

Abstract
Tissue regenerative potential displays striking divergence across phylogeny and ontogeny, but the underlying mechanisms remain enigmatic. Loss of mammalian cardiac regenerative potential correlates with cardiomyocyte cell-cycle arrest and polyploidization as well as the development of postnatal endothermy. We reveal that diploid cardiomyocyte abundance across 41 species conforms to Kleiber’s law—the ¾-power law scaling of metabolism with bodyweight—and inversely correlates with standard metabolic rate, body temperature, and serum thyroxine level. Inactivation of thyroid hormone signaling reduces mouse cardiomyocyte polyploidization, delays cell-cycle exit, and retains cardiac regenerative potential in adults. Conversely, exogenous thyroid hormones inhibit zebrafish heart regeneration. Thus, our findings suggest that loss of heart regenerative capacity in adult mammals is triggered by increasing thyroid hormones and may be a trade-off for the acquisition of endothermy.
In adult zebrafish and neonatal mice, heart regeneration occurs primarily through proliferation of preexisting cardiomyocytes (1, 2). Most mammalian cardiomyocytes undergo polyploidization and permanently withdraw from the cell-cycle postnatally, whereas lower vertebrates, such as newts and zebrafish, generally maintain >95% diploid mononucleated cardiomyocytes even as adults (1, 2). Recent studies in both mice and zebrafish further substantiate the positive correlation between diploid mononucleated cardiomyocyte abundance and heart regenerative capacity (3, 4). We therefore exploited cardiomyocyte ploidy as a proxy for heart regenerative potential and analyzed its variability across vertebrate species (Fig 1A and tables S1 and S2).
Fig. 1. Phylogenetic analysis of vertebrate cardiomyocyte (CM) nucleation and ploidy.
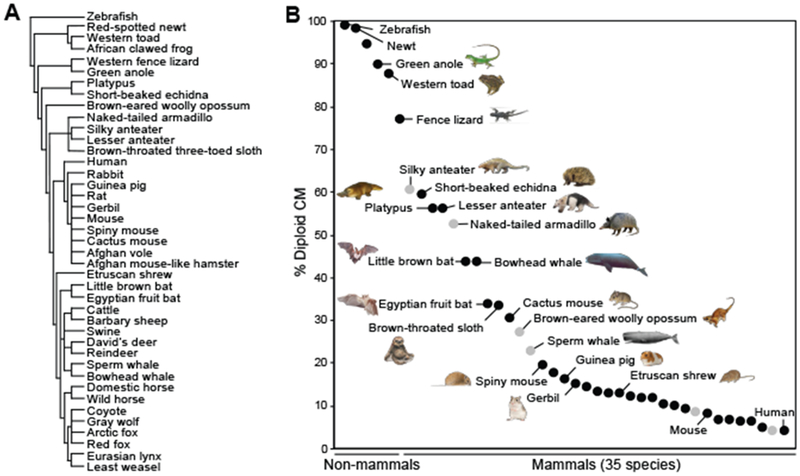
(A) Cladogram of species examined here. (B) Percentages of mononucleated diploid CMs. Each dot represents the value from the adult heart(s) of one species. Black, multiple samples were quantified; gray, only a single specimen was collected and analyzed. The data are listed from left to right according to the value of diploid CMs. Each new species analyzed in the current study is marked with an animal symbol next to it. Unlabeled dots represent species with previously reported CM ploidy values. All data are presented in table S2. Mammalian species with >30% mononucleated (diploid) CMs are first identified in this study.
Our phylogenetic analysis confirmed that cardiomyocytes from adult fish, amphibians, and reptiles are mostly (98.3 to 77.1%) mononucleated and diploid (Fig. 1B), which is consistent with their high proliferative and regenerative capacities (fig. S1) (5, 6). Certain monotreme, edentate, cetacean, and chiropteran species retained high percentages of mononucleated diploid cardiomyocytes in the adult heart. In particular, the short-beaked echidna (Tachyglossus aculeatus), platypus (Ornithorhynchus anatinus), lesser anteater (Tamandua tetradactyla), bowhead whale (Balaena mysticetus), little brown bat (Myotis lucifugus), and Egyptian fruit bat (Rousettus aegyptiacus) possessed 59.3 to 34.1% diploid cardiomyocytes as compared with 9.4% observed in mice (Fig. 1B, fig. S2, and table S2).
