

Abstract
Clozapine is the only medication indicated for treating refractory schizophrenia, due to its superior efficacy among all antipsychotic agents, but its mechanism of action is poorly understood. To date, no studies of human postmortem brain have characterized the gene expression response to clozapine. Therefore, we addressed this question by analyzing expression data extracted from published microarray studies involving brains of patients on antipsychotic therapy. We first performed a systematic review and identified four microarray studies of postmortem brains from antipsychotic-treated patients, then extracted the expression data. We then performed generalized linear model analysis on each study separately, and identified the genes differentially expressed in response to clozapine compared to other atypical antipsychotic medications, as well as their associated canonical pathways. We also found a number of genes common to all four studies that we analyzed: GCLM, ZNF652, and GYPC. In addition, pathway analysis highlighted the following processes in all four studies: clathrin-mediated endocytosis, SAPK/JNK signaling, 3-phosphoinositide synthesis, and paxillin signaling. Our analysis yielded the first comprehensive compendium of genes and pathways differentially expressed upon clozapine treatment in the human brain, which may provide insight into the mechanism and unique efficacy of clozapine, as well as the pathophysiology of schizophrenia.
Keywords: Schizophrenia, Bipolar affective disorder, Antipsychotic, Microarray
1. Introduction
Schizophrenia is a chronic, debilitating psychiatric disorder with a lifetime prevalence of about 1% of the general population (Perala et al., 2007; Saha et al., 2005). Up to one third of schizophrenia patients fail to respond to standard antipsychotic therapy and present with a treatment-refractory form of the disease, which causes a significant loss in quality of life (Conley and Kelly, 2001; Kennedy et al., 2014; Miyamoto et al., 2014). Clozapine, the first atypical (or 2nd-generation) antipsychotic agent, is the only medication indicated for refractory schizophrenia, and has the greatest efficacy despite the development of newer atypical agents (Conley and Kelly, 2001; Leucht et al., 2013; McEvoy et al., 2006; Meltzer, 2012; Siskind et al., 2016). However, it has a number of unique and serious side effects, most notably the rare but life-threatening risk of agranulocytosis. Given these side effects and the difficulty of implementing care (as patients must be enrolled in a centralized registry and followed up with regular blood counts), clozapine is underutilized and currently recommended for patients who have failed therapy with two other antipsychotic medications (Conley and Kelly, 2001; Hasan et al., 2012; Siskind et al., 2016).
Despite a long history of utilization, clozapine’s mechanism of action and the basis of its superior efficacy over other antipsychotics are still poorly understood. Clozapine is known to bind to a broad array of receptors, including dopamine, serotonin, histamine, muscarinic, and adrenergic receptors (Ereshefsky et al., 1989), but it is unclear which of these are most relevant to its efficacy. It is likely that the regulation of downstream gene expression is critical, since clozapine and other antipsychotics usually require several weeks to evoke a stable therapeutic response.
Several groups have applied microarray or RNA-sequencing to postmortem brains of patients treated with antipsychotics including clozapine. However, they do not differentiate between antipsychotic medications, and address questions other than the specific impact of clozapine on gene expression (Aston et al., 2004; Chen et al., 2013; Iwamoto et al., 2005; Mudge et al., 2008; Pietersen et al., 2014a,b; Schmitt et al., 2011; Wu et al., 2012). At present, we are not aware of any studies that have characterized the differential gene expression profile of clozapine in the human brain.
Therefore, we performed a systematic review to identify gene expression studies of brains from patients on antipsychotic therapy, and analyzed their expression data to determine which genes are modulated specifically by clozapine, instead of performing a conventional meta-analysis. As a result, we found four such studies in the literature, and our novel analysis of their expression data identified the genes and pathways in each study that are differentially expressed in response to clozapine compared to other atypical antipsychotics. We also determined which of these genes and pathways are common to all four studies, in order to formulate a consensus of the literature regarding the gene expression profile of clozapine in the human brain. Our analysis highlighted three genes (GCLM, ZNF652, and GYPC) that are modulated by clozapine in all four datasets, as well as four pathways (clathrin-mediated endocytosis, SAPK/JNK signaling, 3-phosphoinositide signaling, and paxillin signaling).
2. Experimental/materials and methods
2.1. Systematic review
We reviewed the literature to identify studies that used microarray or RNA-seq to characterize differential gene expression in the postmortem brains of patients treated with antipsychotic agents including clozapine. We searched three reference databases, PubMed, Embase, and BIOSIS (on May 14, 2015), and the gene expression data repositories GEO and ArrayExpress (on July 17, 2015). Detailed search strategies are listed in Table S1.
2.2. Eligibility criteria for study selection
Studies were selected in accordance with the following eligibility criteria: a) gene expression studies that used microarray or RNA-seq; b) studies that analyzed human postmortem brain; c) studies whose cohorts included at least one patient exposed to clozapine, and at least one patient exposed to an atypical antipsychotic other than clozapine. We excluded abstracts without full articles, review articles, editorials, case reports, and studies not written in English. If results were duplicated in multiple publications, only one was selected. We also excluded studies if we were unable to obtain adequate expression data for analysis; for example, we excluded studies without data available in the public domain when we could not contact the authors after three attempts at correspondence. Two reviewers screened studies independently in two rounds, first with abstracts alone and second with the full text articles retrieved from eligible abstracts; any discrepancy between reviewers was resolved by consensus. Furthermore, to ensure a more comprehensive review, we manually searched for any additional eligible studies in the reference lists of all full-text articles that we screened.
2.3. Data extraction
Information was extracted from eligible studies in a predetermined form. We noted the first author and year of publication, diagnoses and treatments of study participants who had received clozapine or other atypical antipsychotic medications, source of tissue (brain region and brain bank), and microarray platform. Normalized gene expression datasets for eligible studies were retrieved from the NCBI Gene Expression Omnibus (GEO) database or directly from the authors of the original studies. All available phenotypic information for the subjects analyzed in the studies were extracted by two reviewers independently, and any discrepancy was resolved by consensus.
