

Abstract
Objectives:
Extending prior studies of prenatal adversity and depressive symptoms, we tested associations between maternal prenatal major depressive disorder (MDD) and infant cortisol regulation. Based on prior findings by our group, we also tested placenta glucocorticoid (HSD11B2 methylation) and serotonin (SLC6A4 gene expression) signaling as moderators of links between prenatal MDD and infant cortisol.
Methods:
Participants were 153 mother-infant pairs from a low-income, diverse sample (Mean age=26±6). Repeated structured diagnostic interviews were utilized to identify: mothers with (a) prenatal MDD, (b) preconception-only MDD, (c) controls. Placenta samples were assayed for HSD11B2 methylation and SLC6A4 gene expression. Infant salivary cortisol response to a neurobehavioral exam was assessed at one month.
Results:
Daughters of prenatal MDD mothers had 51% higher baseline (Ratio=1.51, 95% CI=1.01-2.27, p=.045) and 64% higher stress responsive cortisol (Ratio=1.64, 95% CI=1.05-2.56, p=.03) than daughters of controls and 75% higher stress-responsive cortisol (Ratio=1.75, 95% CI=1.04-2.94, p=.04) than daughters of preconception-only MDD mothers. HSD11B2 methylation moderated links between prenatal MDD and baseline cortisol (p=.02), with 1% methylation decreases associated with 9% increased baseline cortisol in infants of prenatal MDD mothers (Ratio=1.09, 95% CI=1.01-1.16). SLC6A4 expression moderated links between prenatal MDD and cortisol response among boys alone (p=.007), with tenfold increases in expression associated with threefold increases in stress-responsive cortisol (Ratio=2.87, 95% CI=1.39-5.93) in sons of control mothers.
Conclusions:
Results highlight specificity of associations between prenatal vs. preconception MDD and cortisol regulation and the importance and complexity of placenta glucocorticoid and serotonergic pathways underlying the intergenerational transmission of risk from maternal adversity.
Keywords: depression, cortisol, HSD11B2, pregnancy, serotonin, infant
INTRODUCTION
Exposure to maternal prenatal depression represents one of the most common forms of early adversity. In the United States, approximately one in five infants is exposed to elevated maternal depressive symptoms and one in ten infants is exposed to maternal major depressive disorder (MDD) during pregnancy, with the majority of depressed women remaining untreated during pregnancy (1-3). Exposure to maternal depressive symptoms and prenatal MDD have been associated with adverse physical health and behavioral outcomes across development, including preterm delivery, low birth weight, and alterations in fetal heart rate and activity (4-7) in the fetal period, alterations in affect, brain structure and brain activity in the newborn period (8-12), and alterations in growth, cognition, brain structure, and psychopathology symptoms and disorders in childhood (13-16). However, despite relatively consistent associations across development, mechanisms and moderators of long-term outcomes following exposure to maternal depression remain unclear.
Fetal programming of offspring brain and stress systems is a prominent proposed mechanism underlying links between prenatal adversity and offspring health and behavioral outcomes (13, 17-20).In particular, dysregulation of the fetal hypothalamic pituitary adrenocortical (HPA) axis has been proposed as a final common pathway underlying links between prenatal adversity and long-term health and behavioral outcomes (21, 22). We propose programming of the fetal HPA axis as a mechanism underlying links between prenatal MDD and long-term offspring outcomes. A small number of studies lend credence to this hypothesis. In normative samples, maternal prenatal depressive symptoms were associated with altered basal cortisol levels in newborns (24-27), (adopted) children (28), and adolescents (29), and with alterations in cortisol response to challenge in toddlers (30). Brennan et al. (31) documented associations between both peripartum (pre and postpartum) and lifetime depressive disorders (measured via postpartum structured interviews) and alterations in infant cortisol response to stress at six months. To our knowledge, however, no studies of infant HPA regulation have included prospective measurement of depressive disorders over pregnancy or included a preconception-only depression group. The preconception-only depression group allows for disentangling effects of prenatal depression from effects of lifetime depressive episodes prior to pregnancy (e.g., alterations in the maternal hormonal milieu from prior depressive episodes) and familial/genetic factors. Thus, the first goal of the present study is to investigate the specific influences of prenatal depressive disorder versus preconception history of depressive disorder (measured by gold-standard structured interviews) on offspring cortisol response. We focused on cortisol response in the neonatal period in order to identify the earliest biological pathways to long term health outcomes.
We and others have proposed placental enzymes regulating maternal-fetal glucocorticoid and serotonin transfer as key modulators of fetal programming effects of maternal depression (32-34). The placenta is a unique endocrine and metabolic organ that mediates transmission of environmental signals, nutrition, and endocrine/immune and gas exchange between mother and fetus, and supports fetal brain development through adaptive responses to the maternal environment and protection from environmental insults (35). . One key regulator of fetal glucocorticoid exposure is placental 11 β hydroxysteroid dehydrogenase Type 2 (11β HSD2). 11β HSD2 is an enzyme which converts cortisol to inactive cortisone, protecting the fetus from increasing maternal glucocorticoids over pregnancy (36).
Epigenetic alterations in the HSD11B2 promoter are posited to underlie changes in placental 11β HSD-2 activity and have been highlighted as critical modulators of fetal programming by maternal stress (36). Epigenetic mechanisms involve alterations to DNA that influence gene expression without altering the nucleotide sequence. Methylation is a stable epigenetic mechanism that involves activation or suppression of gene expression through effects on transcription factor binding. Our group has shown associations between socio-economic adversity and altered placental HSD11B2 promoter methylation (37) and between altered HSD11B2 methylation and infant birth weight and quality of movement (38). Further, complementing a seminal study of associations between NR3C1 methylation (another gene in the glucocorticoid signaling pathway), prenatal depression and infant cortisol response (39), we documented an interaction between prenatal anxiety and placental HSD11B2 methylation in relation to newborn muscle tone (32), such that increasing HSD11B2 methylation was associated with increasing hypotonicity only in infants exposed to prenatal anxiety. In the present study, the second goal is to investigate placental HSD11B2 methylation as a modulator of links between prenatal MDD vs. preconception-only MDD and infant cortisol response.
Serotonin signaling pathways have been implicated in both the pathophysiology of MDD and in programming of the fetal brain and HPA axis development. Preclinical studies as well as human studies of prenatal selective serotonin reuptake inhibitor (SSRI) use have highlighted the critical role of serotonin signaling in fetal development (40, 41). There is also emerging evidence for a key role of serotonin synthesis and transport in the placenta in programming fetal brain development (34). SLC6A4, which encodes the serotonin transporter, is also expressed in the placenta. Our group has previously shown increased placental SLC6A4 gene expression in mothers with depression/anxiety (documented by medical chart review) compared to controls (33), while Raikonnen et al. found no direct association between prenatal depressive symptoms and SLC6A4 (42). Inconsistent findings across studies may be due to a moderating as opposed to mediating role of SLC6A4 on links between prenatal depression and infant outcomes. In the present study, the third goal is to investigate SLC6A4 gene expression as a moderator of links between prenatal MDD vs. preconception MDD and infant cortisol response.
The present study is an intensive, prospective study of maternal prenatal versus preconception-only MDD, placental glucocorticoid and serotonin signaling pathways, and infant cortisol response. Sex differences have been documented at every step of fetal programming pathways, including placental regulatory processes (43-46); thus, sex-specific links between prenatal MDD and infant cortisol will be investigated, including sex-specific moderation of these links by placenta glucocorticoid and serotonin pathways will be investigated.
METHODS
Participants
Participants were 153 pregnant mothers (mean age=26±6) and their healthy infants (48% female, mean age=32 days) from the Behavior and Mood in Mothers, Behavior in Infants (BAMBI) study, an intensive, prospective study of prenatal MDD and neonatal neurobehavior and stress response. BAMBI data collection took place between March, 2008 and January, 2013. Maternal exclusion criteria for the present study were as follows: age <18 or >40, psychotropic medication use after pregnancy recognition, steroid medications during pregnancy, psychotic or bipolar disorder, medical conditions during pregnancy (gestational diabetes, hypertension, pre-eclampsia, hyper/hypothyroidism), other high risk perinatal conditions (e.g., non-singleton pregnancy), illicit drug use besides marijuana (meconium confirmed). Infants were singletons born >36 weeks gestational age (GA). Infants with congenital anomalies or serious medical complications were excluded. All participants provided written informed consent, and followed procedures reviewed and approved by Institutional Review Boards at Women and Infants’ Hospital of Rhode Island and Lifespan Hospitals.