Diploid cardiomyocyte abundance did not significantly correlate with bodyweight, heart rate, or blood pressure (fig. S3A and table S3). However, it displayed a robust inverse correlation with standard metabolic rate (Fig. 2A and fig. S3B). According to Kleiber’s law (7), basal metabolic rate (MR) scales with body mass (M) as MR = MR0M0.75, in which MR0 is the standard metabolic rate. Ectothermic animals have a distinct MR0 value from that of endothermic animals: The average value of MR0 is 0.21 W kg−0.75 for fish, amphibians, and reptiles (8), an order of magnitude less than that observed in endothermic eutherians (3.35 W kg−0.75) (9). The change of cardiomyocyte ploidy in vertebrates seemingly occurred in parallel with the major metabolic transition from ectotherms to endotherms (Fig. 2A).
Fig. 2. The percentage of diploid CM inversely correlates with standard metabolic rate, body temperature, and plasma total T4 levels.
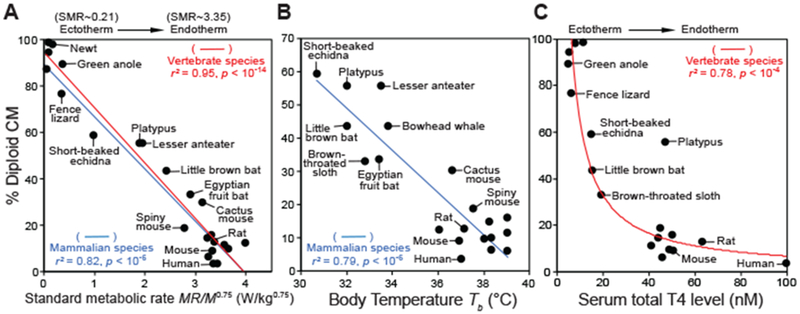
(A) Effect of standard metabolic rate (SMR, in W/kg0.75) on the content of diploid CMs. SMR is defined as MR/M0.75 (MR, metabolic rate; M, body mass). The data are fit by regression lines (red for vertebrate species and blue for mammalian species). (B) Effect of body temperature (Tb, in degrees celsius) on the percentage of diploid CMs. (C) Effect of plasma T4 levels on the frequency of diploid CMs. In (A) to (C), analyses include all species that have published physiological data to our knowledge. r2 denotes the coefficient of determination. For visual clarity, only a subset of ectodermic and endothermic species are labeled. All data are presented in tables S2 to S4.
In addition to mass, another key determinant of metabolic rate is temperature. Standard metabolic rate MR0 is proportional to biochemical reaction rates and therefore varies with temperature as MR0 ~ e−E/kT, where E represents the average activation energy for the reactions, k is the Boltzmann constant, and T is the absolute temperature (10). Data collected from microbes, plants, and animals confirm a universal inverse relationship between MR0 and e−1000/T (10). We evaluated cardiomyocte nucleation as a function of e−1000/T—particularly among mammalian species, for which T is stable throughout most of their lives—and found a strong inverse correlation (fig. S3C). In mammals, T is a simple function of body temperature (Tb) as T = Tb + 273. In the physiological range of mammalian Tb (30 to 39°C), e−1000/T displays a linear relationship with T (fig. S3D). Indeed, when we plotted mammalian diploid cardiomyocyte percentages against their respective body temperatures, a linear relationship was observed (Fig. 2B and figs. S3E and S4).
As the major regulators of energy metabolism and thermogenesis (fig. S5) (11), thyroid hormones are hypothesized to drive the ectotherm-to-endotherm transition (12). We analyzed the relationship between previously reported thyroid hormone levels on cardiomyocyte nucleation and uncovered an inverse correlation between plasma thyroxine (T4) levels and diploid cardiomyocyte content (Fig. 2C and table S4). Altogether, our analyses implicate that animals with lower standard metabolic rates, body temperatures, and serum T4 levels may have more abundant diploid cardiomyocytes.