2.4. Data analysis
We analyzed the gene expression data of each study separately using a uniform approach. We used a generalized linear model approach coupled with empirical Bayes standard errors shrinkage (Smyth, 2004), including coefficients for data heterogeneity as derived from surrogate variable analysis (SVA) (Leek and Storey, 2007), to identify differentially expressed genes between groups of patients treated with clozapine or other atypical antipsychotics. This design-contrast parametrization approach allowed us to capture data heterogeneity, and control (in an unbiased way) for unknown sources of variation using all samples in each dataset. Correction for multiple testing was performed with the Benjamini-Hochberg method. Differentially expressed genes were selected based on false discovery rate (FDR) and ordered according to the moderated t-statistics obtained from our linear model analysis. We then used Ingenuity® Pathway Analysis (IPA® build version 389077M and content version 27821452, release date 2016–06-14, Qiagen, Redwood City) to identify pathways and biological processes associated with the differentially expressed genes.
3. Results
3.1. Systematic review
We first performed a systematic review of the literature to identify gene expression studies ofpostmortem brains from patients on antipsychotic therapy (Fig. 1). Our search of PubMed, Embase, and BIOSIS yielded 5238 publications, of which 1300 were duplicated between the three databases and were excluded. We then screened 3938 abstracts, excluded 3849 based on our eligibility criteria (see Experimental/Materials and methods), and reviewed the remaining 89 publications in full text, of which 83 were excluded. When we reviewed the reference lists of these 89 articles, we identified one additional study that satisfied our eligibility criteria. We also screened 72 studies yielded by a direct search of two gene expression data repositories, GEO and ArrayExpress, but excluded all of them due to our eligibility criteria. Of the remaining studies, two publications by Pietersen et al. (2014a,b) analyzed specific cell types (parvalbumin interneurons and pyramidal neurons) in the superior temporal gyrus from the same cohort of patients, so we considered them as one study and combined the gene expression data into a single dataset for analysis.
Fig. 1.
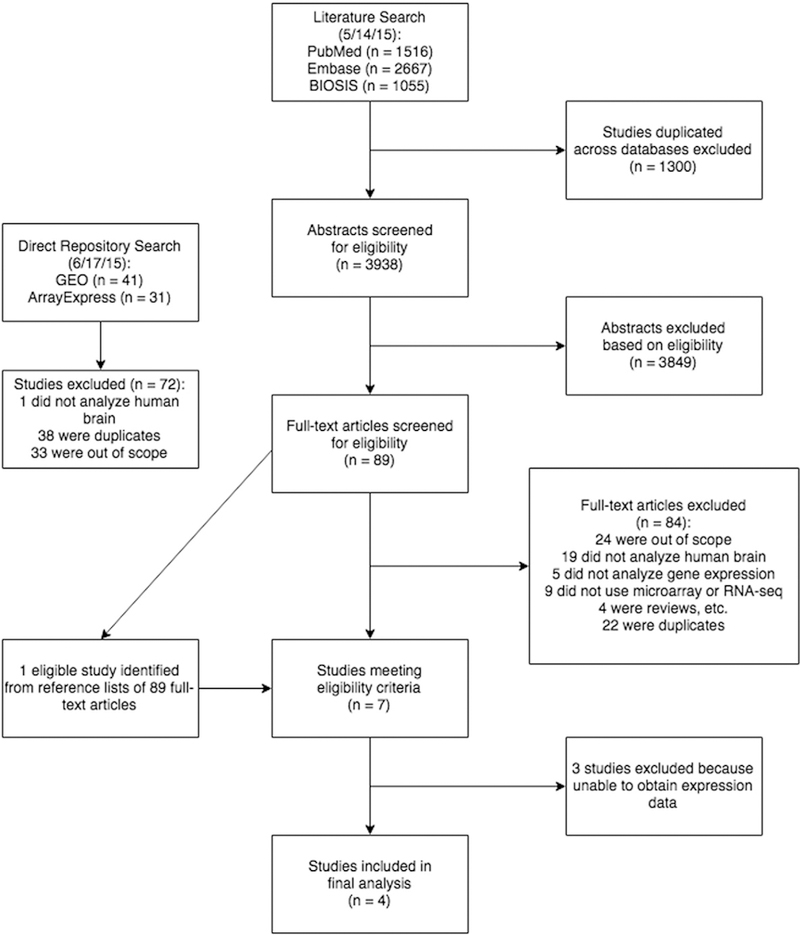
Screening process for eligible studies and resulting sample size.We performed a systematic reviewof three reference databases, PubMed, Embase, and BIOSIS, to identify studies of human postmortembrain that included patients treatedwith clozapine, and usedmicroarray or RNA-seq to characterize differential gene expression. After two rounds of screening by two reviewers, we identified seven studies meeting our predetermined eligibility criteria. Of these, we were able to obtain the expression data for four studies. We also searched the gene expression data repositories GEO and ArrayExpress directly, which yielded no eligible studies
As a result, we identified seven studies that analyzed gene expression in brains of patients treated with antipsychotics including clozapine (Aston et al., 2004; Chen et al., 2013; Iwamoto et al., 2005; Mudge et al., 2008; Pietersen et al., 2014a,b; Schmitt et al., 2011; Wu et al., 2012). We were able to obtain the expression data from four of these seven studies, either from GEO or directly from the authors. The details of the search and review process, including the number of studies excluded for each eligibility criterion, are described in Fig. 1.
3.2. Characteristics of studies
We extracted the following information from the four studies identified in our review, summarized in Table 1. Each study included two or three patients diagnosed with schizophrenia or bipolar affective disorder who had been treated with clozapine, and varying numbers of patients treated with other atypical antipsychotics (risperidone, olanzapine, or quetiapine); however, none had information on duration of disease or treatment. None of the studies had the gene expression profile of clozapine as their objective. Therefore we queried their expression data to address our own question: which genes have differential expression upon treatment with clozapine vs. other atypical antipsychotics?
Table 1.
Characteristics of eligible studies.
