

Abstract
Eradicating triple negative breast cancer (TNBC) resistant to neoadjuvant chemotherapy (NACT) is a critical unmet clinical need. In this study, patient-derived xenograft (PDX) models of treatment-naïve TNBC and serial biopsies from TNBC patients undergoing NACT were used to elucidate mechanisms of chemoresistance in the neoadjuvant setting. Barcode-mediated clonal tracking and genomic sequencing of PDX tumors revealed that residual tumors remaining after treatment with standard front-line chemotherapies, doxorubicin (Adriamycin) combined with cyclophosphamide (AC), maintained the subclonal architecture of untreated tumors yet their transcriptomes, proteomes, and histologic features were distinct from those of untreated tumors. Once treatment was halted, residual tumors gave rise to AC-sensitive tumors with similar transcriptomes, proteomes, and histological features to those of untreated tumors. Taken together, these results demonstrated that tumors can adopt a reversible drug-tolerant state that does not involve clonal selection as an AC resistance mechanism. Serial biopsies obtained from patients with TNBC undergoing NACT revealed similar histologic changes as well as maintenance of stable subclonal architecture, demonstrating that AC-treated PDXs capture molecular features characteristic of human TNBC chemoresistance. Finally, pharmacologic inhibition of oxidative phosphorylation using an inhibitor currently in phase I clinical development delayed residual tumor regrowth. Thus, AC resistance in treatment-naïve TNBC can be mediated by non-selective mechanisms that confer a reversible chemotherapy-tolerant state with targetable vulnerabilities.
One-sentence summary:
Resistance to neoadjuvant chemotherapy in triple-negative breast cancer can be mediated by a reversible chemotherapy-tolerant state.
INTRODUCTION
TNBC comprises 15–20% of breast cancers and is an aggressively metastatic disease for which there are no approved molecularly targeted therapies. Notably, 50% of patients with localized TNBC treated with NACT have substantial residual cancer burden (RCB-II or –III) detected by pathology evaluation of the breast and axillary nodes at the time of surgical resection. This insensitivity to chemotherapy is associated with a 40– 80% risk of recurrence, resulting in distant metastasis and death for most patients (1–4). Delineating mechanisms used by TNBCs to evade NACT is expected to improve therapeutic interventions for patients with newly diagnosed disease, thereby improving long-term prognoses for those patients who respond poorly to NACT. TNBCs exhibit varying degrees of genomic intra-tumor heterogeneity (ITH) at the time of diagnosis (5–7). Studies profiling serial biopsies before and after NACT regimens consisting of various cytotoxic chemotherapies and targeted therapies have demonstrated that TNBCs can exhibit shifts in subclonal architecture or maintain stable genomic architecture after treatment (5, 8–10). However, the functional contributions made by genomically heterogeneous tumor cell populations to therapy resistance and tumor progression remain unclear.
Resistance to therapy can occur through pre-existing (intrinsic) or acquired (adaptive) mechanisms, and these can be due to genomic and/or non-genomic properties of tumor cells. Studies of reversible (non-genomic) chemoresistance mechanisms, including preclinical studies characterizing drug-tolerant states, chemotherapy re-challenge studies in the clinical and pre-clinical settings, and mathematical modeling of adaptive treatment regimes, have demonstrated that chemoresistant tumors can return to a chemo-sensitive state after a drug holiday (11, 12). It is yet unclear whether genomic and/or non-genomic mechanisms functionally contribute to chemoresistance in treatment-naïve TNBC. In the case of lung cancer, resistance to EGFR inhibition can occur through selection of a rare pre-existing subclone harboring a resistance-enabling mutation or through eventual acquisition of the same mutation in initially non-mutant cells (13). Epigenetic modifications have been found to contribute to drug resistance in several cancer types. For example, resistance to EGFR tyrosine kinase inhibitors and carboplatin in drug-tolerant cancer cells was mediated by the histone demethylase KDM5A (14). A recent study revealed that epigenetic plasticity mediated by BRD4 and other chromatin modifiers drives survival of persister TNBC cells after targeted therapy treatment (15). Furthermore, cancer stem-like cells harboring intrinsic mechanisms of drug resistance, including over-expression of drug efflux pumps (16), enhanced DNA repair capacity (17), and mesenchymal-like properties (18) can also account for resistance to therapy. Although enrichment of tumor cells with stem-like properties has been reported in estrogen receptor-positive tumors after endocrine therapy and in metaplastic breast tumors after docetaxel (19, 20), this was not observed in residual TNBCs after NACT (9).
Several studies interrogating human TNBCs before and after therapy have provided insights into potential mechanisms of resistance to chemotherapy. Activation of various signaling pathways (Mitogen-Activated Protein Kinase Kinase (MEK), Transforming Growth Factor Beta 1 (TGF-β) (8, 21), Protein Kinase B alpha (AKT1), Insulin Like Growth Factor 1 (IGF1)), as well as extracellular matrix remodeling and hypoxia (10, 22, 23) are associated with resistance in some TNBC cases. In addition, tumor-infiltrating lymphocyte (TIL) concentration in treatment-naïve TNBC is predictive of response to NACT (24, 25). However, none of this information is being used clinically to stratify patients or to predict outcome in the context of treatment-naïve TNBC. Of note, TNBC is characterized by substantial inter-patient heterogeneity at the genomic, transcriptomic, and histologic levels (26–29). Thus, it is likely that diverse molecular mechanisms contribute to chemotherapy resistance within this patient population.
In this study, we investigated mechanisms of AC resistance using PDX models of treatment-naïve TNBC and serial biopsies obtained from TNBC patients undergoing NACT. Results revealed non-genomic mechanisms of resistance, whereby all treatment-naïve tumor subclones were equally capable of adopting a reversible drug-tolerant state upon exposure to AC. Tumor cells in this drug-tolerant state had distinct transcriptomes, proteomes, and histologic features compared with untreated tumors, but gave rise to tumors with restored drug sensitivity, transcriptomes, proteomes, and histologic features of untreated tumors. Analyses of serial biopsies from TNBC patients undergoing NACT corroborated these histologic and genomic findings. Gene expression profiles of residual tumors in PDXs as well as in residual tumors of breast cancer patients revealed alterations in metabolic gene expression programs. Based on the gene expression signatures of residual tumors, mitochondrial oxidative phosphorylation was identified as a potential dependency in residual tumors, and inhibition of oxidative phosphorylation delayed residual tumor regrowth in PDX models of TNBC. Taken together, these studies revealed that a reversible phenotypic state not mediated by genomic selection can confer AC resistance in treatment-naïve TNBC and that the residual tumor state is a therapeutic window in chemo-refractory TNBC.
RESULTS
PDX models of treatment-naïve TNBC exhibit distinct responses to NACT
PDX models of treatment-naïve TNBC were established in alignment with an ongoing IRB-approved clinical trial at the University of Texas MD Anderson Cancer Center (MDACC) (30, 31). Tumor cells obtained by fine-needle aspiration were engrafted into the fourth mammary fat pads (MFPs) of NOD/SCID mice, and PDX lines were considered established after three consecutive passages (data file S1). All PDX models were established from treatment-naïve primary breast tumor biopsies, with the exception of PIM001-M, which was established from a treatment-naïve chest wall metastasis. PDX models were treated with AC because standard front-line NACT for patients with TNBC at MDACC consists of 4 cycles of AC (1 dose every 21 days). A maximum tolerated dose of 0.5 mg/kg doxorubicin plus 50 mg/kg cyclophosphamide was determined for NOD/SCID and NOD-Rag-Gamma (NRG) mice, and this dose was used throughout our studies (fig. S1A–F) because maximum tumor response was achieved with this dose (fig. S1G). PDX models of treatment-naïve TNBC exhibited diverse responses when treated with AC, including transient responses characterized by reduction in tumor size followed by regrowth, slowed tumor growth in the presence of AC, or no response to AC (Fig. 1). To date, we have not generated a PDX model that exhibits a complete pathological response to AC.
Figure 1. PDX models of treatment-naïve TNBC exhibit diverse responses to AC.

One or two cycles of AC were administered to PDX models of treatment-naïve TNBC starting on day zero (arrows). All PDXs were derived from primary tumors with the exception of PIM001-M, which was derived from the dermal metastasis to the chest wall of the same patient from whom PIM001-P (primary tumor) was derived. Data shown are mean +/− standard error of the mean (SEM; n=3 per group).
Chemoresistance can be modeled in PDXs of treatment-naïve TNBC
Three PDX models, characterized by a reduction in tumor size followed by regrowth after treatment, were selected for in-depth characterization to identify mechanisms of chemotherapy resistance. These models included PIM001-P (Patient-In-Mouse 1, derived from the primary breast tumor of a patient with treatment-naïve metastatic TNBC), PIM001-M (derived from a treatment-naïve synchronous dermal metastasis of the patient whose primary tumor was used to generate PIM001-P), and PIM005 (derived from the breast tumor of a second patient with treatment-naïve TNBC). To determine whether multiple cycles of AC treatment could eradicate tumors, AC was administered every 21 days for 1–8 cycles. In all three PDX models, AC elicited tumor regression, however, tumors regrew once treatment stopped (Fig. 2A). To determine if regrown tumors were sensitive to AC, tumor-bearing mice were treated with a single dose of AC, tumors were allowed to regrow to the starting tumor volume, and AC was re-administered to the mice. Subsequent dosing elicited repeated, albeit reduced, tumor shrinkage after each AC cycle (Fig. 2B–C). Treatment with paclitaxel, a common second-line NACT agent, did not elicit responses in either the naïve or post-AC settings (fig. S1H), so it was not included in subsequent NACT modeling experiments. Together, these results demonstrated that AC treatment was unable to eradicate tumor cells and once treatment was halted, surviving cells gave rise to tumor cells that were again sensitive to AC.