The level of circulating thyroid hormones rises >50-fold in newborn mice shortly after birth (fig. S6A) (13), which coincides with cardiomyocyte cell-cycle exit, binucleation, and loss of regenerative capacity (1). The physiological role of thyroid hormones in cardiomyocyte proliferation has not been established. Thyroid hormones have been reported to either inhibit (14,15), enhance (16), or have no effect on (17) cardiomyocyte proliferation. We found no effect of exogenous triiodothyronine (T3) on cardiomyocyte proliferation in vitro (fig. S6B). In addition, thyroid hormones were recently proposed to promote a burst of mouse cardiomyocyte division at postnatal day 15 (P15) (18); however, others have found no evidence of cardiomyocyte expansion at this stage (19,20).
To determine the role of thyroid hormones in the regulation of cardiomyocyte proliferation in vivo, we injected mice daily with NH3, a specific inhibitor of thyroid hormone receptors (fig. S7A) (21). Cardiomyocytes were identified by means of perinuclear staining of pericentriolar material 1 (PCM1) protein, and proliferation was assessed at P14, when cardiomyocyte binucleation and cell-cycle arrest are largely completed (1). Our results demonstrate that treatment with NH3 enhanced myocyte proliferation by approximately fourfold (fig. S7, B to D), whereas chemical inhibition of other pathways documented to change during the perinatal window did not significantly affect cardiomyocyte proliferation in our assay (fig. S7B and table S5). We further validated the role of thyroid hormone signaling in promoting myocyte postnatal cell-cycle exit by blocking thyroid hormone synthesis with pro-pylthiouracil (PTU) and analyzing mutant mice with global expression of dominant negative (DN) thyroid hormone receptor-α (TRα) (fig. S8) (22). Both conditions similarly resulted in approximately threefold increases in proliferating and diploid cardiomyocytes.
We next generated mutant mice with cardiomyocyte-specific expression of dominant negative TRα (Myh6-Cre; ThraDN/+) (Fig. 3A). Despite a normal bodyweight at P14, the heart weight of Myh6-Cre;ThraDN/+ mice increased by 21% (Fig. 3B and fig. S9B). We also observed a ~2.3-fold increase in total cardiomyocyte numbers as estimated with dissociation (Fig. 3C) and stereological approaches (fig. S9D), retention of ~30% diploid cardiomyocytes (Fig. 3C), and a 47% reduction in myocyte size in mutant hearts (Fig. 3C and fig. S9C). Furthermore, increased numbers of mutant cardiomyocytes expressed proliferation markers Ki67 (3.5-fold), phospho-histone 3 ser10 (pHH3) (twofold), and cleavage furrow-localized Aurora B kinase (8.8-fold) (Fig. 3, D and E, and fig. S9E). Furthermore, EdU incorporation assays from P12 through P14 revealed that thyroid hormone mutants possessed increased numbers of EdU-positive diploid cardiomyocytes, whereas in control mice, all EdU-positive cardiomyocytes were polyploid (Fig. 3F and fig. S9F). Furthermore, we generated Myh6-merCremer;ThraDN/+ mice and induced DN-TRα expression around the perinatal window (fig. S10). Increased prevalence of diploid proliferative cardiomyocytes was also observed in these mutants (fig. S10). Taken together, these data demonstrate that perinatal thyroid hormone signaling has an intrinsic role in cardiomyocyte cell-cycle arrest and polyploidization.
Fig. 3. CM-specific inactivation thyroid hormone receptor enhances CM proliferation in neonatal mice.
