to bench), novel mechanistic hypotheses can be formulated to advance the field of precision oncology. In this study, 11,117 germline DNA variants in 56 genes were tested for association with survival in 295 mRCC patients from the TARGET study. For variants associated with survival, a series of functional experiments were employed to determine novel mechanism by which DNA variation in angiogenesis genes might affect the biology of RCC and the efficacy of sorafenib.
Materials and Methods
TARGET study and patient characteristics
TARGET was a double-blind, randomized, placebo-controlled phase III trial of patients with mRCC who had received prior cytokine therapy (n=903) (2,12). Patients were randomized 1:1 to either 400 mg sorafenib orally twice daily or placebo. Patients remained on study until disease progression, discontinuation due to intolerable toxicity, or death. The TARGET primary endpoint was OS, defined as the time from the date of randomization until the date of death, and OS was also used as the primary endpoint for this genetic study. To avoid the confounding effect of crossover of patients from the placebo arm to the sorafenib arm, the OS data used in this study were recorded before patient crossover. PFS was measured from the date of randomization until the date of progression, as defined by the trial protocol (2). The clinical characteristics and median OS of the 295 genotyped patients were comparable to those of the entire TARGET population (Table 1). All patients provided written informed consent to participate in this genetic analysis, approved by the institutional review board at each center.
Table 1.
Clinical characteristics of genotyped patients in the TARGET study.
| All TARGET Patients (n=903) | Genotyped TARGET Patients (n=295) | |||
|---|---|---|---|---|
| Sorafenib (n=451) | Placebo (n=452) | Sorafenib (n=155) | Placebo (n=140) | |
| Male gender - no. (%) | 315 (70) | 340 (75) | 114 (74) | 108 (77) |
| Median age - year (range) | 58 (19–86) | 59 (29–84) | 59 (19–80) | 58 (31–82) |
| ECOG performance status - no. (%) | ||||
| 0 | 219 (49) | 210 (46) | 83 (54) | 81 (58) |
| 1 | 223 (49) | 236 (52) | 72 (46) | 58 (42) |
| 2 | 7 (2) | 4 (1) | 0 | 0 |
| Missing | 2 (<1) | 2 (<1) | 0 | 1 (<1) |
| Number of metastatic sites - no. (%) | ||||
| 1 | 62 (14) | 63 (14) | 26 (17) | 26 (19) |
| 2 | 131 (29) | 129 (29) | 42 (27) | 40 (29) |
| >2 | 256 (57) | 258 (57) | 87 (56) | 74 (53) |
| Missing | 2 (<1) | 2 (<1) | 0 | 0 |
| Lung or liver metastatic sites - no. (%) | 377 (84) | 382 (85) | 133 (86) | 120 (86) |
| Previous cytokine use - no. (%) | 374 (83) | 368 (81) | 141 (91) | 122 (88) |
| Median duration of disease - year (range) | 2 (<1–19) | 2 (<1–20) | 1.7 (<1–19.5) | 1.5 (<1–16) |
| MSKCC prognostic risk - no. (%) | ||||
| Low | 233 (52) | 228 (50) | 73 (47) | 68 (49) |
| Intermediate | 218 (48) | 223 (49) | 82 (53) | 72 (51) |
| Missing | 0 | 1 (<1) | 0 | 0 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; MSKCC, Memorial Sloan Kettering Cancer Center.
Genotyping of gene variants
A total of 56 genes were selected for genotyping. The genes, their chromosomal location, and their biological function are described in Table S1. Selection criteria for single-nucleotide polymorphisms (SNPs) to be genotyped are described in the Supplementary Methods and Materials. Germline DNA was extracted from peripheral blood (FlexiGene DNA kit, Qiagen) and 1,536 SNPs were genotyped using the Illumina GoldenGate assay. Genotypes were determined using Illumina GenomeStudio software v2011. Variants were excluded if the genotype call rate was <97.5%, the minor allele frequency (MAF) was <1%, or if they deviated from Hardy-Weinberg equilibrium (HWE; P<0.0001). GenTrain scores were derived from Illumina GenomeStudio scatter plots to measure variant detection reliability based on genotypic clustering distributions (13), and only SNPs with GenTrain scores >0.4 were included. Additional variants were obtained through imputation using Impute2 version 2.30 (14). Overall, 11,117 variants were included in the final analyses (Figure S1). Imputation methods are included in the Supplementary Methods.
Statistical analyses
The primary study objective was to identify gene variants associated with OS. Univariate log rank tests were conducted to test each variant-OS association. Likelihood ratio tests were performed to examine associations between OS and each of the following factors: country of origin, gender, age, Eastern Cooperative Oncology Group performance status, time since RCC diagnosis, previous systemic treatment with IL-2 or INF, MSKCC prognostic risk score, number of metastatic sites, and evidence of liver or lung metastases. Cox proportional hazards regression included both genetic variants identified by the log rank tests described above and significant clinical covariates (P<0.05). Additive, dominant, or recessive models were used because the mode of inheritance of risk alleles was not known a priori. To use an inclusive approach to detect variant-OS associations, they were tested in the sorafenib arm, the placebo arm, and both arms combined.
FDR q-values were calculated to account for multiple testing (15). FDR was employed in lieu of correcting the family-wise error rate (i.e., Bonferroni correction) to better account for the correlation among tests resulting from the LD among variants and co-linearity among the three genetic models. Variant-OS associations were regarded statistically significant if P<0.05 and q<0.10. These significance thresholds were used as features to select variants for subsequent functional in vitro studies. An interaction test (Wald test) between the two arms (placebo versus sorafenib) was also run on variants that passed the above thresholds, and the interaction P-value cut-off for significance was set at 0.1. The restricted mean OS was used to report the OS for each genotype, and was selected in lieu of median OS because the median was undefined for many variants, either because more than half of the patients were alive at the date of the final event, or the last event was censored (16).
Cell culture
Human embryonic kidney cells (HEK-293) (ATCC) were cultured in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Human clear-cell mRCC cells (Caki-1) (a kind gift from Dr. William Kim, University of North Carolina, Chapel Hill, NC) were cultured in McCoy’s 5A media (Iwakata and Grace Modification) containing L-glutamine, 10% FBS and 1% penicillin/streptomycin. Human telomerase-immortalized microvascular endothelial cells (TIME) (ATCC) and human liver parenchyma endothelial cells (LPEC) (a kind gift of Dr. Lee Ellis, MD Anderson Cancer Center, Houston, TX) were cultured in endothelial basal media supplemented with the EGM™−2MV BulletKit™. Human umbilical vein endothelial cells (HUVECs) (PromoCell GmbH) were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin. Cell lines used in this study were either purchased within six months of their use as part of in vitro experiments, or were authenticated by STR Mapping using Applied Biosystems GeneMapper IDv3.2.