Assignment to Study Groups.
Mothers were assigned to one of three groups (Prenatal MDD, Preconception-only MDD, Control) based on prenatal and/or lifetime diagnoses of major and minor depressive episodes obtained through structured clinical interviews (See details below); Prenatal MDD group included women who met criteria for major or minor depressive episode at any time during the current pregnancy or in the conception window (within 3 months of conception); Preconception-only MDD group included women with a history of one or more lifetime major or minor depressive episodes prior but not during the current pregnancy/conception window; Controls were free of lifetime and current pregnancy mood disorder diagnoses. Women in the prenatal MDD group included those with (54%) and without (46%) episodes of lifetime preconception MDD. Participants in all groups had no lifetime history of psychotic or bipolar disorder. The sample included 64 mothers with Prenatal MDD, 39 mothers with Preconception-only MDD, and 50 controls.
Procedures
Maternal Interviews.
Participants completed up to five interviews (M=5, SD=1) between 2nd trimester and one month postpartum, including up to 3 interviews during 2nd and 3rd trimester of pregnancy, a post-delivery interview (95% completion), and an interview at one month postpartum (93% completion). Pregnancy interviews were conducted between 18 and 39 weeks gestation (M’s=23±3, 30±1, and 36±1 weeks gestation for the first, second, and third interviews, respectively). At the first maternal interview, women completed the mood, anxiety, and psychotic screen modules of the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) Axis I Disorders-Research Version, Non-patient edition (SCID-I/NP) (47). Major depressive episodes were based on DSM-IV criteria (≥5 symptoms, ≥2 weeks, functional impairment); minor depressive episodes were based on DSM-IV appendix B criteria (2-4 symptoms, ≥2 weeks, functional impairment). At the initial interview, women reported on mood disorders during their lifetime and during the current pregnancy up until the time of the first interview. At each subsequent interview over pregnancy and postpartum, the SCID current mood disorders module was administered to assess for major and minor depressive episodes since the prior assessment. A reliability analysis based on re-rating of 10% of study interviews by independent judges yielded a Cohen’s kappa of .93 for depressive disorders. Mothers also completed the Inventory of Depressive Symptomatology-Self Rated (IDS-SR) (48) at each prenatal interview, which includes four-point Likert ratings of 21 depressive symptoms over the past week.
At study enrollment, participants provided demographic information and completed a socioeconomic status (SES) interview from which education, occupation, income, and Hollingshead four-factor index of SES (49) were extracted. Additionally, participants completed interviews regarding health and pregnancy history, pre-pregnancy weight, and a calendar/anchor-based Timeline Follow Back (TLFB) interview, in which smoking and alcohol use over pregnancy and three months prior to conception were measured on a daily level (50, 51).
Delivery/Birth.
Information regarding maternal and infant health and medical conditions, as well as infant sex, delivery method, gestational age at birth, and birth weight was extracted by medical chart review. Mothers and study staff collected diapers containing meconium for three days post-birth for verification of tobacco and other drug use. Placental tissue was collected (M=1.2, SD=.65 hours after delivery) and preserved for extraction of DNA for HSD11B2 methylation and RNA for SLC6A4 gene expression analyses.
One Month Infant Stress Response Assessment.
Cortisol stress response was elicited using the NICU Network Neurobehavioral Scale (NNNS) (52, 53) administered by certified examiners blind to maternal depression status at M=32 (SD=3) days. The NNNS involves mild stress as the infant is observed and handled during periods of sleep, awake, crying, and non-crying states, and lasted an average of 27±8 minutes. Four saliva samples were collected for cortisol during and after the NNNS exam (baseline, end of NNNS, 20 and 40 minutes post-NNNS) (54) using the sorbette sampling device (Salimetrics LLC, State College, Pennsylvania). At the one month stress assessment, mothers also provided information about their infant’s feeding (breast/bottle, time since most recent feeding). Mothers also completed the Maternal Attachment Inventory (55) to assess attachment to their infants.
Bioassays.
Saliva Cortisol.
Saliva cortisol provides a non-invasive and reliable estimate of free (unbound) cortisol (56). Following collection, infant saliva samples were frozen at −80°C until analysis by Salimetrics using a high-sensitive enzyme immunoassay. The intra and inter-assay coefficients of variation were < 8%.
Meconium analysis reflects cumulative substance use over the third trimester. Meconium was analyzed by the U.S. Drug Testing Laboratories (Des Plaines, IL) for cotinine (nicotine metabolite), cannabinoids, opiates, cocaine, and amphetamines via enzyme-multiplied immunoassay technique (EMIT) and enzyme-linked immunosorbent assay (ELISA) screens as well as tandem gas or liquid chromatography mass spectrometry confirmation (57-60). Samples from all included participants were negative for cocaine, opiates, and amphetamines. Samples with cotinine ≥10 ng/g and cannabinoid (carboxy THC) ≥40 ng/g were considered positive.
Placental HSD11B2 promoter methylation and SLC6A4 gene expression.
Placental tissue free from maternal decidua was excised throughout the placenta and placed immediately in RNAlater solution (Life Technologies, Grand Island, NY) then stored at 4° C. At least 72 hours later, placenta samples were removed from RNAlater, blotted dry, snap-frozen in liquid nitrogen, pulverized to homogeneity, and stored at −80° C until analysis. Placental HSD11B2 methylation. Placental genomic DNA was extracted using the QIAmp DNA Mini kit (Qiagen, Valencia, CA), and assessed for quantity and quality using a ND-1000 Spectophotometer (Nanodrop, Wilmington, DE). DNA samples were sodium bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA). Extent of methylation at the HSD11B2 promoter region was examined with a quantitative pyrosequencing approach (61) as previously described (38) using the PyroMark MD Pyrosequencing System (Qiagen). The region analyzed contains 4 CpGs previously associated with infant behavior (32, 38) with reactions performed in triplicate. Sodium bisulfite-modified, fully-methylated referent positive control and fully-unmethylated (whole genome amplified) negative control DNA (Qiagen) was examined with each batch. SLC6A4 gene expression. Placental RNA was extracted using TRIzol Reagent® (62) and assessed for quantity and quality using a ND-1000 Spectophotometer. The SuperScript® III First Strand Synthesis System Supermix was used to synthesize cDNA from 3 μg of RNA from each sample and a pooled internal control sample using random hexamer primers. Real-time quantitative PCR was carried out using cDNA from each placental sample to determine RNA expression levels. Samples were analyzed for gene expression using an Applied Biosystems 7500 Real-time PCR System (Applied Bio-systems, CA; assay Hs00169010_m1). Each sample was run in triplicate. mRNA expression was determined relative to the multiplexed 18S housekeeping gene, which has stable expression levels in placental tissue regardless of gestational age and delivery method (63) and also relative to a pooled internal control sample using methods of Pfaffl (64).
Statistical Analysis
Preliminary analyses/data summaries.
Between-group comparisons (Prenatal MDD, Preconception-only MDD, Controls) on demographics, pregnancy history, infant, and psychiatric characteristics utilized F, X2, Kruskall-Wallis ANOVA, and Fisher’s tests as appropriate. A trapezoidal rule was applied to the four cortisol measures to produce an integrated cortisol response measure (area under the curve; AUC (65)). Given high magnitude associations between CpG’s (pairwise r’s=.65-.76), methylation of the SLC6A4 promoter was summarized as mean methylation across all 4 CpG sites (37). Continuous depressive symptoms were summarized as the mean IDS score across prenatal interviews.