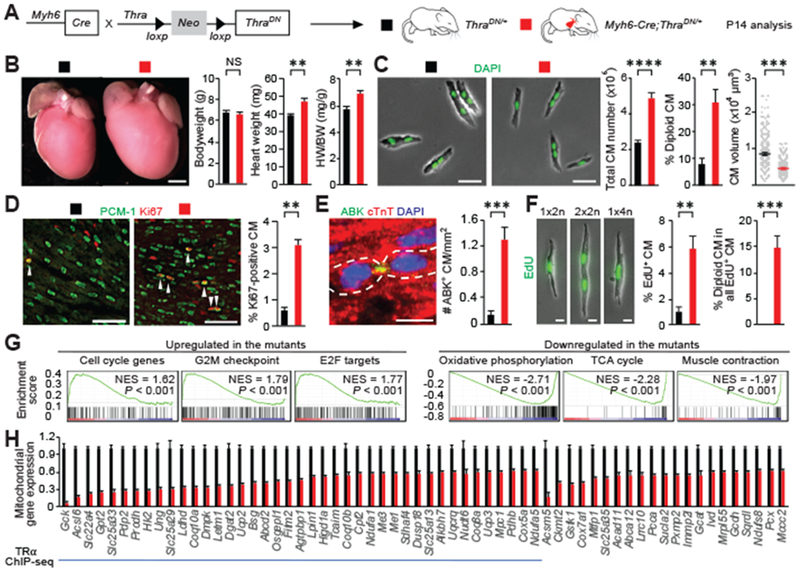
(A) Schematic for generating Myh6-Cre;ThraDN/+ mice with CM-restricted expression of a dominant negative (DN) TRα and analyzing phenotypes at P14. (B) Measurement of the bodyweight (BW) and heart weight (HW) (n = 4 mice). (C) Ventricular CM number, ploidy, and size analysis (n = 3 to 7 mice). (D to F) CM proliferative activity analysis. Representative images and quantifications of (D) proliferating CMs that stained positive for Ki67, (E) Aurora B kinase (ABK) localization at the cleavage furrow, and (F) EdU (n = 4 animals). Arrowheads indicate proliferating CM. In (E), cardiomyocytes undergoing cytokinesis are outlined. In (F), EdU was analyzed in dissociated CMs at P14 from mice injected with EdU at P12 and P13. 1×2n, 2×2n, and 1×4n denote CMs with one diploid nucleus, two diploid nuclei, and one tetraploid nucleus, respectively. (G) Expression profiling. Gene set enrichment analysis of up-regulated and down-regulated pathways in the mutant heart based on the RNA-seq analysis. (H) RNA-seq and ChIP-seq analysis. Mitochondrial genes whose expression is significantly down-regulated in the mutant hearts are shown (n = 3 hearts). The blue line marks mitochondrial genes directly targeted by TRα. Values are reported as mean ± SEM. NS, not significant. **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, (B) 1 mm; (C) 50 μm; (D) 100 μm; (E) and (F) 20 μm.
To identify transcripts regulated by thyroid hormone signaling, we next performed RNA sequencing (RNA-seq) analyses of P14 control and Myh6-Cre;ThraDN/+ hearts. Gene-set enrichment analyses revealed that cell-cycle, E2F target, and G2M checkpoint genes were significantly up-regulated (Fig. 3G and fig. S11). In genes down-regulated in thyroid hormone mutant hearts, we found an enrichment of components involved in oxidative phosphorylation, citrate (TCA) cycle, and cardiac muscle contraction (Fig. 3G and fig. S11). The expression levels of many mitochondrial genes were reduced (Fig. 3H). To identify TRα direct gene targets, we generated a mouse strain termed ThraTAG in which the coding sequence for protein G-tagged TRα1 receptor is knocked into the endogenous Thra locus, allowing for Cre-dependent expression. Chromatin immunoprecipitation-sequencing (ChIP-seq) analyses suggest that a majority of down-regulated mitochondrial genes are direct targets of TRα (Fig. 3H and fig. S12). One of these targets is carnitine palmitoyltransferase 2 (Cpt2), which encodes an enzyme essential for β oxidation of long-chain fatty acids in the mitochondria (23). Both Cpt2 mRNA and protein levels were partially (~40%) depleted in the hearts of Myh6-Cre;ThraDN/+ mice (fig. S13). We further showed that partial depletion of Cpt2 in Myh6-Cre;Cpt2fl/+ heterozygous mutant mice resulted in enhanced cardiomyocyte proliferation (fig. S14) without increased heart size, which is in contrast to the severe pathological hypertrophy observed in homozygous mutants (24). Thus, our findings suggest that reduction of Cpt2, at least in part, contributes to the increased cardiomyocyte proliferation observed in thyroid hormone mutants.