Figure 2. Residual and regrown tumors cannot be eliminated by continued chemotherapy treatment.
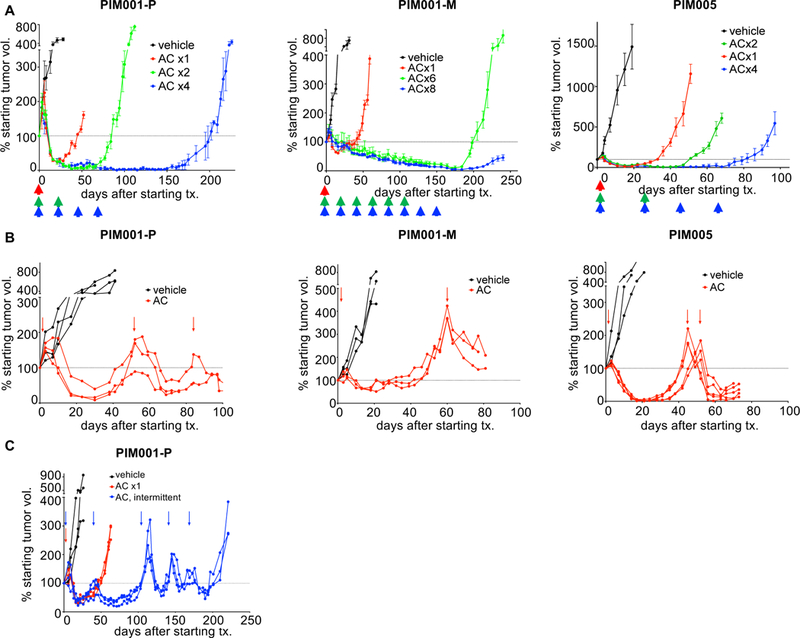
A. To model the schedule of AC treatments administered to patients, AC was administered to mice in regular 21-day intervals (arrows). To enable prolonged dosing without toxic side effects, we used NRG mice for these long-term treatment studies. The horizontal dotted line denotes 100% of the starting tumor volume (measured on day 0). Data shown are mean +/− SEM (n=4 per group).
B. NOD/SCID mice were treated with AC on day 0 and were only re-dosed with AC when tumors regrew to the starting tumor size (arrows). Data shown are mean +/− SEM.
C. NRG mice bearing PIM001-P tumors were treated with a total of 5 cycles of AC, and each subsequent dose was administered only when tumors re-grew to the starting tumor size (arrows). Data shown are mean +/− SEM.
Residual tumors adopt distinct histological features and are not enriched for cells with stem-like properties
Histologic features of residual tumors after AC treatment were distinct from those of vehicle-treated and regrown PIM001-P, PIM001-M, and PIM005 tumors (Fig. 3A). Residual tumors showed desmoplasia characterized by increased stromal component and tumor cell pleomorphism, including irregular nuclear and cytoplasmic size and shape. In residual tumors, alpha-smooth muscle actin (α-SMA)-positive fibroblasts and collagen replaced epithelial membrane antigen (EMA)-positive tumor cells (fig. S2). In contrast, the histologic features of regrown tumors closely resembled those of vehicle-treated tumors, demonstrating the reversibility of the phenotype (Fig. 3 and fig. S2). In addition, subpopulations of residual tumor cells stained positive for Ki67 or phosphorylated histone H3 (pHH3), indicating that they were cycling (fig. S3).
Figure 3. Residual tumors adopt a distinct histologic state that is reverted in regrown tumors.

A. Replicate FFPE tumor samples were assembled into TMAs (triplicate 1 mm punches), and stained with H&E.
B. FFPE primary tumor samples obtained from a TNBC patient (ART-57) before, during, and after completion of NACT were stained with H&E and imaged. An image of her metastatic relapse to the chest wall is shown in the bottom panel. Chemotherapy effects on fibrosis and tumor cell morphology are shown with arrows. PaCT, panitumumab + carboplatin + paclitaxel (Taxol). RCB, residual cancer burden assessed by examination of the surgical biopsy. No tx, no treatment was administered between surgery and metastatic relapse. Volumetric reduction after AC treatment was assessed by ultrasound.
Scale bars are 200 µm for parts A&B.
Rare populations of CD44high/CD24low and ganglioside GD2-positive cells were detected in vehicle-treated and residual tumor cells that had been depleted of mouse stroma (fig. S4), and AC treatment did not significantly enrich for these cell populations (fig. S5A–B). Residual tumors were not enriched for mammosphere-forming capabilities in vitro (fig. S5C) or tumor-initiating capabilities in vivo, as assessed by a limiting dilution transplantation assay (fig. S5D). Finally, residual tumors exhibited a reduction in vimentin-positive cells (fig. S6A–B) and a reduced EMT pathway activation score (32) (fig. S6C) relative to vehicle-treated tumors. These results demonstrated that AC treatment did not significantly enrich for cells with stem-like or mesenchymal properties.
Histologic changes associated with AC treatment are similar in PDXs and patient tumors
To determine if the therapy-induced changes observed in PDX tumors were similar to those observed in patient tumors, we examined serial biopsies from TNBC patients enrolled on an ongoing neoadjuvant clinical trial (ARTEMIS) at MDACC (31). Tumor tissue was obtained from five patients with treatment-naïve TNBC before treatment, after four cycles of neoadjuvant chemotherapy consisting of AC, and in two cases (ART-6 and ART-57) also after a three-month course of targeted therapy (atezolizumab + paclitaxel, and panitumumab + carboplatin + paclitaxel, respectively). ART-57 developed a chest wall metastasis, which was also evaluated. Four (ART-11, −14, −57, and −119) out of five biopsies obtained after AC treatment exhibited similar histologic changes to those observed in residual PDX tumors (Fig. 3B and fig. S7). Fibrosis and a marked reduction in tumor cellularity were evident in the ART-57 mid-treatment biopsy, and the post-treatment surgical biopsy exhibited marked chemotherapy effects on cytologic appearance, including irregular nuclear shape and size and loss of chromatin detail (Fig. 3B). Moreover, the patient’s ipsilateral chest wall metastasis that arose 11 months after surgery in the absence of continued treatment had a similar appearance to the pre-treatment primary tumor biopsy, indicating that chemotherapy-induced histologic changes were reversible (Fig 3B). Similarly, ART-11, −14, and −119 exhibited an overall reduction in tumor cellularity and altered tumor cell morphology after AC, including giant-cell cytology, squamoid cytoplasms, and altered cell shape, respectively (fig. S7). ART-14 and −119 also exhibited increased fibrous stroma after AC treatment. These histologic changes are consistent with those observed in human breast tumors after treatment and are associated with partial response to AC and poor overall survival (33). One sample, ART-6, exhibited primary resistance characterized by continued tumor growth in the presence of AC and negligible histologic changes throughout NACT (fig. S7). Together, these results indicated that PDXs recapitulate key histologic changes associated with chemotherapy treatment in tumors of patients with TNBC.
Transcriptomes of vehicle-treated and regrown tumors are highly similar but distinct from those of residual tumors
In order to identify gene expression changes that accompany AC treatment, whole-transcriptome sequencing (RNA-seq) was performed on vehicle-treated, residual, and regrown tumors (data file S2). Vehicle-treated and regrown tumors were size-matched. RNA-seq data were computationally purified of mouse sequences (34), and gene expression alterations in the human tumor cell compartment were determined by differential expression analysis (data file S2). Notably, the transcriptomes of vehicle-treated and regrown tumors were highly similar (Fig. 4A–B and fig. S8A). Thus, most gene expression changes observed in residual tumors reverted as the tumors regrew after AC treatment. Residual tumors exhibited hundreds of alterations in gene expression compared to vehicle-treated and regrown tumors. A total of 97 genes were deregulated in residual tumors across all three PDX models, and the 54 genes altered compared to vehicle-treated tumors fell into pathways involving the cytoskeleton and development (Fig. 4C). Pathway analysis of genes that were deregulated in residual tumors compared to vehicle-treated tumors revealed consistent alterations in cell-cell adhesion, extracellular matrix remodeling, and inflammatory pathways across all three PDX models (fig. S8B). Gene Set Enrichment Analysis (GSEA) of hallmark pathways in residual tumor signatures revealed that although each PDX had a distinct hallmark activation profile, several pathways, including glycolysis, MYC signaling, p53 signaling, heme metabolism, mTORC1 signaling, EMT, and hypoxia, were altered in residual tumors of multiple PDX models (fig. S9).
Figure 4. Shifts in the transcriptome of residual tumors are reversible.
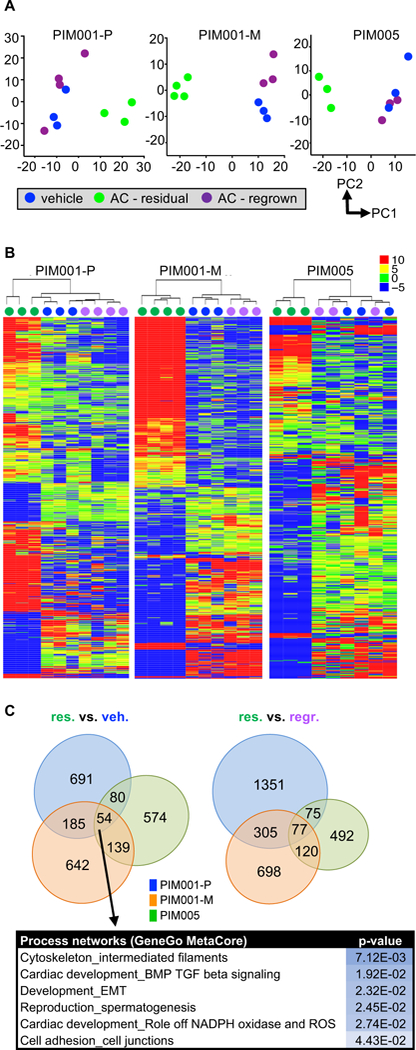
Vehicle (blue; day 0), residual (green; AC-treated day 21), and regrown (purple; AC-treated day 50) tumors were subjected to RNAseq.