VEGFR-3 phosphorylation assays
The effect of T494A on VEGFR-3 expression was tested using an SDS-PAGE gel by immunoprecipitation and western blotting. VEGFR-3 WT and T494A were expressed in HUVECs upon retroviral transfection. The constructs were Strep-tagged to allow separation from the endogenous VEGFR-3 in the HUVECs. Streptactin-sepharose precipitated samples were run on an 8% SDS-PAGE and western blotted with anti-VEGFR-3 antibodies (AF743, R&D Systems) as previously reported (17,18).
The effect of T494A on VEGFR-3 phosphorylation was investigated in VEGF-C-stimulated HUVECs. After a 2 h starvation, 494T and 494A retrovirally-transfected cells were stimulated with mature VEGF-C (ΔNΔC-VEGF-C) at a final concentration of 50 ng/mL for 20 min. Strep-tagged VEGFR-3 proteins in the lysates were precipitaed with streptactin sepharose. They were then loaded onto an 8% SDS-PAGE gel and western blotted for phospho-tyrosines (4G10) and total VEGFR-3. A Student’s t test was used to compare means from cells with the WT versus T494A cells (n=3).
VEGFR-3 pulse-chase experiments to evaluate VEGFR-3 processing
VEGFR-3 transfected HUVECs were starved for 2 h in met-/cys-deficient DMEM without serum, after which 1 mL of labelling medium (met-/cys-deficient DMEM supplemented with 20 µL EasyTag EXPRESS 35S/ml) was added for 2 h. Cells were washed with warm PBS and 5 mL of chase medium was added (DMEM 10% FCS+1 mM cold L-methionine+2 mM cold L-cysteine). At the indicated time points, cell plates were place on ice, the medium was removed, and the cells washed with PBS before being lysed. Analysis was performed by precipitating Strep-tagged VEGFR-3 from the samples with streptactin sepharose. The precipitated samples were resolved on a reducing SDS-PAGE, and signals captured with a phosphoimager plate and a Typhoon scanner. The 125 kDa gel band gel appears at 130 kDa band due to the double strep tag.
Viability assays in sorafenib-treated cells transfected with FLT4 rs307826 (A>G, T494A)
FLT4 cDNA with the WT allele (A allele) was introduced in a pCMV6-XL5 expression vector, and the variant rs307826 G allele (leading to T494A) was introduced using site-directed mutagenesis. Sanger sequencing was performed to confirm mutagenesis. VEGFR-3 WT and T494A were expressed in HUVECs upon retroviral transfection, while HEK-293 cells were transfected with using Lipofectamine® 2000. After 24 h, cells were treated with ascending concentrations of sorafenib from 0.5–50 µM (in DMSO 0.1%) based upon pharmacologically relevant concentrations of sorafenib in humans (6–15 µM) (19,20). After 72 h, cells were either stimulated with VEGF-C in DMEM media (200 ng/mL) or DMEM alone for 1 h prior to the addition of AlamarBlue®, which was used to assess cell viability. Three independent experiments in triplicate were performed at each concentration in VEGF-C-stimulated and unstimulated HUVEC and HEK-293 cells. IC50 values were obtained using a four-parameter non-linear regression model to 1.0 to assess log10 sorafenib concentration versus average percent viability. A one-way ANOVA, followed by pairwise comparisons and a Dunnett’s correction for multiple testing, was used to assess differences between IC50 values.
Luciferase activity assays of intronic variants
All constructs containing variants in VEGFA, ITGAV, and WWOX were synthesized and inserted into the pGL4.26 [luc2/minP/Hygro] plasmid upstream of its minimal promoter. Site-directed mutagenesis was used to introduce all variants. Sanger sequencing was used to confirm mutagenesis. Caki-1 cells were transfected using Lipofectamine® LTX and a Renilla HSV-TK plasmid control reporter. TIME cells were transfected using TransIT-2020 transfection reagent and a Renilla SV40 plasmid control reporter. LPEC cells were transfected using magnetofection with CombiMag magnetic nanoparticles, TransIT-2020, and Renilla SV40. Cells were lysed 40 h after transfection and luciferase assays were performed. Four independent experiments were conducted in triplicate for each construct. Luciferase activity was calculated as the Firefly to Renilla luciferase ratio, normalized to the empty vector. A one-way ANOVA followed by pairwise comparisons was used, with Dunnett’s correction for multiple testing.
Creation of isogenic cell lines of VEGFA rs58159269 (T>C)
A custom transcription activator-like effector nuclease (TALEN) pair, engineered to allow single base pair editing of rs58159269, was designed and inserted into a pTAL.CMV.T7.v2 plasmid backbone (Cellectis Bioresearch). A donor plasmid containing 250 bp of DNA sequence upstream and downstream of rs58159269, as well as the desired nucleotide substitution, was designed (Cellectis Bioresearch), synthesized and cloned into a pUC57-Amp expression vector (GeneWiz, Inc.). TIME cells were transfected with the TALEN pair (10 µg) and donor plasmid (2:1 ratio of TALENs to donor plasmid) using TransIT-2020 transfection reagent 48 h prior to fluorescence-activated cell sorting (FACS) for isolation of single cell colonies. Following expansion of single cell colonies, DNA was extracted and Sanger sequencing used to verify the appropriate point variant (T>C) at the rs58159269 locus. Three clones for each genotype of TT or CC were selected and determined by Sanger sequencing not to harbor off-target variants in potential region that are targets for the TALEN constructs.
Cell proliferation and sorafenib cytotoxicity assays in isogenic cell lines of VEGFA rs58159269 (T>C)
TT and CC isogenic endothelial cell lines were plated in 96 well plates at 3,000 cells/well in EGM-2 MV medium (Lonza). After 8 h, cells were quiesced overnight in EBM-2 medium (Lonza) with 1% FBS (Omega Scientific) in addition to hydrocortisone, and ascorbic acid (Lonza) at the same concentration as EGM-2 MV medium. For experiments with sorafenib, media was removed and replaced with EGM-2 MV containing sorafenib 0–10 µM in DMSO (Biotang, Inc.) after 24 h. Untreated cells contained DMSO alone at 1:1000. After 72 h of proliferation at 37 oC 5% CO2, and 95% humidity, viable cells were stained using Cyquant Direct Cell Proliferation Assay (Life Technologies). Images were acquired using the EVOS FLc microscope (Life Technologies) using a 10x objective and counts were accomplished with FIJI particle analyzer. A two-tailed, unpaired t-test was used to analyze differences in cell proliferation and cytotoxicity between TT and CC cells.