Hypotheses regarding MDD group effects on infant cortisol response (baseline, AUC) were tested using normal linear regression modeling, with MDD group as the independent variable (IV), and infant baseline and AUC cortisol as the dependent variables. Specifically, we tested two a priori linear contrasts: (a) Control group vs. Prenatal MDD group; (b) Preconception-only MDD group vs. Prenatal MDD group, with Prenatal MDD group as the reference. Hypotheses regarding moderation of MDD group effects on infant cortisol response (baseline, AUC) by HSD11B2 promoter methylation and SLC6A4 gene expression were investigated using normal linear regression modeling and the same a priori group contrasts (Control group vs. Prenatal MDD group; Preconception-only MDD group vs. Prenatal MDD group). Finally, a parallel set of regression analyses were conducted with continuous depressive symptoms (mean of prenatal depressive symptom assessments) as the IV, including main effects and interactions with HSD11B2 methylation and SLC6A4 expression.
Both baseline and AUC cortisol were transformed to the logarithmic scale prior to regression modeling. However, model findings are interpreted in the original cortisol scale by exponentiating linear regression coefficients and associated 95% confidence interval endpoints. Outcome summaries in Figures 1 and 2 are also presented in the original scale for ease of visual display. All associations were investigated using unstandardized regression coefficients in the overall sample and also stratified by offspring sex. Sex by MDD group interactions were tested for each set of analyses. Actual sample sizes for each regression model depend on joint missingness in the cortisol outcome and model covariates, and are noted for each MDD group within the overall and sex-stratified samples in Tables 2-4.
Figure 1.
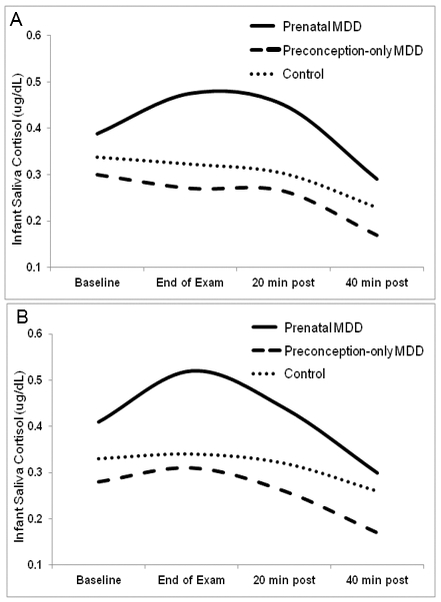
Cortisol response to the NICU Network Neurobehavioral Scale (NNNS) in three groups of one-month old neonates: Prenatal MDD (neonates exposed to mothers with prenatal major depressive disorder), Preconception MDD (neonates exposed to mothers with MDD prior to but not during pregnancy), and Controls for (A) overall sample and (B) daughters only.
Note. Four saliva samples were collected for cortisol during and after the NNNS (baseline, end of NNNS, 20 and 40 minutes following the NNNS). Although baseline and area under the curve (AUC) cortisol were modeled in the logarithmic scale for statistical analyses, results are presented with raw mean cortisol values for ease of visual display.
Figure 2.
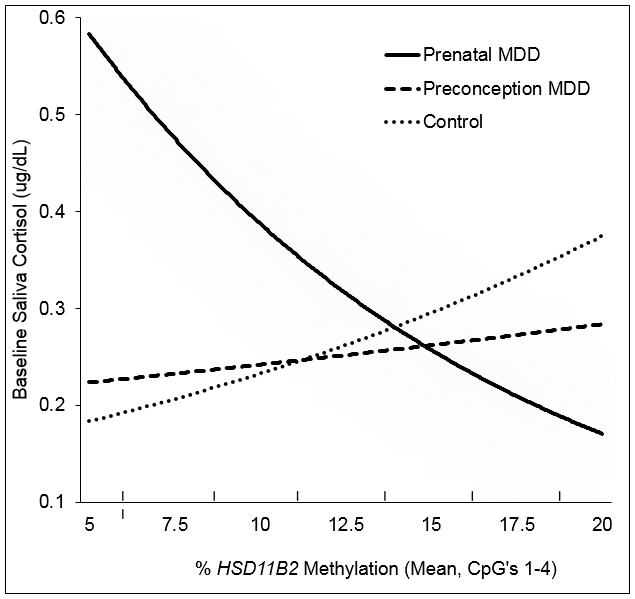
Model-based interaction between prenatal depression group and placental HSD11B2 methylation on baseline cortisol at one month.
Note. Although baseline cortisol was modeled in the logarithmic scale for statistical analyses, fitted values are presented in the original cortisol scale. Fitted values are presented controlling for education, and time since feeding.
Table 2a.
Maternal prenatal depression group and baseline cortisol at one month for the overall sample (n=139), and stratified by daughters (n=63) and sons (n=76).1
| All |
Daughters |
Sons |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictor variables | β | SE | p | β | SE | p | β | SE | p |
| Baseline Cortisol2 | |||||||||
| Intercept | −.13 | .37 | .71 | 1.26 | .50 | .01 | −1.09 | .50 | .03 |
| Time since feeding (min) 2 | −.30 | .09 | .001 | −.68 | .13 | <.001 | −.05 | .13 | .72 |
| Education3 | −.34 | .15 | .02 | −.30 | .21 | .17 | −.48 | .20 | .02 |
| Control vs. Prenatal MDD4 | −.20 | .16 | .23 | −.42 | .21 | .045 | .03 | .24 | .88 |
| Preconception-only MDD vs. Prenatal MDD4 | −.24 | .18 | .19 | −.40 | .24 | .10 | .00 | .26 | 1.00 |
Per group cell sizes for the overall sample were n’s=57, 36, 46 for Prenatal MDD, Preconception-only MDD, and Control, respectively; per group cell sizes for daughters were n’s =21, 18, and 24, respectively; per group cell sizes for sons were n’s=36, 18, and 22. Unstandardized regression coefficients are presented.
Natural log transformed.
≤ high school degree vs. > high school degree.
MDD=major depressive disorder; contrasts are based on categorical MDD groups.
Table 4.
Interaction of prenatal depression group and placental SLC6A4 gene expression for AUC1 cortisol at one month for the overall sample (n=103) and stratified by daughters (n=50) and sons (n=53).2
| All |
Daughters |
Sons |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictor variables | β | SE | P | β | SE | p | β | SE | p |
| AUC Cortisol1,3 | |||||||||
| Intercept | 2.67 | .37 | <.001 | 3.16 | .60 | <.001 | 2.47 | .45 | <.001 |
| Time since feeding (min) 3 | .12 | .09 | .21 | .04 | .15 | .79 | .15 | .12 | .20 |
| Education4 | −.36 | .16 | .02 | −.23 | .25 | .36 | −.54 | .20 | .01 |
| Control vs. Prenatal MDD5 | −.16 | .16 | .33 | −.43 | .25 | .09 | −.02 | .21 | .93 |
| Preconception-only MDD vs. Prenatal MDD5 | −.34 | .19 | .08 | −.52 | .29 | .08 | −.23 | .26 | .36 |
| SLC6A4 Gene Expression3 | −.13 | .12 | .29 | −.03 | .23 | .90 | −.16 | .13 | .24 |
| [Control vs. Prenatal MDD5] * SLC6A4 | .26 | .18 | .14 | −.17 | .30 | .57 | .61 | .22 | .007 |
| [Preconception vs. Prenatal MDD5] * SLC6A4 | .18 | .23 | .44 | .06 | .42 | .89 | .31 | .27 | .26 |
AUC=Area Under the Curve;
Per group cell sizes for the overall sample were n’s=45, 23, 35 for Prenatal MDD, Preconception-only MDD, and Control, respectively; per group cell sizes for daughters were n’s =17, 13, and 20, respectively; per group cell sizes for sons were n’s=28, 10, and 15. Unstandardized regression coefficients are presented.
Natural log transformed;
≤ high school degree vs. > high school degree;
MDD=major depressive disorder; contrasts are based on categorical MDD groups.