We extracted information from each of the four microarray studies identified by our systematic review, including the diagnosis of patients (schizophrenia [SZ] or bipolar affective disorders [BPAD]) and the specific antipsychotic drugs (APDs) they received. For three of the studies, we obtained their expression data from the public domain through the GEO accession numbers provided in the publications; for the other, we received the data directly from the authors.
| Study | Subjects (age, sex, APD) | Tissue source | Microarray platform |
|---|---|---|---|
| Iwamoto et al., 2005 (GSE12649) | 34 controls | Prefrontal cortex (Stanley Array Collection) | Affymetrix Human Genome 133A Array |
| SZ patient (31, M, clozapine) | |||
| SZ patient (40, M, clozapine) | |||
| SZ patient (32, F, risperidone) | |||
| SZ patient (53, M, risperidone) | |||
| BPAD patient (64, M, clozapine) | |||
| BPAD patient (29, M, risperidone) | |||
| BPAD patient (33, F, risperidone) | |||
| BPAD patient (41, F, risperidone) | |||
| BPAD patient (42, M, risperidone) | |||
| BPAD patient (45, M, risperidone) | |||
| BPAD patient (51, M, risperidone) | |||
| BPAD patient (35, F, olanzapine) | |||
| BPAD patient (45, M, olanzapine) | |||
| BPAD patient (56, M, olanzapine) | |||
| BPAD patient (19, M, quetiapine) | |||
| BPAD patient (43, F, quetiapine) | |||
| BPAD patient (44, F, quetiapine) | |||
| Mudge et al., 2008 (GSE12297) | 6 controls | Cerebellar cortex (Maryland Brain Collection) | NCGR Human RefSeq 7 08 (GPL7100) |
| SZ patient (31, M, clozapine) | |||
| SZ patient (32, M, clozapine) | |||
| SZ patient (33, M, olanzapine) | |||
| SZ patient (45, M, olanzapine) | |||
| SZ patient (46, M, olanzapine) | |||
| SZ patient (62, M, olanzapine) | |||
| SZ patient (37, M, risperidone) | |||
| Chen et al., 2013 (data provided by authors) | 12 controls | Right premotor cortex (Stanley Foundation Brain Collection) | Affymetrix GeneChip U133 plus 2.0 |
| BPAD patient (30, M, clozapine) | |||
| BPAD patient (50, M, clozapine) | |||
| BPAD patient (34, M, risperidone) | |||
| Pietersen et al., 2014a,b (GSE46509, GSE37981) | 9 controls | – Superior temporal gyrus: parvalbumin neurons – Superior temporal gyrus: pyramidal neurons (Harvard Brain Tissue Resource Center) |
Affymetrix Human X3P Array |
| SZ patient (36, M, clozapine) | |||
| SZ patient (55, M, clozapine) | |||
| SZ patient (56, M, olanzapine) | |||
Of note, we found that each study had been performed in a different brain region: Chen et al. (2013) in premotor cortex (PMC); Pietersen et al. (2014a,b) in superior temporal gyrus (STG), specifically parvalbumin interneurons and pyramidal neurons therein; Mudge et al. (2008) in cerebellar cortex (CC); and Iwamoto et al. (2005) in prefrontal cortex (PFC). Also, each had used a different microarray platform. Due to these and other disparities between studies (e.g. unavailable information on dosing and duration of antipsychotic therapy), we performed generalized linear model analysis on each study separately.
3.3. Differentially expressed genes with false discovery rate < 0.10, and associated pathways, comparing patients on clozapine vs. other atypical antipsychotics
We hypothesized that comparing clozapine to other atypical antipsychotics would provide the most insight into clozapine’s mechanism of action, since its therapeutic efficacy is superior among all antipsychotics (including other atypical agents). Also, typical and atypical agents have different receptor binding properties (Meltzer et al., 1989), while there is no significant difference in efficacy between atypical and typical agents (Leucht et al., 2013), thus we did not compare clozapine to typical antipsychotics. We did not compare clozapine to control, since comparing healthy controls to patients with schizophrenia or bipolar disorder would introduce a confounding factor in the gene expression analysis.
Our analysis of differentially expressed genes with FDR<0.10 (Fig. 2) yielded 2251 genes (listed in Tables S2a and S2b): 678 upregulated and 691 downregulated genes in the study by Chen et al. (2013) in PMC; two upregulated and three downregulated genes in Pietersen et al. (2014a,b) in STG; 272 upregulated and 195 downregulated genes in Mudge et al. (2008) in CC; and 253 upregulated and 177 downregulated genes in Iwamoto et al. (2005) in PFC. The lower number of differentially expressed genes in Pietersen et al. (2014a, b) may be due to their analysis of specific neuronal subtypes in the STG, as opposed to whole tissue like the other three studies (Arion et al., 2015). Twenty-five of these genes are present in more than one study (Table 2a). This analysis also highlighted the following genes implicated in a genome-wide association study (GWAS) by the Psychiatric Genomics Consortium (2014) that linked 108 loci to schizophrenia: NT5C2 (PFC), DOC2A (PMC), MAN2A1 (PMC), RFTN2 (PMC), and TMTC1 (PMC) are upregulated; OSBPL3 (CC), C2orf82 (PMC), CSMD1 (PMC), PARD6A (PMC), SBNO1 (PMC), SHJSA8 (PMC), and TRJM8 (PMC) are downregulated.
Fig. 2.
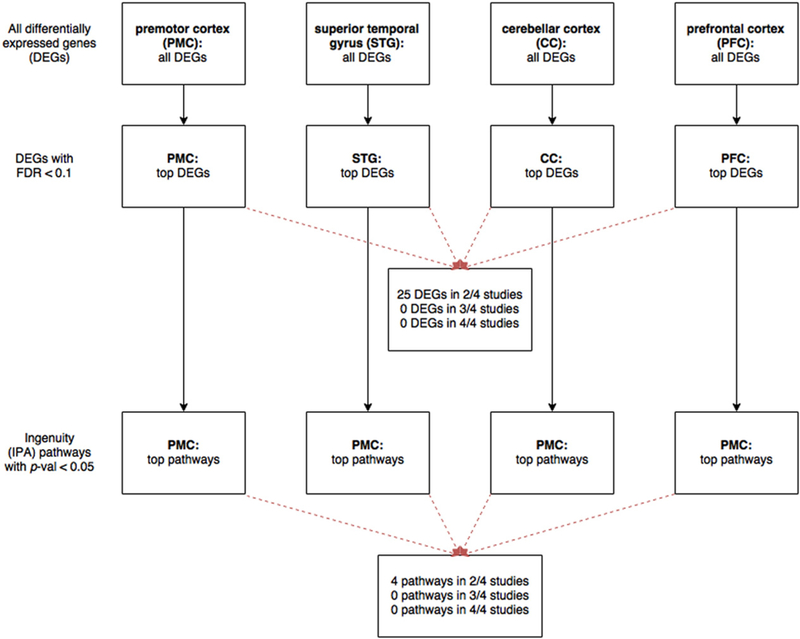
Workflow of analysis to identify differentially expressed genes with FDR< 0.10, and their associated pathways.We identified four studies by our systematic review: Chen et al. (inpremotor cortex, PMC), Pietersen et al. (superior temporal gyrus, STG),Mudge et al. (cerebellar cortex, CC), and Iwamoto et al. (prefrontal cortex, PFC). After extracting the expression data, we analyzed differentially expressed genes (DEGs) mediated by clozapine vs. other atypical antipsychotic medications. For each study, we ranked the DEGs by false discovery rate (FDR) and selected those with FDR < 0.10.We also performed Ingenuity pathway analysis (IPA) on the geneswith FDR b 0.10 in each study, and identified canonical pathwayswith p-value <0.05
Table 2a.
