

SUMMARY
The diversity of mRNA lifetimes in bacterial cells is difficult to reconcile with the relaxed cleavage-site specificity of RNase E, the endonuclease most important for governing mRNA degradation. This enzyme has generally been thought to locate cleavage sites by searching freely in three dimensions. However, our results now show that its access to such sites in 5′-monophosphorylated RNA is hindered by obstacles, such as bound proteins or ribosomes or coaxial sRNA base pairing, that disrupt the path from the 5′ end to those sites and prolong mRNA lifetimes. These findings suggest that RNase E searches for cleavage sites by scanning linearly from the 5′-terminal monophosphate along single-stranded regions of RNA and that its progress is impeded by structural discontinuities encountered along the way. This discovery has major implications for gene regulation in bacteria and suggests a general mechanism by which other prokaryotic and eukaryotic regulatory proteins can be controlled.
Keywords: RNA decay, RNA processing, RNase G, ribonuclease, 5′ terminus, ribosome, uORF, SgrS, yigL, phosphosugar stress
Graphical Abstract
eTOC blurb
Richards and Belasco show that mRNA lifetimes in E. coli are controlled by various obstacles that hinder RNase E access to downstream cleavage sites. Their findings suggest that this regulatory endonuclease searches for cutting sites in 5′-monophosphorylated RNA by one-dimensional diffusion from the 5′ end along single-stranded regions.
INTRODUCTION
Messenger RNA degradation is among the most important mechanisms for controlling gene expression in all organisms. Along with transcription, it directly impacts protein synthesis by governing the concentration of every mRNA in a cell.
mRNA lifetimes vary widely, ranging from minutes to days in higher eukaryotes and from seconds to as long as an hour in bacteria (Belasco, 2010). The half-life of a given transcript depends not only on its identity but also on cell type, growth conditions, and environmental signals (Hui et al., 2014). Moreover, different segments within the same transcript can have distinct lifetimes, especially in bacteria.
In Escherichia coli, the ribonuclease with the greatest influence on mRNA lifetimes is RNase E, an essential endonuclease that is also important for the maturation of rRNA and tRNA and is present in most other bacteria, including a variety of pathogens (Mackie, 2013b; Hui et al., 2014). RNase E is a large enzyme comprising an N-terminal domain that is the source of its catalytic activity and a membrane-associated C-terminal domain that serves as a scaffold for the assembly of a multienzyme complex known as the RNA degradosome.
RNase E cuts RNA with only modest sequence specificity in single-stranded regions that are AU-rich while shunning regions that are C-rich (McDowall et al., 1994; Lin-Chao et al., 1994; Kaberdin, 2003; Del Campo et al., 2015; Richards and Belasco, 2016; Chao et al., 2017). It can gain access to internal cleavage sites in primary transcripts either directly or by a 5′-end-dependent mechanism. Cleavage by direct access needs no prior modification of the transcript (Deana et al., 2008). By contrast, 5′-end-dependent access requires prior conversion of the 5′-terminal triphosphate to a monophosphate, a two-step process involving a diphosphorylated intermediate from which the β phosphate is removed by the RNA pyrophosphohydrolase RppH to generate a monophosphorylated 5′ end that accelerates subsequent internal cleavage by RNase E (Mackie, 1998; Celesnik et al., 2007; Deana et al., 2008; Luciano et al., 2017). Regardless of the access mechanism, the initial endonucleolytic event generates two products: (i) a 5′ fragment that lacks a protective 3′-terminal stem-loop and is therefore susceptible to rapid 3′-exonucleolytic degradation and (ii) a 3′ fragment that is monophosphorylated and therefore vulnerable to further rapid attack by RNase E.
The preference of this endonuclease for monophosphorylated substrates is a consequence of a discrete 5′-end-binding pocket on the surface of the protein (Callaghan et al., 2005; Garrey et al., 2009). This pocket, which is distinct from the catalytic center, can bind a 5′ end that is monophosphorylated but apparently not one bearing a triphosphate, diphosphate, or hydroxyl (Jiang and Belasco, 2004; Luciano et al., 2017). The productive engagement of a 5′ monophosphate there requires at least two unpaired 5′-terminal nucleotides and typically results in a 10-100 fold increase in the rate of cleavage at sites that are sometimes far downstream (Richards and Belasco, 2016).
E. coli cells also contain a closely related nonessential endonuclease, RNase G, which comprises a single structural domain homologous to the catalytic domain of RNase E (McDowall et al., 1993; Li et al., 1999; Wachi et al., 1999). RNase G shares many of the properties of RNase E, including its modest cleavage-site specificity and preference for monophosphorylated substrates (Jiang et al., 2000; Tock et al., 2000; Richards and Belasco, 2016). Because of the similarities between these two endonucleases, it is not unusual for both to participate in the degradation of a given mRNA in E. coli (Ow et al., 2003; Luciano et al., 2012). However, the influence of RNase E is usually greater due to its much higher cellular concentration (Lee et al., 2002).
Because the cleavage-site specificity of RNase E is so relaxed, mRNAs typically contain many sites where this enzyme can cut (Clarke et al., 2014; Chao et al., 2017), no one of which is usually essential for rapid decay. Nevertheless, the lifetimes of E. coli mRNAs can differ greatly from one another, as can the degradation rates of monophosphorylated mRNA decay intermediates, whose reported half-lives range from ≤1 min to 15 min or more (Båga et al., 1988; Richards et al., 2012; Lodato et al., 2012). Furthermore, it is not uncommon for mRNA lifetimes to be prolonged in a translation-independent manner by the binding of a trans-acting regulatory factor such as an sRNA or protein that impedes RNase E cleavage by a mechanism that remains unclear (Papenfort et al., 2013; Fröhlich et al., 2013; Michaux et al., 2017; Zhang et al., 2018). Together, these observations suggest that mRNA longevity is determined not by the number or intrinsic quality of internal cleavage sites but rather by the ease with which RNase E can gain access to them. However, the RNA characteristics that govern access are poorly understood.
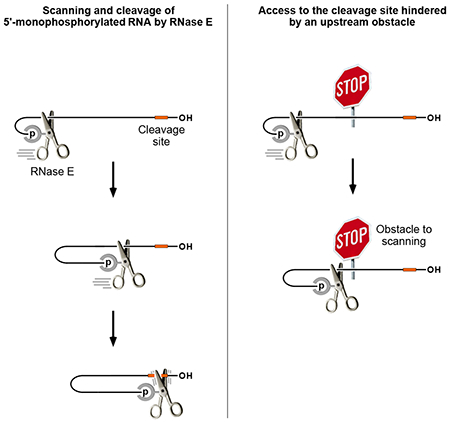
In an effort to address this conundrum, we decided to re-evaluate the common assumption that RNase E bound to a monophosphorylated 5′ end is spatially unconstrained as it seeks potential cleavage sites downstream. Our results show that the ability of this endonuclease to access such sites can be hindered by a variety of intervening structural obstacles. This discovery suggests that, rather than searching freely in three dimensions, RNase E tracks linearly along single-stranded regions of RNA as it scans from the 5′ end for sites of cleavage. By clarifying the molecular mechanisms that govern access by RNase E, these discoveries reveal previously unrecognized parameters that control gene expression post-transcriptionally.
RESULTS
Reporter transcript for testing potential obstacles to scanning by RNase E
The mechanism by which RNase E locates cleavage sites in monophosphorylated RNA is not known. Although it is generally assumed to search in three dimensions, it remains possible that this endonuclease instead scans linearly from 5′ to 3′. Only the latter mechanism should be susceptible to hindrance by obtacles encountered along the way.
To investigate whether 5′-monophosphate-assisted cleavage by RNase E is influenced by structural features between the 5′ end and cutting sites, we devised a reporter transcript (AC3) whose 5′ untranslated region (UTR) contained several RNase E cleavage sites flanking a locus where potential obstacles to communication between the 5′ monophosphate and downstream cleavage sites could be inserted. This reporter was derived from E. coli yeiP mRNA, which encodes a paralog of elongation factor EF-P and is rapidly degraded by a 5′-end-dependent mechanism (half-life of 1.2 min in wild-type cells versus 7.2 min in ΔrppH cells, where the 5′ triphosphate of the full-length transcript cannot be converted to a monophosphate) (Richards et al., 2012). The yeiP 5′ UTR is unstructured and contains multiple sites whose cleavage by RNase E and RNase G is greatly enhanced (≥13-fold) by prior conversion of the 5′-terminal triphosphate to a monophosphate (Richards et al., 2012; Richards and Belasco, 2016). To create AC3, an additional copy of the 24-nt yeiP 5′ UTR segment upstream of the ribosome-binding site, an RNase E/G-resistant (AC)3 spacer, a BglII restriction site, and an (AC)5 spacer were fused in tandem to the 5′ end of yeiP mRNA. As a result, the 5′ UTR of AC3 contained duplicate copies of most of the yeiP cleavage sites on either side of a BglII site where structural impediments could be inserted (Figure 1A).
Figure 1. Protection of downstream mRNA cleavage sites in E. coli by discontinuities that result from protein binding.
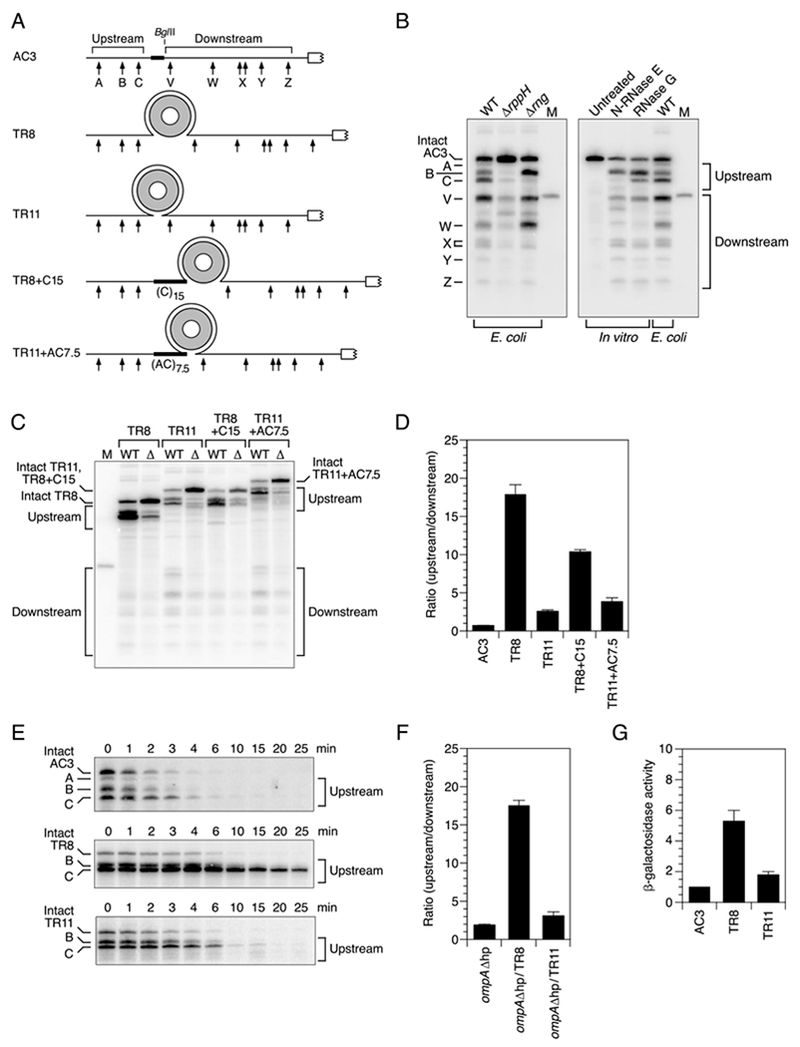
(A) 5′ UTR of reporter mRNAs. Arrows, cleavage sites; gray ring, TRAP 11-mer; white rectangle, beginning of the protein coding region; broad black lines, heteropolymeric (AC)3 or (AC)7.5 or homopolymeric (C)15 spacers. The regions encompassing upstream sites A, B, and C and downstream sites W, X, and Y are identical in sequence. See also Figure S1.