Consistent with reduced expression of many mitochondrial genes, Myh6-Cre;ThraDN/+ mutant hearts have less mitochondrial DNA, an increased ratio of reduced to oxidized glutathione, and diminished levels of reactive oxygen species (fig. S15). Previous studies have implicated that increasing reactive oxygen species in the postnatal heart, associated with mitochondrial biogenesis, is a major inhibitor of cardiomyocyte proliferation and heart regeneration (25, 26). Thus, our results identify thyroid hormone signaling as an upstream regulator of this pathway.
We next characterized the hearts of adult Myh6-Cre;ThraDN/+ mutant mice and their regenerative capacity (Fig. 4A). Adult thyroid hormone mutant hearts were 37% larger than controls (Fig. 4B and fig. S16) and contained an increased number (approximately twofold) of cardiomyocytes as shown with both dissociation (Fig. 4C) and stereological methods (fig. S16). Mutant cardiomyocytes are smaller, and ~19% of them remain diploid (Fig. 4C). To assess the cardiac regenerative potential of these adult mutants, we performed cardiac surgery to induce ischemia reperfusion (IR) injury. First, cardiomyocyte viability was assessed, and no significant difference was found between the mutant and control hearts 24 hours after ischemia (fig. S17). Second, cardiomyocyte proliferation was examined 10 days after injury. Mutant mice possess a 3- to 10-fold increase in proliferating cardiomyocytes after cardiac injury (Fig. 4, D and E, and fig. S18). Injection of EdU during the first 10 days after injury revealed a 10-fold increase in EdU-positive diploid cardiomyocytes 28 days after injury, further suggesting that complete cardiomyocyte division is increased in thyroid hormone mutant hearts (Fig. 4F and fig. S18). Third, cardiac functions were evaluated by means of echocardiography, which revealed that mutant mice exhibited significant improvement of systolic functions after injury by 11%, whereas control mice did not (Fig. 4G and fig. S19A). Last, analysis of cardiac fibrosis 1 month after injury revealed a ~62% reduction of scar area in mutant hearts (Fig. 4H and fig. S19B). Taken together, these results show that adult cardiomyocytes with defects in thyroid hormone receptor activation retain significant proliferative and regenerative potential.
Fig. 4. CMs deficient in thyroid hormone signaling retain significant regenerative potential in adult mice.
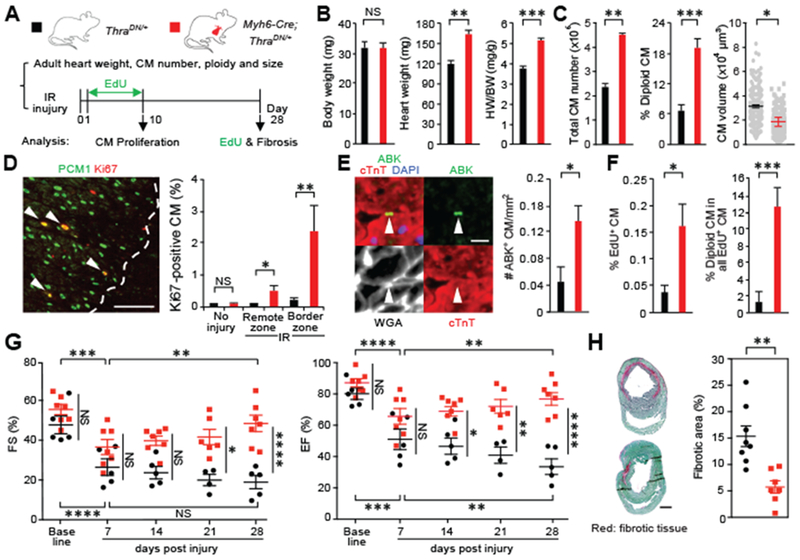
(A) Schematic of the experimental plan to analyze adult hearts and assess regenerative response after myocardial ischemia-reperfusion (IR) injury. (B) Measurement of the bodyweight (BW) and heart weight (HW) (n = 5 mice). (C) Analysis of ventricular CM number, ploidy, and cell size in adult mice (n = 3 animals). (D and E) CM proliferation analysis 10 days after injury. Quantification and representative images of CMs that are stained positive for Ki67 and Aurora B kinase (ABK) localization at the cleavage furrow in the hearts with no injury (n = 3 or 4 animals) or the hearts after IR injury (n = 4 or 5 animals). Dash lines mark the border of injury, whereas arrowheads show proliferating CMs. (F) Analysis of the number and ploidy of EdU-incorporated CMs. EdU is injected daily in the first 10 days after injury. Dissociated CMs are analyzed 28 days after IR (n = 4 animals). (G) Measurement of cardiac functions by means of echocardiography. The fraction shortening (FS) and ejection faction (EF) data of individual animals are presented (n = 5 to 7 animals). (H) Fibrosis analysis 28 days after injury (n = 7 or 8 animals). Values are reported as mean ± SEM. NS, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, (D) 100 μm; (E) 20 μm; (H) 1 mm.