A. RNA-seq data were analyzed by principal component analysis, and the first two principal components (PC1&2) are plotted for each PDX model. Principal components were calculated using log2-transformed TPM values for the 500 genes with the highest variance between samples, considering only genes with at least 20 reads in at least one sample. The mean was set as zero.
B. Within each PDX model, genes significantly altered (log2FC≥0.5, FDR<0.05, Benjamini-Hochberg test, sum of TPMs across all samples ≥100) in any pairwise comparison (vehicle-vs-regrown, residual-vs-vehicle, residual-vs-regrown) are displayed in a heat map organized by hierarchical clustering. The color scale refers to TPMs.
C. Genes significantly differentially expressed, as defined in B, in residual tumors compared to vehicle-treated tumors were compared between three PDX models. The list includes significantly altered process networks (GeneGo Metacore) regulated by the 54 genes significantly differentially expressed in residual tumors compared to vehicle-treated tumors across all three PDX models.
Next, patient gene expression data were mined to determine if the expression changes induced by AC treatment of PDX tumors were similar to those reported for patient tumors after NACT. Analysis of tumors from 89 TNBC patients post-NACT revealed that subsets of tumors have activation of MEK and/or TGF-β signaling (8). MEK pathway activation was found to be modulated in both PIM001-M and PIM005 when the same MEK (35) and TGF-β (36) scoring system was applied to the expression data from PDX residual tumors (fig. S10A). PDX expression data were also evaluated in the context of microarray data from 39 matched pre- and post-NACT breast tumor pairs collected in the I-SPY-1 trial (23). Post-NACT surgical biopsies clustered separately from pre-treatment biopsies, and seven pathways were altered in residual tumors (fig. S10B). Several of these pathways, including glycolysis, p53 signaling, heme metabolism, and mTORC1 signaling, were also deregulated in residual tumors of multiple PDX models (fig. S9). Thus, the reversible gene expression alterations observed in residual PDX tumors shared similarities with gene expression changes observed in residual tumors of chemotherapy-treated breast cancer patients.
Proteomes of residual tumors differ from those of vehicle-treated and regrown tumors
Vehicle-treated, residual, and regrown tumors were subjected to reverse-phase protein array (RPPA) to identify alterations in protein amounts and activation states that accompany a cycle of NACT treatment (data file S3). Whereas numerous proteins were differentially expressed and/ or activated in residual tumors relative to vehicle-treated and regrown tumors, there were no significant differences between vehicle-treated and regrown tumors (fig. S11A–B). Several proteins were enriched in residual tumors relative to matched vehicle-treated tumors across multiple PDX models, including those involved in cytoskeleton structure and activated fibroblasts (HSP27-pS82, Myosin-11, Caveolin-1, Collagen-VI, PDGFR-β, fibronectin), unfolded protein response (XBP1), fatty acid metabolism (ACC1), and interferon signaling (IRF1) (fig. S11C). Many of these cellular processes were altered in residual tumors at the transcriptomic level as well (Fig. 4C and fig. S8B). It is important to note that stromal infiltrates and desmoplasia contributed to the distinct proteome identified in residual tumors, because the majority of antibodies on the RPPA panel do not distinguish between human and mouse proteins. For example, IHC confirmed that FASN and ACC1-pS79 were increased in tumor cells, whereas fibronectin and PDGFR-β were increased in the stromal component of residual PIM001-P tumors (fig. S11D). Together, these results revealed that residual tumors entered a transient phenotypic state characterized by both tumor cell and stromal cell alterations, which was reversed after discontinuation of AC.
Barcode-mediated clonal tracking during a cycle of NACT in vivo reveals lack of clonal selection in residual tumors
We next sought to identify the population(s) of pre-treatment tumor clones that conferred tumor cell survival and regrowth after AC treatment. High complexity barcode-mediated clonal tracking was used to quantitatively track clonal populations of PDX tumor cells (Fig. 5A). Tumors from the three PDX models were harvested, dissociated into single cells, depleted of mouse stroma (fig. S4), and transduced with a pooled lentiviral library of more than 50 million unique DNA barcodes at a low multiplicity of infection (MOI) to ensure that the majority of transduced cells received a single viral integrant (one unique barcode). To eliminate non-transduced cells, cells were cultured as spheroids and exposed to brief drug selection (fig. S12A). Next, barcoded tumor cells were engrafted into the MFPs of recipient mice, and a reference cell pellet was frozen (Fig. 5A and fig. S12B). To avoid high rates of necrosis that typically occur in large tumors, we compared size-matched treatment-naive and regrown tumors after AC treatment. When barcoded tumors reached 150 mm3, mice were euthanized and their untreated MFP tumors were collected. The remaining barcoded mice were treated with AC, and mice were euthanized and tumors collected at the tumor volume nadir 21 days after AC treatment (residual tumors) as well as when tumors regrew to 150 mm3 (regrown tumors) (Fig. 5A). Genomic DNA was extracted, and genome-integrated barcodes were quantified by Illumina next-generation sequencing.
Figure 5. Residual tumors maintain the clonal architecture and genomic complexity of pre-treatment tumors.
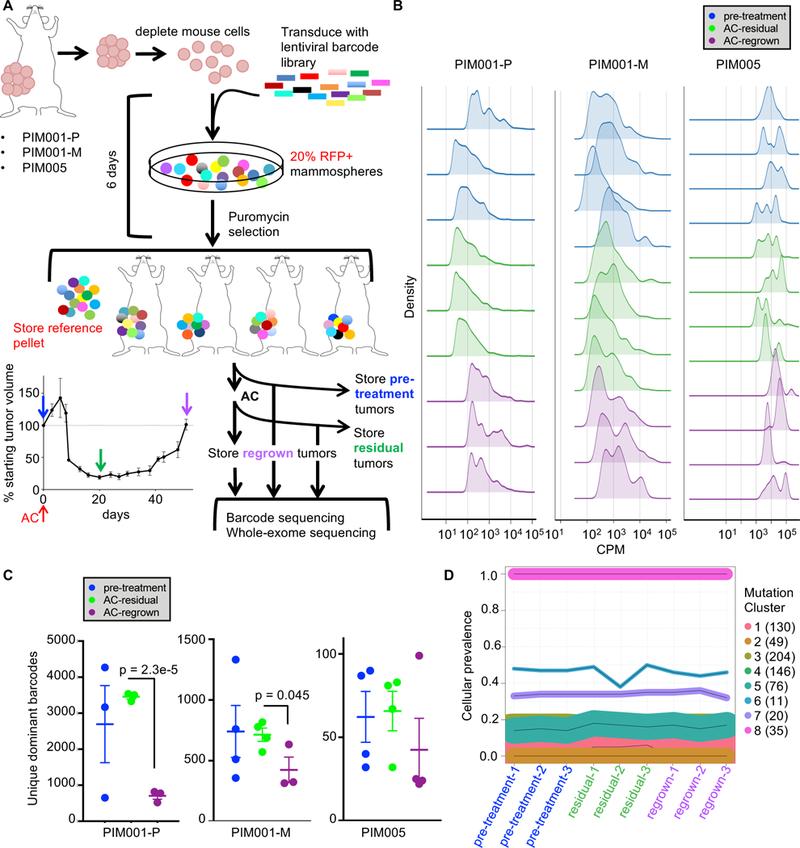
A. Lentiviral barcodes were introduced into freshly dissociated tumor cells from three PDX models, then after brief ex vivo culture engrafted into the MFPs of NOD/SCID mice. DNA extracted from tumors was subjected to high-throughput barcode sequencing.
B. Density plots show the overall distribution of the top 95% most frequent barcodes in each sample. CPM, counts per million.
C. The top 95% most abundant barcodes were quantified in each sample, thus excluding barcodes detected at extremely low frequencies (two-tailed T-tests comparing residual to regrown). Data shown are mean +/− SEM.
D. Line plots of estimated cellular prevalence of mutation clusters in PIM001-P as modeled by PyClone analysis of WES data are shown. Each line represents a mutation cluster, and the thickness of the line is proportional to the number of mutations within that cluster. The number of mutations comprising each cluster is shown in parentheses.
We observed high concordance of barcodes detected in technical replicate library preparations, but due to the high complexity of the library and low MOI used, there was no correlation of the barcodes detected between replicate mice (fig. S12C), so downstream analyses focused on comparisons of barcode quantities and distribution patterns between mice. The high complexity of the barcode library enabled detection of prevalent as well as extremely rare barcoded lineages in each sample. In PIM001-P, an average of 240,322 unique barcodes was observed in the pre-implantation reference cell pellet, whereas an average of 24,442 unique barcodes was observed in vehicle-treated tumors. This corresponded to a tumor-initiating cell (TIC) frequency of 10.17% (fig. S12B&D). The total quantity of unique clones was not significantly altered in any of the AC-treated PDX tumor samples (fig. S12D). Thus, although AC treatment resulted in substantially reduced tumor cell number, tens of thousands of uniquely barcoded tumor cells persisted after AC treatment.
Because the majority of barcodes were maintained in very low quantities, we focused downstream analyses on dominant clones (defined as uniquely barcoded lineages) that substantially contributed to the tumor bulk by quantifying the top 95% most abundant barcodes in each sample. AC-treated tumors harvested in both the residual and regrown states revealed maintenance of both rare and high-abundance clones (Fig. 5B). Notably, the number of dominant barcodes, as well as the Shannon Diversity index, remained unchanged between residual tumors and pre-treated tumors (Fig. 5B–C and fig. S12E). The lack of barcode enrichment in residual tumors demonstrated that all pre-treated tumor clones had similar fitness for surviving AC treatment. By contrast, regrown tumors tended to harbor fewer dominant barcodes and a lower Shannon Diversity Index relative to pre-treated and residual tumors (Fig. 5B–C and fig. S12E). Together, these results demonstrated that AC treatment induced non-selective killing of tumor clones and that residual tumors cell could experience a selective bottleneck as they regrew. This selective regrowth of tumor subclones could be due to inherent properties, genomic and/or phenotypic, harbored within these clones, or due to stochastic outgrowth of tumor cell subpopulations from the residual state.