Potential confounders from prior literature (demographic characteristics, pregnancy history, infant characteristics; See Table 1) were evaluated for associations with MDD group and cortisol outcomes. Only one covariate (maternal education) showed significant associations with both MDD group and cortisol outcome (p<.05) and was included as a confounder in all regression models. Time since feeding was also included in the model based on associations with baseline and AUC cortisol in our prior work (66).
Table 1.
Maternal and infant characteristics for the full sample and by prenatal depression group
| Total (n=153) Mean (SD), N (%2) |
Prenatal MDD1 (n=64) Mean (SD), N (%2) |
Preconception-only MDD1 (n=39) Mean (SD), N (%2) |
Controls (n=50) Mean (SD), N (%2) |
|
|---|---|---|---|---|
| Maternal Characteristics | ||||
| Maternal age (years) | 26 (6) | 26 (6) | 29 (5) | 25 (5) ** |
| Race (% non-Hispanic White) | 71 (46%) | 22 (34%) | 26 (67%) | 23 (46%) ** |
| Education (% ≤ HS degree) | 58 (38%) | 31 (48%) | 7 (18%) | 20 (40%) ** |
| Low SES3 | 50 (33%) | 27 (42%) | 7 (18%) | 16 (33%) * |
| Gravida4 | 2 (2) | 3 (2) | 2 (1) | 2 (1) * |
| Depressive Symptoms5 | 17 (10) | 24 (11) | 14 (5) | 10 (5) *** |
| Prenatal anxiety6 | 24 (16%) | 16 (25%) | 7 (19%) | 1 (2%) ** |
| Postpartum MDD7 | 16 (10%) | 12 (19%) | 2 (5%) | 2 (4%) * |
| Prenatal substance use8 | 13 (8%) | 7 (11%) | 4 (10%) | 2 (4%) |
| Pre-pregnancy BMI9 | 26 (6) | 26 (8) | 27 (5) | 25 (5) |
| Attachment to infant10 | 3.9 (.5) | 3.8 (.6) | 4.0 (.4) | 3.9 (.4) |
| Infant Characteristics | ||||
| Sex (% female) | 73 (48%) | 28 (44%) | 19 (49%) | 26 (52%) |
| Delivery Mode (% vaginal) | 111 (73%) | 49 (77%) | 25 (64%) | 37 (74%) |
| Gestational age at birth (wks) | 40 (1) | 40 (1) | 40 (1) | 40 (1) |
| Infant birthweight percentile11 | 45 (25) | 42 (26) | 47 (24) | 46 (26) |
| Infant Apgar at 5 min ≥ 9 | 138 (90%) | 56 (90%) | 38 (97%) | 44 (88%) |
NOTE: p<.05;
p<.01,
p<.001.
MDD=major depressive disorder;
Percentages are calculated based on available data for each variable.
Based on a score of 4 or 5 on the Hollingshead Index;
Number of pregnancies (including current);
Number of depressive symptoms measured on the Inventory for Depressive Symptoms, Self Report version (IDS-SR) averaged over three pregnancy interviews;
Diagnosed anxiety disorder during pregnancy measured by structured clinical interview;
Diagnosed depressive episode during the postpartum period measured by structured clinical interview;
>1 cigarette or drink per week, or meconium positive for nicotine or marijuana metabolites;
BMI=Body Mass Index.
Measured by Maternal Attachment Inventory completed one month postpartum;
Birth weight percentile for gestational age.
RESULTS
Sample Characteristics
Descriptive statistics for the overall sample and stratified by MDD group are presented in Table 1. Participants ranged in age from 18-40 (M=26, SD=5) and were racially and ethnically diverse: 26% Hispanic, 16% African American, 12% Multiracial/Other; 46% Non-Hispanic White). The sample was primarily low socioeconomic status: 85% of mothers had an annual income < $30,000/year and 56% were unemployed. Fifty-eight percent of mothers were unmarried; 58% of pregnancies were unplanned. For infants, average gestational age was 40±1 weeks; average birth weight was 3,366±449 grams; average Agpar score was 9.
Mothers in the prenatal MDD group had more pregnancies, increased depressive symptoms, and were more likely to have been diagnosed with an anxiety disorder in pregnancy or a postpartum depressive episode vs. those in the Preconception-only MDD and Control groups. Mothers in the Preconception-only MDD group were older, less diverse, more educated, and of higher SES than those in the Prenatal MDD and Control groups. Mothers in the three depression groups did not differ in substance use, pre-pregnancy body mass index, or attachment to their infants at one month. Infants from the three maternal depression groups did not differ significantly in GA at birth, birth weight percentile, Apgar scores >8, or mode of delivery.
Mothers in the Prenatal MDD group experienced an average of 1 depressive episode during pregnancy (range: 1-3 episodes; 78% major, 22% minor depressive episodes). Median episode duration was 12 weeks (Inter-Quartile Range (IQR)=6-19) for major episodes and 5 weeks (IQR=3-7) for minor episodes. Fifty four percent of mothers in the Prenatal MDD group had experienced one or more episodes of depression prior to pregnancy. By definition, all mothers in the Preconception-only MDD group had experienced at least one depressive episode prior to pregnancy. Prenatal and Preconception-only MDD groups did not differ in terms of proportions of mothers experiencing two or more prior depressive episodes, prenatal or preconception anxiety disorders.
Effects of MDD group on infant cortisol response
Baseline cortisol (Med=.23, IQR=.14-.36, skewness=3.12) and AUC cortisol (Med=15.8, IQR=9.7-28.7, skewness=2.06) were successfully symmetrized via a logarithmic transformation (ln baseline: M=−1.44, SD=.83, skew=.47; ln AUC: M=2.82, SD=.72, skew=.19). Table 2 and Figure 1 show MDD group effects on infant baseline and AUC cortisol response for the overall sample and stratified by daughters and sons.
After entry of relevant covariates (maternal education, time since feeding), significant differences in AUC cortisol emerged between the Preconception-only and Prenatal MDD groups (β=−.37, SE=.18, p=.04) in the overall sample, with infants in the Prenatal MDD group showing 45% increased AUC cortisol vs. infants in the Preconception-only MDD group (Ratio=1.45, 95% CI= 1.02-2.05). No significant group differences emerged for baseline cortisol in the overall sample.
Although the interaction of infant sex and MDD group contrasts did not attain significance for either baseline or AUC cortisol (p’s>.10), stratifying by infant sex revealed significant differences in baseline cortisol between Control and Prenatal MDD groups for daughters only (β=−.42, SE=.21, p=.045); daughters of mothers with Prenatal MDD had 51% higher baseline cortisol than daughters of Controls (Ratio=1.51, 95% CI=1.01-2.27). Although differences were not statistically significant (p=.095), daughters of Prenatal MDD mothers showed 49% higher baseline cortisol vs. girls in the Preconception MDD group (Ratio=1.49, 95% CI=.93-2.36). Significant Control vs. Prenatal MDD (β=−.50, SE=.23, p=.03) and Preconception-only vs. Prenatal MDD (β=−.56, SE=.26, p=.04) differences also emerged for AUC cortisol, with daughters in the Prenatal MDD group showing 64% increased AUC cortisol vs. Controls (Ratio=1.64, 95% CI=1.05-2.56) and 75% increased AUC cortisol vs. daughters in the Preconception-only MDD group (Ratio=1.75, 95% CI=1.04-2.94). No significant effects of MDD group emerged for sons for either baseline or AUC cortisol (p’s>.49).
No significant main effects of continuous depressive symptoms emerged in the overall and stratified samples for baseline or AUC cortisol.
Interaction of MDD group and HSD11B2 promoter methylation on infant cortisol response.
HSD11B2 methylation was symmetrically distributed (M=12.8, SD=3.3, skew=−.14, range 5-20), similar to prior studies (37). No significant main effects of MDD group or continuous depressive symptoms emerged on HSD11B2 methylation. Table 3 shows MDD group by HSD11B2 methylation interactions for infant baseline cortisol response for the overall and sex-stratified samples.
Table 3.