Endothelial cell tube formation assays in isogenic cell lines
TT and CC isogenic endothelial cell lines were plated, grown and quiesced, as described above for proliferation studies. Matrigel (EMD Millipore) was set into 96 well plates according to the manufactures instructions. Cell viability was assessed using Trypan Blue (Bio-Rad). Cells were then added to each well at a final concentration of 50,000 cells/150 µl in EGM-2 MV media with 2.5% FBS. Images were acquired using the EVOS FLc microscope (Life Technologies) using a 4x objective, and number of branch points (nodes) was determined using FIJI angiogenesis analyzer. A two-tailed, unpaired t-test with Welch’s correction was used to analyze differences in endothelial tube formation between TT and CC cells.
Results
A total of 11,117 germline variants were tested for association with overall survival (OS) in 295 patients, and a study schematic of patients and variants is provided in Figure S1. The clinical characteristics of patients are shown in Table 1. Using both P<0.05 and false discovery rate (FDR) q<0.10 as the thresholds for statistical significance, seven variants were associated with OS (Table 2). Because the Memorial Sloan Kettering Cancer Center (MSKCC) prognostic risk score (P=0.05) and the number of metastatic sites (P=0.001) were associated with OS, they were included in multivariate models testing associations between variants and OS. The MSKCC score is used often in outcome studies in RCC (15–17).
Table 2. Gene variants associated with OS.
Five variants were associated with OS in patients from the sorafenib arm. Two variants were associated with OS in both arms combined. All these seven variants passed the cut-off for statistical significance (P<0.05 and FDR q<0.10). The interaction P-value (Wald test) in both arms combined is also reported.
| Variant ID | Chr | Gene | Alleles | Feature | MAF | HR (95% CI) | P-value | FDR q-value | Interaction P-value |
|---|---|---|---|---|---|---|---|---|---|
| Sorafenib Arm, n=155 | |||||||||
| rs1885657 | 6 | VEGFA | T>C | Intron | 0.17 | 17.3 (5.7–52.7) | 1.4×10−4 | 0.08 | 0.17 |
| rs200809375* | 10 | - | A>ATG | 7.5 kb 3’ of NRP1 | 0.22 | 6.8 (2.6–17.5) | 2.7×10−4 | 0.08 | 0.87 |
| rs6719561* | 2 | - | C>T | 1.8 kb 3’ of UGT1A9 | 0.34 | 3.8 (1.6–9.5) | 3.3×10−4 | 0.08 | 0.006 |
| rs8047917 | 16 | WWOX | T>A | Intron | 0.08 | 4.1 (1.9–8.8) | 3.3×10−4 | 0.08 | 0.84 |
| rs3816375 | 2 | ITGAV | A>G | Intron | 0.38 | 5.9 (2.1–16.4) | 4.9×10−4 | 0.05 | 0.10 |
| Both arms combined, n=295 | |||||||||
| rs307826 | 5 | FLT4 | A>G | Exon, missense (T494A) | 0.10 | 13.8 (3.0–62.6) | 1.2×10−4 | 0.09 | 0.09 |
| rs3024987* | 6 | VEGFA | C>T | Intron | 0.11 | 3.0 (1.7–5.4) | 8.3×10−5 | 0.09 | 0.26 |
Denotes imputed variants. Abbreviations: Chr, chromosome; MAF, minor allele frequency.
Variants associated with OS in the sorafenib arm
Three variants located in genes were associated with OS (Figure 1A–C). VEGFA rs1885657 (T>C, HR=17.3; 95% CI, 5.7–52.7; P=1.4×10−4), ITGAV rs3816375 (A>G, HR=5.9; 95% CI, 2.1–16.4; P=4.9×10−4), and WWOX rs8047917 (T>A, HR=4.1; 95% CI, 1.9–8.8; P=3.3×10−4) were associated with shorter OS. The mean OS was 270 (range, 221–319) days for VEGFA rs1885657 CC patients versus 336 (34–377) days for TT/TC patients; 387 (34–497) days for ITGAV rs3816375 GG patients versus 371 (35–463) days for AA/AG patients; and 307 (34–377) days for WWOX rs8047917 TA patients versus 355 (35–497) days for TT patients (no AA patients were identified).
Figure 1. Kaplan-Meier plots of genetic variants associated with OS.
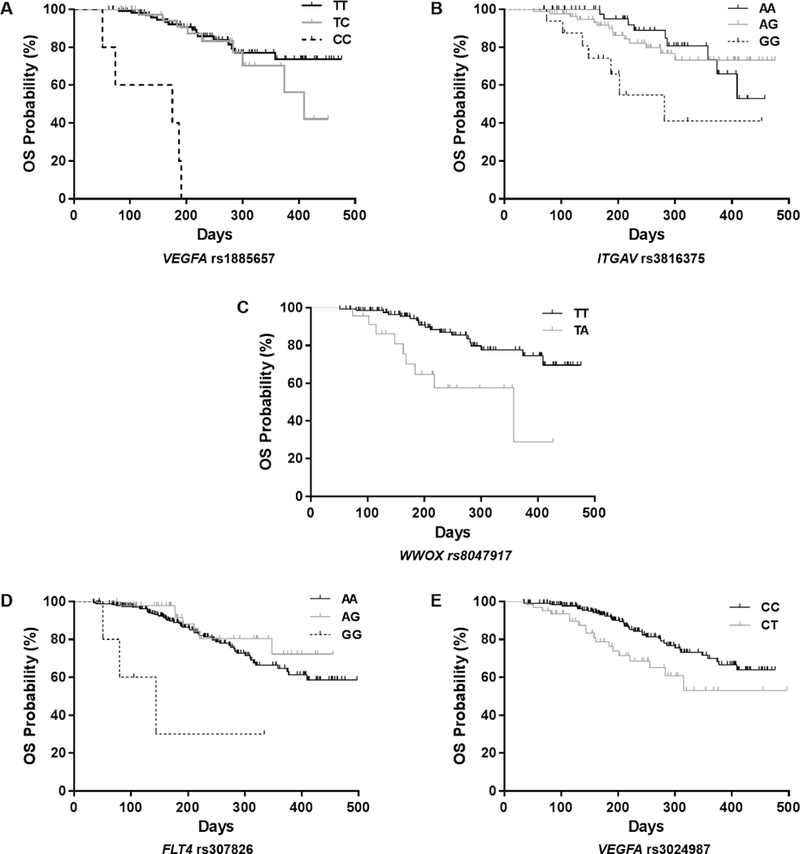
Vertical bars on the survival curves indicate censored observations. A–C) Associations observed in the sorafenib arm. D–E) Associations observed in both arms combined. Vertical bars on the survival curves indicate censored observations.
Two gene-flanking variants were associated with OS. Both rs6719561 (3’ of UGT1A9, C>T, HR=3.8; 95% CI, 1.6–9.5; P=3.3×10−4) and rs200809375 (3’ of NRP1, ATG insertion, HR=6.8; 95% CI, 2.6–17.5; P=2.7×10−4) were associated with shorter OS. The mean OS was 322 (84–453) days for rs6719561 TT patients versus 384 (34–497) days for CC/CT patients; and 322 (34–497) days for patients with an rs200809375 ATG insertion versus 377 (129–455) days for patients without the ATG insertion. None of the significant associations in the sorafenib arm were also significant in both arms combined. No significant associations were found in the placebo arm.