Differentially expressed genes with FDR <0.10 that are present in more than one study. Of the genes with false discovery rate (FDR) <0.10 that are differentially expressed in response to clozapine vs. other atypical antipsychotics, we present those that are in more than one study. The four studies had been performed in the following brain regions: Chen et al. in premotor cortex (PMC), Pietersen et al. in superior temporal gyrus (STG), Mudge et al. in cerebellar cortex (CC), and Iwamoto et al. in prefrontal cortex (PFC). For each gene, we list the log2 fold change (logFC) and FDR for each brain region.
| Gene ID | Gene name | logFC | FDR |
|---|---|---|---|
| ACVR1B | Activin A receptor type IB | −1.08 (PMC), 0.24 (CC) | 0.05 (PMC), 0.08 (CC) |
| ARMCX2 | Armadillo repeat containing X-linked 2 | 0.35 (PMC), 0.31 (PFC) | 0.07 (PMC), 0.05 (PFC) |
| CBX7 | Chromobox homolog 7 | −0.22 (PMC), 0.29 (PFC) | 0.07 (PMC), 0.06 (PFC) |
| CSNK2A2 | Casein kinase 2 alpha prime polypeptide | −0.31 (CC), 0.29 (PFC) | 0.09 (CC), 0.043 (PFC) |
| CXADR | Coxsackie virus and adenovirus receptor | 0.50 (PMC), −0.34 (PFC) | 0.02 (PMC), 0.05 (PFC) |
| DLEC1 | Deleted in lung and esophageal cancer 1 | −0.46 (CC), 0.27 (PFC) | 0.04 (CC), 0.02 (PFC) |
| DTNA | Dystrobrevin alpha | 1.02 (PMC), 0.80 (PFC) | 0.04 (PMC), 0.02 (PFC) |
| FTH1 | Ferritin heavy polypeptide 1 | 0.55 (PMC), 0.46 (PFC) | 0.07 (PMC), 0.07 (PFC) |
| GNAL | Guanine nucleotide binding protein (G protein) alpha activating activity polypeptide olfactory type | 1.35 (PMC), 0.50 (CC) | 0.04 (PMC), 0.08 (CC) |
| HEBP2 | Heme binding protein 2 | −0.55 (PMC), 0.65 (PFC) | 0.01 (PMC), 0.003 (PFC) |
| LUC7L2 | LUC7-like 2 (S. cerevisiae) | 1.23 (PMC), −0.35 (PFC) | 0.02 (PMC), 0.09 (PFC) |
| MAP3K2 | Mitogen-activated protein kinase kinase kinase 2 | 0.40 (PMC), 0.34 (PFC) | 0.06 (PMC), 0.07 (PFC) |
| MAP4K5 | Mitogen-activated protein kinase kinase kinase kinase 5 | 0.47 (PMC), −45.68 (STG) | 0.05 (PMC), 0.10 (STG) |
| MFSD4 | Major facilitator superfamily domain containing 4 | 0.58 (PMC), 0.422 (CC) | 0.03 (PMC), 0.04 (CC) |
| PAPLN | Papilin proteoglycan-like sulfated glycoprotein | 0.38 (PMC), 0.34 (CC) | 0.03 (PMC), 0.09 (CC) |
| PCDHGC4 | Protocadherin gamma subfamily C4 | −0.55 (PMC), 0.58 (CC) | 0.02 (PMC), 0.08 (CC) |
| PCP4 | Purkinje cell protein 4 | −0.80 (PMC), −0.66 (PFC) | 0.03 (PMC), 0.10 (PFC) |
| PITPNA | Phosphatidylinositol transfer protein alpha | −0.31 (PMC), −0.30 (PFC) | 0.05 (PMC), 0.03 (PFC) |
| RNF150 | Ring finger protein 150 | 0.71 (PMC), −0.55 (CC) | 0.01 (PMC), 0.06 (CC) |
| RPL31 | Ribosomal protein L31 | 1.34 (PMC), 0.55 (PFC) | 0.004 (PMC), 0.04 (PFC) |
| RPP25 | Ribonuclease P/MRP 25 kDa subunit | −0.49 (PMC), 0.37 (PFC) | 0.02 (PMC), 0.03 (PFC) |
| SELP | Selectin P (granule membrane protein 140 kDa antigen CD62) | 0.31 (PMC), −0.18 (PFC) | 0.03 (PMC), 0.09 (PFC) |
| TM7SF2 | Transmembrane 7 superfamily member 2 | 0.42 (PMC), −0.37 (PFC) | 0.01 (PMC), 0.06 (PFC) |
| USP53 | Ubiquitin specific peptidase 53 | −0.71 (PMC), 0.06 (CC) | 0.02 (PMC), 0.06 (CC) |
| VNN1 | Vanin 1 | −0.24 (PMC), 0.30 (CC) | 0.07 (PMC), 0.05 (CC) |
We performed pathway analysis of the differentially expressed genes with FDR < 0.10, and identified canonical pathways with p-value <0.05 (listed in Table S2c). A number of pathways are present in more than one study (Table 2b): amyloid processing (in PMC and PFC), aryl hydrocarbon receptor signaling (PMC and PFC), cAMP-mediated signaling (PMC and CC), and hepatic fibrosis and hepatic stellate cell activation (CC and PFC). Since these pathways are modulated by clozapine in multiple brain regions, they may underlie more global mechanisms mediated by clozapine.
Table 2b.