Interaction of prenatal depression group and placental HSD11B2 methylation for baseline cortisol at one month for the overall sample (n=94) and stratified by daughters (n=42) and sons (n=52).1
| All |
Daughters |
Sons |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictor variables | B | SE | p | β | SE | p | β | SE | p |
| Baseline Cortisol2 | |||||||||
| Intercept | .78 | .64 | .22 | 1.87 | .77 | .02 | .23 | 1.00 | .82 |
| Time since feeding (min)2 | −.27 | .10 | .01 | −.64 | .15 | <.001 | −.08 | .14 | .58 |
| Education3 | −.53 | .18 | .003 | −.64 | .26 | .02 | −.53 | .24 | .03 |
| Control vs. Prenatal MDD4 | −1.80 | .73 | .02 | −2.58 | .89 | .006 | −1.09 | 1.20 | .37 |
| Preconception-only MDD vs. Prenatal MDD4 | −1.45 | .80 | .07 | −1.39 | .89 | .13 | −.92 | 1.37 | .51 |
| HSD11B2 Methylation | −.08 | .04 | .02 | −.05 | .04 | .21 | −.10 | .06 | .09 |
| [Control vs. Prenatal MDD4] * HSD11B2 | .13 | .05 | .02 | .16 | .06 | .01 | .08 | .09 | .38 |
| [Preconception vs. Prenatal MDD4] * HSD11B2 | .10 | .06 | .10 | .07 | .07 | .35 | .08 | .10 | .43 |
Per group cell sizes for the overall sample were n’s=43, 21, 30 for Prenatal MDD, Preconception-only MDD, and Control, respectively; per group cell sizes for daughters were n’s=16, 10, and 16, respectively; per group cell sizes for sons were n’s=27, 11, and 14. Unstandardized regression coefficients are presented.
Natural log transformed.
≤ high school degree vs. > high school degree.
MDD=major depressive disorder; contrasts are based on categorical MDD groups.
A significant interaction of Control vs. Prenatal MDD groups and HSD11B2 promoter methylation emerged for baseline cortisol (β=.13, SE=.05, p=.02), such that significant associations emerged between HSD11B2 methylation and baseline cortisol in the Prenatal MDD group (β=−.08, SE=.04, p=.02), but not in the Control group (β=.05, SE=.04, p=.26). No association emerged for the Preconception-only MDD group (β=.02, SE=.05, p=.74). Exponentiation of methylation effects in the Prenatal MDD group revealed a 9% increase in baseline cortisol per one-percentile decrease in HSD11B2 methylation levels (Ratio=1.09, 95% CI=1.01-1.16). Consistent with Figure 2, region of significance analyses across the 5% to 20% range of observed methylation levels revealed that Control vs. Prenatal MDD differences were only evident at lower levels of methylation; group contrasts were no longer significant above 11% methylation, and were nullified at 14% methylation. No significant interaction emerged between MDD groups and HSD11B2 methylation and AUC cortisol.
Although the interaction of sex X MDD groups X HSD11B2 did not attain significance for baseline or AUC cortisol (p’s>.50), stratifying baseline cortisol analyses by infant sex revealed qualitatively similar MDD group by HSD11B2 methylation interactions for both sexes that only attained significance among daughters (Control vs. Prenatal MDD contrast: β=.16, SE=.06, p=.01). Paralleling MDD group findings, an interaction between continuous depressive symptoms and HSD11B2 emerged for baseline cortisol in daughters only (β=−.008, SE=.004, p=.03) with effects of continuous depressive symptoms only evident at lower levels of methylation and nullified at 14% methylation levels.
Interaction of MDD group and SLC6A4 gene expression on infant cortisol response.
SLC6A4/18S gene expression was skewed (Med=1.03, IQR=.57-1.92, skew=4.42), but was successfully symmetrized via a logarithmic transformation (M=.03, SD=.94, skew=.13) similar to prior studies (33). No significant main effects of MDD group or continuous depressive symptoms emerged on SLC6A4 gene expression. No significant interactions emerged between MDD groups and SLC6A4 gene expression on baseline cortisol in overall or stratified samples. Table 4 shows MDD group by SLC6A4 expression interactions on AUC cortisol for overall and sex-stratified samples.
For AUC cortisol, a statistically significant interaction of infant sex by (Control vs. Prenatal MDD groups) by SLC6A4 expression levels emerged (β=−.78, SE=.36, p=.03), such that AUC cortisol differed among sons in the Control vs. Prenatal MDD groups (β=.61, SE=.22, p=.007), but not among daughters. Specifically, a significant association between SLC6A4 expression and infant AUC cortisol for sons emerged in the Control group (β=.46, SE=.16, p=.007), but not in the Prenatal MDD group (β=−.16, SE=.13, p=.24). No association was observed in the Preconception-only MDD group (β=.15, SE=.23, p=.51). Exponentiation of SLC6A4 effect for control sons revealed that a tenfold increase in expression levels from .1 to 1.0, (approximate sample minimum to median), was associated with a nearly threefold increase in AUC cortisol (Ratio=2.87, 95% CI=1.39-5.93). Region of significance testing suggested that Control vs. Prenatal MDD contrasts were only evident at very low or high levels of expression; group contrasts were no longer significant for expression levels in the region .34-2.5, and were nullified at the sample median of 1.0 (Ratio =.98, 95% CI=.65-1.49). Specifically, AUC cortisol was about 4X higher in sons exposed to Prenatal MDD than Control sons at expression levels of .1 (sample minimum) and 4X higher in Controls than sons exposed to Prenatal MDD at expression levels of 10 (sample maximum) (Ratio=4.11, 95% CI=1.55-10.90).
No significant interaction of continuous depressive symptoms and SLC6A4 on AUC or baseline cortisol emerged for the overall or sex-stratified samples.
DISCUSSION
Exposure to maternal depression is one of the most common forms of early adversity, with known links to long-term health and behavioral outcomes. However, mechanisms remain unclear. To our knowledge, the present study is the first to reveal unique associations between prenatal major depressive disorder (MDD) versus preconception history of MDD on offspring cortisol response in the neonatal period. Our study is also the first to investigate placental glucocorticoid (epigenetic regulation of HSD11B2) and serotonin (SLC6A4 gene expression) signaling pathways as moderators of links between prenatal MDD exposure and offspring cortisol. As hypothesized, we found that both placental HSD11B2 methylation and SLC6A4 gene expression moderated the influence of prenatal MDD on infant cortisol regulation. All findings were influenced by fetal sex. Effects of prenatal MDD on infant cortisol and modulating effects of HSD11B2 were strongest for newborn daughters, while moderating effects of SLC6A4 emerged only in sons.
Our study revealed unique programming effects of prenatal depressive disorder on newborn glucocorticoid regulation, with newborn daughters exposed to prenatal MDD showing approximately fifty to seventy-five percent increases in baseline and stress-responsive cortisol versus daughters of controls and daughters of mothers with preconception MDD only. Although significant differences in stress-responsive cortisol between preconception and prenatal MDD emerged in the overall sample (daughters and sons), overall effects were driven by stronger effects in daughters and only weak effects in sons. Results support our hypothesis that prenatal depressive disorders versus prior history of maternal depression, and potentially, genetic/familial liability to depression, program the fetal HPA axis. Our focus on the neonatal period, the lack of maternal attachment differences between groups at one month, and the lack of influence of postnatal MDD on infant cortisol (covariate analyses), also point to an influence of prenatal versus postpartum MDD in programming the newborn HPA axis. Results extend prior findings of links between prenatal depressive symptoms and increased urine basal cortisol in the immediate newborn period (first postnatal week) in several normative samples (24-27) as well as increased saliva cortisol response to challenge at 6 months in relation to peripartum depressive disorders (31). Future research is needed focused on timing of depressive episodes during pregnancy in relation to infant cortisol regulation.