Adult zebrafish possess the ability to completely regenerate their hearts (5); thus, we investigated how exogenous thyroid hormones could affect zebrafish cardiomyocyte proliferation and regeneration. T3 treatment resulted in a 45% reduction in cardiomyocyte proliferation, impaired regeneration accompanied with prominent scars, and a fivefold increase of cardiomyocyte binucleation (fig. S20). These results demonstrate that exogenous thyroid hormones inhibit zebrafish cardiomyocyte proliferation and regeneration after myocardial injury and implicate an evolutionarily conserved function of thyroid hormone in regulating cardiomyocyte proliferation.
Our findings support a model (fig. S21) in which loss of cardiac regenerative capacity is driven by increasing levels of circulating thyroid hormones. This process coincides with the metabolic increase and thermogenesis that occurs during the ectotherm-to-endotherm transition in both phylogeny (Fig. 2) and ontogeny. Newborn mice, like many other mammalian neonates, are poikilothermic at birth and later increase heat production to regulate body temperature (27), coinciding with increasing levels of thyroid hormones in circulation. The essential role of thyroid hormones in driving thermogenesis is evident in adult mice devoid of thyroid hormone receptors (28) or those treated with thyroid hormone synthesis inhibitor PTU (fig. S22), which have dramatically decreased basal body temperatures compared with those observed in monotremes. Elevated metabolic rate and the evolution of endothermy likely offered early mammals survival advantages as they inhabited a temporally wide nocturnal niche and expanded into regions of colder climates previously unexplored by reptiles. Altogether, our study suggests that loss of mammalian cardiac regenerative potential may be a trade-off for thyroid hormone-dependent acquisition of endothermy during animal development and evolution. It will be of key importance to identify other pathways, in addition to thyroid hormone signaling, regulating the logic of regenerative diversity to further understand why this capacity is lost in adult mammals.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Julius, B. Black, and J. Chen for discussions and suggestions. We also thank Huang laboratory members for data analyses; C. White for the contribution of the original concept and initial nucleation analyses; B. Gillet and S. Hughes of the PSI deep-sequencing facility for their help in ChIP-seq; R. Gibeaux and R. Heald for providing the hearts of African clawed frogs; J. Wilson-Rawls, K. Kusumi, and C. Xu for providing the hearts of green anoles; S. Ray, N. Pak, M. Brecht, S. Erdman, and E. Boyden for providing the hearts of Etruscan shrew; the Museum of Vertebrate Zoology, University of California, Berkeley for various mammalian, reptilian, and amphibian tissues (MVZ:Mamm:155186, MVZ:Mamm:155186, MVZ:Mamm:157801, MVZ:Mamm:190247, MVZ:Mamm:193692, MVZ:Herp:142847, MVZ:Herp:107011, MVZ: Herp:265556, and MVZ:Herp:265557); and the Museum of Southwestern Biology (MSB:Mamm:140338 and MSB:Mamm:211353) and the Museum of the North, University of Alaska for the toad and whale samples (UAM:Mamm:53932, UAM:Mamm:66458, UAM: Mamm:97497, UAM:Mamm:115643, UAM:Herp:14, and UAM:Herp:9).