Variants associated with OS in both arms combined
Two variants located in genes were associated with OS (Figure 1D–E). FLT4 rs307826 (A>G, HR=13.8; 95% CI, 3.0–62.6; P=1.2×10−4) and VEGFA rs3024987 (C>T, HR=3.0; 95% CI, 1.7–5.4; P=8.3×10−5) were associated with shorter OS. The mean OS was 194 (51–334) days for FLT4 rs307826 GG patients versus 394 (34–497) days for AA/AG patients; and 348 (39–497) days for VEGFA rs3024987 CT patients versus 403 (34–409) days for CC patients (no TT genotypes were found). Neither of the significant associations in both arms combined were significant either in the sorafenib arm or the placebo arm.
Variants associated with progression-free survival (PFS)
As an exploratory analysis, variants that were significantly associated with OS (Table 2, Figure 1A–E) were also tested for their association with PFS (Figure S2). VEGFA rs1885657 (HR=4.00, 95% CI 1.57–10.21; P=0.004), rs6719561 (3’ of UGT1A9, HR=1.94, CI 1.00–3.76; P=0.05), WWOX rs8047917 (HR=1.77, CI 1.03–3.04; P=0.04), and FLT4 rs307826 (HR=2.92, CI 1.19–7.19; P=0.019) were also associated with PFS. However, ITGAV rs3816375, NRP1 rs200809375, and VEGFA rs3024987 were not associated with PFS (P>0.05).
FLT4 rs307826 (A>G, T494A) increases VEGFR-3 phosphorylation and processing
FLT4 rs307826 is an A>G change that leads to a T494A amino acid substitution in VEGFR-3. FLT4 rs307826 GG was associated with shorter OS in both arms combined (Figure 1D). We tested the hypothesis that T494A might affect the function of VEGFR-3. VEGFR-3 is expressed as a 170 kDa precursor that matures to the 190 kDa fully-glycosylated cell-surface VEGFR-3, which is then proteolytically cleaved into 125 kDa C-terminal fragment and a shorter N-terminal fragment (21). In transfected HUVECs, the T494A VEGFR-3 and the wild-type (WT) VEGFR-3 both showed the same polypeptide bands, indicating similar glycosylation and proteolytic processing (Figure S3). In transfected HUVECs, T494A leads to increased phosphorylation of the fully-processed (190 kDa band) and the proteolytically-processed C-terminal (125 kDa band) forms of VEGFR-3 (22), when compared to wild-type (WT) VEGFR-3 (Figure 2A). This effect was potentiated by VEGF-C stimulation. This result was also confirmed in independent experiments (Figure S3).
Figure 2. Effects of FLT4 rs307826 (A>G, T494A) on VEGFR-3 phosphorylation and post-translational processing.
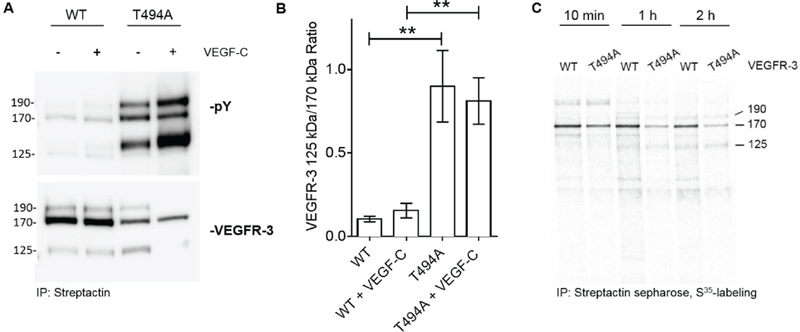
A) VEGFR-3 phosphorylation with and without VEGF-C stimulation in HUVECs transfected with either the A (WT) or the G (T494A) allele. Notably, the major precursor of VEGFR-3 (170 kDa) matures to a 190 kDa fully glycosylated cell-surface VEGFR-3, which is then proteolytically cleaved into 125 kDa C-terminal fragment, and a shorter N-terminal fragment (not visible). Please note that pY indicates phosphorylated VEGFR-3 forms (top gel). B) Quantification of the 125:170 kDa VEGFR-3 ratio bands in the experiments reported in the A panel. The mean±SEM is shown. ** P≤ 0.01. C) Pulse-chase analysis of metabolically-labeled and streptactin sepharose-precipitated VEGFR-3 polypeptides.
To test the effect of T494A on VEGFR-3 post-translational processing, a pulse chase experiment was conducted in HUVECs. Increased VEGFR-3 125 kDa to 170 kDa ratios were observed in cells with T494A compared to WT cells, with and without VEGF-C stimulation (P<0.01) (Figure 2B). At both the 1 h and 2 h pulse chase time points, a clear decrease in VEGFR-3 levels was evident in T494A cells (Figure 2C).
FLT4 rs307826 (A>G, T494A) reduces sorafenib cytotoxicity
Based on the association between FLT4 rs307826 (A>G) and OS (Figure 1D) and results from the VEGFR-3 phosphorylation experiments, we tested the hypothesis that rs307826 might influence the cytotoxicity of sorafenib. In HUVECs, T494A cells were more resistant than WT cells both in the absence of VEGF-C stimulation (mean IC50 11.33 versus 3.80 µM, respectively; P<0.0001) and in the presence of VEGF-C stimulation (mean sorafenib IC50 7.61 versus 2.80 µM, respectively; P<0.0001) (Figure 3A). These results were replicated in human embryonic kidney (HEK-293) cells, where T494A cells were more resistant than WT cells both in the absence of VEGF-C stimulation (mean IC50 15.45 versus 7.58 µM, respectively; P<0.0001) and in the presence of VEGF-C stimulation (mean sorafenib IC50 7.67 versus 2.02 µM, respectively; P<0.0001) (Figure 3B).
Figure 3. Effects of FLT4 rs307826 (A>G, T494A) on sorafenib cytotoxicity.