As hypothesized, placental 11β HSD2, moderated effects of prenatal MDD group on infant cortisol. Specifically, we found significant associations between HSD11B2 methylation and infant baseline cortisol for infants of mothers who experienced prenatal MDD. In contrast, alterations in HSD11B2 did not influence infant cortisol when mothers did not experience a depressive episode during pregnancy. Effects of prenatal depression were most pronounced at low levels of methylation (likely linked to higher levels of expression) and no longer evident at higher levels of methylation (lower levels of expression). A prior study from our group also demonstrated an interaction between methylation of placental HSD11B2 with maternal psychopathology (chart-documented anxiety) in predicting newborn muscle tension (32). Both studies showing effects of methylation in the psychopathology vs. control group; however, decreased methylation was associated with decreased hypotonicity (32) vs. increased cortisol in the present study. Contrasting directions of results may be due to differential outcomes (cortisol vs. muscle tone). Future studies are needed to reconcile differential findings for neurobehavioral vs. neuroendocrine outcomes; however, both studies highlight the importance of HSD11B2 activity in modulating effects of maternal prenatal psychopathology on the infant.
Increased maternal glucocorticoids have been posited as a key mechanism underlying transmission of maternal adversity to the fetus in both human and animal models. Maternal glucocorticoids have independently been linked to risk for child and adult psychopathology/affective states, health outcomes, as well as brain development (67-69). For example, Buss and colleagues (67) revealed links between maternal prenatal cortisol and both increased affective problems and altered right amygdalar volume (potential biomarker of depression) in 7-year-old girls; they further showed that amygdalar volume mediated the link between prenatal glucocorticoid exposure and increased childhood affective problems. Negative associations between HSD11B2 methylation and infant cortisol levels in the depressed group in the present study suggest a compensatory mechanism to protect the fetus from increased maternal glucocorticoids from prenatal MDD.
Placental 11β HSD2 activity has been shown to increase over pregnancy then to taper close to gestation (70, 71). In the present study, methylation of placental HSD11B2 was measured at birth. Because methylation tends to represent a longer-term process, it is possible that HSD11B2 methylation levels at birth represent the culmination of all changes over the course of pregnancy, or, alternatively, only a portion of the prenatal period. Future studies and technological advances are needed to elucidate the time course of placental HSD11B2 methylation across pregnancy in relation to birth methylation levels and in relation to the timing of depressive episodes over pregnancy.
Similar to HSD11B2 methylation, we found that placental SLC6A4 gene expression moderated effects of prenatal MDD group on infant cortisol, but for sons only. Further, results suggest a greater influence of prenatal depression on infant cortisol when available placental serotonin was low. Our study highlights the importance of placental serotonin signaling in modulating prenatal programming of the fetal HPA axis by maternal adversity. Results extend a prior study by our group demonstrating direct effects of chart-documented maternal depression and anxiety on placental SLC6A4 gene expression (33), and recent work by Raikonnen and colleagues in a normative sample demonstrating associations between SLC6A4 gene expression and decreased infant regulatory behaviors (72), but no direct association between prenatal depressive symptoms and SLC6A4 (42). Our study suggests that discrepancies in direct effects of prenatal depression on SLC6A4 between past studies may be due to a moderating effect of placental SLC6A4 and infant sex on links between prenatal depression and infant outcomes.
Effects of prenatal MDD on infant cortisol and moderating effects of placental HSD11B2 emerged most strongly for newborn daughters, whereas direct and moderating effects of SLC6A4 were evident only for sons. Sex differences have been shown in maternal glucocorticoid regulation (depending on fetal sex), in placental structure and function, the placental transcriptome and epigenome, and in offspring outcomes following prenatal stress (44-46, 73). It has been proposed that while males are at greater risk for mortality and morbidity (viability) from prenatal exposure to adversity, emerging evidence suggests that females’ adaptive flexibility in the face of prenatal adversity increases vulnerability to a reactive endophenotype that is linked to risk for affective disorders (44). Our study highlights the possibility in humans that differing placental signaling pathways (e.g., glucocorticoid versus serotonin) may modulate effects of prenatal insults for daughters and sons. Longitudinal extensions of our study may reveal insight into consistent sex differences in depressive symptoms and disorders (females>males). We can speculate that if placental SLC6A4 expression parallels expression of SLC6A4 and serotonin in the fetal brain that the positive associations between SLC6A4 expression and infant cortisol in boys may be related to decreased “serotonergic vulnerability” to depression (74) in boys, whereas altered glucocorticoid levels and signaling pathways may be linked to increase vulnerability to depression in girls (67).
Given the complex nature of the findings and small group sizes for sex-stratified analyses, replication of these findings in an independent and larger cohort is needed. An additional limitation of the present study was that we included HSD11B2 methylation but not gene expression, and SLC6A4 gene expression but not methylation. Relatedly, our study focused on two specific genes involved in placental glucocorticoid and serotonergic signaling. Clearly, other placental genes along these and other pathways are likely to be involved in the intergenerational transmission of adversity. Third, mothers in the preconception MDD group differed on several demographic characteristics from mothers in the prenatal MDD and control groups (education, age, race/ethnicity). Although relevant confounders were statistically controlled, prenatal and preconception MDD groups did not differ in number of past depressive episodes or comorbid anxiety, and our analyses revealed programming differences between prenatal MDD and both preconception MDD and controls, it is possible that these demographic characteristics also influenced prenatal programming pathways. Finally, although the preconception MDD group was included to control for heightened genetic/familial risk for depression in the prenatal MDD group, our study did not include a genetically-sensitive design or allelic variation in the serotonin transporter.
Conclusions
This intensive, prospective study provides evidence for unique programming effects of prenatal major depressive disorder on neonatal cortisol response. Exposure to prenatal, but not preconception-only MDD led to increased baseline and stress-responsive cortisol--especially in newborn daughters. We found initial evidence that placental glucocorticoid and serotonergic pathways moderated effects of prenatal MDD on infant cortisol. Modulating effects were most pronounced for daughters for HSD11B2, and emerged for sons only for SLC6A4. Results support placental glucocorticoid and serotonergic signaling pathways as promising modulators of the influence of and maternal adversity on offspring, and, potentially, key modifiable therapeutic targets for identification of and intervention with high risk offspring.
Table 2b.
Maternal prenatal depression group and AUC1 cortisol at one month for the overall sample (n=114), and stratified by daughters (n=56) and sons (n=58) 2.
| All |
Daughters |
Sons |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Predictor variables | β | SE | p | β | SE | p | β | SE | p |
| AUC Cortisol1,3 | |||||||||
| Intercept | 2.52 | .34 | <.001 | 3.29 | .55 | <.001 | 1.99 | .44 | <.001 |
| Time since feeding (min)3 | .16 | .09 | .06 | .00 | .14 | 1.00 | .28 | .11 | .02 |
| Education4 | −.32 | .14 | .02 | −.21 | .23 | .37 | −.42 | .19 | .03 |
| Control vs. Prenatal MDD5 | −.21 | .15 | .18 | −.50 | .23 | .03 | .02 | .22 | .92 |
| Preconception-only MDD vs. Prenatal MDD5 | −.37 | .18 | .04 | −.56 | .26 | .04 | −.18 | .26 | .49 |
AUC=Area Under the Curve.
Per group cell sizes for the overall sample were n’s=48, 26, 40 for Prenatal MDD, Preconception-only MDD, and Control, respectively; per group cell sizes for daughters were n’s =18, 15, and 23, respectively; per group cell sizes for sons were n’s=30, 11, and 17. Unstandardized regression coefficients are presented.
Natural log transformed.
≤ high school degree vs. > high school degree.
MDD=major depressive disorder; contrasts are based on categorical MDD groups.
Acknowledgments
Conflicts of Interest and Source of Funding: No conflicts of interest were declared. Preparation of this manuscript was supported by the National Institutes of Health (R01 MH079153 to L.R.S.). We gratefully acknowledge the families who contributed to this study and the Maternal-Infant Studies Laboratory staff for their assistance with data collection.