(B) Endonucleolytic cleavage of the 5′ UTR of AC3 in vivo and in vitro. Equal amounts of total cellular RNA from isogenic wild-type (WT), ΔrppH, or Δrng strains of E. coli containing AC3 mRNA were examined by Northern blotting to detect cleavage within the AC3 5′ UTR. Alternatively, monophosphorylated AC3 RNA synthesized by in vitro transcription was partially digested with purified N-RNase E (20 nM) or RNase G (80 nM) and analyzed similarly. The blots were probed with a radiolabeled oligonucleotide complementary to an AC3 segment downstream of site Z (Table S2). M, boundary marker between the upstream and downstream cleavage sites.
(C) Cleavage within the 5′ UTR of reporter mRNAs in vivo. Equal amounts of total cellular RNA from isogenic wild-type (WT) and ΔrppH (Δ) strains of E. coli containing each reporter mRNA (TR8, TR11, TR8+C15, or TR11+AC7.5) and TRAP were analyzed by Northern blotting as in panel (B). See also Figure S1.
(D) Relative abundance of 5′ UTR cleavage products in wild-type cells. The sum of the intensities of the bands in panel (C) resulting from cleavage upstream of the obstacle insertion site (A + B + C) was divided by the corresponding sum for cleavage downstream (V + W + X + Y + Z). Data from three biological replicates were used to calculate each mean and standard deviation. See also Figures S2 and S4.
(E) Decay rates of reporter mRNAs and their upstream cleavage products in E. coli. Transcription was arrested by adding rifampicin to wild-type cells that contained AC3, TR8, or TR11 and TRAP, and equal amounts of total cellular RNA isolated at time intervals thereafter were examined by Northern blotting. See also Figures S1, S2, and S4.
(F) Context independence of the effect of discontinuities on downstream cleavage. The TRAP-binding site of TR8 or TR11 was inserted between the two principal RNase E cleavage sites within the 5′ UTR of ompAΔhp (an ompA mRNA derivative bearing an unpaired 5′ end), and cleavage upstream versus downstream of the insertion site was compared in wild-type cells containing TRAP. Data from three biological replicates were used to calculate each mean and standard deviation. See also Figure S3.
(G) Effect of structural discontinuities in the 5′ UTR on gene expression, β-galactosidase levels were compared in wild-type cells containing both TRAP and a lacZ translational fusion of AC3, TR8, or TR11 and normalized to AC3-lacZ. Data from three biological replicates were used to calculate each mean and standard deviation.
The cleavage of AC3 and its derivatives was examined in E. coli strains lacking the chromosomal yeiP gene to avoid confusing their cleavage products with those of the endogenous yeiP transcript. In rppH+ cells (“WT”), the 5′ UTR of AC3 was cleaved at all the expected sites, as evidenced by the appearance of the corresponding decay intermediates on a Northern blot (Figure 1B and Figure S1A). The molar ratio of decay intermediates produced by endonucleolytic cleavage upstream versus downstream of the BglII site was 0.7 ± 0.1. As anticipated, cleavage at all of these sites was greatly diminished in ΔrppH cells, as was the percentage of intact AC3 that was monophosphorylated (Figure S1B), evidence that most of the cleavage observed for this transcript in rppH+ cells is 5′-monophosphate-assisted. Cleavage at similar sites was observed when monophosphorylated AC3 was synthesized by in vitro transcription and then treated with either of two purified enzymes: the catalytic N-terminal domain of E. coli RNase E (N-RNase E; amino acid residues 1-529, a reliable and more tractable surrogate for the full-length protein) or its homolog RNase G (Figure 1B). An analogous set of bands was also observed when AC3 was extracted from E. coli cells containing RNase E but lacking RNase G (Δrng); however, their intensity was somewhat altered, suggesting that both endonucleases ordinarily contribute to cleavage at those sites in E. coli.
Protection by a bound protein in E. coli
We next introduced a binding site for TRAP, the tryptophan operon RNA-binding attenuation protein of Bacillus subtilis. In B. subtilis, this protein attenuates transcription of the trpEDCFBA tryptophan biosynthesis operon when tryptophan is abundant (Gollnick et al., 2005). TRAP is a multimer of 11 identical subunits that assemble to form a ring. In the presence of tryptophan, TRAP binds selectively to single-stranded RNA sites comprising up to 11 G/UAG trinucleotide repeats separated by spacers of 2-3 nucleotides. In the complex of the protein with 11 repeats, the RNA ligand wraps around the outside of the protein ring, such that each TRAP subunit binds one G/UAG trinucleotide and the entry and exit points are adjacent to one other (Antson et al., 1999). Sites containing fewer (just 5-10) G/UAGNN repeats also bind TRAP with high affinity (Babitzke et al., 1996).
To create a large structural discontinuity in the 5′ UTR of AC3, we replaced the (AC)3 spacer immediately upstream of the BglII site with a truncated TRAP-binding site comprising just eight GAG trinucleotides spaced two nucleotides apart so that the RNA entry and exit points in the complex of the resulting transcript (TR8) with TRAP would be distant from one another (Figure 1A, Table S1). This transcript was produced together with TRAP in E. coli cells growing in a tryptophan-rich medium, and the effect of the discontinuity on reporter cleavage was determined by Northern blotting (Figure 1C). The molar ratio of decay intermediates produced by cleavage upstream versus downstream of the TRAP-binding site was 18 ± 1, a value much greater than the ratio of 0.7 ±0.1 measured for AC3 (Figure 1D). A large ratio was also observed for TR8 in cells containing RNase E but lacking RNase G (Figure S1C). As expected for a transcript degraded by a 5′-end-dependent mechanism, cleavage at each of these sites and the percentage of intact TR8 that was monophosphorylated (Figure S1B) were both markedly reduced in ΔrppH cells.
These findings suggest that TRAP binding can interfere selectively with 5′-monophosphate-assisted cleavage downstream of the protein-binding site. If so, the shortest of the upstream cleavage products should be relatively long-lived despite its monophosphorylated 5′ end due to the inefficiency of cleavage at the downstream sites. Consistent with this prediction, TR8 cleavage product C had a much longer half-life in E. coli (10 ± 1 min) than did either the corresponding cleavage product of AC3 (1.2 ± 0.1 min) or the full-length TR8 transcript (2.1 ± 0.1 min) (Figure 1E), whose decay rate was RppH-dependent (Figure S1D).
A very different result was obtained when a complete TRAP-binding site comprising 11 GAG trinucleotides spaced two nucleotides apart was inserted (TR11) so that, upon TRAP binding, the RNA entry and exit points in the complex would be directly adjacent to one other (Figure 1A, Table S1). In this case, the ratio of decay intermediates produced by cleavage upstream versus downstream of the TRAP-binding site (2.6 ± 0.2) was much lower than for TR8 and differed only a few-fold from that of AC3 (Figure 1C, D). Moreover, decay intermediate C of TR11 was only slightly more stable than the full-length transcript (half-life of 1.8 ± 0.2 min versus 1.2 ± 0.1 min) (Figure 1E) and much less stable than the corresponding intermediate of TR8.
The selective protection of the downstream cleavage sites of TR8 required TRAP (Figure S2A) and, importantly, was not a result of altering the single-stranded character of the downstream region, which was indistinguishable in AC3, TR8+TRAP, and TR11+TRAP when probed chemically with two different reagents (Figure S2B). Protection of the downstream cleavage sites of TR8 also was not due to the proximity of the protein-binding site to the monophosphorylated 5′ end of the long-lived TR8 decay intermediate, as the spacing between them (5 nt) was not only ample for 5′-monophosphate-assisted cleavage (Richards and Belasco, 2016) but also identical in TR8 and TR11. This conclusion was confirmed by showing that increasing that distance by inserting 15 inert C nucleotides there to generate TR8+C15 (Figure 1A, Table S1) had little effect on cleavage downstream of the protein-binding site, which was nearly as inefficient for TR8+C15 as for TR8 (Figure 1C, D). Nor was the protective effect of TRAP binding to TR8 due to the greater spatial separation of the upstream and downstream sites in TR8 versus TR11, as cleavage at the downstream sites in TRAP-associated TR11 was not significantly impeded by increasing the corresponding distance in that transcript by 15 nucleotides (equal in length to the structural discontinuity created by TRAP binding to TR8) to generate TR11+AC7.5 (Figure 1A, C, D; Table S1). We conclude that TRAP binding impedes downstream cleavage only when the structural discontinuity it creates is large, irrespective of the spacing between cleavage sites. This combination of properties is difficult to reconcile with a mechanism in which RNase E identifies cleavage sites by unconstrained looping from the 5′ end.
The TRAP-binding sites of TR8 and TR11 also had disparate effects when inserted between cleavage sites in the 5′ UTR of an entirely different transcript whose degradation is RppH-dependent: ompAΔhp, which lacks the 5′-terminal stem-loop that ordinarily occludes the 5′ end of ompA mRNA (Figure 1F, Figure S3, Table S1). When bound by TRAP, the TR8 site increased the ratio of upstream to downstream cleavage products in ompAΔhp by a factor of 9.0 ± 0.7, whereas the TR11 site increased this ratio only 1.6 ± 0.3 fold, demonstrating the context independence of this phenomenon.
The influence of TRAP binding on gene expression was tested by using the original set of reporters to construct lacZ translational fusions. In the presence of TRAP, β-galactosidase synthesis from a TR8-lacZ fusion was 5.3 ± 0.7 times greater than from a control fusion (AC3-lacZ), whereas β-galactosidase synthesis from TR11-lacZ was only 1.8 ± 0.2 times greater than from AC3-lacZ (Figure 1G). These results show that the large discontinuity in TR8 impacts not only the rate of mRNA degradation but also the consequent level of protein synthesis.
Protection by a bound protein in vitro
To determine whether the influence of TRAP binding on 5′-monophosphate-assisted cleavage is an intrinsic property of RNase E or whether other cellular factors are required, we tested whether this effect could be reconstituted in vitro with purified components. Monophosphorylated AC3, TR8, and TR11 RNAs were synthesized by in vitro transcription, combined with purified TRAP, and partially digested with purified N-RNase E. As observed in E. coli, cleavage downstream of the protein-binding site was severely and selectively impeded in monophosphorylated TR8, but not AC3 or TR11, despite efficient cleavage at the upstream sites of all three (Figure 2). Cleavage of the triphosphorylated counterparts of all three transcripts was much slower, as expected for a 5′-monophosphate-assisted process. The greater shielding of the downstream sites in monophosphorylated TR8 versus TR11 was not due to a difference in occupancy of the TRAP-binding site, as evidenced by gel-shift analysis under the same conditions, which showed both binding sites to be completely and specifically bound by TRAP (Figure S4). The influence of TRAP was equally dramatic when TR8 was treated with purified RNase G (Figure S5). We conclude that the protective effect of a bound protein on 5′-monophosphate assisted cleavage at downstream sites is an intrinsic property of both of these endonucleases and that no extrinsic energy source such as ATP is required.
Figure 2. Ability of a discontinuity to protect downstream sites from cleavage by purified N-RNase E.