Clonal architecture is conserved throughout a cycle of AC treatment
Whole-exome sequencing (WES) was conducted to determine if reproducible alterations in genomic architecture occurred in replicate mice after AC treatment (data files S4–S6). After subtraction of mouse sequences and mutations present in the germline of the patient whose tumor was used to generate PIM001-P, the mutant allele frequency (MAF) of each somatic non-silent mutation was calculated in pre-treated, residual, and regrown tumors (fig. S13A). The spectrum of MAFs was highly correlated between replicate tumors within each treatment group, indicating that the architecture of genomic subclones was stably transplantable and reproducible between mice (fig. S13B). Significant alterations in MAFs (|∆MAF|≥0.05) were not observed between the three treatment groups (fig. S13C–D). All mutations present in pre-treated tumors were also detected in residual and regrown tumors, and de novo mutations were not detected after AC. Similarly, cancer cell frequencies estimated using PyClone (37) to normalize for copy number status (data file S6) and tumor purity were consistent between treatment groups, revealing stable subclonal architecture throughout a cycle of AC treatment (Fig. 5D and fig. S14). These results indicated that genomic alterations did not account for the survival and repopulation capacity of residual tumor cells after AC. Furthermore, the barcoded subclones that became enriched in regrown tumors recapitulated the genomic, transcriptomic, proteomic, and histologic features of the treatment-naïve state, suggesting that the selective bottleneck in regrown tumors did not functionally impact the tumor cell population.
Genomic architecture is maintained in patient TNBCs after AC.
Next, we analyzed the first WES data available from the breast tumors of two patients with treatment-naïve TNBC (ART-57 and ART-6) enrolled on an ongoing clinical trial (ARTEMIS(31)) at MD Anderson Cancer Center (data files S4, S7, and S8). ART-57 had RCB-II at the time of surgery, whereas ART-6 had RCB-III (Fig. 6A). We compared biopsies obtained before treatment and after four cycles of AC (mid-treatment, Fig. 6A) using PyClone (37) to estimate subclone prevalence after normalization for tumor purity and local copy number status. This analysis did not identify subclone enrichment or acquisition of new mutations after AC treatment in the tumors of either patient (Fig. 6, fig. S15, and data file S7). These results are consistent with findings made in our PDX models. By contrast, surgical biopsies obtained from these patients after they underwent additional combination therapies that included chemotherapy, targeted therapy, or immune therapy (atezolizumab + paclitaxel for ART-6 and panitumumab + carboplatin + paclitaxel for ART-57) revealed subclone expansion and evolution of likely de novo mutations (Fig. 6 and fig. S15).
Figure 6. Subclone analysis of serially biopsied human TNBCs reveals lack of subclone enrichment after AC.

A. Serial biopsies from two TNBC patients were analyzed by WES. The tumors’ volumetric changes in response to four cycles of AC treatment are indicated. PDT, atezolizumab + Abraxane. PaCT, panitumumab + carboplatin + paclitaxel (Taxol).
B. Line plots of estimated cellular prevalence of mutation clusters modeled by PyClone are shown. Each line represents a mutation cluster, and the thickness of the line is proportional to the number of mutations within the cluster. The number of mutations comprising each cluster is shown in parentheses.
C. These plots display the prevalence of subclones throughout treatment. Subclonal architecture was reconstructed based on PyClone results.
Residual tumors exhibit a dependency on mitochondrial oxidative phosphorylation
Due to the adaptive nature of chemoresistance observed during a cycle of NACT, we investigated phenotypic features present in the residual tumor state that could serve as potential therapeutic targets. GSEA of RNA-seq data revealed that glycolysis was down-regulated in residual tumors of all three PDX models (fig. S9) as well as in post-NACT residual tumors of a cohort of 39 patients (23) with breast cancer (fig. S10B). This reduction in glycolysis after NACT suggested that oxidative phosphorylation could serve as a compensatory metabolic pathway in residual tumors. Notably, mitochondrial oxidative phosphorylation was the most significantly up-regulated pathway in residual PIM001-P tumors compared to vehicle-treated PIM001-P tumors. To determine if residual tumors exhibited increased oxidative phosphorylation and/ or reduced glycolysis, we measured oxygen consumption rate (OCR; a readout for oxidative phosphorylation) and extracellular acidification rate (ECAR; a readout for glycolysis) in PIM001-P tumor cells that had been isolated from untreated and AC-treated residual tumors and depleted of mouse stroma. Residual tumor cells exhibited higher OCR and lower ECAR than untreated tumors (fig. S16A). To test the dependency of residual tumors on mitochondrial respiration, we conducted preclinical trials with IACS-010759, an inhibitor of the mitochondrial electron transport chain complex I (38). As a single agent, IACS-010759 delayed tumor growth compared with vehicle, although it was not as effective as AC treatment (Fig. 7). Moreover, IACS-010759 delayed the regrowth of AC-treated residual tumors in all three PDX models (Fig. 7).
Figure 7. The residual tumor state is targetable by inhibition of oxidative phosphorylation.
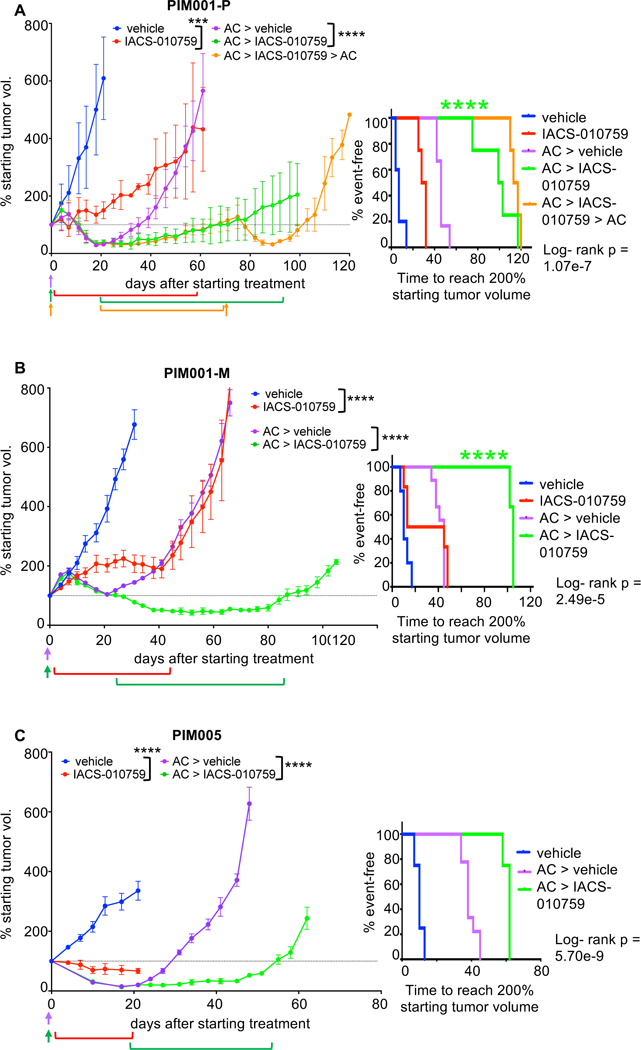
A. NOD/SCID mice bearing PIM001-P tumors were treated with an inhibitor of oxidative phosphorylation (IACS-010759, per os [p.o.], quaque die [q.d.]) or vehicle in the treatment-naïve or in the residual setting after AC treatment (> in the figure indicates sequential treatments). Days of IACS-010759 treatment are indicated by brackets. Days of AC treatment are indicated by arrows. ***ANOVA p-value <0.001 (day 21), ****ANOVA p-value <0.0001 (day 61). Data shown are mean +/− SEM (n=4–6 per group). The right panel is a Km curve of the time for each mouse’s tumor to reach 200% of the starting tumor volume (measured on day 0), and the log-rank p-value is shown. Testing for interaction of treatment effects using a hazards model (data file S9) shows synergy in the AC + IACS-010759 sequential combination (****p<0.0001).
B. As above, mice bearing PIM001-M tumors were treated with the indicated agents. ****ANOVA p-value <0.0001 (day 31 & day 66). Data shown are mean +/− SEM (n=3–9 per group). Testing for interaction of treatment effects using a hazards model (data file S9) shows synergy in the AC + IACS-010759 sequential combination (****p<0.0001).
C. As above, mice bearing PIM005 tumors were treated with the indicated agents. ****ANOVA p-value <0.0001 (day 21 & day 48). Data shown are mean +/− SEM (n=4–8 per group).
Using the Additive Hazards Model (39), the combination of AC and IACS-010759 was found to be synergistic, rather than additive, when IACS-010759 treatment followed AC treatment (Fig. 7 and data file S9). Of note, PIM001-P residual tumors that became resistant to IACS-010759 remained sensitive to AC (Fig. 7A). A reduction in hypoxia was observed in tumors treated with IACS-010759, confirming that the drug was bioavailable and engaged its target (fig. S16B). These results demonstrated that the efficacy of IACS-010759 was enhanced in the post-AC residual setting compared with the treatment-naïve setting and suggests that a sequential regimen consisting of AC followed by IACS-010759 could prolong duration of responses to chemotherapy in TNBCs.