Acronyms:
- MDD
major depressive disorder
- NNNS
NICU Network Neurobehavioral Scale
REFERENCES
- 1.Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005;106:1071–83. [DOI] [PubMed] [Google Scholar]
- 2.Marcus SM, Flynn HA, Blow FC, Barry KL. Depressive symptoms among pregnant women screened in obstetrics settings. J Womens Health (Larchmt). 2003;12:373–80. [DOI] [PubMed] [Google Scholar]
- 3.Le Strat Y, Dubertret C, Le Foll B. Prevalence and correlates of major depressive episode in pregnant and postpartum women in the United States. J Affect Disord. 2011;135:128–38. [DOI] [PubMed] [Google Scholar]
- 4.Grigoriadis S, VonderPorten EH, Mamisashvili L, Tomlinson G, Dennis CL, Koren G, Steiner M, Mousmanis P, Cheung A, Radford K, Martinovic J, Ross LE. The impact of maternal depression during pregnancy on perinatal outcomes: a systematic review and meta-analysis. J Clin Psychiatry. 2013;74:e321–41. [DOI] [PubMed] [Google Scholar]
- 5.Grote NK, Bridge JA, Gavin AR, Melville JL, Iyengar S, Katon WJ. A meta-analysis of depression during pregnancy and the risk of preterm birth, low birth weight, and intrauterine growth restriction. Arch Gen Psychiatry. 2010;67:1012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allister L, Lester BM, Carr S, Liu J. The effects of maternal depression on fetal heart rate response to vibroacoustic stimulation. Dev Neuropsychol. 2001;20:639–51. [DOI] [PubMed] [Google Scholar]
- 7.Monk C, Sloan RP, Myers MM, Ellman L, Werner E, Jeon J, Tager F, Fifer WP. Fetal heart rate reactivity differs by women’s psychiatric status: an early marker for developmental risk? J Am Acad Child Adolesc Psychiatry. 2004;43:283–90. [DOI] [PubMed] [Google Scholar]
- 8.Salisbury AL, Wisner KL, Pearlstein T, Battle CL, Stroud L, Lester BM. Newborn neurobehavioral patterns are differentially related to prenatal maternal major depressive disorder and serotonin reuptake inhibitor treatment. Depress Anxiety. 2011;28:1008–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salisbury AL, O’Grady KE, Battle CL, Wisner KL, Anderson GM, Stroud LR, Miller-Loncar CL, Young ME, Lester BM. The Roles of Maternal Depression, Serotonin Reuptake Inhibitor Treatment, and Concomitant Benzodiazepine Use on Infant Neurobehavioral Functioning Over the First Postnatal Month. Am J Psychiatry. 2016;173:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones NA, Field T, Fox NA, Lundy B, Davalos M. EEG activation in 1-month-old infants of depressed mothers. Dev Psychopathol. 1997;9:491–505. [DOI] [PubMed] [Google Scholar]
- 11.Rifkin-Graboi A, Bai J, Chen H, Hameed WB, Sim LW, Tint MT, Leutscher-Broekman B, Chong YS, Gluckman PD, Fortier MV, Meaney MJ, Qiu A. Prenatal maternal depression associates with microstructure of right amygdala in neonates at birth. Biol Psychiatry. 2013;74:837–44. [DOI] [PubMed] [Google Scholar]
- 12.Davis EP, Glynn LM, Schetter CD, Hobel C, Chicz-Demet A, Sandman CA. Prenatal exposure to maternal depression and cortisol influences infant temperament. J Am Acad Child Adolesc Psychiatry. 2007;46:737–46. [DOI] [PubMed] [Google Scholar]
- 13.Stein A, Pearson RM, Goodman SH, Rapa E, Rahman A, McCallum M, Howard LM, Pariante CM. Effects of perinatal mental disorders on the fetus and child. Lancet. 2014;384:1800–19. [DOI] [PubMed] [Google Scholar]
- 14.Gentile S Untreated depression during pregnancy: Short- and long-term effects in offspring. A systematic review. Neuroscience. 2015. [DOI] [PubMed] [Google Scholar]
- 15.Pearson RM, Evans J, Kounali D, Lewis G, Heron J, Ramchandani PG, O’Connor TG, Stein A. Maternal depression during pregnancy and the postnatal period: risks and possible mechanisms for offspring depression at age 18 years. JAMA Psychiatry. 2013;70:1312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandman CA, Buss C, Head K, Davis EP. Fetal exposure to maternal depressive symptoms is associated with cortical thickness in late childhood. Biol Psychiatry. 2015;77:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gelman PL, Flores-Ramos M, Lopez-Martinez M, Fuentes CC, Grajeda JP. Hypothalamic-pituitary-adrenal axis function during perinatal depression. Neurosci Bull. 2015;31:338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beijers R, Buitelaar JK, de Weerth C. Mechanisms underlying the effects of prenatal psychosocial stress on child outcomes: beyond the HPA axis. Eur Child Adolesc Psychiatry. 2014;23:943–56. [DOI] [PubMed] [Google Scholar]
- 19.Sandman CA, Davis EP, Buss C, Glynn LM. Exposure to prenatal psychobiological stress exerts programming influences on the mother and her fetus. Neuroendocrinology. 2012;95:7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandman CA, Davis EP, Buss C, Glynn LM. Prenatal programming of human neurological function. Int J Pept. 2011;2011:837596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cottrell EC, Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 2009;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seckl JR. Glucocorticoids, developmental ‘programming’ and the risk of affective dysfunction. Prog Brain Res. 2008;167:17–34. [DOI] [PubMed] [Google Scholar]
- 23.McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153:2093–101. [PubMed] [Google Scholar]
- 24.Diego MA, Field T, Hernandez-Reif M, Cullen C, Schanberg S, Kuhn C. Prepartum, postpartum, and chronic depression effects on newborns. Psychiatry. 2004;67:63–80. [DOI] [PubMed] [Google Scholar]
- 25.Field -T. Infants of depressed mothers. Infant-Behavior-and-Development. 1995;18:1–13. [DOI] [PubMed] [Google Scholar]
- 26.Lundy B, Jones NA, Field T, Nearing G, Davalos M, Pietro P, Schanberg S, Kuhn C. Prenatal depression effects on neonates. Infant Behavior and Development. 1999;22:119–29. [Google Scholar]
- 27.Field T, Diego M, Hernandez-Reif M, Vera Y, Gil K, Schanberg S, Kuhn C, Gonzalez-Garcia A. Prenatal maternal biochemistry predicts neonatal biochemistry. Int J Neurosci. 2004;114:933–45. [DOI] [PubMed] [Google Scholar]
- 28.Laurent HK, Leve LD, Neiderhiser JM, Natsuaki MN, Shaw DS, Harold GT, Reiss D. Effects of prenatal and postnatal parent depressive symptoms on adopted child HPA regulation: independent and moderated influences. Dev Psychol. 2013;49:876–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Donnell KJ, Glover V, Jenkins J, Browne D, Ben-Shlomo Y, Golding J, O’Connor TG. Prenatal maternal mood is associated with altered diurnal cortisol in adolescence. Psychoneuroendocrinology. 2013;38:1630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laurent HK, Ablow JC, Measelle J. Risky shifts: how the timing and course of mothers’ depressive symptoms across the perinatal period shape their own and infant’s stress response profiles. Dev Psychopathol. 2011;23:521–38. [DOI] [PubMed] [Google Scholar]
- 31.Brennan PA, Pargas R, Walker EF, Green P, Newport DJ, Stowe Z. Maternal depression and infant cortisol: influences of timing, comorbidity and treatment. J Child Psychol Psychiatry. 2008;49:1099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conradt E, Lester BM, Appleton AA, Armstrong DA, Marsit CJ. The roles of DNA methylation of NR3C1 and 11beta-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics. 2013;8:1321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponder KL, Salisbury A, McGonnigal B, Laliberte A, Lester B, Padbury JF. Maternal depression and anxiety are associated with altered gene expression in the human placenta without modification by antidepressant use: implications for fetal programming. Dev Psychobiol. 2011;53:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonnin A, Levitt P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience. 2011;197:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maccani MA, Marsit CJ. Epigenetics in the placenta. Am J Reprod Immunol. 2009;62:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Togher KL, O’Keeffe MM, Khashan AS, Gutierrez H, Kenny LC, O’Keeffe GW. Epigenetic regulation of the placental HSD11B2 barrier and its role as a critical regulator of fetal development. Epigenetics. 2014;9:816–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Appleton AA, Armstrong DA, Lesseur C, Lee J, Padbury JF, Lester BM, Marsit CJ. Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS One. 2013;8:e74691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsit CJ, Maccani MA, Padbury JF, Lester BM. Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome. PLoS One. 2012;7:e33794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. [DOI] [PubMed] [Google Scholar]