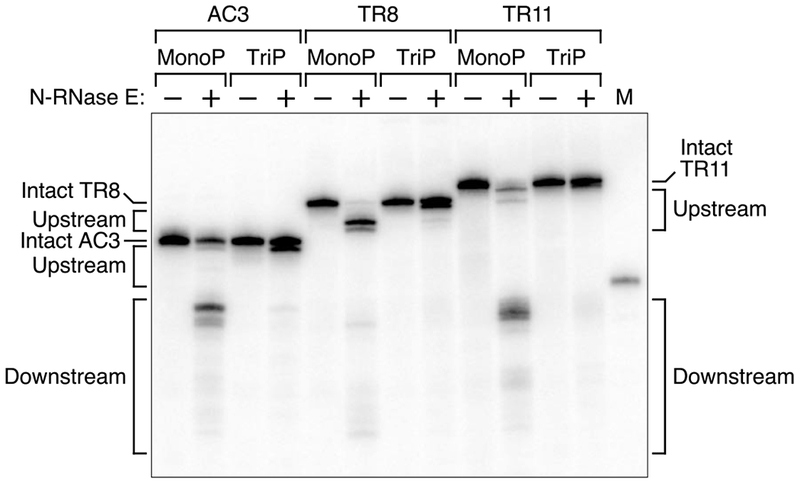
Monophosphorylated (MonoP) or triphosphorylated (TriP) AC3, TR8, or TR11 RNAs that had been synthesized by in vitro transcription and mixed with TRAP were treated for 4 min with equal amounts of purified N-RNase E (20 nM). The reaction products were then analyzed by Northern blotting as in Figure 1. M, boundary marker between the upstream and downstream cleavage sites. See also Figures S2, S4, and S5.
Protection by ribosomes
While protein binding to mRNA is commonplace in E. coli, ribosome binding to mRNA is universal. To ascertain whether a bound ribosome can inhibit monophosphate-assisted cleavage at downstream sites, we introduced into AC3 a six-codon upstream open reading frame (uORF) preceded by each of four Shine-Dalgarno elements (Figure 3A, Table S1). These elements differed in their degree of complementarity to the 3′ end of 16S rRNA, from low (SD-low: ACGA) to medium (SD-med: AGGA) to high (SD-high: AGGAG) to very high (SD-ultra: UAAGGAGG). The impediment posed by these Shine-Dalgarno elements to cleavage downstream of the uORF was well correlated with their predicted affinity for ribosomes (Ringquist et al., 1992). Thus, compared to AC3, SD-low had only a modest effect on the ratio of upstream versus downstream cleavage (ratio = 1.5 ± 0.1), SD-med had an intermediate effect (ratio = 2.0 ± 0.1), and SD-high and SD-ultra had large effects (ratios of 3.7 ± 0.3 and 5.8 ± 0.2, respectively) (Figure 3B). The protective influence of SD-ultra apparently resulted from a high level of ribosome occupancy that impeded scanning to the downstream cleavage sites and not from direct occlusion of those sites by bound ribosomes, as the spacing between the uORF and all but one of the downstream cleavage sites (17-42 nt for W, X, Y, and Z) was greater than the downstream footprint of a ribosome at a stop codon (10 nt (Woolstenhulme et al., 2015)) and as protection by SD-ultra was not diminished when this spacing was increased by 20 nt in SD-ultra+AC10 (ratio = 6.7 ± 0.8) (Figure 3B, Table S1).
Figure 3. Effect of ribosome binding on downstream cleavage in E. coli.
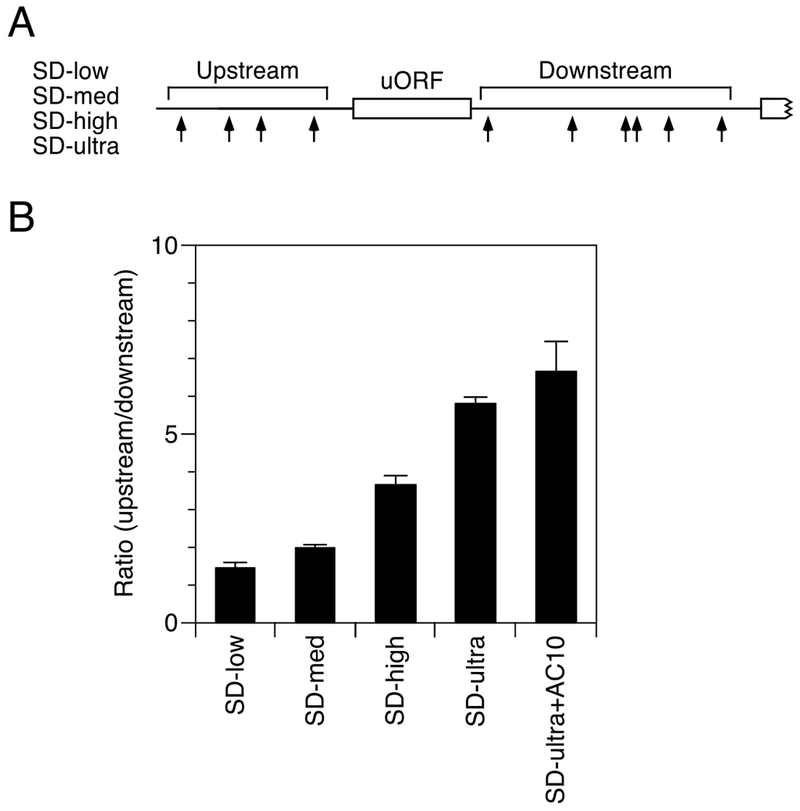
(A) 5′ UTR of reporter mRNAs containing an upstream open reading frame. Arrows, cleavage sites; white rectangles, protein coding regions.
(B) Relative abundance of 5′ UTR cleavage products for SD-low, SD-med, SD-high, SD-ultra, and SD-ultra+AC10 in wild-type cells. SD-ultra+AC10 is identical to SD-ultra except for the presence of a heteropolymeric (AC)10 spacer 4 nt downstream of the uORF. The sum of the intensities of the bands resulting from cleavage upstream of the uORF was divided by the corresponding sum for cleavage downstream. Data from three biological replicates were used to calculate each mean and standard deviation.
Lack of protection by orthogonal base pairing or a protein bound peripherally
In every experiment described so far, the obstacle to RNase E lay directly on the path from the 5′ end to a cleavage site. To determine whether downstream cleavage is inhibited by an obstacle at a peripheral location, we tested the effect of a protein bound at the top of a stem-loop orthogonal to the path. Either of two hairpins was introduced into the reporter. One (HpTR8) was a 10-bp stem topped by a 62-nt loop that comprised a 38-nt TRAP-binding site, like that in TR8, flanked on each side by 12 unpaired nucleotides (Figure 4A, Table S1). The other (HpGNRA) was the same stem topped by a simple GCAA loop. Neither stem-loop posed a significant obstacle to downstream cleavage by RNase E in E. coli (Figure 4B) or in vitro (Figure S6) despite the presence of TRAP, which bound the large hairpin loop of HpTR8 tightly and selectively (Figure S4). These results indicate that RNase E is capable of bypassing obstacles in base-paired regions orthogonal to the single-stranded path from the monophosphorylated 5′ end to a cleavage site. They also show that, like the small discontinuity in TR11, the bottom of a stem-loop orthogonal to that path poses only a minor impediment to scanning by this enzyme.
Figure 4. Meager effect of orthogonal base pairing on downstream cleavage in E. coli.
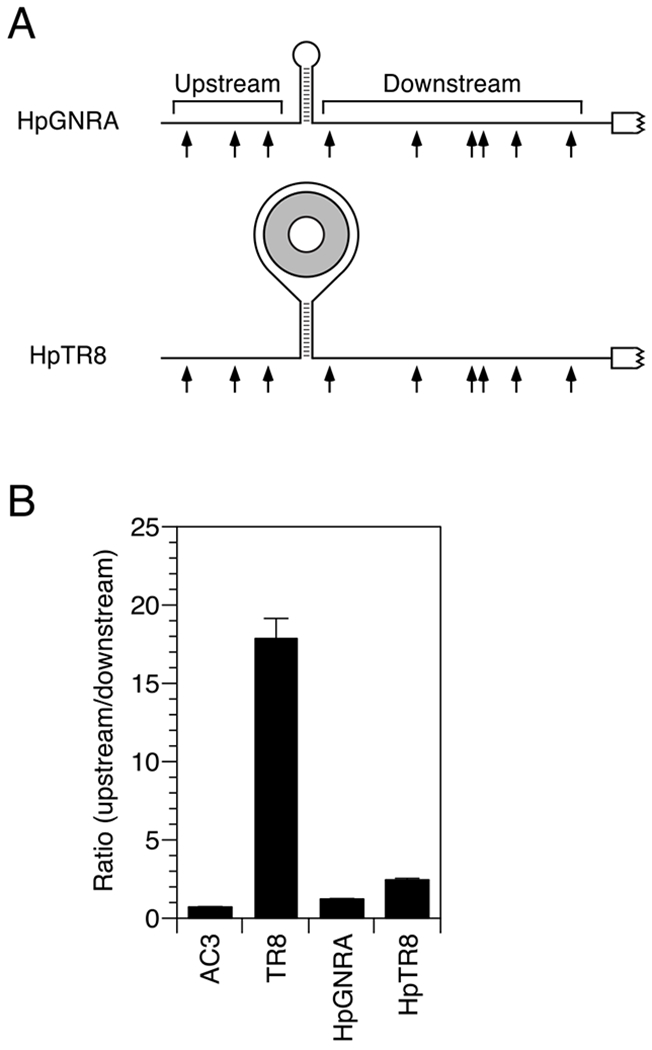
(A) 5′ UTR of reporter mRNAs containing a stem-loop structure. Arrows, cleavage sites; gray ring, TRAP 11-mer.
(B) Relative abundance of 5′ UTR cleavage products for HpGNRA and HpTR8 in wild-type cells. The sum of the intensities of the bands resulting from cleavage upstream of the stem-loop was divided by the corresponding sum for cleavage downstream. Data from three biological replicates were used to calculate each mean and standard deviation. See also Figures S4 and S6.
Inaccessibility of cleavage sites distal to coaxial base pairing
The ability of RNase E to bypass a protein obstacle at the top of a stem-loop raised the possibility that it might also bypass potential cleavage sites at such locations. To address this question, we tested a well known RNase E cleavage site (the hexamer GUAUUU) in different structural contexts. In one case, it was situated near the middle of a 25-nt loop atop a 10-bp stem (HpHX1) (Figure 5A, Table S1). The large size of this loop was intended to maximize the conformational flexibility that might be needed for RNase E to cut there. The other reporter was similar except that the complementary stem segments flanking the loop were removed (HX1). In E. coli, cleavage of the GUAUUU hexamer was conspicuous in the latter context but barely detectable in the context of a hairpin loop (Figure 5B). These findings suggest that, when seeking cleavage sites in monophosphorylated RNA, RNase E scans primarily along single-stranded regions, bypassing orthogonal base-paired regions and any potential cleavage sites they may contain.
Figure 5. Effect of intramolecular coaxial base pairing on distal cleavage in E. coli.
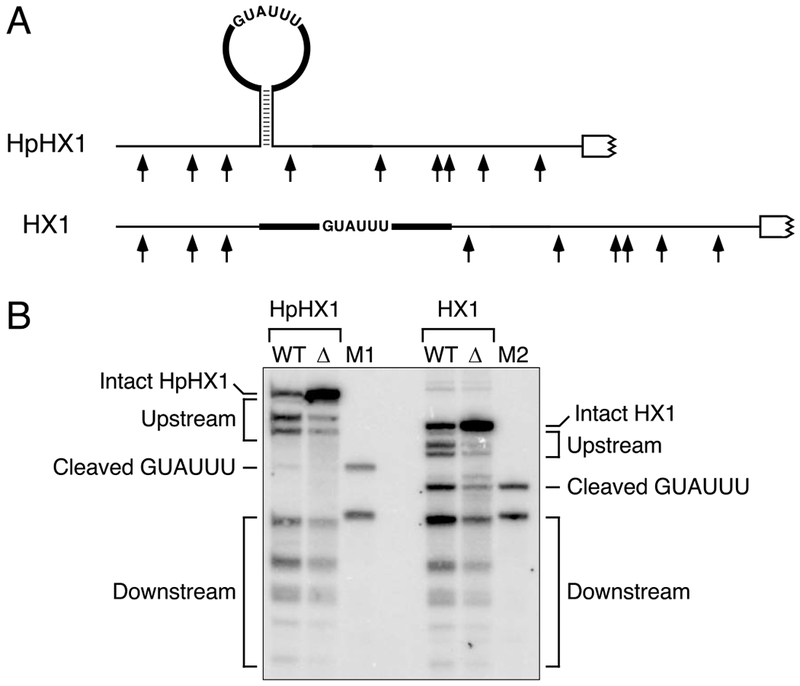
(A) 5′ UTR of reporter mRNAs containing an RNase E cleavage site (GUAUUU) in a hairpin loop (HpHX1) or in an otherwise identical sequence context without the flanking base-paired stem (HX1). Broad black lines, heteropolymeric (AC)n spacers that were identical in both transcripts.