Due to the dynamic nature of the residual tumor state, we sought to determine whether epigenetic mechanisms contribute to AC resistance. Using residual tumor gene expression signatures, we used the Virtual Inference of Protein-activity by Enriched Regulon (VIPER) algorithm (40) to identify differentially activated epigenetic regulatory proteins in residual PDX tumors compared to vehicle-treated tumors. This analysis revealed that the activity of numerous epigenetic regulatory proteins is predicted to be altered in residual tumors (data file S10). HDAC7, HDAC10, and SIRT7 were the epigenetic regulators predicted to have the highest up-regulation of activity across the three PDX models. Regulators such as these are promising contributors to the residual tumor state, and dual targeting of both metabolic and epigenetic programs may provide durable responses in TNBC.
DISCUSSION
Elucidating the molecular basis of chemoresistance in TNBC and identifying functional vulnerabilities of chemoresistant tumor cells is a critical unmet clinical need requiring the development of models that faithfully recapitulate the human disease and its responses to therapy. In this study, PDX models of treatment-naïve TNBC were generated and subjected to AC treatment, recapitulating molecular features observed in the tumors of TNBC patients undergoing NACT. Although TNBC is a disease with extensive inter-patient heterogeneity, and it is widely assumed that treatment regimens will eventually need to be tailored to the unique properties of an individual patient’s tumor, there may be common properties of the residual tumor state that are targetable across patients, including a dependency on oxidative phosphorylation, for example.
A major goal of this study was to characterize residual tumors that survived AC to determine if chemotherapy resistance was due to intrinsic (genomic) and/or acquired (non-genomic) mechanisms, and to determine if resistant tumors had specific vulnerabilities that could be targeted therapeutically. We found that adaptive mechanisms accounted for AC resistance in all three PDX models tested. Our data revealed that all tumor subclones were equally capable of adopting a drug-tolerant state characterized by distinct histologic features, transcriptomes, and proteomes. Reversibility was a key feature of this drug-tolerant state, and the histologic, genomic, and transcriptomic features of pre-treated tumors were restored when residual tumor cells repopulated the tumors after treatment. Barcoding revealed that not all clones were equally capable of repopulating the tumor once treatment stopped. Although this regrowth was not accompanied by genomic selection, phenotypic properties not rooted in genomic features may have endowed a subset of clones with the capacity to preferentially re-populate the tumor.
Features of the residual state documented in our study that recapitulate observations made in tumors of TNBC patients undergoing NACT include (A) lack of enrichment in cells with stem-like or tumor-initiating properties (9); (B) chemotherapy-induced histologic features that are reversible after treatment (also in (33)); (C) transcriptomic alterations similar to those identified in a cohort of residual TNBCs after NACT(8) as well as signatures generated from matched pre- and post-NACT TNBC biopsies(10, 23); and (D) lack of clonal selection consistent with findings in TNBC patients identified in our study as well as in subsets of patients in other studies using in situ analysis (9) and next-generation sequencing analyses (5, 10) of matched pre- and post-NACT TNBC biopsies. Finally, clinical studies demonstrating that chemotherapy re-challenge of breast cancers after disease progression elicits repeated responses in some patients (41, 42) are consistent with the regained chemo-sensitivity in regrown tumors observed in our AC-treated PDX models. A recent study using single-cell sequencing analyzed clonal architecture in matched pre- and post-NACT biopsies from 4 TNBC patients who harbored residual disease after NACT (10). Although the NACT regimen in this study (up-front combination of anthracycline with a taxane followed by a combination of VEGF inhibitor and chemotherapy) is not directly comparable to ours, three of these four patients exhibited shifts in clonal architecture, whereas one patient exhibited maintenance of clonal architecture after NACT, indicating that clonal architecture can remain stable or change during NACT treatment of TNBC. Thus, the PDX models reported in this study provide an opportunity to study the drug-tolerant state and adaptive mechanisms of AC resistance. It is also clear that clonal selection can accompany chemoresistance in the setting of treatment-naïve TNBC in subsets of patients (5, 10) which should be characterized in the appropriate PDX models.
Transcriptomic analyses revealed that mitochondrial oxidative phosphorylation was the most significantly up-regulated pathway in residual PIM001-P tumors and that glycolysis was down-regulated in residual tumors across three PDX models. Furthermore, our analysis of residual tumors of patients with breast cancer (23) revealed down-regulation of glycolysis, and oxidative phosphorylation is up-regulated in TNBC cell lines harboring MYC and MCL1 amplifications, which are frequently observed in chemoresistant TNBCs (43). Although alterations in MYC and MCL1 copy numbers, expression, and amounts of protein were not observed in residual PDX tumors relative to their untreated counterparts, we demonstrated that IACS-010759 (38), an inhibitor of mitochondrial oxidative phosphorylation, delayed the regrowth of AC-treated residual tumors, and additive hazards modeling revealed synergy when IACS-010759 followed AC treatment. Increased oxidative phosphorylation can contribute to therapy resistance in chronic myeloid leukemia, colon cancer, and prostate cancer cells (44, 45), as well as in models of residual pancreatic cancer (46). In addition, up-regulated OXPHOS contributes to cancer stem-like cell phenotypes associated with chemoresistance in breast cancer cells (43). Given that AC-resistant residual tumors maintained the ITH of untreated tumors, it is not unexpected that IACS-010759 as a single targeted agent would be insufficient to eradicate all residual tumor cells. Several mechanisms could account for the incomplete response of tumors to IACS-010759 and the eventual regrowth of tumor cells in its presence, including outgrowth of tumor subclones not dependent on oxidative phosphorylation, activation of compensatory metabolic programs that bypass the requirement for oxidative phosphorylation, or clonal selection of tumor cells with acquired mutations in mitochondria complex I, the drug target. As preclinical studies identified well-tolerated doses of IACS-010759, it is currently in a Phase I clinical dose-escalation study (NCT03291938) for patients with acute myeloid leukemia and solid malignancies, including breast cancer. Combination of IACS-010759 with additional chemotherapies and/ or targeted therapies will necessitate careful management of toxicity profiles. Our data suggest that sequential administration of AC followed by IACS-010759 could prolong TNBC responses in the neoadjuvant setting and delay time to disease recurrence.
Given that tumor-infiltrating lymphocytes have been shown to impact how tumors respond to NACT (22, 23), a limitation of our study is the use of mice lacking an intact immune system. In addition, the results obtained with AC may not be generalizable to other NACT regimens, for example those that also include taxanes. Finally, breast cancer patients undergo surgery after NACT to remove any residual disease at the primary site, and tumor regrowth generally occurs in metastases that have seeded before or during therapy. It will be important to assess whether metastatic lesions also adopt a drug-tolerant state during therapy because this is expected to impact therapeutic decisions moving forward.
MATERIALS AND METHODS
Study design
Endpoints for animal experiments were selected in accordance with IACUC-approved criteria. Statistical analyses were conducted when applicable and are included in figure legends. The objective of this study was to identify molecular alterations that occur upon AC treatment of PDX tumors. We performed next-generation sequencing, monitored tumor behavior in mice, and conducted targeted therapy treatments of PDX models in this study. For each animal experiment, mice were randomized based on tumor volume. Investigators were blinded to treatment groups when monitoring tumor volumes and health status of mice. The number of replicates included in each experiment is indicated in each figure legend displaying summary data (otherwise, individual data points for each replicate are displayed in figures).
Study approval
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (NIH) Institutional Animal Care and Use Committee (IACUC). The protocol was approved by the IACUC at MD Anderson Cancer Center (protocol 0000978-RN01). Mice were euthanized when they became moribund or when they reached defined study end points. Animals were euthanized as dictated by the Association for Assessment and Accreditation of Laboratory Animal Care International and IACUC euthanasia endpoints. Informed consent was obtained from all human participants, and all relevant ethical regulations were followed as approved by the Institutional Review Board (IRB) ant MD Anderson. Patient biopsies for PDX generation, histologic analysis, and next-generation sequencing were obtained through approved IRB protocols at the University of Texas M.D. Anderson Cancer Center (protocols 2011–0007 and 2014–0185).
Patient samples for histologic and genomic analyses
For histologic analyses, patient biopsies were fixed in formalin, then embedded in paraffin blocks. Sections were then stained with hematoxylin and eosin. Biopsies for WES were immediately placed into RNA-later (Thermo Fisher Scientific) and stored at −80°C until DNA was extracted.
Generation of PDX models
PDX models were established (30), and details of each PDX line are outlined in data file S1. Briefly, the fourth mammary fat pads (MFPs) of 3- to 5-week-old NOD/SCID mice (NOD.CB17-Prkdcscid/NcrCrl, Charles River, NCI Colony) were pre-humanized with GFP-labeled immortalized human mammary stromal fibroblasts 3–4 weeks before tumor cell engraftment. Fine needle aspirates (FNA) were obtained from the breast tumors of patients diagnosed with TNBC who had not yet received any therapeutic intervention. Tumor cells were maintained on ice and brought to the laboratory within one hour. Cells were pelleted by centrifugation at 800 g, washed with DME:F12 supplemented with 5% bovine calf serum (BCS), and resuspended in red blood cell lysing buffer (Sigma R7757). Cells were filtered through a 70–100 µm sterile filter. Tumor cells were mixed with EG fibroblasts (data file S1) and 1/3 volume Matrigel (Corning) and injected into pre-humanized MFPs. Cells in Matrigel were maintained on ice until engraftment.
When tumors reached approximately 1000 mm3, they were harvested and dissociated into single cells and organoids by mechanical mincing followed by digestion with 3 mg/mL collagenase (Roche) and 0.6 mg/mL hyaluronidase (Sigma) supplemented with 1.3% bovine serum albumin (BSA) (Sigma) in DME:F12 medium containing antibiotics (100 units/mL penicillin, 100 µg/mL streptomycin, 0.25 µg/mL amphotericin B). Tumor digests were incubated on a rotating platform for 4 hours at 37°C. Digested PDX tumor cells were processed as described above. One million viable tumor cells were resuspended in 50% volume Matrigel and injected into non-pre-humanized MFPs of NOD/SCID mice.