Canonical pathways identified in more than one study, as determined by pathway analysis of differentially expressed genes with FDR <0.10. We performed Ingenuity pathway analysis (IPA) on the genes with false discovery rate (FDR) <0.10 that are differentially expressed in response to clozapine vs. other atypical antipsychotics. In each of the four studies, we present pathways with p-value <0.05. In addition to the p-value, we list the z-score: positive values indicate that the pathway is activated, negative values indicate that it is inhibited, and “Unknown” indicates that the Ingenuity database had inadequate data to determine activation or inhibition.
| Brain region (study) | Canonical pathway | z-Score | p-Value |
|---|---|---|---|
| Premotor cortex (Chen et al.) | Aryl hydrocarbon receptor signaling | 2.45 | p = 0.02 |
| cAMP-mediated signaling | 2.50 | p = 0.02 | |
| Amyloid processing | Unknown | p = 0.05 | |
| Superior temporal gyrus (Pietersen et al.) | None | ||
| Cerebellar cortex (Mudge et al.) | Hepatic fibrosis/hepatic stellate cell activation | Unknown | p = 0.02 |
| cAMP-mediated signaling | 2.00 | p = 0.05 | |
| Prefrontal cortex (Iwamoto et al.) | Amyloid processing | Unknown | p < 0.01 |
| Hepatic fibrosis/hepatic stellate cell activation | Unknown | p = 0.01 | |
| Aryl hydrocarbon receptor signaling | 2.00 | p = 0.05 | |
3.4. Differentially expressed genes in top 10 percentile by t-statistic, and associated pathways, in order to find consensus between all four studies
Although our analysis yielded many interesting genes that may underlie mechanisms specific to clozapine compared to other atypical antipsychotics, we sought to narrow down the list of 2251 genes to formulate a consensus of the field on this subject. Therefore we identified the genes differentially expressed in response to clozapine (vs. other atypical antipsychotics) that are present in multiple studies. However, as can be seen in Table 2a, no such genes with FDR <0.10 are present in more than two studies. This lack of consensus likely reflects the heterogeneity of the data we obtained from the literature, such as the differences in brain region and microarray platform among the four studies.
Therefore we used a different cutoff to identify differentially expressed genes present in all four studies (Fig. 3). When we ranked the genes by moderated t-statistic and selected those in the top 10 percentile for upregulation and the top 10 percentile for downregulation, we found two genes upregulated in all four studies, GCLM (glutamate-cysteine ligase modifier) and ZNF652 (zinc finger protein 652), and one gene downregulated in all four studies, GYPC (glycophorin C) (Table 3a). However, since genes with differential expression in two or three studies could also yield interesting data regarding clozapine’s mechanism of action, we provide supplementary data on all genes identified by t-statistic that were significantly up-or down-regulated in at least two studies (Tables S4a and S4b).
Fig. 3.
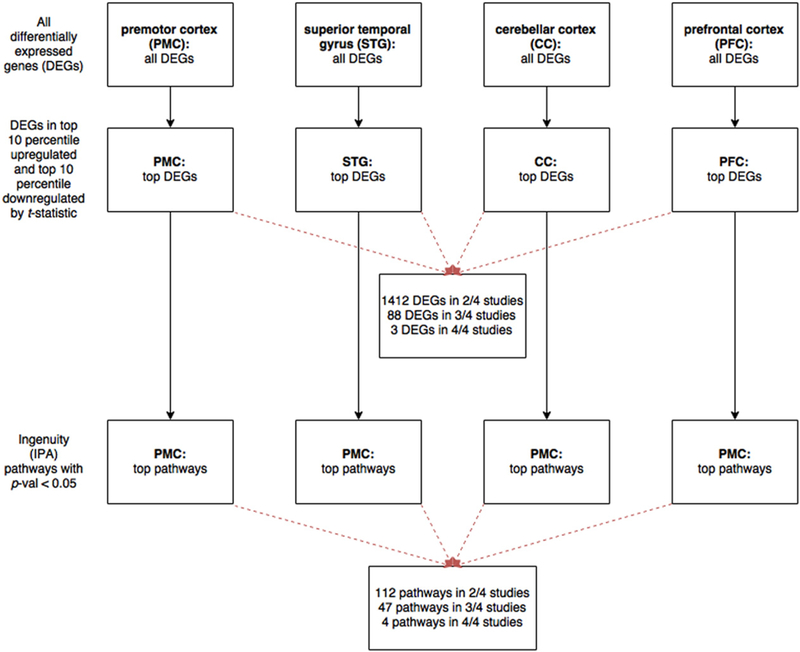
Workflow of analysis to identify differentially expressed genes bymoderated t-statistic, and their associated pathways.We again analyzed differentially expressed genes (DEGs) in response to clozapine vs. other atypical antipsychotics in the four studies by our systematic review: Chen et al. (in premotor cortex, PMC), Pietersen et al. (superior temporal gyrus, STG), Mudge et al. (cerebellar cortex, CC), and I wamoto et al. (prefrontal cortex, PFC). For each study, we ranked the DEGs by moderated t-statistic, and selected the top 10 percentile of upregulated genes and the top 10 percentile of downregulated genes.We also performed Ingenuity pathway analysis (IPA) on these genes and identified canonical pathways with p-value <0.05.
Table 3a.
Differentially expressed genes, identified by moderated t-statistic, that are present in all four studies. Of the top 10 percentile each of upregulated and downregulated genes, we present the genes that are in all four studies: Chen et al. (in premotor cortex, PMC), Pietersen et al. (superior temporal gyrus, STG), Mudge et al. (cerebellar cortex, CC), and Iwamoto et al. (prefrontal cortex, PFC). For each gene, we list the log2 fold change (logFC) and t-statistic for each brain region.
| Gene ID |
Gene name | logFC in each brain region |
t-Statistic in each brain region |
|---|---|---|---|
| GCLM | Glutamate-cysteine ligase modifier | PMC: 0.83 | PMC: 4.43 |
| STG: 4.75 | STG: 1.64 | ||
| CC: 0.27 | CC: 2.83 | ||
| PFC: 0.19 | PFC: 1.85 | ||
| GYPC | Glycophorin C | PMC: – 0.20 | PMC: – 3.02 |
| STG: –10.26 | STG: –1.57 | ||
| CC: – 0.35 | CC: – 2.67 | ||
| PFC: −0.16 | PFC: – 2.28 | ||
| ZNF652 | Zinc finger protein 52 | PMC: 1.63 | PMC: 3.10 |
| STG: 8.17 | STG: 1.57 | ||
| CC: 0.24 | CC: 2.51 | ||
| PFC: 0.90 | PFC: 5.85 | ||
Pathway analysis revealed that the genes selected by t-statistic are associated with four pathways that are present in all four studies (Table 3b): clathrin-mediated endocytosis signaling, SAPK/JNK signaling, 3-phosphoinositide biosynthesis, and paxillin signaling. Since these pathways are regulated by clozapine in four distinct brain regions, they may underlie more global effects mediated by clozapine and may also provide directions toward further study of the pathophysiology of schizophrenia. In addition, our analysis yielded a number of pathways in three out of four brain regions (Table S3) that are already linked to schizophrenia and thus promising for further investigation (Jia et al., 2010; Lencz et al., 2013; Mamdani et al., 2013; Need et al., 2009; Pouget et al., 2016; Sakurai, 2016; Takahashi and Sakurai, 2013).