(B) Cleavage within the GUAUUU hexamer of HpHX1 and HX1 in vivo. Equal amounts of total cellular RNA from isogenic wild-type (WT) and ΔrppH (Δ) strains of E. coli containing each reporter mRNA were analyzed by Northern blotting. The bands corresponding to cleavage of the GUAUUU hexamer are indicated. M1 and M2, markers to identify the GUAUUU cleavage products.
If RNase E is able to scan efficiently only along single-stranded regions, its access to potential cleavage sites should be blocked not only by intramolecular base pairing that is coaxial with the path from the 5′ end to those sites but also by any intermolecular coaxial base pairing that it may encounter along the way. This prediction was tested by introducing a unique 20-nt RNA segment into the reporter and determining the effect of a fully complementary oligoribonucleotide bearing a 5′ hydroxyl (OLIGO20) on cleavage of the resulting transcript (RB20) in vitro (Figure 6A, Table S1). In the absence of OLIGO20, purified N-RNase E readily cleaved monophosphorylated RB20 on both sides of the insert (Figure 6B, lanes 1 and 2). By contrast, OLIGO20 selectively impeded cleavage downstream of where it bound despite annealing far from the monophosphorylated 5′ ends of the degradation intermediates it protected (Figure 6B, lanes 3 and 4). No such protection was observed for a monophosphorylated control transcript (AC10) containing a 20-nt segment to which OLIGO20 could not bind (Figure 6B, lanes 5-8). We conclude that coaxial base pairing along the path from a monophosphorylated 5′ end to a cleavage site blocks RNase E access to that site, irrespective of whether the base pairing is intramolecular or intermolecular.
Figure 6. Effects of intermolecular coaxial base pairing on distal cleavage in vitro.
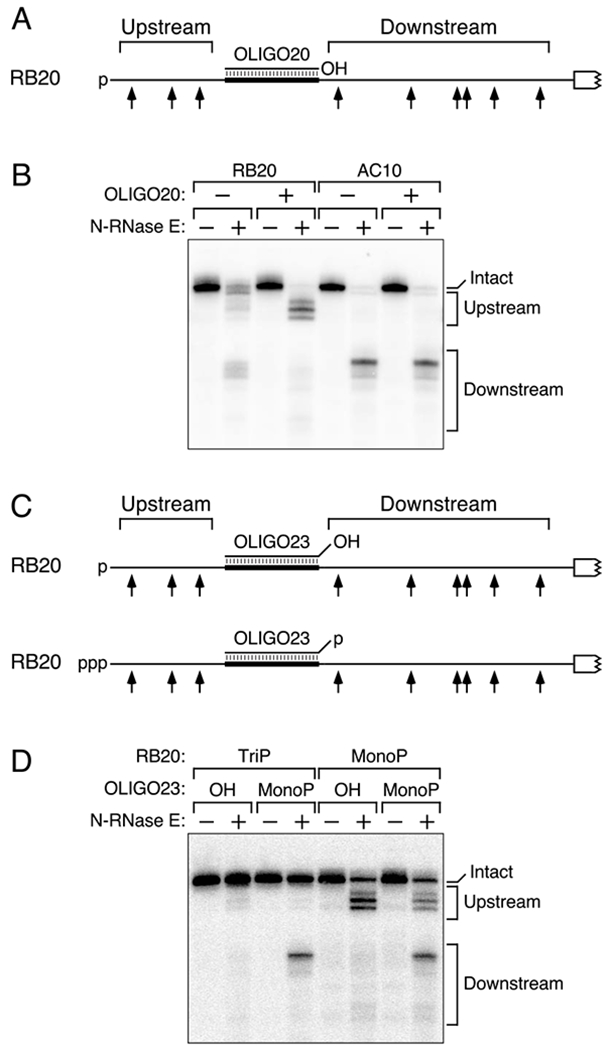
(A) 5′ UTR of RB20 RNA base paired with OLIGO20. Broad black line, 5′ UTR element complementary to OLIGO20. p, 5′ monophosphate; OH, 5′ hydroxyl.
(B) Effect of OLIGO20 on the cleavage of monophosphorylated RB20 and AC10 by purified N-RNase E. Monophosphorylated RB20 or AC10 RNA that had been synthesized by in vitro transcription and annealed or not annealed with OLIGO20 bearing a 5′ hydroxyl was treated for 16 min with purified N-RNase E (20 nM). The reaction products were then analyzed by Northern blotting.
(C) 5′ UTR of RB20 RNA base paired with OLIGO23. Broad black line, 5′ UTR element complementary to all but the first three nucleotides of OLIGO23. p, 5′ monophosphate; ppp, 5′ triphosphate; OH, 5′ hydroxyl.
(D) Effect of OLIGO23 on the cleavage of RB20 by purified N-RNase E. Triphosphorylated (TriP) or monophosphorylated (MonoP) RB20 RNA that had been synthesized by in vitro transcription and annealed with OLIGO23 bearing a 5′ hydroxyl (OH) or monophosphate (MonoP) was treated for 4 min with purified N-RNase E (20 nM). The reaction products were then analyzed by Northern blotting.
We next examined the effect of duplex formation by a similar oligoribonucleotide (OLIGO23) bearing three additional, noncomplementary nucleotides at its 5′ terminus (Figure 6C). As observed for OLIGO20, coaxial base pairing by 5′-hydroxylated OLIGO23 allowed N-RNase E cleavage upstream but not downstream of the oligonucleotide binding site in monophosphorylated RB20 (Figure 6D, lanes 5 and 6). Conversely, when annealed to triphosphorylated RB20, 5′-monophosphorylated OLIGO23 directed cleavage downstream but not upstream of its binding site (Figure 6D, lanes 3 and 4), an outcome consistent with the orthogonal orientation of the base-paired region relative to the path from the 5′ monophosphate of OLIGO23 to the downstream sites and its coaxial orientation relative to the upstream sites. As expected, cleavage occurred readily on both sides of the oligonucleotide-binding site when both RNAs were monophosphorylated (Figure 6D, lanes 7 and 8) and was almost undetectable when neither RNA was monophosphorylated (Figure 6D, lanes 1 and 2). The ability of a monophosphorylated oligonucleotide to stimulate RNase E cleavage of a complementary transcript to which it binds is consistent with prior reports of trans-activation of RNase E (Bandyra et al., 2012; Mackie, 2013a; Richards and Belasco, 2016), but the regiospecificity of this phenomenon relative to the location of the 5′ monophosphate was not previously recognized. Furthermore, the potential for RNase E to cut either upstream or downstream of the base-paired region, depending on which of the two RNA 5′ ends is monophosphorylated, corroborates the conclusion (Figure S2B) that a large discontinuity can impede cleavage regioselectively without altering the structure of either flanking region in a way that would make it intrinsically resistant to cleavage.
Importance of coaxial base pairing for mRNA protection during phosphosugar stress
sRNAs often upregulate gene expression by prolonging mRNA lifetimes without affecting translation, but the mechanism by which they do so is poorly understood. To test the importance of coaxial base pairing for this phenomenon, we investigated the mechanism by which an sRNA stabilizes a message that is key for the response of E. coli cells to stress. E. coli yigL, the second gene of the pldB-yigL operon, encodes a phosphosugar phosphatase crucial for detoxifying phosphosugars (Papenfort et al., 2013). Its expression is upregulated during phosphosugar stress by a post-transcriptional mechanism involving SgrS, an sRNA whose synthesis is induced by this stress (Vanderpool and Gottesman, 2004; Papenfort et al., 2013; Sun and Vanderpool, 2013). SgrS binds the dicistronic pldB-yigL transcript 75 nt upstream of yigL and selectively protects the yigL translational unit from rapid degradation (Figure 7A), allowing it to accumulate as a long-lived mRNA processing product despite the monophosphate at the 5′ end of the yigL fragment, 94 nt upstream of the SgrS-binding site (Papenfort et al., 2013). Stabilization by SgrS is not dependent on yigL translation, an observation consistent with the substantial distance between the SgrS-binding site and the yigL ribosome-binding site. Instead, as the complementary pldB-yigL site to which SgrS binds is otherwise susceptible to RNase E attack, it has been proposed that cleavage there may be critical for yigL mRNA degradation and that SgrS protects yigL by directly masking that site (Papenfort et al., 2013). However, mRNAs typically contain many RNase E cleavage sites (Clarke et al., 2014; Chao et al., 2017), and it is not clear why this particular site would be more important than any other. Alternatively, the stabilizing effect of SgrS may be a consequence of coaxial base pairing by this sRNA so as to block scanning by RNase E from the 5′ monophosphate of yigL to downstream cleavage sites.
Figure 7. Stabilization of yigL mRNA by coaxial base pairing with SgrS in E. coli.
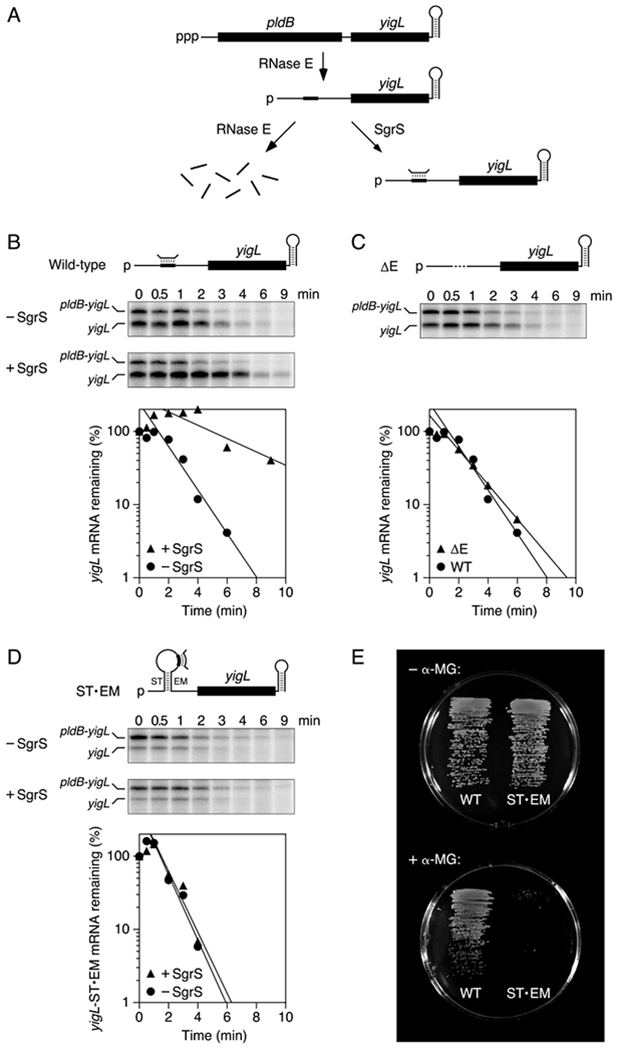
(A) SgrS-dependent pathway for the degradation of the dicistronic pldB-yigL transcript by RNase E in E. coli. Bent line, SgrS; broad line, SgrS-binding site; black rectangles, pldB and yigL translational units; ppp, 5′ triphosphate; p, 5′ monophosphate.