Chemotherapy treatment of PDX models
All chemotherapy treatment studies of PDX models used mice between passages 3–8. For studies involving treatment of tumor-bearing mice with one dose of AC, NOD/SCID mice (NOD.CB17-Prkdcscid/NcrCrl, Charles River, NCI Colony) were used. For studies involving two or more doses of AC, NRG mice (Nod.Cg-Rag1tm1Mom/IL2rgtm1wjl/SzJ, The Jackson Laboratory) were used. No differences in tumor take rate, growth rate, or histologic characteristics were observed between NOD/SCID and NRG mice.
Adriamycin (‘A’, doxorubicin, ChemiTek) powder was solubilized in sterile water for injection immediately before administration to mice. Cyclophosphamide (‘C’, Baxter) was purchased from the MDACC pharmacy, and powder was solubilized in sterile water for injection immediately before administration to mice. The solutions were protected from light and were made fresh before injecting each cohort of animals. Each solution was administered by intraperitoneal (i.p.) injection separately at a dose volume of 5–10 ml/kg. Paclitaxel (ChemieTek) powder was solubilized in a sterile solution of 50% ethanol + 50% Kolliphor (Sigma-Aldrich) at a concentration of 20 mg/ml. This solution was diluted 1:20 in sterile saline solution immediately before i.p. injection of mice at a dose volume of 10 ml/kg (corresponding to a 10 mg/kg dose).
To identify a dose of AC that would be tolerated in PDX models and that would allow for long-term survival and analysis of resistance patterns, we first tested a dose of AC commonly used in the published literature for short-term studies (47, 48), 100 mg/kg C + 2 mg/kg A to treat non-tumor-bearing NOD/SCID mice aged 10 weeks (to match the approximate age of mice bearing 150 mm3 tumors). Due to observed toxicities, we conducted dose reduction studies (fig. S1) to identify a dose that would allow mice to maintain health and overall condition for at least 60 days (a time sufficient to evaluate the overall response to AC). Overall health and body weight were monitored 2–3 times weekly. Animals were euthanized if they were in moribund condition or lost greater than 20% of their starting body weight. Chemotherapy-induced toxicity was monitored by analyzing blood cell counts and concentrations of serum liver enzymes at the MDACC Research Animal Support Facility (RASF). Serial retro-orbital bleeding of mice was conducted to obtain 50 µl of blood, which was centrifuged at 600 g for 5 minutes. Serum was submitted to the RASF for clinical chemistry analysis on the Roche Integra 400+. Serum concentrations of AST (aspartate aminotransferase) and ALT (alanine aminotransferase) were measured according to the International Federation of Clinical Chemistry, but without pyridoxal-5’-phosphate. Serum AP (alkaline phosphatase) was quantified using a standard colorimetric assay measuring absorbance at 409 nm. Terminal total blood draws were conducted to obtain 200 µl of blood to submit to the RASF for white blood cell counts using the System 120 Siemens ADVIA Hematology instrument. This instrument was also used to measure hematocrit (the ratio of red blood cells to total blood volume) to monitor anemia induced by chemotherapy.
Dose reduction studies revealed that up to two cycles of 50 mg/kg C + 0.5 mg/kg A would allow mice to maintain good health for up to 65 days after initiation of dosing in NOD/SCID mice. Therefore, all AC treatments of NOD/SCID mice involved one or two cycles of 50 mg/kg C + 0.5 mg/kg A. Treatment of PIM001-P tumor-bearing mice with this dose elicited as much reduction in tumor size as did higher doses of AC (fig. S1G). For long-term treatment studies using more than two doses of AC, NRG mice were dosed with 50 mg/kg C + 0.5 mg/kg A (see figure legends for dose frequency). NRG mice tolerated up to 8 cycles of this dose concentration.
Barcode-mediated clonal tracking throughout chemotherapy treatment
Tumors were barcoded to enable high-complexity lineage tracing. We first confirmed whether PIM001-P tumor cells would maintain viability ex vivo for the time required for viral transduction and antibiotic selection. After mouse cell depletion by magnetic activated cell sorting (Miltenyi mouse cell depletion kit), viable human tumor cells were plated in mammosphere conditions (MammoCult, StemCell Technologies) in ultra-low-attachment 96-well plates (Corning Costar) at a density of 1000 cells/well. Viability was monitored by Cell-Titer-Glo (Promega) luminescence assays according to the manufacturer’s protocol and imaged using a ClarioStar luminescence plate reader (BMC Lab Tech). We observed that cells maintained viability for approximately 2 weeks after tumor digestion, but that viability began decreasing after 6 days in mammosphere conditions.
PDX tumors were harvested, pooled, dissociated into single cells, and depleted of mouse stroma by magnetic-activated cell sorting according to the manufacturer’s protocol. Depletion of mouse cells was validated by flow cytometry analysis of cells stained with an antibody against mouse-specific MHC class I (anti-H2kd clone SF1–1.1, BioLegend). After mouse cell depletion, 30 million viable human tumor cells were plated in mammosphere conditions (MammoCult, StemCell Technologies) in ultra-low-attachment plates (Corning Costar) at a density of 1.25×106 cells/ml. Immediately after plating, the pooled lentiviral barcode library was added to cells (Cellecta, CellTracker 50M packaged lentiviral barcode library, Cat. # BC13X13–30M-V) at an MOI of 0.2 to ensure that each infected cell received only one single barcode, along with 10 µg/ml polybrene (Sigma-Aldrich). Cells were maintained at 5% CO2. Medium was replaced with fresh MammoCult 24 hours after addition of lentivirus. Cells were maintained in mammosphere conditions for an additional 48 hours. At that time (36 hours after tumor digestion), medium was refreshed, and puromycin was added to a final concentration of 2.5 µg/fcell killing. Cells were maintained in puromycin-containing medium for a total of 36 hours. An aliquot of cells was used to confirm appropriate transduction efficiency by flow cytometry to detect RFP (present in the lentiviral backbone) on day 5 after lentiviral transduction.
A total of 72 hours after tumor cell digestion, barcoded cells were pelleted, washed, resuspended in fresh MammoCult, and counted. One million viable barcoded cells in MammoCult were combined with 50% Matrigel (Corning 354234) and engrafted into the non-pre-humanized MFPs of NOD/SCID mice. Two pre-implantation reference pellets of one million cells each were snap-frozen at that time.
Time point matching of vehicle-treated tumors with AC-treated regrown tumors (harvested 50 days after initiating treatment) was not possible due to limitations in allowable animal tumor burden. Thus, downstream studies were designed to compare AC-treated tumors with pre-treated tumors harvested on the day of treatment initiation (day 0), representing the subclonal architecture present at the time of treatment initiation.
Barcoded mammary tumors were monitored until they reached ~150 mm3 to avoid necrosis and loss of subsets of barcodes (approximately 6 weeks after implantation), at which time 3–4 mice were euthanized and each mammary tumor was snap-frozen (vehicle-treated samples). The remaining 6 barcoded mice each received one dose of AC. Twenty-one days after the dose of AC, at the tumor size nadir, a subset of mice was euthanized and the residual tumors were collected. The remaining mice were monitored until 50 days after the dose of AC, at which point tumors re-grew to 100% of the initial tumor size. At that time, regrown tumors were collected. For each tumor collection, total tumors were resected and snap-frozen without dividing the sample to avoid loss of barcodes.
Mammosphere formation and limiting dilution transplantation assays
PDX tumors (vehicle-treated tumors and residual tumors harvested 21 days after the first dose of AC) were resected, and cells were dissociated and depleted of mouse stroma as described above. Viable human cells were counted using a Cell-O-Meter (Nexcelom) and plated at 500, 1000, or 2000 cells per well in ultra-low attachment 96-well plates in mammosphere conditions as described above with medium including 1% methylcellulose. The medium was refreshed with 20% volume of fresh medium every four days, and mammospheres were counted 20–24 days after plating without passaging cells.
To facilitate the limiting dilution transplantation assay, we used a sub-line of PIM001-P that had been labeled with a bioluminescent and fluorescent marker. Briefly, this sub-line was generated by ex vivo transduction of PIM001-P tumor cells with a lentivirus encoding Click beetle red luciferase and mCherry, followed by fluorescence-activated cell sorting and re-engraftment into NOD/SCID mice. Mice were treated with vehicle or AC, and tumors were harvested in the vehicle-treated or residual state. PIM1-CBRluc tumors were resected, and cells were dissociated and depleted of mouse stroma as described above. Viable cells were counted using a Cell-O-Meter (Nexcelom) and immediately engrafted into the 4th mammary fat pads (both right and left sides) of NOD/SCID mice as described above. Bioluminescence imaging (BLI; IVIS Lumina and IVIS Spectrum, Perkin Elmer) was performed and was used to monitor tumor growth every 4 weeks until tumor resection (18–22 weeks later), at which time resected tumors were imaged by BLI ex vivo to confirm their identity as PIM1-CBRluc tumors.
Treatment of PDX models with oxidative phosphorylation inhibitor
For the oxidative phosphorylation inhibitor study, NOD/SCID mice were engrafted with PIM001-P, PIM001-M, or PIM005 tumors as described above. Once tumors reached 100–150 mm3, mice were randomized into treatment groups based on tumor volume (determined by measurement with digital calipers). Mice were treated with an oxidative phosphorylation inhibitor (IACS-010759 (38), 5 mg/kg, 10 ml/kg, per os (p.o.), quaque die (q.d.), n=6), vehicle for IACS-010759 (0.5% methylcellulose, 10 mL/kg, p.o., q.d., n=6), or a single dose of AC (i.p., n=20) (initiation of treatments = day 0). IACS-010759 was formulated as previously described(38) and were stored for up to one week at 4°C with constant stirring. Body weight and tumor size were monitored 2–3 times weekly. AC-treated mice were re-randomized based on tumor size at the tumor size nadir (20 days after the dose of AC). AC-treated mice then began either IACS-010759 (n=10) or vehicle (0.5% methylcellulose, n=10) treatment (p.o., q.d.) on day 21 after the dose of AC. Of PIM001-P mice treated with AC followed by IACS-010759, a subset (n=5) were treated with an additional dose of AC on day 69, and IACS-010759 treatment was halted.