Table 3b.
Canonical pathways identified in all four studies, as determined by pathway analysis of differentially expressed genes identified by t-statistic. We performed Ingenuity pathway analysis (IPA) on the differentially expressed genes identified by moderated t-statistic in each of the four studies, and selected canonical pathways with p-value <0.05. Here we present those pathways that are in all four studies. In addition to the p-value, we list the z-score: positive values indicate that the pathway is activated, negative values indicate that it is inhibited, and “Unknown” indicates that the Ingenuity database had inadequate data to determine activation or inhibition.
| Canonical pathway | z-Score | p-Value |
|---|---|---|
| Clathrin-mediated endocytosis signaling | PMC: Unknown | PMC: p < 0.01 |
| STG: Unknown | STG: p = 0.02 | |
| CC: Unknown | CC: p = 0.03 | |
| PFC: Unknown | PFC: p < 0.01 | |
| SAPK/JNK signaling | PMC: 1.18 | PMC: p < 0.01 |
| STG: 0.41 | STG: p = 0.03 | |
| CC: 0.22 | CC: p = 0.03 | |
| PFC: 0.85 | PFC: p < 0.01 | |
| 3-Phosphoinositide biosynthesis | PMC: Unknown | PMC: p = 0.01 |
| STG: Unknown | STG: p = 0.02 | |
| CC: Unknown | CC: p = 0.01 | |
| PFC: Unknown | PFC: p = 0.02 | |
| Paxillin signaling | PMC: 2.24 | PMC: p = 0.03 |
| STG: – 0.22 | STG: p = 0.01 | |
| CC: 1.00 | CC: p = 0.05 | |
| PFC: 1.29 | PFC: p = 0.02 | |
4. Discussion
Through a systematic review of the literature, we identified four microarray studies that contained the primary data necessary to analyze gene expression changes elicited by clozapine in the human brain. As none of the original studies characterized the differential gene expression profile of clozapine, we extracted and queried their expression data to address our own question of how clozapine modulates gene expression. Also, since we found that each study had been performed in a different brain region with a different microarray platform, we analyzed each dataset separately with generalized linear model analysis. As a result, we identified genes differentially expressed in response to clozapine vs. other atypical agents.
After each studies was analyzed independently, we identified genes and pathways that were common to multiple studies. We found three differentially expressed genes that are present in all four studies: GCLM and ZNF652 are upregulated, while GYPC is downregulated, by clozapine compared to other atypical antipsychotics in premotor cortex, superior temporal gyrus, cerebellar cortex, and prefrontal cortex. Our pathway analysis showed the canonical pathways associated with the differentially expressed genes in each dataset, including four pathways that are present in all four studies: clathrin-mediated endocytosis signaling, SAPK/JNK signaling, 3-phosphoinositide biosynthesis, and paxillin signaling. Interestingly, these pathways may be relevant to the pathophysiology of schizophrenia and bipolar disorder through their implications in protein trafficking, neuronal migration and survival, brain morphogenesis, axodendritic architecture, and synaptic function (Law et al., 2012; Morris and Pratt, 2014; Rosse et al., 2009; Schubert et al., 2012). Since schizophrenia affects many brain regions (Honea et al., 2005; Lawrie and Abukmeil, 1998; Levitt et al., 2010), we believe changes in gene expression across multiple brain regions may be informative about more global processes modulated by clozapine.
There are a number of limitations with our study. Foremost is the absence of studies in the literature that have characterized the differential gene expression mediated by clozapine specifically, which necessitated our novel approach of analyzing primary data from studies that involved patients on antipsychotic medications including clozapine. As such, our systematic review yielded a sample of four independent studies, each with two or three patients treated with clozapine. Moreover, these studies do not contain information on dose, duration, and therapeutic response to the antipsychotic treatments, or other factors that were not possible to control for in our analysis. Also, we cannot exclude the possibility that differences in gene expression changes between clozapine and other atypical antipsychotics could be due to differences between patients requiring clozapine and those who do not. Future studies could address this by analyzing differential gene expression and treatment response in patients before and after clozapine therapy. These aspects underscore the value of optimizing the accuracy of our results by analyzing each dataset separately and uniformly, and applying surrogate variable analysis (Leek and Storey, 2007) to adjust for unknown sources of variation. We believe our gene-list-based study represents one of the best approaches possible to accurately analyzing the heterogeneous body of evidence available in the field to yield biologically meaningful results, as it overcomes the intrinsic limitations associated with small effect and sample sizes, as was shown by Mootha et al. (2003) in their gene set enrichment analysis. In addition, future studies could also apply this approach to microarray data from animal models and compare with our results from human postmortem brain.
Our analysis does not determine whether each gene contributes to clozapine’s therapeutic efficacy or its side effects, but this assessment is possible for many genes from their known functions and associations with schizophrenia. For example, we found that all four studies showed that GCLM is upregulated by clozapine, which is consistent with the hypothesis that the pathogenesis of schizophrenia involves a dysregulated response to oxidative stress (Landek-Salgado et al., 2016). Thus, modulation of GCLM may possibly indicate an antipsychotic mechanism distinguishing clozapine from other atypical agents.
GCLM is the regulatory subunit of glutamate-cysteine ligase (GCL), the first rate-limiting enzyme in the synthesis of glutathione (GSH). GSH is a major mediator of the brain antioxidant response, and has been shown to be reduced in peripheral blood, cerebrospinal fluid, and postmortem brain of schizophrenia patients (O’Donnell et al., 2014; Steullet et al., 2016). GSH-deficient mouse models show behavioral and electrophysiological phenotypes relevant to schizophrenia that are reversed by N-acetyl cysteine, a GSH precursor found to protect against parvalbumin interneuron dysfunction in an animal model (Cabungcal et al., 2014). These animal studies offer insight into possible mechanisms by which oxidative stress may disrupt dopaminergic synapses, NMDA receptors, and interneuron function, all supporting a role for GSH in the pathophysiology of schizophrenia. Interestingly, oxidative stress is linked to several processes highlighted in our pathway analysis, such as NRF2-mediated oxidative stress response, eNOS signaling, and JNK signaling. Furthermore, polymorphisms in GCLM specifically have been associated with schizophrenia in human case-control studies (Tosic et al., 2006), while knockout mouse models of GCLM show elevated oxidative stress in the brain and a behavioral phenotype relevant to schizophrenia (Steullet et al., 2016). In addition, ZNF652 may be related to GCLM as it was shown in a ChIP-X enrichment analysis to be a potential binding site of NRF2 (Rouillard et al., 2016), a major regulator of genes involved in antioxidant responses including GCLM.