Funding: This work is supported by JSPS Overseas Research Fellowships (to K.H.), a UCSF-IRACDA postdoctoral fellowship to A.P.), a NIGMS IMSD fellowship and Hillblom fellowship (to S.C.), an ARC fellowship (to F.G.), the Agence Nationale de Recherche Thyromut2 program, ANR-15-CE14–0011-01 (to F.F.), Intramural Research Program of the NIH, National Institute of Environmental Health Sciences 1ZIAES102745 (to G. H.), NIH (R01HL13845) Pathway to Independence Award (R00HL114738), Edward Mallinckrodt Jr. Foundation, March of Dimes Basil O’Conner Scholar Award, American Heart Association Beginning Grant-in-Aid, American Federation for Aging Research, Life Sciences Research Foundation, Program for Breakthrough Biomedical Research, UCSF Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research Seed Grant, UCSF Academic Senate Committee on Research, REAC Award (Harris Fund), Department of Defense, and Cardiovascular Research Institute (to G.N.H.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: RNA-seq and ChIP-seq datasets were deposited in the Gene Expression Omnibus (GEO) database under accession nos. GSE125596 and GSE125414, respectively. All other data are available in the manuscript or supplementary materials.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/364/6436/184/suppl/DC1
Materials and Methods
References (29–65)
MATERIALS SUPPLEMENTARY http://science.sciencemag.org/content/suppl/2019/03/06/science.aar2038.DC1
REFERENCES AND NOTES
- 1.Xin M, Olson EN, Bassel-Duby R, Nat. Rev. Mol. Cell Biol 14, 529–541 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matrone G, Tucker CS, Denvir MA, Cell. Mol. Life Sci 74, 1367–1378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patterson M et al. , Nat. Genet 49, 1346–1353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González-Rosa JM et al. , Dev. Cell 44, 433–446.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poss KD, Wilson LG, Keating MT, Science 298, 2188–2190 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Rumyantsev PP, Growth and Hyperplasia of Cardiac Muscle Cells, Carlson BM, Ed. (Harwood, 1991). [Google Scholar]
- 7.Kleiber M, Hilgardia 6, 315–353 (1932). [Google Scholar]
- 8.Makarieva AM et al. , Proc. Natl. Acad. Sci. U.S.A 105, 16994–16999 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt-Nielsen K, Bolis L, Taylor CR, Eds., Comparative Physiology: Primitive Mammals (Cambridge Univ. Press, 1980). [Google Scholar]
- 10.Gillooly JF, Brown JH, West GB, Savage VM, Charnov EL, Science 293, 2248–2251 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Hulbert AJ, Biol. Rev. Camb. Philos. Soc 75, 519–631 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Little AG, Seebacher F, J. Exp. Biol 217, 1642–1648 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Friedrichsen S et al. , Endocrinology 144, 777–784 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Chattergoon NN, Giraud GD, Thornburg KL, Endocrinol J. 192, R1–R8 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Yang X et al. , J. Mol. Cell. Cardiol 72, 296–304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segar JL, Volk KA, Lipman MH, Scholz TD, Exp. Physiol 98, 722–733 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Svensson Holm AC, Lindgren I, Osterman H, Altimiras J, Physiol. Rep 2, e12182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naqvi N et al. , Cell 157, 795–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soonpaa MH et al. , Cell 163, 781–782 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Alkass K et al. , Cell 163, 1026–1036 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Nguyen NH et al. , J. Med. Chem 45, 3310–3320 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Quignodon L, Vincent S, Winter H, Samarut J, Flamant F, Mol. Endocrinol 21, 2350–2360 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Ceccarelli SM, Chomienne O, Gubler M, Arduini A, J. Med. Chem 54, 3109–3152 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Pereyra AS et al. , J. Biol. Chem 292, 18443–18456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puente BN et al. , Cell 157, 565–579 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tao G et al. , Nature 534, 119–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagerspetz K, Helgol. Wiss. Meeresunters 14, 559–571 (1966). [Google Scholar]
- 28.Gauthier K et al. , Mol. Cell. Biol 21, 4748–4760 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.