In all studies, mice were euthanized when they showed signs of poor health or when tumors reached 1000–1500 mm3. Tumor growth curves and Km plots were generated using Graphpad Prism. Log-rank p-values for Km plots were calculated using the R package for survival analysis. The Additive Hazards Model (39) was used to determine if there was synergy when IACS-010759 treatment followed AC treatment (Fig. 7).
Statistics
Two-tailed unpaired t tests were used when comparing two groups. When comparing three groups, ANOVA tests were used with Tukey’s multiple comparisons tests to calculate adjusted p values. P values <0.05 were considered statistically significant. For RNA-seq analyses, gene expression was evaluated using DESeq2 and differentially expressed genes were selected based on FDR <0.05 using Benjamini-Hochberg tests. Log-rank p-values for Km plots were calculated using an R package for survival analysis. The Additive Hazards Model (39) was used to assess drug synergy in pharmacologic studies. Normal-based 95% confidence intervals were calculated for each statistical test.
Supplementary Material
Fig. S1. Establishing a tolerated NACT regime for administration to immune-compromised mice.
Fig. S2. Histologic characterization of the desmoplastic response in residual tumors.
Fig. S3. Residual tumors maintain subpopulations of cycling cells.
Fig. S4. Depletion of mouse cells from freshly dissociated PDX tumors.
Fig. S5. Residual tumors are not enriched for cancer stem-like cell properties.
Fig. S6. Assessment of EMT status of the residual tumor state.
Fig. S7. Histologic features in pre- and post-AC treated TNBC patient biopsies.
Fig. S8. RNA sequencing of three PDX models throughout chemotherapy treatment.
Fig. S9. GSEA of residual tumor signatures across three PDX models.
Fig. S10. Mining available gene expression data from post-NACT residual breast tumors from patients.
Fig. S11. Reversible shifts in the proteome of residual tumors.
Fig. S12. Barcoding to monitor clonal dynamics during AC treatment in PDXs.
Fig. S13. WES to monitor genomic evolution during AC treatment in PIM001-P.
Fig. S14. Modeling of genomic subclonal architecture in PIM001-P.
Fig. S15. Genomic analysis of serially biopsied human TNBCs.
Fig. S16. Assessment of drug target engagement of PIM001-P tumors treated with the oxidative phosphorylation inhibitor.
Data file S1. PDX Characteristics.
Data file S2. RNA sequencing data from PDXs.
Data file S3. RPPA data from PDXs.
Data file S4. WES sample summary.
Data file S5. PDX tumor mutation data.
Data file S6. PDX tumor copy number data.
Data file S7. Patient tumor mutation data.
Data file S8. Patient tumor copy number data.
Data file S9. IACS010759 synergy calculations.
Data file S10. Prediction of epigenetic regulator activity.
Data file S11. Individual data points.
Acknowledgments
The authors are grateful to the patients who provided tumor biopsies for PDX model establishment, genomic analysis, and histologic analysis. PDX models and derivatives were obtained from the Cazalot Breast Cancer Model Resource at The University of Texas MD Anderson Cancer Center (MDACC). This resource was established through a generous gift from the Cazalot family and from funds from the MDACC Breast Cancer Moon Shot Program. The MDACC Breast Cancer Moonshot Program is thanked for supporting sequencing of patient tumor samples. The MDACC Center for Co-Clinical Trials in vivo pharmacology group conducted formulation and dosing of IACS-010759 for PDX treatment studies. Rebekah Gould Hubbar procured H&E-stained slides from patients enrolled on ARTEMIS. Ningping Feng provided insight and assistance for preclinical studies. Jared Burks provided guidance for quantitative IHC analyses. Yongying Jiang and the Pharmaceutical Science Facility conducted pharmaco-kinetic analyses for preclinical studies.
Funding
Funding sources that supported this work include The Cancer Prevention and Research Institute of Texas RP150148 (to HPW), RP160710 (to WFS, HPW, and JTC), and RP170668 (to JTC); The National Institutes of Health CA209978 (to JTC), U01 CA217858 Cancer Target Discovery and Development (CTD2), R35 CA197745 (Outstanding Investigator Award), S10 OD012351 (Shared Instrumentation Grant), and S10 OD021764 (Shared Instrumentation Grant), P30 CA013696 (to ACalifano); and The American Cancer Society 130423-PF-17–067-01-TBG (to GVE). Experiments performed in this study used the MDACC Research Animal Support Facility for blood toxicity studies, Functional Proteomics Core Facility for RPPA, the Sequencing and Microarray Facility for WES and RNA-seq, Flow Cytometry and Cellular Imaging Core Facility for Vectra 3 imaging and flow cytometry, and the Characterized Cell Line Core for STR profiling, all funded by National Cancer Institute Cancer Center Support Grant CA016672.
Footnotes
Competing interests
S.L.M. receives research funding as principal investigator for ongoing clinical trials at her institution from Novartis, EMD Serono, Roche/Genentech Seattle Genetics. Oncothyreon, Pfizer, Bayer, and Lily. MJA is chief scientific officer and shareholder of DarwinHealth, Inc. ACalifano is founder, equity holder, consultant, and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc. All other authors declare they have no competing interests.
Data and materials availability
RNA sequencing and whole-exome sequencing data generated in this study have been deposited in dbGAP with accession code phs001742.v2.
References and notes
- 1.Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L, Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J Clin Oncol 26, 1275–1281 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, Bonnefoi H, Cameron D, Gianni L, Valagussa P, Swain SM, Prowell T, Loibl S, Wickerham DL, Bogaerts J, Baselga J, Perou C, Blumenthal G, Blohmer J, Mamounas EP, Bergh J, Semiglazov V, Justice R, Eidtmann H, Paik S, Piccart M, Sridhara R, Fasching PA, Slaets L, Tang S, Gerber B, Geyer CE Jr., Pazdur R, Ditsch N, Rastogi P, Eiermann W, von Minckwitz G, Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014). [DOI] [PubMed] [Google Scholar]
- 3.von Minckwitz G, Untch M, Blohmer JU, Costa SD, Eidtmann H, Fasching PA, Gerber B, Eiermann W, Hilfrich J, Huober J, Jackisch C, Kaufmann M, Konecny GE, Denkert C, Nekljudova V, Mehta K, Loibl S, Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 30, 1796–1804 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Symmans WF, Wei C, Gould R, Yu X, Zhang Y, Liu M, Walls A, Bousamra A, Ramineni M, Sinn B, Hunt K, Buchholz TA, Valero V, Buzdar AU, Yang W, Brewster AM, Moulder S, Pusztai L, Hatzis C, Hortobagyi GN, Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J Clin Oncol 35, 1049–1060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, Li Y, Ju YS, Ramakrishna M, Haugland HK, Lilleng PK, Nik-Zainal S, McLaren S, Butler A, Martin S, Glodzik D, Menzies A, Raine K, Hinton J, Jones D, Mudie LJ, Jiang B, Vincent D, Greene-Colozzi A, Adnet PY, Fatima A, Maetens M, Ignatiadis M, Stratton MR, Sotiriou C, Richardson AL, Lonning PE, Wedge DC, Campbell PJ, Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 21, 751–759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan R, Giuliany, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S, The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, Menzies A, Stebbings LA, Leroy C, Jia M, Rance R, Mudie LJ, Gamble SJ, Stephens PJ, McLaren S, Tarpey PS, Papaemmanuil E, Davies HR, Varela I, McBride DJ, Bignell GR, Leung K, Butler AP, Teague JW, Martin S, Jonsson G, Mariani O, Boyault S, Miron P, Fatima A, Langerod A, Aparicio SA, Tutt A, Sieuwerts AM, Borg A, Thomas G, Salomon AV, Richardson AL, Borresen-Dale AL, Futreal PA, Stratton MR, Campbell PJ, Breast C Cancer Working Group of the International Cancer Genome, The life history of 21 breast cancers. Cell 149, 994–1007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sanchez V, Kurupi R, Moore PD, Pinto JA, Doimi FD, Gomez H, Horiuchi D, Goga A, Lehmann BD, Bauer JA, Pietenpol JA, Ross JS, Palmer GA, Yelensky R, Cronin M, Miller VA, Stephens PJ, Arteaga CL, Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 4, 232–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez-Farre X, Munoz M, Russnes HG, Helland A, Rye IH, Borresen-Dale AL, Maruyama R, van Oudenaarden A, Dowsett M, Jones RL, Reis-Filho J, Gascon P, Gonen M, Michor F, Polyak K, Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell reports 6, 514–527 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, Crosetto N, Foukakis T, Navin NE, Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA, Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res 78, 2127–2139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gatenby RA, Silva AS, Gillies RJ, Frieden BR, Adaptive therapy. Cancer Res 69, 4894–4903 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HC, Krishnamurthy Radhakrishna V, Siravegna G, Hu H, Raoof S, Lockerman E, Kalsy A, Lee D, Keating CL, Ruddy DA, Damon LJ, Crystal AS, Costa C, Piotrowska Z, Bardelli A, Iafrate AJ, Sadreyev RI, Stegmeier F, Getz G, Sequist LV, Faber AC, Engelman JA, Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nature medicine 22, 262–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J, A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, Alvarez MJ, Kendsersky ND, Pelz CR, Johnson-Camacho K, Dobrolecki LE, Chin K, Aswani AJ, Wang NJ, Califano A, Lewis MT, Tomlin CJ, Spellman PT, Adey A, Gray JW, Sears RC, Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nature communications 9, 3815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK, A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A 101, 14228–14233 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN, Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA, The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC, Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 106, 13820–13825 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand A, Sahin, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, He X, Weigman V, Fan C, Palazzo J, Hortobagyi GN, Nolden LK, Wang NJ, Valero V, Gray JW, Perou CM, Mills GB, Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 69, 4116–4124 