As a result of our analysis, we present here the first comprehensive compendium of differentially expressed genes modulated by clozapine in the human brain, which could help clarify the mechanism of this antipsychotic medication. Our findings may lead to a better understanding of this established but uniquely effective antipsychotic, and help elucidate the pathophysiology of schizophrenia.
Supplementary Material
Acknowledgments
Role of funding
This work was supported by the National Institute of Mental Health MH-094268 Silvio O. Conte center, MH-092443, MH-105660 (A.S.); National Institute on Drug Abuse DA-040127 (A.S.); foundation grants from Stanley, S-R, RUSK, NARSAD, Maryland Stem Cell Research Fund (A.S.); and the Johns Hopkins Institute for Clinical and Translational Research (ICTR) which is funded in part by Grant Number UL1 TR 001079 from the National Center for Advancing Translational Sciences (NCATS), and NIH Roadmap for Medical Research (L.M.).
Abbreviations:
- CC
cerebellar cortex
- PFC
prefrontal cortex
- PMC
premotor cortex
- STG
superior temporal gyrus
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.schres.2016.12.017.
Conflict of interest
We report no conflicts of interest.
References
- Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, Cacace AM, Zaczek R, Albright CF, Tseng G, Lewis DA, 2015. Distinctive transcriptome alterations of pre-frontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol. Psychiatry 20 (11), 1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston C, Jiang L, Sokolo VBP, 2004. Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J. Neurosci. Res. 77 (6), 858–866. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Counotte DS, Lewis EM, Tejeda HA, Piantadosi P, Pollock C, Calhoon GG, Sullivan EM, Presgraves E, Kil J, Hong LE, Cuenod M, Do KQ, O’Donnell P, 2014. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 83 (5), 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wang N, Zhao X, Ross CA, O’Shea KS, McInnis MG, 2013. Gene expression alterations in bipolar disorder postmortem brains. Bipolar Disord. 15 (2), 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley RR, Kelly DL, 2001. Management of treatment resistance in schizophrenia. Biol. Psychiatry 50 (11), 898–911. [DOI] [PubMed] [Google Scholar]
- Ereshefsky L, Watanabe MD, Tran-Johnson TK, 1989. Clozapine: an atypical antipsychotic agent. Clin. Pharm. 8 (10), 691–709. [PubMed] [Google Scholar]
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Thibaut F, Möller HJ, World Federation of Societies of Biological Psychiatry (WFSBP) Task Force on Treatment Guidelines for Schizophrenia, 2012. World Federation ofSocieties of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J. Biol. Psychiatry 13, 318–378.22834451 [Google Scholar]
- Honea R, Crow TJ, Passingham D, Mackay CE, 2005. Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am. J. Psychiatry 162 (12), 2233–2245. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Kato T, 2005. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet. 14 (2), 241–253. [DOI] [PubMed] [Google Scholar]
- Jia P, Wang L, Meltzer HY, Zhao Z, 2010. Common variants conferring risk of schizophrenia: a pathway analysis of GWAS data. Schizophr. Res. 122 (1–3), 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JL, Altar CA, Taylor DL, Degtiar I, Hornberger JC, 2014. The social and economic burden of treatment-resistant schizophrenia: a systematic literature review. Int. Clin. Psychopharmacol. 29 (2), 63–76. [DOI] [PubMed] [Google Scholar]
- Landek-Salgado MA, Faust TE, Sawa A, 2016. Molecular substrates of schizophrenia: homeostatic signaling to connectivity. Mol. Psychiatry 21 (1), 10–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Wang Y, Sei Y, O’Donnell P, Piantadosi P, Papaleo F, Straub RE, Huang W, Thomas CJ, Vakkalanka R, Besterman AD, Lipska BK, Hyde TM, Harrison PJ, Kleinman JE, Weinberger DR, 2012. Neuregulin 1-ErbB4-PI3K signaling in schizophrenia and phosphoinositide 3-kinase-p1108 inhibition as a potential therapeutic strategy. Proc. Natl. Acad. Sci. U. S. A. 109 (30), 12165–12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie SM, Abukmeil SS, 1998. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br. J. Psychiatry 172,110–120. [DOI] [PubMed] [Google Scholar]
- Leek JT, Storey JD, 2007. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3 (9), 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencz T, Guha S, Liu C, Rosenfeld J, Mukherjee S, DeRosse P, John M, Cheng L, Zhang C, Badner JA, Ikeda M, Iwata N, Cichon S, Rietschel M, Nöthen MM, Cheng AT, Hodgkinson C, Yuan Q, Kane JM, Lee AT, Pisante A, Gregersen PK, Pe’er I, Malhotra AK, Goldman D, Darvasi A, 2013. Genome-wide association study implicates NDST3 in schizophrenia and bipolar disorder. Nat. Commun. 4,2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, Samara M, Barbui C, Engel RR, Geddes JR, Kissling W, Stapf MP, Lässig B, Salanti G, Davis JM, 2013. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet 382 (9896), 951–962. [DOI] [PubMed] [Google Scholar]
- Levitt JJ, Bobrow L, Lucia D, Srinivasan P, 2010. A selective review of volumetric and morphometric imaging in schizophrenia. Curr. Top. Behav. Neurosci. 4, 243–281. [DOI] [PubMed] [Google Scholar]
- Mamdani F, Martin MV, Lencz T, Rollins B, Robinson DG, Moon EA, Malhotra AK, Vawter MP, 2013. Coding and noncoding gene expression biomarkers in mood disorders and schizophrenia. Dis. Markers 35 (1), 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY, Rosenheck RA, Swartz MS, Perkins DO, Keefe RS, Davis CE, Severe J, Hsiao JK, Investigators, C.A.T.I.E., 2006. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am. J. Psychiatry 163 (4), 600–610. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, 2012. Clozapine: balancing safety with superior antipsychotic efficacy. Clin. Schizophr. Relat. Psychoses 6 (3), 134–144. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Matsubara S, Lee JC, 1989. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharmacol. Exp. Ther. 25, 238–246. [PubMed] [Google Scholar]