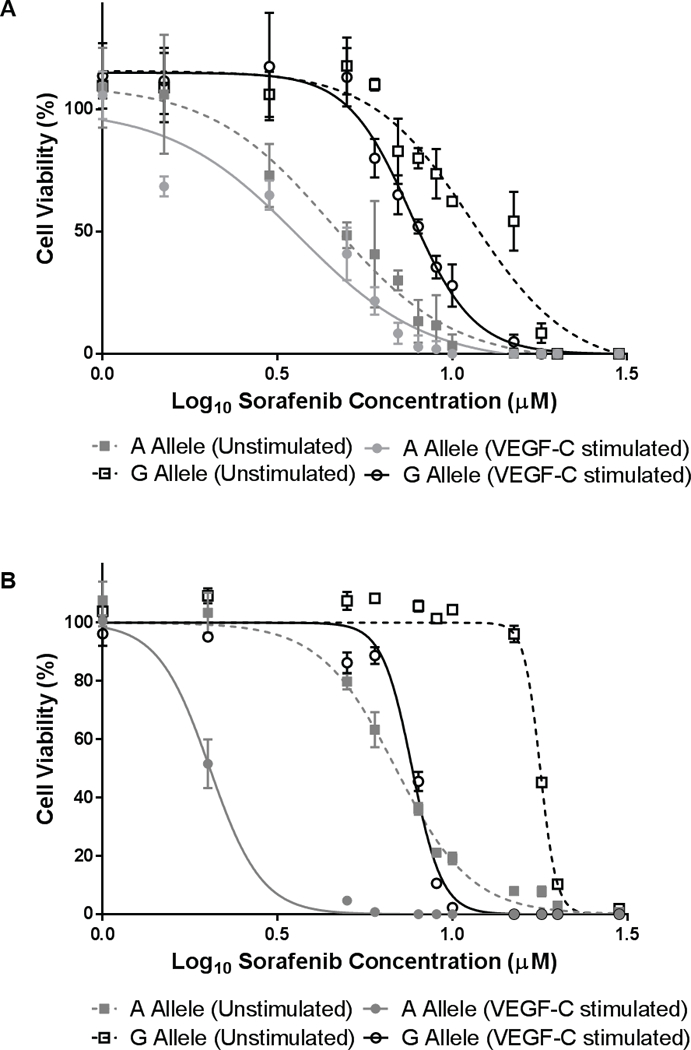
A) HUVECs were transfected with either the A or the G allele, and then treated with sorafenib with and without VEGF-C stimulation. B) Results were replicated in HEK-293 cells. All experiments were conducted in triplicate (n=9 at for each concentration). Relative fluorescence units (RFUs) were generated to determine the percentage of viable cells present. The mean±SEM of RFUs is shown.
Intronic variants associated with OS affect transcriptional activity in luciferase assays
With the exception of FLT4 rs307826, all other gene variants associated with shorter OS (Table 2) are intronic. Because they were of unknown significance, we tested their effect on transcriptional regulation using luciferase assays in human endothelial (LPEC, TIME) and RCC (Caki-1) cells.
VEGFA rs1885657 C increased luciferase activity by an average of 48% in LPECs (P=0.0116), 57% in TIME cells (P<0.0001), and 30% in Caki-1 cells (P=0.0166), when compared to the reference T allele. rs58159269 and rs943070 were also tested because in perfect linkage disequilibrium (LD) with rs1885657 (r2 ≥ 1.0 for both variants) (Table S2). rs58159269 C and rs943070 G also increased luciferase activity. The “triple variant” construct (rs1885657 C/rs58159269 C/rs943070 G) had the greatest increase in luciferase activity (70–99%) in all cell lines (P<0.0001) (Figure 4A). VEGFA rs3024987 T increased luciferase activity in Caki-1 cells (34%, P=0.0032), TIME cells (38%, P=0.0002), and LPECs (32%, P=0.0001), when compared to the reference C allele (Figure 4B).
Figure 4. Effects of intronic variants on luciferase activity.
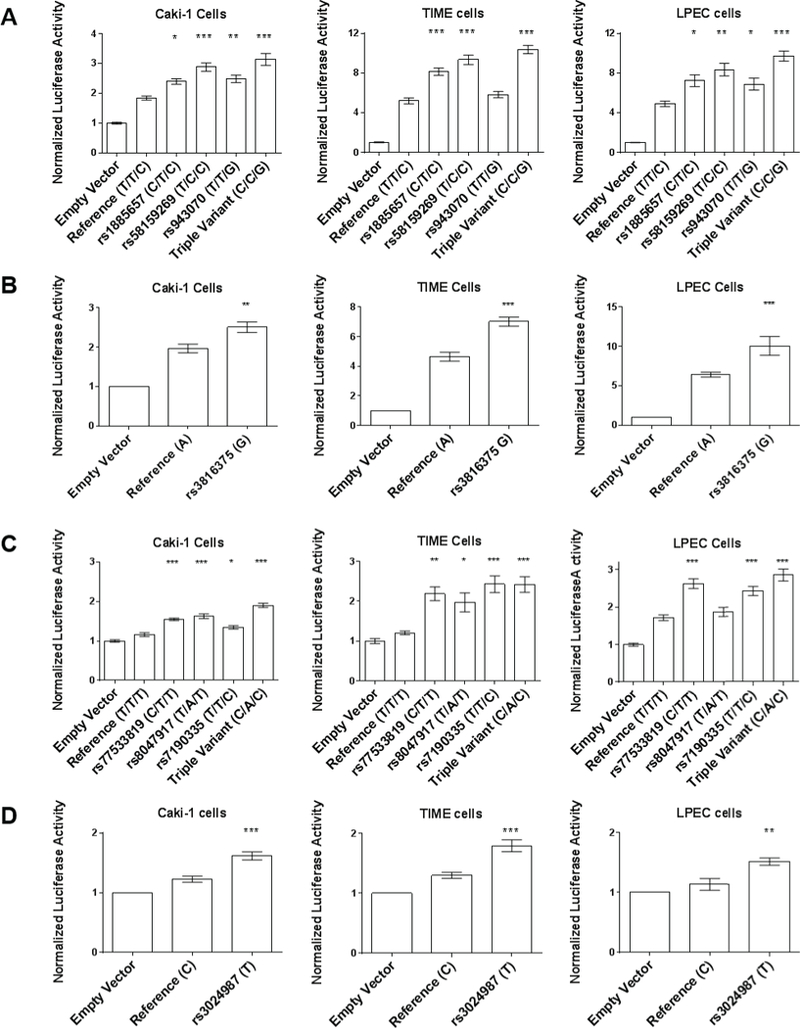
A) VEGFA rs1885657 C, rs58159269 C, rs943070 G, and a “triple variant” construct (C/C/G). B) VEGFA rs3024987 T. C) WWOX rs77533819 C, rs8047917 A, rs7190335 C, and a “triple variant” construct (C/A/C). D) ITGAV rs3816375 G. Luciferase activity was tested in Caki-1, TIME and LPEC lines (from left to right). The mean±SEM of luciferase activity of the experiments, conducted in quadruplicate (n=12 for each construct), is shown. *P<0.05, **P<0.01, ***P<0.001.