(B) Protection of wild-type yigL mRNA by SgrS in E. coli. Transcription in cells lacking or containing SgrS was arrested by treatment with rifampicin, and equal amounts of total cellular RNA isolated at time intervals thereafter were examined by S1 analysis. (Top) Diagram of the yigL 5′ UTR; (middle) gel images; (bottom) semilogarithmic graph of the amount of yigL mRNA remaining as a function of time. Representative experiments are shown.
(C) Rapid degradation of yigLΔE mRNA in E. coli cells lacking SgrS. Dashed line, deleted site of RNase E cleavage and SgrS binding. The graph compares the decay rates of yigLΔE and wild-type yigL in the absence of SgrS. A representative experiment is shown.
(D) Inability of SgrS to protect yigL-ST•EM mRNA from rapid degradation in E. coli. ST and EM, complementary RNA segments inserted upstream and downstream of the SgrS binding site. Representative experiments are shown. See also Figure S7.
(E) Importance of coaxial base pairing with the yigL 5′ UTR for the ability of SgrS to protect E. coli from α-methylglucoside toxicity. Isogenic E. coli cells whose chromosome contained either a wild-type yigL allele (left) or a yigL-ST•EM allele (right) and in which SgrS was expressed from its native promoter were grown on minimal glycerol medium lacking (top) or containing (bottom) α-methylglucoside (α-MG). To avoid masking the effect of differences in yigL Mrna stability on YigL production and α-methylglucoside resistance, synthesis of the PtsG glucose transporter was rendered insensitive to repression by SgrS in both of these strains.
To distinguish between these hypotheses, we first deleted the RNase E cleavage site in question and determined the effect of this deletion on the decay rate of yigL mRNA. In E. coli cells containing the wild-type pldB-yigL operon, SgrS produced under the control of an arabinose-inducible PBAD promoter had the expected effect on the stability of the yigL mRNA fragment, increasing its half-life from 1.0 ± 0.1 min in the absence of the sRNA to 3.3 ± 0.8 min in its presence (Figure 7B). A 21-nt deletion that removed the RNase E cleavage site along with the rest of the SgrS-binding site surrounding it resulted in a mutant message (yigLΔE) with a half-life of 1.3 ± 0.1 min, similar to that of its wild-type counterpart in cells lacking SgrS (Figure 7C). The finding that this RNase E cleavage site is dispensable for the rapid degradation of yigL mRNA in the absence of SgrS is inconsistent with direct masking of a crucial cleavage site by SgrS but compatible with the hypothesis that SgrS protects downstream sites by coaxial base pairing.
To corroborate the importance of coaxial base pairing by SgrS, we inserted a pair of complementary GC-rich RNA segments (ST and EM) 15-nt in length on either side of the SgrS-binding site so as to position the bound sRNA peripherally within the 115-nt loop of a hairpin orthogonal to the path from the 5′ monophosphate to the yigL coding region. As predicted, this modification (ST•EM) neutralized the protective influence of SgrS, rendering yigL mRNA constitutively short-lived regardless of whether SgrS was present or absent (Figure 7D). By contrast, inserting either RNA segment individually (ST or EM) had little effect on the rate of yigL degradation and its modulation by SgrS (Figure S7A). Gel-shift analysis with in vitro transcripts showed that the yigL 5′ UTR is still able to bind SgrS when the sRNA-binding site is located in the ST•EM hairpin loop but not when that site has been deleted (yigLΔE) (Figure S7B). Together, these findings indicate that SgrS prolongs the lifetime of monophosphorylated yigL mRNA by coaxial base pairing that protects downstream sites from RNase E cleavage.
The failure of SgrS to protect yigL-ST•EM mRNA from degradation should make E. coli cells that produce this yigL mRNA variant more susceptible to the harmful effects of phosphosugar stress. To test this hypothesis, we compared the ability of the phosphosugar phosphatase levels encoded by yigL and yigL-ST•EM to protect E. coli from the toxicity of α-methylglucoside, a non-metabolizable monosaccharide whose phosphorylation induces phosphosugar stress. Isogenic strains bearing either yigL allele on the E. coli chromosome grew well in the absence of phosphosugar stress. However, in the presence of α-methylglucoside, the cells that produced yigL mRNA grew robustly whereas those that produced yigL-ST•EM mRNA grew very slowly if at all (Figure 7E), demonstrating the physiological importance of mRNA stabilization by coaxial base pairing.
DISCUSSION
It has long been recognized that RNase E plays a crucial role in governing mRNA lifetimes in bacterial cells, yet the features of transcripts that account for their disparate rates of cleavage by this endonuclease have remained obscure. Particularly vexing has been the difficulty of reconciling the diversity of bacterial mRNA half-lives with the relaxed cleavage-site specificity of RNase E. Even the discovery of the stimulatory effect of monophosphorylated RNA 5′ ends on cleavage by this enzyme failed to resolve this mystery, as the lifetimes of decay intermediates bearing a 5′ monophosphate are themselves quite diverse. Our present results now reveal that the ability of RNase E to cleave at internal sites in monophosphorylated RNA can be hindered by structural impediments well upstream of those sites. As a result, the longevity of such RNAs in E. coli depends not only on the presence of an unpaired 5′ end and potential cleavage sites but also on any obstacles that may lie between them. The protective effect of such an obstacle results from the structural discontinuity that it creates and not merely from a change in the overall distance between the 5′ monophosphate and the site of cleavage. These findings are inconsistent with a mechanism in which RNase E searches for cutting sites by unconstrained looping from the monophosphorylated 5′ terminus. Instead, they suggest that this enzyme identifies cleavage sites in monophosphorylated RNA by linear scanning from the 5′ end along RNA segments that are single-stranded.
Obstacles able to obstruct scanning by RNase E include coaxial base pairing and bound proteins or ribosomes that create a large structural discontinuity directly on the single-stranded path from the monophosphorylated 5′ end to the cleavage site. Obstacles orthogonal or peripheral to that path (e.g., an orthogonal stem-loop or a protein that is either bound to the top of such a stem-loop or completely encircled by the RNA) have a much smaller effect, presumably because the diminutive size of the associated discontinuities makes them easier to bypass. The fact that small discontinuities such as these pose only a minor impediment to downstream cleavage implies that the step size of RNase E as it scans RNA in search of cleavage sites is greater than one nucleotide.
Although a stem-loop positioned internally within monophosphorylated RNA has only a modest effect on RNase E cleavage at downstream sites to which it is orthogonal, its base-paired stem can substantially impede cleavage within the loop because the two are coaxial. Such protection is distinct from that of a stem-loop located at the extreme 5′ end, which can greatly hinder cleavage throughout the message by depriving RNase E of the unpaired 5′-terminal nucleotides that it needs to bind a monophosphate there (Bouvet and Belasco, 1992; Mackie, 1998; Richards and Belasco, 2016).
Our findings suggest that duplex formation between an sRNA and mRNA can either inhibit or facilitate mRNA cleavage downstream of the site where the sRNA binds, depending on which of the two RNAs is monophosphorylated. If only the mRNA is monophosphorylated, mRNA sites downstream of the intermolecular duplex will be protected from 5′-monophosphate-assisted RNase E cleavage because the base pairing is coaxial with the path from the monophosphorylated 5′ terminus to those sites. In contrast, binding by a monophosphorylated sRNA can stimulate cleavage of a triphosphorylated transcript downstream, but not upstream, of the sRNA binding site because the base-paired region is orthogonal to the path from the sRNA 5′ end to the downstream mRNA sites but coaxial with the path to the upstream sites.
Even large discontinuities that eclipse downstream cleavage sites can be bypassed by RNase E at a low but detectable rate, presumably as a result of cleavage by direct access, structural heterogeneity in the RNA substrate, or a macromolecule that associates only transiently with the RNA. The latter phenomenon could explain why the impediment to downstream cleavage posed by the strong ribosome-binding site in SD-ultra is less than that posed by TRAP bound to TR8, as a tightly bound protein may remain in place longer than a bound ribosome, which will eventually initiate translation and move on, thereby exposing distal sites. The residence time of a ribosome on mRNA may be particularly brief when translating a short uORF, such as that in SD-low, SD-med, SD-high, and SD-ultra.
The scanning mechanism implied by our observations appears to be an innate property of both RNase E and its paralog RNase G, as the purified catalytic domain of each can be impeded by an intervening obstacle without the involvement of any accessory factors. The selective navigability of single-stranded but not double-stranded regions of RNA suggests that RNase E might utilize the single-stranded RNA-binding channel between its 5′-monophosphate-binding pocket and catalytic center (Callaghan et al., 2005) to migrate along unpaired segments of RNA in a manner reminiscent of a monorail. Scanning of monophosphorylated RNA by RNase E appears to be an isoenergetic process, as it does not require an energy source such as ATP. Therefore, although it begins at the 5′ end, it must proceed from there as a random walk because an intrinsically unidirectional mechanism is forbidden by the principle of microscopic reversibility. While our current evidence does not rule out a modulatory role in vivo for ancillary factors such as the RNase E-associated RNA helicase RhlB (Py et al., 1996; Miczak et al., 1996), the efficacy of coaxial base pairing as an obstacle to scanning in E. coli limits the potential impact of any such influence.
Our results do not address the mechanism by which RNase E locates cleavage sites in triphosphorylated RNA by direct access. Furthermore, they differ from a previous report that RNase E searches for cleavage sites in monophosphorylated RNA by scanning from 3′ to 5′ (Feng et al., 2002). That earlier conclusion was based in part on the faster rate of cleavage by purified RNase E, but not RNase G, at sites 15-25 nt, rather than 5 nt, from the 5′ end of monophosphorylated oligonucleotides containing multiple sites. However, this preference likely resulted not from directionality but rather from differences in the minimum 5′-terminal spacing required by these two enzymes: ≥7 nt for RNase E versus ≥5 nt for RNase G (Richards and Belasco, 2016).
The ability of obstacles upstream of cleavage sites to impede RNase E attack is likely to be of broad importance for understanding the diversity of mRNA lifetimes in bacteria, as (i) orthologs of RNase E are encoded in ~80% of sequenced bacterial genomes (Hui et al., 2014), (ii) the kinds of obstacles that we have identified (e.g., ribosomes) are ubiquitous, and (iii) monophosphorylated intermediates are invariably produced during mRNA degradation as a result of endonucleolytic cleavage or 5′-terminal phosphate removal by RppH. Moreover, the translational unit(s) in a polycistronic mRNA that are shielded in this manner need not include the one(s) closest to the 5′ end of the primary transcript, as illustrated by the selective stabilization of the yigL segment of the discistronic pldB-yigL transcript. Our data have specifically implicated this mechanism in mRNA protection by an sRNA induced during phosphosugar stress, enabling increased synthesis of a detoxifying enzyme, and it is likely to play a key role in other instances of mRNA stabilization by sRNA binding. It also likely accounts for the marked protective effect of a short uORF with a strong ribosome-binding site on a monophosphorylated mRNA (espADB) encoding translocon proteins important for the virulence of enterohemorrhagic E. coli (Lodato et al., 2012). More generally, our findings may help to explain the stabilizing influence of ribosomes associated with the principal open reading frames of bacterial transcripts (Deana and Belasco, 2005; Dreyfus, 2009) as well as the protection afforded by bacterial cold-shock proteins (Michaux et al., 2017; Zhang et al., 2018).