- 40.Oberlander TF. Fetal serotonin signaling: setting pathways for early childhood development and behavior. J Adolesc Health. 2012;51:S9–16. [DOI] [PubMed] [Google Scholar]
- 41.Velasquez JC, Goeden N, Bonnin A. Placental serotonin: implications for the developmental effects of SSRIs and maternal depression. Front Cell Neurosci. 2013;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reynolds RM, Pesonen AK, O’Reilly JR, Tuovinen S, Lahti M, Kajantie E, Villa PM, Laivuori H, Hamalainen E, Seckl JR, Raikkonen K. Maternal depressive symptoms throughout pregnancy are associated with increased placental glucocorticoid sensitivity. Psychol Med. 2015:1–8. [DOI] [PubMed] [Google Scholar]
- 43.Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015;39:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandman CA, Glynn LM, Davis EP. Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J Psychosom Res. 2013;75:327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenfeld CS. Sex-Specific Placental Responses in Fetal Development. Endocrinology. 2015;156:3422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stress. 2011;14:348–56. [DOI] [PubMed] [Google Scholar]
- 47.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Non-Patient Edition (SCID-I/NP). New York: Biometrics Research, New York State Psychiatric Institute; 2002. [Google Scholar]
- 48.Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The Inventory of Depressive Symptomatology (IDS): psychometric properties. Psychol Med. 1996;26:477–86. [DOI] [PubMed] [Google Scholar]
- 49.Gottfried AW. Measures of socioeconomic status in child development research: Data and recommendations. Merrill Palmer Q. 1985;31:85–92. [Google Scholar]
- 50.Sobell LC, Sobell MB. Timeline Followback: A technique for assessing self-reported alcohol consumption In: Litten R, Allen J, editors. Measuring alcohol consumption: Psychosocial and biochemical methods. New Jersey: Humana Press; 1992. [Google Scholar]
- 51.Sobell LC, Buchan G, Cleland P, Sobell MB, Fedoroff I, Leo GI, editors. The reliability of the Timeline Followback (TLFB) method as applied to drug, cigarette, and the cannabis use. 30th meeting of the Association for Advancement of Behavior Therapy; 1996; New York, NY. [Google Scholar]
- 52.Lester BM, Tronick EZ. History and description of the Neonatal Intensive Care Unit Network Neurobehavioral Scale. Pediatrics. 2004;113:634–40. [PubMed] [Google Scholar]
- 53.Lester BM, Tronick EZ, Brazelton TB. The Neonatal Intensive Care Unit Network Neurobehavioral Scale procedures. Pediatrics. 2004;113:641–67. [PubMed] [Google Scholar]
- 54.Goldberg S, Levitan R, Leung E, Masellis M, Basile VS, Nemeroff CB, Atkinson L. Cortisol concentrations in 12- to 18-month-old infants: stability over time, location, and stressor. Biol Psychiatry. 2003;54:719–26. [DOI] [PubMed] [Google Scholar]
- 55.Muller ME. A questionnaire to measure mother-to-infant attachment. J Nurs Meas. 1994;2:129–41. [PubMed] [Google Scholar]
- 56.Egliston KA, McMahon C, Austin MP. Stress in pregnancy and infant HPA axis function: conceptual and methodological issues relating to the use of salivary cortisol as an outcome measure. Psychoneuroendocrinology. 2007;32:1–13. [DOI] [PubMed] [Google Scholar]
- 57.Lewis D, Moore C, Becker J, Leikin J. Prevalence of meta-hydroxybenzoylecgonine (m-OH-BZE) in Meconium Samples. Bulletin of the International Association of Forensic Toxicologists. 1995;25:33–6. [Google Scholar]
- 58.Lewis DE, inventor Forensically Acceptable Determinations of Gestational Fetal Exposure to Drugs and Other Chemical Agents patent U.S. Patent Number 5,532,131 1996.
- 59.Moore C, Lewis D, Becker J, Leikin J. The determination of 11-nor-delta 9-tetrahydrocannabinol-9-carboxylic acid (THCCOOH) in meconium. J Anal Toxicol. 1996;20:50–1. [DOI] [PubMed] [Google Scholar]
- 60.Dempsey D, Moore C, Deitermann D, Lewis D, Feeley B, Niedbala RS. The detection of cotinine in hydrolyzed meconium samples. Forensic Sci Int. 1999;102:167–71. [DOI] [PubMed] [Google Scholar]
- 61.Dupont JM, Tost J, Jammes H, Gut IG. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004;333:119–27. [DOI] [PubMed] [Google Scholar]
- 62.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical biochemistry. 1987;162:156–9. [DOI] [PubMed] [Google Scholar]
- 63.Patel P, Boyd CA, Johnston DG, Williamson C. Analysis of GAPDH as a standard for gene expression quantification in human placenta. Placenta. 2002;23:697–8. [DOI] [PubMed] [Google Scholar]
- 64.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology. 2003;28:916–31. [DOI] [PubMed] [Google Scholar]
- 66.Stroud LR, Papandonatos GD, Rodriguez D, McCallum M, Salisbury AL, Phipps MG, Lester B, Huestis MA, Niaura R, Padbury JF, Marsit CJ. Maternal smoking during pregnancy and infant stress response: test of a prenatal programming hypothesis. Psychoneuroendocrinology. 2014;48:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buss C, Davis EP, Shahbaba B, Pruessner JC, Head K, Sandman CA. Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc Natl Acad Sci U S A. 2012;109:E1312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stinson LJ, Stroud LR, Buka SL, Eaton CB, Lu B, Niaura R, Loucks EB. Prospective evaluation of associations between prenatal cortisol and adulthood coronary heart disease risk: the New England family study. Psychosom Med. 2015;77:237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stroud LR, Papandonatos GD, Shenassa E, Rodriguez D, Niaura R, LeWinn KZ, Lipsitt LP, Buka SL. Prenatal glucocorticoids and maternal smoking during pregnancy independently program adult nicotine dependence in daughters: a 40-year prospective study. Biol Psychiatry. 2014;75:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McTernan CL, Draper N, Nicholson H, Chalder SM, Driver P, Hewison M, Kilby MD, Stewart PM. Reduced placental 11beta-hydroxysteroid dehydrogenase type 2 mRNA levels in human pregnancies complicated by intrauterine growth restriction: an analysis of possible mechanisms. J Clin Endocrinol Metab. 2001;86:4979–83. [DOI] [PubMed] [Google Scholar]
- 71.Murphy VE, Clifton VL. Alterations in human placental 11beta-hydroxysteroid dehydrogenase type 1 and 2 with gestational age and labour. Placenta. 2003;24:739–44. [DOI] [PubMed] [Google Scholar]
- 72.Raikkonen K, Pesonen AK, O’Reilly JR, Tuovinen S, Lahti M, Kajantie E, Villa P, Laivuori H, Hamalainen E, Seckl JR, Reynolds RM. Maternal depressive symptoms during pregnancy, placental expression of genes regulating glucocorticoid and serotonin function and infant regulatory behaviors. Psychol Med. 2015;45:3217–26. [DOI] [PubMed] [Google Scholar]
- 73.DiPietro JA, Costigan KA, Kivlighan KT, Chen P, Laudenslager ML. Maternal salivary cortisol differs by fetal sex during the second half of pregnancy. Psychoneuroendocrinology. 2011;36:588–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jans LA, Riedel WJ, Markus CR, Blokland A. Serotonergic vulnerability and depression: assumptions, experimental evidence and implications. Mol Psychiatry. 2007;12:522–43. [DOI] [PubMed] [Google Scholar]