WWOX rs8047917 A increased luciferase activity in TIME cells (63%, P=0.0174), and Caki-1 cells (40%, P<0.0001), but not LPECs, when compared to the reference T allele. rs77533819 and rs7190335 were also tested because in high LD with rs8047917 (r2 ≥ 0.96 for both variants) (Table S2). The “triple variant” construct (rs77533819 C/rs8047917 A/rs7190035 C) had the greatest increase in luciferase activity (64–101%) in all cell lines (P<0.0001) (Figure 4C).
ITGAV rs381637 G increased luciferase activity in Caki-1 cells (57%, P<0.0001), TIME cells (51%, P<0.0001), and LPECs (27%, P=0.005), when compared to the reference C allele (Figure 4D).
VEGFA rs58159269 (T>C) increases endothelial cell proliferation
Out of the intronic variants that increased luciferase activity (Figure 4A–D), the effects of VEGFA rs58159269 were tested in angiogenesis assays. Among the three VEGFA variants tested in the luciferase assays (Figure 4A), rs58159269 is in perfect LD with rs1885657, which was the VEGFA variant associated with OS in the sorafenib arm (Figure 1A), was predicted in silico to affect VEGFA (Table S2) and had the strongest luciferase activity in all three cell lines (Figure 4A). Thus, there was clear rationale to test the effect of rs58159269 on endothelial cell proliferation. In isogenic TIME cells transfected with either the rs58159269 reference T allele or variant C allele, endothelial cell proliferation was significantly increased in cells with the C cells, when compared to cells with the T allele (176±11 versus 103±6 viable cells at 72 h, n=16, P<0.0001) (Figure 5A).
Figure 5. Effects of VEGFA rs58159269 (T>C) on endothelial cell proliferation and tube formation.
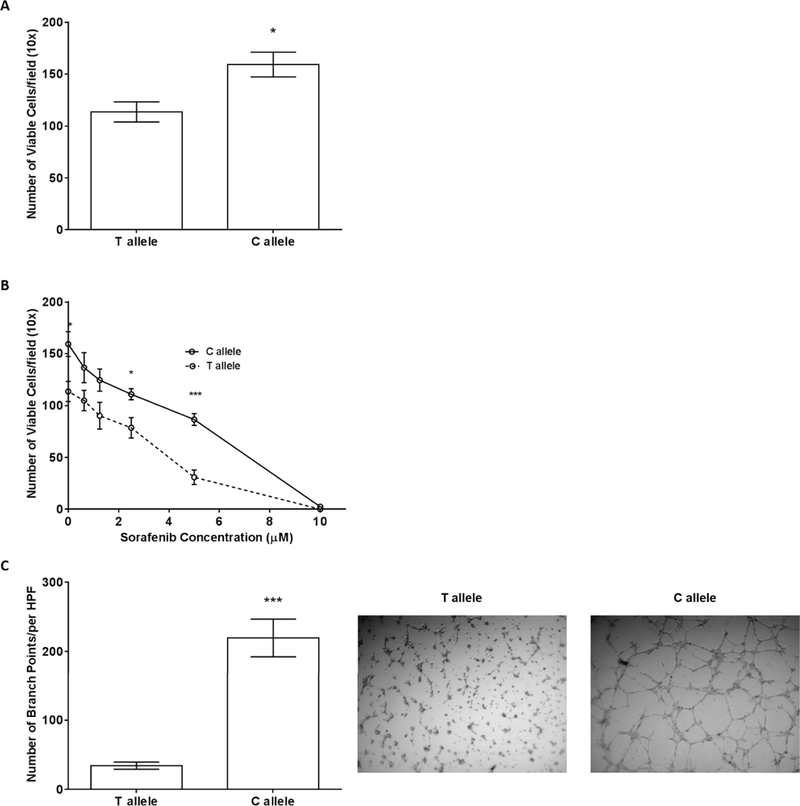
A) Endothelial cell proliferation for VEGFA rs58159269 in the T versus C cells at 72 h. Increased cell viability was demonstrated for the variant C allele (n=16, P=0.02). B) Endothelial cell proliferation for VEGFA rs58159269 in the T versus C cells at 72 h in the presence of increasing concentrations of sorafenib (0–10 μM). The C allele conferred resistance to sorafenib compared to the T allele (n=8 per concentration, P<0.0001 at 5 μM). C) Matrigel® endothelial tube formation assay at 4 h revealed an increase in tube formation for the C cells (n=20, P<0.0001). In each panel, the mean±SEM is shown. HPF, high power field. *P<0.05, ***P<0.001.
VEGFA rs58159269 (T>C) reduces sorafenib cytotoxicity
Whether rs58159269 (T>C) might affect differential response to sorafenib was further characterized using a proliferation assay. In the same isogenic TIME cells described previously, cell proliferation was significantly increased in cells with the variant rs58159269 C allele after sorafenib 2.5 µM (n=8, P=0.01) and 5 µM (n=8, P<0.0001), when compared to cells with the reference. And, while TIME cells transfected with either the reference T or variant C allele were responsive to sorafenib, the IC50 of cells with the C allele was nearly double the IC50 of cells with the T allele (IC50 6.43 versus 3.50 µM, P=0.17) (Figure 5B).
VEGFA rs58159269 (T>C) increases endothelial cell tube formation
The hypothesis that rs58159269 C would affect endothelial tube formation was also tested. Using the same isogenic TIME cells previously described, endothelial tube formation assays revealed a significant increase branch points in cells with the rs58159269 variant C allele, when compared to cells with the T allele (219.50±27.21 versus 34.35±5.21 branch points, respectively, n=16, P<0.0001) (Figure 5C).
Discussion
This study represents the most comprehensive genetic analysis of sorafenib outcomes in any tumor type. The most compelling discovery of this study is the clinical identification and subsequent experimental validation of FLT4 rs307826 as a negative determinant of OS. Patients with the FLT4 rs307826 GG genotype experienced significantly shorter OS than AA/AG patients in both arms combined (Figure 1D). FLT4 encodes VEGFR-3, a transmembrane kinase receptor that is a target of sorafenib, which mediates lymphangiogenesis and plays a crucial role in vasculature growth and remodeling (23–25). Inhibition of VEGFR-3 can suppress vascular network formation (26), and preclinical models have shown that VEGFR-3 blockade can inhibit lymphatic metastasis (27).
FLT4 rs307826 (A>G) results in a threonine to alanine amino acid substitution (T494A). Our experimental results clearly indicate that T494A not only increases VEGFR-3 phosphorylation, but also VEGFR-3 membrane trafficking. T494A occurs in the 5th immunoglobulin (Ig)-like domain of the extracellular domain of VEGFR-3 (Figure S4) (17), which contributes important homotypic interactions that are essential for VEGFR-3 dimerization and activation.