The possibility that nucleic-acid binding proteins might utilize passive one-dimensional diffusion to help them locate their target elements has been debated for some time (Berg et al., 1981; Halford, 2009; Mechetin and Zharkov, 2014). However, this theoretical and empirical discussion has focused predominantly on whether linear diffusion may sometimes be preferred over other search mechanisms and not on any regulatory consequences that it may have. Our findings now reveal the regulatory potential of such a mechanism by showing that the rate of degradation of monophosphorylated RNA by RNase E, and therefore the level of gene expression, depends not only on an unpaired 5′ end and one or more internal cleavage sites but also on any of a variety of structural obstacles that may lie between them. In addition to having the potential to explain much of the diversity of mRNA lifetimes in E. coli, this discovery has important implications for the control of other processes governing prokaryotic or eukaryotic gene expression in which a regulatory factor must locate a site of action on a nucleic acid target that is distinct from its initial binding site.
STAR METHODS
EXPERIMENTAL MODEL AND SUBJECT DETAILS
RNA cleavage by RNase E was examined in E. coli cells during exponential growth and in vitro with purified transcripts and proteins.
METHOD DETAILS
Strains and growth conditions
Analysis of the cleavage and degradation of mRNA was performed in the E. coli K-12 strain BW25113 (Datsenko and Wanner, 2000), as well as in isogenic derivatives bearing an in-frame deletion of the rppH and/or rng coding region. In addition, the strains used to analyze the cleavage of plasmid-encoded AC3 or ompAΔhp and derivatives thereof lacked the chromosomal yeiP gene or the chromosomal ompA and micA genes, respectively, and often contained a plasmid encoding TRAP (pTRAP-H6), while those used to analyze the degradation of plasmid-encoded pldB-yigL and its derivatives lacked the chromosomal yigL and sgrS genes and instead contained a plasmid encoding SgrS under the control of a PBAD promoter (pPBAD-SgrS) or the corresponding empty vector (pPBAD-empty). Each chromosomal deletion mutant other than ΔmicA and ΔsgrS was constructed by P1 transduction of the corresponding kan-disrupted gene from the Keio collection (Baba et al., 2006) followed by excision of the kan gene. The ΔmicA mutant was constructed by P1 transduction of the micA::cat allele from E. coli strain G897 (Udekwu et al., 2005), and the ΔsgrS mutant was constructed by P1 transduction of the sgrS::kan allele from E. coli strain CV104 (Vanderpool and Gottesman, 2004).
Cultures for analyzing the cleavage and degradation of AC3 and its derivatives were grown in MOPS medium containing 0.2% glucose (Neidhardt et al., 1974), whereas those for examining the cleavage of ompAΔhp and its derivatives were grown in LB containing 0.2% glucose. TRAP synthesis and binding were enabled by supplementing the growth medium with IPTG (1 mM) and L-tryptophan (100 μg/ml). Cultures for analyzing the degradation of yigL mRNA and its derivatives were grown in LB, and 0.2% arabinose was added to induce SgrS synthesis 15 min before rifampicin addition. In every case, the cells were grown at 37°C to mid-exponential phase before harvesting.
To examine the effect of the ST•EM insertions on the response of E. coli to phosphosugar stress, the ST and EM 15-mers (see below) were introduced into the chromosome of BW25113 by two-step allelic exchange with a nonreplicating plasmid bearing a counterselectable sacB gene (Edwards et al., 1998) and verified by DNA sequencing. In addition, the strains used for this experiment contained a low-copy plasmid (pPtsGmut) bearing an SgrS-insensitive variant of the E. coli ptsG glucose transporter gene. The resulting pair of isogenic strains encoding either wild-type pldB-yigL or the pldB-yigL ST•EM variant were grown to late exponential phase in LB, and equal numbers of cells (determined by absorbance at 650 nm) were then streaked on MOPS minimal agar containing 0.4% glycerol and either 0% or 0.04% (w/v) α-methylglucoside and grown for three days at room temperature.
Plasmids
A reporter for testing the effect of potential obstacles on the cleavage of monophosphorylated mRNA in vivo was constructed by duplicating much of the 5′ UTR of a plasmid-borne copy of the E. coli yeiP gene. Specifically, a DNA fragment comprising the first 24 nt of the yeiP 5′ UTR (the portion preceding the Shine-Dalgarno element) followed by the sequence AGATCTACACACACAC (BglII site underlined) was inserted immediately upstream of the yeiP transcription initiation site in the medium-copy plasmid pYeiP1 (Richards et al., 2012) to generate pYeiPdup. DNA fragments encoding various possible obstacles were then inserted between the upstream copy of the 5′ UTR and the BglII site (Tables S1 and S3). The corresponding templates for in vitro transcription were then constructed by replacing the yeiP promoter with a T7 ϕ2.5 promoter (Coleman et al., 2004) and introducing a T→G substitution at position 2 of the transcribed region. Translational fusions to lacZ were constructed by fusing the promoter, 5′ UTR, and first 20 codons of AC3, TR8, or TR11 in-frame to codons 9-1024 of lacZ in a derivative of plasmid pPM30 (Meacock and Cohen, 1980) that encodes kanamycin resistance.
Plasmid pOMPAΔhp is a deriviative of pOMPA+4 (Emory et al., 1992) in which (i) the first 68 transcribed nucleotides of ompA+4 were changed to AGACA so as to remove the 5′-terminal RNA stem-loop and (ii) the region encoding an internal 5′ UTR stem-loop (CCGTGTTATCTCGTTGGAGATATTCATGG) was replaced by the spacer GCACACACACAC. The TR8 and TR11 variants of pOMPAΔhp were generated by inserting 8 or 11 GAG repeats, each separated by AT, after the second nucleotide of this spacer.
Plasmid pTRAP-H6 is a derivative of pPlacRppH6 (Deana et al., 2008) in which the rppH coding region has been replaced with the B. subtilis mtrB coding region and an extension encoding a carboxy-terminal hexahistidine tag (GHHHHHH).
Plasmid pPldB-YigL was constructed by inserting the entire E. coli pldB-yigL operon (from 232 bp upstream of the pldB coding region to 212 bp downstream of the yigL coding region) between the EcoRI and PstI sites of pBR322fd (Richards et al., 2012). Plasmid pPldB-YigLΔE is a derivative from which the SgrS binding site (ACGCAATGCGCTCAGTCGCGC) and the RNase E cleavage site within it were deleted. Plasmid pPldB-YigL-ST contains the 15-nt ST sequence AGATCGCCCACAGGC inserted immediately after position 848 of the pldB coding region. Plasmid pPldB-YigL-EM contains the complementary 15-nt EM sequence GCCTGTGGGCGATCA inserted immediately after position 963 of the pldB coding region. Plasmid pPldB-YigL-ST•EM contains both the ST insert and the EM insert.
Plasmid pPBAD-SgrS is a derivative of pPlacRppH6 (Deana et al., 2008) in which the lacl, Plac, and rppH regions have been replaced with the araC-PBAD region of plasmid pBAD/His (Thermo Fisher Scientific) followed by the transcribed region of E. coli sgrS positioned at the transcription initiation site of the PBAD promoter. The negative-control plasmid pPBAD-empty is a derivative of pPBAD-SgrS from which the sgrS sequence has been deleted.
Plasmid pPtsGmut encodes a ptsG mRNA variant insensitive to repression by SgrS. It was constructed from the low-copy plasmid pPM30 (Meacock and Cohen, 1980) by replacing the small EcoRI-BamHI fragment with a bla promoter joined to a promoterless E. coli ptsG gene in which the region immediately preceding the Shine-Dalgarno element had been mutated (TACTCAGGAG → TAGTCAGGAG) to impair SgrS binding (Kawamoto et al., 2006).
Analysis of 5′ UTR cleavage in E. coli
Total cellular RNA was isolated from E. coli strains bearing a reporter plasmid (Table S1), as well as the TRAP expression plasmid pTRAP-H6 when required, after growth at 37°C to mid-exponential phase. To extract RNA, a culture sample (10-15 ml) was rapidly chilled by pipetting it into a 50-ml centrifuge tube filled with crushed ice, and the bacteria were pelleted by centrifugation at 6,000 × g for 10 min at 0°C. The cell pellet was resuspended at 0°C in 125 μl of ice-cold buffer A (0.3 M sucrose, 10 mM sodium acetate, pH 4.5, 10 mM EDTA), transferred to a chilled microfuge tube, mixed with 250 μl of room-temperature buffer B (2% SDS, 10 mM sodium acetate, pH 4.5), and heated to 70°C for 3 min to lyse the cells. The cell lysate was extracted three times with 250 μl of hot, unneutralized, water-saturated phenol by vigorously agitating the mixture and then heating it to 70°C for 3 min. After each phenol extraction, the mixture was chilled rapidly in a dry ice/ethanol bath and then centrifuged at 21,000 × g for 5 min before transferring the aqueous layer to a new microfuge tube. Finally, the RNA was recovered by ethanol precipitation, washed with 70% ethanol, air-dried, dissolved in water (30-70 μl) to a concentration of 0.5-2.0 μg/μl, and stored at −20°C.
To resolve and detect the various decay intermediates, mRNA was subjected to site-specific cleavage with either of two 10-23 deoxyribozymes (Santoro and Joyce, 1997), DZyeiP69 (used to cut AC3-related reporters in yeiP codon 12) or DZompA149 (used to cut ompAΔhp-related reporters in ompA codon 6) (Table S2) (Richards et al., 2012). Equal amounts of total RNA (10 or 15 μg) were combined with 600 pmol of DZyeiP69 or DZompA149 in a total volume of 36 μl, heated to 85°C for 5 min, and then slowly cooled to room temperature. A buffer containing 500 mM Tris-HCl (pH 7.5), 100 mM MgCl2, and 100 mM dithiothreitol (4 μl) was added, and deoxyribozyme cleavage was allowed to proceed for 4 hr at 37°C. The reaction was quenched with 3 mM EDTA (210 μl), and the products were recovered by ethanol precipitation, subjected to electrophoresis on a 7.5% polyacrylamide – 8 M urea gel, and analyzed by Northern blotting. In each case, RNA was electroblotted from the gel to an Immobilon-Ny+ membrane and probed with a radiolabeled oligonucleotide (Table S2) that annealed between the 3′-most RNase E cleavage site and the site of deoxyribozyme cleavage. Radioactive bands were visualized on a Typhoon Trio imager (GE Healthcare) and quantified by using ImageQuant TL software (GE Healthcare).
Analysis of the phosphorylation state and decay rate of mRNA in E. coli
The percentage of AC3, TR8, and TR11 transcripts that were monophosphorylated at steady state was determined by PABLO (Luciano et al., 2017) on the basis of their ability to undergo splinted ligation to a DNA oligonucleotide (X22) in the presence of a complementary bridging oligonucleotide (Yyeip) and the 10-23 deoxyribozyme DZyeiP54 (Table S2).
mRNA half-lives were measured by blocking further RNA synthesis and monitoring degradation. At time intervals after inhibiting transcription with rifampicin (200 μg/ml), total RNA was extracted from E. coli cells that contained a plasmid encoding an mRNA of interest (see above). The decay of AC3, TR8, and TR11 mRNA was analyzed by subjecting equal amounts of total RNA from each sample (10 μg) to site-specific cleavage with DZyeiP54, which cuts in yeiP codon 7, followed by gel electrophoresis (7.5% polyacrylamide – 8 M urea) and Northern blotting with a radiolabeled probe (Table S2) that annealed between cleavage site Z and the site of deoxyribozyme cleavage. Radioactive bands were visualized and quantified as described above, and mRNA half-lives were calculated by linear regression analysis. The decay of wild-type pldB-yigL, pldB-yigLΔE, and pldB-yigL-ST•EM mRNA was examined by S1-protection analysis, as follows. Samples of total RNA from each time point (15 μg) were combined with equal amounts of a radiolabeled loading control (a 120-bp double-stranded cat DNA fragment lacking complementarity to any cellular transcript) and hybridized for 4 hr at 45°C with a cognate 5′-end-labeled DNA probe extending from yigL codon 33 to pldB codon 272 and bearing a noncomplementary 48-nt 3' extension (4 μM) in a solution (20 μl) containing 40 mM PIPES (pH 6.4), 400 mM NaCl, 1 mM EDTA, and 80% (v/v) formamide. Each sample was then treated with S1 nuclease (40 U) for 2 hr at 37°C in a solution (200 μl total volume) containing 40 mM sodium acetate (pH 4.5), 300 mM NaCl, and 2 mM ZnSO4 before quenching the reaction with EDTA (3.75 mM). The products of S1 digestion were extracted with phenol-chloroform, precipitated with ethanol, denatured, and analyzed by electrophoresis on a 5% polyacrylamide – 8 M urea gel. Radioactive bands were visualized and quantified as described above, and mRNA half-lives were calculated by linear regression analysis after normalizing the intensity of the yigL band to that of the loading control in the same lane.