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, Sanders ME, Granja-Ingram NM, Smith JJ, Meszoely IM, Salter J, Dowsett M, Stemke-Hale K, Gonzalez-Angulo AM, Mills GB, Pinto JA, Gomez HL, Arteaga CL, Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nature medicine 18, 1052–1059 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esserman LJ, Berry DA, Cheang MC, Yau C, Perou CM, Carey L, DeMichele A, Gray JW, Conway-Dorsey ME, Lenburg, Buxton MB, Davis SE, van’t Veer LJ, Hudis C, Chin K, Wolf D, Krontiras H, Montgomery L, Tripathy D, Lehman C, Liu MC, Olopade OI, Rugo HS, Carpenter JT, Livasy C, Dressler L, Chhieng D, Singh B, Mies C, Rabban J, Chen YY, Giri D, Au A, Hylton N, Investigators IST, Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res Treat 132, 1049–1062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magbanua MJ, Wolf DM, Yau C, Davis SE, Crothers J, Au A, Haqq CM, Livasy C, Rugo HS, Investigators IST, Esserman L, Park JW, van ‘t Veer LJ, Serial expression analysis of breast tumors during neoadjuvant chemotherapy reveals changes in cell cycle and immune pathways associated with recurrence and response. Breast Cancer Res 17, 73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, Budczies J, Huober J, Klauschen F, Furlanetto J, Schmitt WD, Blohmer JU, Karn T, Pfitzner BM, Kummel S, Engels K, Schneeweiss A, Hartmann A, Noske A, Fasching PA, Jackisch C, van Mackelenbergh M, Sinn P, Schem C, Hanusch C, Untch M, Loibl S, Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. The lancet oncology 19, 40–50 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, Wolff AC, Wood WC, Davidson NE, Sledge GW, Sparano JA, Badve SS, Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol 32, 2959–2966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA, Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation 121, 2750–2767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawson SJ, Rueda OM, Aparicio S, Caldas C, A new genome-driven integrated classification of breast cancer and its implications. EMBO J 32, 617–628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH, Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 21, 1688–1698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pareja F, Geyer FC, Marchio C, Burke KA, Weigelt B, Reis-Filho JS, Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2, 16036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, Brisken C, Bult CJ, Cai S, Clarke RB, Dowst H, Ellis MJ, Gonzalez-Suarez E, Iggo RD, Kabos P, Li S, Lindeman GJ, Marangoni E, McCoy A, Meric-Bernstam F, Piwnica-Worms H, Poupon MF, Reis-Filho J, Sartorius CA, Scabia V, Sflomos G, Tu Y, Vaillant F, Visvader JE, Welm A, Wicha MS, Lewis MT, Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer metastasis reviews 35, 547–573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yam C, Hess KR, Litton JK, Yang WT, Piwnica-Worms H, Mittendorf EA, Ueno NT, Lim B, Murthy RK, Damodaran SK, Helgason T, Huo L, Thompson AM, Gilcrease M, Santiago L, Candelaria RP, Rauch G, Adrada B, Symmans WF, Moulder SL, A randomized, triple negative breast cancer enrolling trial to confirm molecular profiling improves survival (ARTEMIS). Journal of Clinical Oncology 35, TPS590–TPS590 (2017). [Google Scholar]
- 32.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA, Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107, 15449–15454 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher ER, Wang J, Bryant J, Fisher B, Mamounas E, Wolmark N, Pathobiology of preoperative chemotherapy: findings from the National Surgical Adjuvant Breast and Bowel (NSABP) protocol B-18. Cancer 95, 681–695 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Powell E, Shao J, Yuan Y, Chen HC, Cai S, Echeverria GV, Mistry N, Decker KF, Schlosberg C, Do KA, Edwards JR, Liang H, Piwnica-Worms D, Piwnica-Worms H, p53 deficiency linked to B cell translocation gene 2 (BTG2) loss enhances metastatic potential by promoting tumor growth in primary and metastatic sites in patient-derived xenograft (PDX) models of triple-negative breast cancer. Breast Cancer Res 18, 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N, (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A 106, 4519–4524 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J, TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Cote A, Shah SP, PyClone: statistical inference of clonal population structure in cancer. Nat Meth 11, 396–398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, Al-Atrash G, Asara J, Bardenhagen J, Carrillo CC, Carroll C, Chang E, Ciurea S, Cross JB, Czako B, Deem A, Daver N, de Groot JF, Dong JW, Feng N, Gao G, Gay J, Do MG, Greer J, Giuliani V, Han J, Han L, Henry VK, Hirst J, Huang S, Jiang Y, Kang Z, Khor T, Konoplev S, Lin YH, Liu G, Lodi A, Lofton T, Ma H, Mahendra M, Matre P, Mullinax R, Peoples M, Petrocchi A, Rodriguez-Canale J, Serreli R, Shi T, Smith M, Tabe Y, Theroff J, Tiziani S, Xu Q, Zhang Q, Muller F, DePinho RA, Toniatti C, Draetta GF, Heffernan TP, Konopleva M, Jones P, Di Francesco ME, Marszalek JR, An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nature medicine 24, 1036–1046 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Rod NH, Lange T, Andersen I, Marott JL, Diderichsen F, Additive interaction in survival analysis: use of the additive hazards model. Epidemiology 23, 733–737 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A, Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48, 838–847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falkson G, Gelman R, Glick J, Falkson CI, Harris J, Reinduction with the same cytostatic treatment in patients with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 12, 45–49 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Yonemori K, Katsumata N, Kaneko M, Uno H, Matsumoto K, Kouno T, Shimizu C, Ando M, Takeuchi M, Fujiwara Y, Prediction of response to repeat utilization of anthracycline in recurrent breast cancer patients previously administered anthracycline-containing chemotherapeutic regimens as neoadjuvant or adjuvant chemotherapy. Breast Cancer Res Treat 103, 313–318 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sanchez V, Sanders ME, Lee T, Gomez H, Lluch A, Perez-Fidalgo JA, Wolf MM, Andrejeva G, Rathmell JC, Fesik SW, Arteaga CL, MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell metabolism 26, 633–647 e637 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yadav N, Kumar S, Marlowe T, Chaudhary AK, Kumar R, Wang J, O’Malley J, Boland PM, Jayanthi S, Kumar TK, Yadava N, Chandra D, Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell death & disease 6, e1969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, Giannoni E, Schrader T, Puhr M, Chiarugi P, Taddei ML, Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 7, 61890–61904 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF, Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde JG, Rebucci M, Tran-Perennou C, Sastre-Garau X, Sigal-Zafrani B, Delattre O, Dieras V, Poupon MF, A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res 13, 3989–3998 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Zielske SP, Spalding AC, Lawrence TS, Loss of tumor-initiating cell activity in cyclophosphamide-treated breast xenografts. Translational oncology 3, 149–152 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen X, Chang JT, Planning bioinformatics workflows using an expert system. Bioinformatics 33, 1210–1215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Callari M, Sammut SJ, De Mattos-Arruda L, Bruna A, Rueda OM, Chin SF, Caldas C, Intersect-then-combine approach: improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med 9, 35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goode DL, Hunter SM, Doyle MA, Ma T, Rowley SM, Choong D, Ryland GL, Campbell IG, A simple consensus approach improves somatic mutation prediction accuracy. Genome Med 5, 90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jia P, Li F, Xia J, Chen H, Ji H, Pao W, Zhao Z, Consensus rules in variant detection from next-generation sequencing data. PLoS One 7, e38470 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A, ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC bioinformatics 7 Suppl 1, S7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giorgi FM. Aracne.networks: ARACNe-inferred gene networks from TCGA tumor datasets. R package version 1.4.0 (2017).
- 55.Califano A, Alvarez MJ, The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer 17, 116–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Establishing a tolerated NACT regime for administration to immune-compromised mice.
Fig. S2. Histologic characterization of the desmoplastic response in residual tumors.
Fig. S3. Residual tumors maintain subpopulations of cycling cells.
Fig. S4. Depletion of mouse cells from freshly dissociated PDX tumors.
Fig. S5. Residual tumors are not enriched for cancer stem-like cell properties.
Fig. S6. Assessment of EMT status of the residual tumor state.
Fig. S7. Histologic features in pre- and post-AC treated TNBC patient biopsies.
Fig. S8. RNA sequencing of three PDX models throughout chemotherapy treatment.
Fig. S9. GSEA of residual tumor signatures across three PDX models.
Fig. S10. Mining available gene expression data from post-NACT residual breast tumors from patients.
Fig. S11. Reversible shifts in the proteome of residual tumors.
Fig. S12. Barcoding to monitor clonal dynamics during AC treatment in PDXs.
Fig. S13. WES to monitor genomic evolution during AC treatment in PIM001-P.
Fig. S14. Modeling of genomic subclonal architecture in PIM001-P.
Fig. S15. Genomic analysis of serially biopsied human TNBCs.
Fig. S16. Assessment of drug target engagement of PIM001-P tumors treated with the oxidative phosphorylation inhibitor.
Data file S1. PDX Characteristics.
Data file S2. RNA sequencing data from PDXs.
Data file S3. RPPA data from PDXs.
Data file S4. WES sample summary.
Data file S5. PDX tumor mutation data.
Data file S6. PDX tumor copy number data.
Data file S7. Patient tumor mutation data.
Data file S8. Patient tumor copy number data.
Data file S9. IACS010759 synergy calculations.
Data file S10. Prediction of epigenetic regulator activity.
Data file S11. Individual data points.