- Miyamoto S, Jarskog LF, Fleischhacker WW, 2014. New therapeutic approaches for treatment-resistant schizophrenia: a look to the future. J. Psychiatr. Res. 58, 1–6. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Riddersträle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34 (3), 267–273. [DOI] [PubMed] [Google Scholar]
- Morris BJ, Pratt JA, 2014. Novel treatment strategies for schizophrenia from improved understanding of genetic risk. Clin. Genet. 86 (5), 401–411. [DOI] [PubMed] [Google Scholar]
- Mudge J, Miller NA, Khrebtukova I, Lindquist IE, May GD, Huntley JJ, Luo S, Zhang L, van Velkinburgh JC, Farmer AD, Lewis S, Beavis WD, Schilkey FD, Virk SM, Black CF, Myers MK, Mader LC, Langley RJ, Utsey JP, Kim RW, Roberts RC, Khalsa SK, Garcia M, Ambriz-Griffith V, Harlan R, Czika W, Martin S, Wolfinger RD, Perrone-Bizzozero NI, Schroth GP, Kingsmore SF, 2008. Genomic convergence analysis of schizophrenia: mRNA sequencing reveals altered synaptic vesicular transport in post-mortem cerebellum. PLoS One 3 (11), e3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Need AC, Ge D, Weale ME, Maia J, Feng S, Heinzen EL, Shianna KV, Yoon W, Kasperaviciüte D, Gennarelli M, Strittmatter WJ, Bonvicini C, Rossi G, Jayathilake K, Cola PA, McEvoy JP, Keefe RS, Fisher EM, St Jean PL, Giegling I, Hartmann AM, Möller HJ, Ruppert A, Fraser G, Crombie C, Middleton LT, St Clair D, Roses AD, Muglia P, Francks C, Rujescu D, Meltzer HY, Goldstein DB, 2009. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 5 (2), e1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Do KQ, Arango C, 2014. Oxidative/Nitrosative stress in psychiatric disorders: are we there yet? Schizophr. Bull. 40 (5), 960–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perala J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsä E, Pirkola S, Partonen T, Tuulio-Henriksson A, Hintikka J, Kieseppä T, Härkänen T, Koskinen S, Lönnqvist J, 2007. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch. Gen. Psychiatry 64 (1), 19–28. [DOI] [PubMed] [Google Scholar]
- Pietersen CY, Mauney SA, Kim SS, Lim MP, Rooney RJ, Goldstein JM, Petryshen TL, Seidman LJ, Shenton ME, McCarley RW, Sonntag KC, Woo TU, 2014a. Molecular profiles of pyramidal neurons in the superior temporal cortex in schizophrenia. J. Neurogenet. 28 (1 −2), 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietersen CY, Mauney SA, Kim SS, Passeri E, Lim MP, Rooney RJ, Goldstein JM, Petreyshen TL, Seidman LJ, Shenton ME, Mccarley RW, Sonntag KC, Woo TU, 2014b. Molecular profiles of parvalbumin-immunoreactive neurons in the superior temporal cortex in schizophrenia. J. Neurogenet. 28 (1–2), 70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget JG, Gonsalves VF, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Spain SL, Finucane HK, Raychaudhuri S, Kennedy JL, Knight J, 2016. Genome-wide association studies suggest limited immune Gene enrichment in schizophrenia compared to 5 autoimmune diseases. Schizophr. Bull. 42 (5), 1176–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, Formstecher E, Boeckeler K, Zhao Y, Kremerskothen J, White MD, Camonis JH, Parker PJ, 2009. An aPKC-Exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biol. 7 (11), e1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma’ayan A, 2016. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016, baw100. 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Chant D, Welham J, McGrath J, 2005. A systematic review of the prevalence of schizophrenia. PLoS Med. 2 (5), e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, 2016. The role of cell adhesion molecules in brain wiring and neuropsychiatric disorders. Mol. Cell. Neurosci. 10.1016/j.mcn.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014n. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 (7510), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Leonardi-Essmann F, Durrenberger PF, Parlapani E, Schneider-Axmann T, Spanagel R, Arzberger T, Kretzschmar H, Herrera-Marschitz M, Gruber O, Reynolds R, Falkai P, Gebicke-Haerter PJ, 2011. Regulation of immune-modulatory genes in left superior temporal cortex of schizophrenia patients: a genome-wide microarray study. World J. Biol. Psychiatry 12 (3), 201–215. [DOI] [PubMed] [Google Scholar]
- Schubert KO, Focking M, Prehn JH, Cotter DR, 2012. Hypothesis review: are clathrin-mediated endocytosis and clathrin-dependent membrane and protein trafficking core pathophysiological processes in schizophrenia and bipolar disorder? Mol. Psychiatry 17 (7), 669–681. [DOI] [PubMed] [Google Scholar]
- Siskind D, McCartney L, Goldschlager R, Kisely S, 2016. Clozapine v. first- and second-generation antipsychotics in treatment-refractory schizophrenia: systematic review and meta-analysis. Br. J. Psychiatry 10.1192/bjp.bp.115.177261. [DOI] [PubMed] [Google Scholar]
- Smyth GK, 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3,3. [DOI] [PubMed] [Google Scholar]
- Steullet P, Cabungcal JH, Monin A, Dwir D, O’Donnell P, Cuenod M, Do KQ, 2016. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: a “central hub” in schizophrenia pathophysiology? Schizophr. Res. 176 (1), 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Sakurai T, 2013. Roles of glial cells in schizophrenia: possible targets for therapeutic approaches. Neurobiol. Dis. 53,49–60. [DOI] [PubMed] [Google Scholar]
- Tosic M, Ott J, Barral S, Bovet P, Deppen P, Gheorghita F, Matthey ML, Parnas J, Preisig M, Saraga M, Solida A, Timm S, Wang AG, Werge T, Cuenod M, Do KQ, 2006. Schizophrenia and oxidative stress: glutamate cysteine ligase modifier as a susceptibility gene. Am. J. Hum. Genet. 79 (3), 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Wang X, Beveridge NJ, Tooney PA, Scott RJ, Carr VJ, Cairns MJ, 2012. Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLoS One 7 (4), e36351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.