Analysis of 5′ UTR cleavage in vitro
RNA substrates for cleavage by purified RNase E were synthesized by in vitro transcription with T7 RNA polymerase (Luciano et al., 2017), to generate either full-length AC3 (Figure 1B) or truncated RNAs ending at codon 12. To enable the synthesis of these A-initiated RNAs, a DNA template was prepared by PCR amplification of a plasmid segment containing a T7 ϕ2.5 promoter and a G substitution at the second position of the transcript. Triphosphorylated RNA was synthesized for 10 hr at 37°C in a 127-μ1 reaction mixture containing 40 mM Tris-HCl (pH 7.9), 6 mM MgCl2, 10 mM NaCl, 2 mM spermidine, 10 mM dithiothreitol, 0.3 units/μl rRNasin, 1.5 mM ATP, 1 mM GTP, 1 mM CTP, 1 mM UTP, 10-50 ng/μl DNA template, and 5 units/μl T7 RNA polymerase. Monophosphorylated RNA was synthesized in the presence of a 50-fold molar excess of AMP (12.5 mM) over ATP (0.25 mM). The resulting transcripts were treated with TURBO DNase (0.05 units/μl) for 1 hr at 37°C in the buffer supplied by the manufacturer and then purified by gel electrophoresis and elution of the band of interest.
N-RNase E (amino acids 1-529 of E. coli RNase E) and E. coli RNase G, each bearing carboxy-terminal hexahistidine and Myc epitope tags (GGAAAHHHHHHVAAEQKLISEEDLNGAARSA), were overexpressed in E. coli from plasmids pNRNE7529 and pRNG7001, respectively (Richards and Belasco, 2016). The enzymes were then purified from cell extracts by elution from TALON beads with a buffer containing 20 mM Tris-HCl (pH 7.5), 0.25 M imidazole, and 0.5 M NaCl, concentrated/exchanged into a buffer containing 20 mM Tris.Cl (pH 7.5), 0.5 M NaCl, 10 mM dithiothreitol, and 20% (v/v) glycerol, and stored at −80°C. B. subtilis TRAP bearing a C-terminal hexahistidine tag was overexpressed from plasmid pTRAP-H6 in E. coli. After heating the cell extract to 100°C for 10 min, the denatured proteins were removed by centrifugation, and the supernatant was passed over TALON beads. TRAP was eluted in a buffer containing 12 mM sodium phosphate (pH 7.2), 50 mM sodium chloride, 0.1 mM EDTA, and 250 mM imidazole. Fractions containing TRAP were concentrated/exchanged into the same buffer lacking imidazole and stored at −80°C.
Cleavage reactions were carried out at 30°C in a buffer (25 mM Bis-tris propane (pH 8.0), 100 mM NaCl, 15 mM MgCl2 containing in vitro transcribed RNA (37 nM) and purified N-RNase E (20 nM or 32 nM enzyme subunits) or RNase G (80 nM or 240 nM enzyme subunits). The cleavage reactions of TR8, TR11, and HpTR8 also contained TRAP (12.8 μM) and L-tryptophan (2 mM), while those of RB20 and AC10 also contained OLIGO20 or OLIGO23 (78 nM, Table S2) to which the transcripts had been pre-annealed for 1 hr at 50°C. Reactions samples (0.33 pmol) were quenched with a three-fold molar excess of EDTA before or at fixed times after ribonuclease addition so as to achieve partial digestion. They were then mixed with total RNA (5 μg) from E. coli strain BW25113 ΔyeiP (Richards et al., 2012), ethanol precipitated, subjected to electrophoresis on a 7.5% polyacrylamide – 8 M urea gel, and analyzed by Northern blotting with a radiolabeled probe (Table S2) that annealed downstream of cleavage site Z.
Structural probing of RNA
The structure of the 5′ UTR of AC3, TR8+TRAP, and TR11+TRAP was probed by alkylation with dimethyl sulfate (DMS) (Brunei and Romby, 2000) or 2-methylnicotinic acid imidazolide (NAI) (Spitale et al., 2013). NAI was synthesized by adding a solution of 1,1′-carbonyldiimidazole (162 mg) in anhydrous DMSO (0.5 ml) dropwise to a suspension of 2-methylnicotinic acid (137 mg) in anhydrous DMSO (0.5 ml) (Spitale et al., 2013). Full-length AC3, TR8, and TR11 RNAs were synthesized by in vitro transcription of a PCR-amplified DNA template (Table S2), as described above. The RNAs were then dissolved at a concentration of 6 nM in a solution (50 μl) containing total RNA from E. coli strain BW25113 ΔyeiP (100 ng/μl), 40 mM HEPES (pH 7.5), 100 mM KC1, 0.5 mM MgCl2, TRAP (88 μg/ml; TR8 and TR11 only), and L-tryptophan (200 μg/ml). DMS (0.2% v/v) or NAI (6% v/v) was added, and the reaction mixtures were incubated at 22°C for 15 min before being quenched with dithiothreitol (0.5 M). The reaction products were passed through a column of Sephadex G-25 (0.6 ml), and the high molecular weight fraction was ethanol precipitated. Reverse transcription of the alkylated RNA samples and their unalkylated counterparts was then performed at 55°C with a 5′-end-labeled primer complementary to the yeiP coding region (Table S2) and Superscript III reverse transcriptase, according to the manufacturer’s instructions. The primer extension products were subjected to electrophoresis on a 6% polyacrylamide – 8 M urea gel beside a set of ladders generated by using the fmol DNA Cycle Sequencing System to sequence a TR11 DNA template with the same primer and each of the four 2′, 3′-dideoxynucleoside triphosphates. The gel was dried, and radioactive bands were visualized with a Typhoon Trio imager (GE Healthcare).
Electrophoretic mobility shift assays
To detect binding by TRAP, RNA (TR8, TR11, AC19, AC26.5, HpGNRA, or HpTR8; 0.02 μM) that had been synthesized by in vitro transcription (see above) was incubated with TRAP (12.8 μM) at 30°C in a 10-μ1 solution containing 25 mM Bis-tris propane (pH 8.0), 20 mM NaCl, 15 mM MgCl2, 1 mM dithiothreitol, 2 mM L-tryptophan, and 0.1 mg/ml xylene cyanol. After 15 min, glycerol was added to a final concentration of 4% (v/v), and the samples were subjected to electrophoresis on a nondenaturing 10% polyacrylamide gel containing 375 mM Tris-HCl (pH 8.8), 5% glycerol, and 1 mM EDTA. The RNA bands were then visualized by Northern blotting as described above.
To detect binding by SgrS, RNA (wild-type pldB-yigL, pldB-yigLΔE, or pldB-yigL-ST•EM; 10 nM) was cooled on ice and then combined with SgrS (50 nM) and E. coli Hfq (600 nM; a gift from Sarah Woodson, Johns Hopkins University) (Santiago-Frangos et al., 2016) in a 15-μ1 solution containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 50 mM KCl, 50 mM NH4Cl, 0.5 mM EDTA, 5% glycerol, and 1 mg/ml E. coli tRNA. The mixture was incubated for 30 min at 37°C, and the Hfq was then digested by adding proteinase K (1.25 mg/ml) and incubating the mixture for an additional 15 min at 37°C. Glycerol (4% v/v) and bromophenol blue (50 μg/ml) were added, and the samples were subjected to electrophoresis on a nondenaturing 4.5% polyacrylamide gel containing containing 89 mM Tris-borate (pH 8.3), 2 mM EDTA, and 5% glycerol. The RNA bands were visualized by Northern blotting as described above.
β-galactosidase assays
Cultures of E. coli containing lacZ fusions of AC3, TR8, and TR11 were grown at 37°C to midexponential phase, and β-galactosidase activity in permeabilized cells was assayed by measuring the hydrolysis rate of o-nitrophenyl-β-galactoside (ONPG) (Miller, 1992).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification
Band intensities were quantified by using ImageQuant TL software (GE Healthcare). They were normalized either to one another when calculating the ratio of cleavage upstream versus downstream of an obstacle (Figure 1D, legend), to total RNA loaded in the gel lane when measuring half-lives by Northern blotting, or to a pre-mixed internal DNA standard when measuring half-lives by S1-protection analysis.
Statistical Analysis
The histograms in Figures 1, 3, 4, and S2 depict mean values and standard deviations of three biological replicates. In Figures 7 and S7, mRNA half-lives were calculated from the slope of the best-fit line of a semi-log plot, as detemined by linear regression, and the standard deviation of each half-life was calculated from the standard deviation of the slope.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| BW25113 | Datsenko and Wanner, 2000 | N/A |
| BW25113 ΔrppH | Luciano et al., 2017 | N/A |
| BW25113 ΔyeiP | Luciano et al., 2017 | N/A |
| BW25113 ΔyeiP ΔrppH | Luciano et al., 2017 | N/A |
| BW25113 Δrng | This study | N/A |
| BW25113 Δrng ΔrppH | This study | N/A |
| BW25113 ΔompA micA::cat | This study | N/A |
| BW25113 ΔompA ΔrppH micA::cat | This study | N/A |
| BW25113 ΔyigL sgrS::cat | This study | N/A |
| BW25113 yigL-ST•EM | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| N-RNase E | This study | N/A |
| RNase G | This study | N/A |
| TRAP | This study | N/A |
| RppH | Foley et al., 2015 | N/A |
| Phusion High-Fidelity DNA Polymerase | Thermo Fisher Scientific | F530S |
| T7 RNA polymerase | Thermo Fisher Scientific | EP0111 |
| T4 DNA ligase | Luciano et al., 2017 | N/A |
| T4 polynucleotide kinase | New England Biolabs | M0201L |
| S1 nuclease | Thermo Fisher Scientific | EN0321 |
| rRNasin | Promega | N251B |
| SuperScript III reverse transcriptase | Thermo Fisher Scientific | 18080-044 |
| fmol DNA Cycle Sequencing System | Promega | Q4100 |
| TURBO DNase | Thermo Fisher Scientific | AM2239 |
| Hfq | Santiago-Frangos et al., 2016 | N/A |
| Proteinase K | New England Biolabs | P8102S |
| Rifampicin | Sigma-Aldrich | R3501-5G |
| L-Tryptophan | Fisher | BP395-100 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | GoldBio | I2481 |
| 2-Nitrophenyl β-D-galactopyranoside (ONPG) | Sigma-Aldrich | N1127 |
| L-(+)-Arabinose | Sigma-Aldrich | A3256 |
| Methy α-D-glucopyranoside (α-methylglucoside) | Sigma-Aldrich | M9376 |
| Dimethyl sulfate | Sigma-Aldrich | D186309 |
| 2-Methylnicotinic acid | Fisher | AC397320050 |
| 1,1′-Carbonyldiimidazole | Fisher | AC151810100 |
| Dimethyl sulfoxide | Fisher | BP231-100 |
| ATP, [γ-32P] | PerkinElmer | NEG035C005MC |
| ATP | Roche | 11140965001 |
| GTP | Roche | 11140957001 |
| CTP | Roche | 11140922001 |
| UTP | Roche | 11140949001 |
| AMP | Sigma-Aldrich | A1752 |
| Phenol | Fisher | A92-500 |
| TALON Metal Affinity Resin | Clontech | 635503 |
| Sephadex G-25 | GE Healthcare | 17-0031-02 |
| Immobilon-Ny+ | Millipore | INYC00010 |
| Oligonucleotides | ||
| Oligonucleotides used in these studies | Table S2 | N/A |
| Recombinant DNA | ||
| Plasmids used in these studies | Table S3 | N/A |
| Software and Algorithms | ||
| ImageQuant TL | GE Healthcare | 29000737 |
Highlights.