In our mechanistic experiments, T494A increased basal and VEGF-C-stimulated phosphorylation of VEGFR-3 (Figure 2A), suggesting its importance as a driver of increased receptor signaling. T494A may also increase membrane trafficking of VEGFR-3. Increased formation of the proteolytically-processed C-terminal fragment (i.e., increased 125:170 kDa band ratio) in the stimulation assays indicate faster maturation of VEGFR-3 in cells with T494A (Figure 2B). Pulse chase experiments revealed faster decrease of VEGFR-3 specific bands with T494A, which could indicate more rapid endocytosis (Figure 2C). One plausible mechanistic hypothesis could be that T494A renders VEGFR-3 more susceptible to internalization and signaling activation, which is important because VEGFR-3 internalization is crucial to the full activation of downstream signaling pathways (28,29).
Evidence from our clinical and experimental results suggest that, in mRCC patients with T494A, increased VEGFR-3 phosphorylation and membrane trafficking lead to potentiated VEGFR-3 signaling, which gives rise to a more aggressive form of RCC due to more rapid metastatic spread as a result of increased lymphangiogenesis and angiogenesis. In our study, patients with rs307826 (T494A) had shorter OS. A similar effect is also observed on PFS. Similar to ours, other studies of mRCC patients treated with angiogenesis inhibitors (sunitinib and pazopanib) (30–32), patients with rs307826 (T494A) experienced shorter survival. These clinical studies (now including ours) are all consistent and demonstrate that FLT4 rs307826 (T494A) to be a negative determinant of survival in mRCC. One limitation of these clinical studies (including ours) is that they did not establish whether the effect of rs307826 is predictive. In our study, the effect of FLT4 rs307826 is observed in both arms combined and not in each of the two arms, probably due to a reduced sample size leading to loss of power. The sunitinib studies did not have a placebo arm (16, 17), and the preliminary report on the pazopanib study does not mention the results of the placebo arm (28). Our experimental data suggest that rs307826 (T494A) reduces sorafenib cytotoxicity in vitro (Figure 3), and this could imply a possible predictive effect, as suggested by the interaction P-value of 0.09. Therefore, the potential of FLT4 rs307826 (T494A) as a predictive marker of other angiogenesis inhibitors should be explored more extensively in future studies. This would be of particular importance, as the limited sample size of our study precludes definitive conclusions on the predictive nature of the genetic variants, in particular when they tend to have a low allele frequency.
In addition to FLT4, this paper also provides evidence for VEGFA as a genetic determinant of survival in mRCC. VEGFA encodes for VEGF-A, the most potent pro-angiogenic ligand (33). In the present study, two intronic variants in VEGFA were associated with shorter OS: rs1885657 in sorafenib-treated patients (Figure 1A) - also associated with PFS - and rs3024987 in both arms (Figure 1E). To our knowledge, very few studies have evaluated the germline determinants of sorafenib outcome (30, 31). One study in Chinese patients treated with sorafenib has identified rs1570360 as a VEGFA variant associated with increased PFS (32). rs1570360 is not in LD (using a cut-off of r2<0.6 in Asians from the 1,000 Genomes Project) with any of the two VEGFA variants associated with shorter OS in our study (Table 2).
For both of these intronic variants, luciferase assay data in both endothelial and RCC cell lines demonstrated that the rs1885657 variant C allele and the rs3024987 variant G allele were associated with increased transcriptional activity (Figure 4A–B). Although luciferase assays test the activity of a transfected construct and not the entire gene, these results are suggestive of increased regulatory activity of these variants on VEGFA expression. Increased VEGFA expression might confer increased angiogenic potential to the tumor, so we tested this hypothesis in preclinical models of angiogenesis that leveraged isogenic endothelial cells. Instead of assessing the effects of rs1885657 in these angiogenesis models, VEGFA rs58159269 was assessed because it is a variant in perfect LD with rs1885657, in silico predictions of its effects on VEGFA expression (Table S2), and its stronger effect on luciferase activity (Figure 4A). In our experiments rs58159269 potentiate cell proliferation and tube formation, and also demonstrated increased resistance to the cytotoxic effects of sorafenib (Figure 5). These experimental results implicate a mechanism where tumors in patients with either VEGFA rs1885657 or rs58159269 might be more resistant to sorafenib due to a more aggressive angiogenic phenotype. Because preliminary evidence suggests that VEGFA rs58159269 also conferred resistance to sunitinib (Figure S5), the clinical effects of either VEGFA rs1885657 or rs58159269 should be also tested in mRCC patients treated with other angiogenesis inhibitors (e.g., sunitinib or pazopanib).
Additional potentially intriguing variant include ITGAV rs3816375 and WWOX rs8047917, both of which are intronic variants that associated with shorter OS in sorafenib-treated patients. ITGAV codes the αv integrin subunit, which can heterodimerize with multiple β subunits. Integrin αvβ3 is often upregulated in both tumor and endothelial cells, and has been shown to regulate angiogenesis (34,35). The increased luciferase activity of ITGAV rs3816375 G (Figure 4B) generates the hypothesis that rs3816375 could be contributing to ITGAV overexpression, increased angiogenesis, and potentially reduced efficacy of sorafenib treatment. For WWOX rs8047917, the increased luciferase assay appears to be discordant with the well-described tumor suppressor properties of WWOX (36), and additional experimental evaluation is needed.
The evidence from the clinical effects on shorter OS, combined with a mechanistic demonstration of their biological and pharmacological effects, proposes FLT4 rs307826 and VEGFA rs1885657 as two biomarkers for mRCC and sorafenib treatment. Their identification and functional validation is an important step towards personalization of sorafenib treatment in mRCC. In addition, if these variants alter the tumor phenotype leading to increased and abnormal lymphangiogenesis and angiogenesis, these characteristics might hamper the access and homing of immune cells in the tumor microenvironment. Predicting which tumors respond better to vasculature normalization induced by anti-angiogenesis agents might open new venues of combinatorial therapies with checkpoint inhibitors. As a general mechanism, a mutual interplay between vascular normalization and induction of type 1 T-helper lymphocytes in the microenvironment has been recently demonstrated (36).
These identified gene variants should be evaluated in future clinical studies to assess their impact in patients with other types of malignances treated with sorafenib (e.g., unresectable HCC, refractory thyroid carcinoma, or FLT3-mutated AML), but also in patients with mRCC who are treated with other angiogenesis inhibitors (e.g., sunitinib and pazopanib), as well as with checkpoint inhibitors.
Supplementary Material
Statement of Significance.
Clinical and mechanistic data identify germline genetic variants in VEGFA and FLT4 as markers of survival in patients with metastatic renal cell carcinoma.
Acknowledgments:
NIH/NCI R21CA178550-01 (NK-D and FI), NIH/NCI R21CA139280-01 (FI), NIH/NCI K07CA140390-01 (FI), Cancer Research Foundation Young Investigator Award (FI), NIGMS T32GM086330 (DJC), American Foundation for Pharmaceutical Education Fellowship (DJC), the Jane and Aatos Erkko Foundation (KKA), European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme under grant agreement No 743155 (KKA), Jenny and Antti Wihuri Foundation (KKA), the Academy of Finland Centre of Excellence Program 2014–2019 (271845 and 307366) (KKA), and the Sigrid Juselius Foundation (KKA). We would like to acknowledge Dr. Habibul Ahsan and Dr. Muhammad Kibriya of the University of Chicago genotyping core, as well as Mr. Jason Luo of the University of North Carolina-Chapel Hill Mammalian Genotyping Core. We would like to thank Dr. Kurt Ballmer-Hofer for his advice and assistance. Finally, we would like to acknowledge Ms. Jessie Bishop, Mrs. Anna Crollman, Dr. Lana Crona, and Ms. Sara Pettaway, and Dr. William Scott for their assistance in editing and formatting this paper.
Footnotes
Conflict of Interest Statement:
Dr. Peña is an employee of Bayer and owns stock in Bayer. All other authors have no conflicts of interest pertaining to this work and nothing to disclaim.
References
- 1.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004;64:7099–109 [DOI] [PubMed] [Google Scholar]
- 2.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34 [DOI] [PubMed] [Google Scholar]
- 3.Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24:2505–12 [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, Jonasch E, Agarwal N, Bhayani S, Bro WP, Chang SS, et al. Kidney Cancer, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN 2017;15:804–34 [DOI] [PubMed] [Google Scholar]
- 5.Sabo E, Boltenko A, Sova Y, Stein A, Kleinhaus S, Resnick MB. Microscopic analysis and significance of vascular architectural complexity in renal cell carcinoma. Clin Cancer Res 2001;7:533–7 [PubMed] [Google Scholar]
- 6.Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90 [DOI] [PubMed] [Google Scholar]
- 7.Murakami M, Zhao S, Zhao Y, Chowdhury NF, Yu W, Nishijima K, et al. Evaluation of changes in the tumor microenvironment after sorafenib therapy by sequential histology and 18F-fluoromisonidazole hypoxia imaging in renal cell carcinoma. Int J Oncol 2012;41:1593–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy DA, Makonnen S, Lassoued W, Feldman MD, Carter C, Lee WM. Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43–9006). Am J Pathol 2006;169:1875–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rini BI, Small EJ. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol 2005;23:1028–43 [DOI] [PubMed] [Google Scholar]
- 10.Yuen JS, Sim MY, Siml HG, Chong TW, Lau WK, Cheng CW, et al. Inhibition of angiogenic and non-angiogenic targets by sorafenib in renal cell carcinoma (RCC) in a RCC xenograft model. Br J Cancer 2011;104:941–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azam F, Mehta S, Harris AL. Mechanisms of resistance to antiangiogenesis therapy. Eur J Cancer 2010;46:1323–32 [DOI] [PubMed] [Google Scholar]
- 12.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 2009;27:3312–8 [DOI] [PubMed] [Google Scholar]
- 13.Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, et al. Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 2003;68:69–78 [DOI] [PubMed] [Google Scholar]
- 14.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007;39:906–13 [DOI] [PubMed] [Google Scholar]
- 15.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–84 [DOI] [PubMed] [Google Scholar]
- 16.Royston P, Parmar MK. Restricted mean survival time: an alternative to the hazard ratio for the design and analysis of randomized trials with a time-to-event outcome. BMC Med Res Methodol 2013;13:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leppanen VM, Tvorogov D, Kisko K, Prota AE, Jeltsch M, Anisimov A, et al. Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc Natl Acad Sci U S A 2013;110:12960–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tvorogov D, Anisimov A, Zheng W, Leppanen VM, Tammela T, Laurinavicius S, et al. Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding and receptor dimerization. Cancer Cell 2010;18:630–40 [DOI] [PubMed] [Google Scholar]
- 19.Rahmani M, Nguyen T, Dent P, Grant S. The multikinase inhibitor sorafenib induces apoptosis in highly imatinib mesylate-resistant bcr/abl+ human leukemia cells in association with signal transducer and activator of transcription 5 inhibition and myeloid cell leukemia-1 down-regulation. Mol Pharmacol 2007;72:788–95 [DOI] [PubMed] [Google Scholar]
- 20.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 2008;7:3129–40 [DOI] [PubMed] [Google Scholar]
- 21.Pajusola K, Aprelikova O, Pelicci G, Weich H, Claesson-Welsh L, Alitalo K. Signalling properties of FLT4, a proteolytically processed receptor tyrosine kinase related to two VEGF receptors. Oncogene 1994;9:3545–55 [PubMed] [Google Scholar]
- 22.Pajusola K, Aprelikova O, Armstrong E, Morris S, Alitalo K. Two human FLT4 receptor tyrosine kinase isoforms with distinct carboxy terminal tails are produced by alternative processing of primary transcripts. Oncogene 1993;8:2931–7 [PubMed] [Google Scholar]
- 23.Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res 2010;16:3548–61 [DOI] [PubMed] [Google Scholar]
- 24.Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A 1995;92:3566–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 2010;140:460–76 [DOI] [PubMed] [Google Scholar]
- 26.Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008;454:656–60 [DOI] [PubMed] [Google Scholar]
- 27.Roberts N, Kloos B, Cassella M, Podgrabinska S, Persaud K, Wu Y, et al. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res 2006;66:2650–7 [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010;465:483–6 [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Pasula S, Song H, Tessneer KL, Dong Y, Hahn S, et al. Temporal and spatial regulation of epsin abundance and VEGFR3 signaling are required for lymphatic valve formation and function. Sci Signal 2014;7:ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beuselinck B, Karadimou A, Lambrechts D, Claes B, Wolter P, Couchy G, et al. Single-nucleotide polymorphisms associated with outcome in metastatic renal cell carcinoma treated with sunitinib. Br J Cancer 2013;108:887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Donas J, Esteban E, Leandro-Garcia LJ, Castellano DE, del Alba AG, Climent MA, et al. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol 2011;12:1143–50 [DOI] [PubMed] [Google Scholar]
- 32.Xu C, Ball H, Bing N, Sternberg C, Xue Z, McCann L, et al. Association of genetic markers in angiogenesis- or exposure-related genes with overall survival in pazopanib treated patients with advanced renal cell carcinoma. J Clin Oncol 2011;29:303. [DOI] [PubMed] [Google Scholar]
- 33.Rini BI. Vascular endothelial growth factor-targeted therapy in metastatic renal cell carcinoma. Cancer 2009;115:2306–12 [DOI] [PubMed] [Google Scholar]
- 34.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994;264:569–71 [DOI] [PubMed] [Google Scholar]
- 35.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010;10:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Del Mare S, Salah Z, Aqeilan RI. WWOX: its genomics, partners, and functions. J Cell Biochem 2009;108:737–45 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.