A monophosphorylated 5′ end stimulates mRNA degradation by the endonuclease RNase E.
Its ability to cut is hindered by obstacles between the 5′ end and cleavage sites.
Effective obstacles include bound ribosomes, proteins, and sRNAs.
RNase E appears to scan RNA linearly from the 5′ end along single-stranded regions.
ACKNOWLEDGMENTS
We are grateful to Sarah Woodson for purified Hfq, Cari Vanderpool and Gerhart Wagner for E. coli strains lacking SgrS or MicA, and Dan Luciano, Monica Hui, and Rose Levenson-Palmer for helpful discussions. This research was supported by grants to J.G.B. from the National Institutes of Health (R01GM035769 and R01GM123124).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Antson AA, Dodson EJ, Dodson G, Greaves RB, Chen X, and Gollnick P (1999). Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature 401, 235–242. [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitzke P, Yealy J, and Campanelli D (1996). Interaction of the trp RNA-Binding attenuation protein (TRAP) of Bacillus subtilis with RNA: effects of the number of GAG repeats, the nucleotides separating adjacent repeats, and RNA secondary structure. J. Bacteriol 178, 5159–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Båga M, Göransson M, Normark S, and Uhlin BE (1988). Processed mRNA with differential stability in the regulation of E. coli pilin gene expression. Cell 52, 197–206. [DOI] [PubMed] [Google Scholar]
- Bandyra KJ, Said N, Pfeiffer V, Gorna MW, Vogel J, and Luisi BF (2012). The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol. Cell 47, 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belasco JG (2010). All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat. Rev. Mol. Cell Biol. 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg OG, Winter RB, and von Hippel PH (1981). Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 20, 6929–6948. [DOI] [PubMed] [Google Scholar]
- Bouvet P, and Belasco JG (1992). Control of RNase E-mediated RNA degradation by 5′-terminal base pairing in E. coli. Nature 360, 488–491. [DOI] [PubMed] [Google Scholar]
- Brunei C, and Romby P (2000). Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol. 318, 3–21. [DOI] [PubMed] [Google Scholar]
- Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, and Luisi BF (2005). Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 437, 1187–1191. [DOI] [PubMed] [Google Scholar]
- Celesnik H, Deana A, and Belasco JG (2007). Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell 27, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Y, Li L, Girodat D, Förstner KU, Said N, Corcoran C, Śmiga M, Papenfort K, Reinhardt R, Wieden HJ, et al. (2017). In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell 65, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JE, Kime L, Romero AD, and McDowall KJ (2014). Direct entry by RNase E is a major pathway for the degradation and processing of RNA in Escherichia coli. Nucleic Acids Res. 42, 11733–11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman TM, Wang G, and Huang F (2004). Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 φ2.5 promoter. Nucleic Acids Res. 32, el4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A, and Belasco JG (2005). Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 19, 2526–2533. [DOI] [PubMed] [Google Scholar]
- Deana A, Celesnik H, and Belasco JG (2008). The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451, 355–358. [DOI] [PubMed] [Google Scholar]
- Del Campo C, Bartholomäus A, Fedyunin I, and Ignatova Z (2015). Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLoS Genet. 11, e1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus M (2009). Killer and protective ribosomes. Prog. Mol. Biol. Transl. Sci 85, 423–466. [DOI] [PubMed] [Google Scholar]
- Edwards RA, Keller LH, and Schifferli DM (1998). Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207, 149–157. [DOI] [PubMed] [Google Scholar]
- Emory SA, Bouvet P, and Belasco JG (1992). A 5′-terminal stem-loop structure can stabilize mRNA in Escherichia coli. Genes Dev. 6, 135–148. [DOI] [PubMed] [Google Scholar]
- Feng Y, Vickers TA, and Cohen SN (2002). The catalytic domain of RNase E shows inherent 3′ to 5′ directionality in cleavage site selection. Proc. Natl. Acad. Sci. USA 99, 14746–14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley PL, Hsieh PK, Luciano DJ, and Belasco JG (2015). Specificity and evolutionary conservation of the Escherichia coli RNA pyrophosphohydrolase RppH. J. Biol. Chem 290, 9478–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich KS, Papenfort K, Fekete A, and Vogel J (2013). A small RNA activates CFA synthase by isoform-specific mRNA stabilization. EMBO J. 32, 2963–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrey SM, Blech M, Riffell JL, Hankins JS, Stickney LM, Diver M, Hsu YH, Kunanithy V, and Mackie GA (2009). Substrate binding and active site residues in RNases E and G: role of the 5′-sensor. J. Biol. Chem 284, 31843–31850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollnick P, Babitzke P, Antson A, and Yanofsky C (2005). Complexity in regulation of tryptophan biosynthesis in Bacillus subtilis. Annu. Rev. Genet 39, 47–68. [DOI] [PubMed] [Google Scholar]
- Halford SE (2009). An end to 40 years of mistakes in DNA-protein association kinetics? Biochem. Soc. Trans 37, 343–348. [DOI] [PubMed] [Google Scholar]
- Hui MP, Foley PL, and Belasco JG (2014). Messenger RNA degradation in bacterial cells. Annu. Rev. Genet 48, 537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, and Belasco JG (2004). Catalytic activation of multimeric RNase E and RNase G by 5′-monophosphorylated RNA. Proc. Natl. Acad. Sci. USA 101, 9211–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Diwa A, and Belasco JG (2000). Regions of RNase E important for 5′-end-dependentRNA cleavage and autoregulated synthesis. J. Bacteriol 182, 2468–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaberdin VR (2003). Probing the substrate specificity of Escherichia coli RNase E using a novel oligonucleotide-based assay. Nucleic Acids Res. 31, 4710–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto H, Koide Y, Morita T, and Aiba H (2006). Base-pairing requirement for RNA silencing by a bacterial small RNA and acceleration of duplex formation by Hfq. Mol. Microbiol 61, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Lee K, Bernstein JA, and Cohen SN (2002). RNase G complementation of rne null mutation identifies functional interrelationships with RNase E in Escherichia coli. Mol. Microbiol 43, 1445–1456. [DOI] [PubMed] [Google Scholar]
- Li Z, Pandit S, and Deutscher MP (1999). RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J. 18, 2878–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Chao S, Wong T-T, McDowall KJ, and Cohen SN (1994). Effects of nucleotide sequence on the specificity of rne-dependent and RNase E-mediated cleavages of RNA I encoded by the pBR322 plasmid. J. Biol. Chem 269, 10797–10803. [PubMed] [Google Scholar]
- Lodato PB, Hsieh PK, Belasco JG, and Kaper JB (2012). The ribosome binding site of a mini-ORF protects a T3SS mRNA from degradation by RNase E. Mol. Microbiol 86, 1167–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano DJ, Hui MP, Deana A, Foley PL, Belasco KJ, and Belasco JG (2012). Differential control of the rate of 5′-end-dependent mRNA degradation in Escherichia coli. J. Bacteriol 194, 6233–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano DJ, Vasilyev N, Richards J, Serganov A, and Belasco JG (2017). A novel RNA phosphorylation state enables 5′ end-dependent degradation in Escherichia coli. Mol. Cell 67, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie GA (1998). Ribonuclease E is a 5′-end-dependent endonuclease. Nature 395, 720–723. [DOI] [PubMed] [Google Scholar]
- Mackie GA (2013a). Determinants in the rpsT mRNAs recognized by the 5′-sensor domain of RNase E. Mol. Microbiol 89, 388–402. [DOI] [PubMed] [Google Scholar]
- Mackie GA (2013b). RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol 11, 45–57. [DOI] [PubMed] [Google Scholar]
- McDowall KJ, Hernandez RG, Lin-Chao S, and Cohen SN (1993). The ams-1 and rne-3071 temperature-sensitive mutations in the ams gene are in close proximity to each other and cause substitutions within a domain that resembles a product of the Escherichia coli mre locus. J. Bacteriol 175, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowall KJ, Lin-Chao S, and Cohen SN (1994). A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J. Biol. Chem 269, 10790–10796. [PubMed] [Google Scholar]
- Meacock PA, and Cohen SN (1980). Partitioning of bacterial plasmids during cell division: a cis-acting locus that accomplishes stable plasmid inheritance. Cell 20, 529–542. [DOI] [PubMed] [Google Scholar]
- Mechetin GV, and Zharkov DO (2014). Mechanisms of diffusional search for specific targets by DNA-dependent proteins. Biochemistry (Mosc) 79, 496–505. [DOI] [PubMed] [Google Scholar]
- Michaux C, Holmqvist E, Vasicek E, Sharan M, Barquist L, Westermann AJ, Gunn JS, and Vogel J (2017). RNA target profiles direct the discovery of virulence functions for the cold-shock proteins CspC and CspE. Proc. Natl. Acad. Sci. USA 114, 6824–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miczak A, Kaberdin VR, Wei CL, and Lin-Chao S (1996). Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. USA 93, 3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH (1992). A Short Course in Bacterial Genetics (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Neidhardt FC, Bloch PL, and Smith DF (1974). Culture medium for enterobacteria. J. Bacteriol 119, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow MC, Perwez T, and Kushner SR (2003). RNase G of Escherichia coli exhibits only limited functional overlap with its essential homologue, RNase E. Mol. Microbiol 49, 607–622. [DOI] [PubMed] [Google Scholar]
- Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, and Vogel J (2013). Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py B, Higgins CF, Krisch HM, and Carpousis AJ (1996). A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381, 169–172. [DOI] [PubMed] [Google Scholar]
- Richards J, and Belasco JG (2016). Distinct requirements for 5′-monophosphate-assisted RNA cleavage by Escherichia coli RNase E and RNase G. J. Biol. Chem 291, 5038–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J, Luciano DJ, and Belasco JG (2012). Influence of translation on RppH-dependent mRNA degradation in Escherichia coli. Mol. Microbiol 86, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringquist S, Shinedling S, Barrick D, Green L, Binkley J, Stormo GD, and Gold L (1992). Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol. Microbiol 6, 1219–1229. [DOI] [PubMed] [Google Scholar]
- Santiago-Frangos A, Kavita K, Schu DJ, Gottesman S, and Woodson SA (2016). C-terminal domain of the RNA chaperone Hfq drives sRNA competition and release of target RNA. Proc. Natl. Acad. Sci. USA 113, E6089–E6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro SW, and Joyce GF (1997). A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, and Chang HY (2013). RNA SHAPE analysis in living cells. Nat. Chem. Biol 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, and Vanderpool CK (2013). Physiological consequences of multiple-target regulation by the small RNA SgrS in Escherichia coli. J. Bacteriol 195, 4804–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tock MR, Walsh AP, Carroll G, and McDowall KJ (2000). The CafA protein required for the 5′-maturation of 16 S rRNA is a 5′-end-dependent ribonuclease that has context-dependent broad sequence specificity. J. Biol. Chem 275, 8726–8732. [DOI] [PubMed] [Google Scholar]
- Udekwu KI, Darfeuille F, Vogel J, Reimegard J, Holmqvist E, and Wagner EG (2005). Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 19, 2355–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderpool CK, and Gottesman S (2004). Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol. Microbiol 54, 1076–1089. [DOI] [PubMed] [Google Scholar]
- Wachi M, Umitsuki G, Shimizu M, Takada A, and Nagai K (1999). Escherichia coli cafA gene encodes a novel RNase, designated as RNase G, involved in processing of the 5′ end of 16S rRNA. Biochem. Biophys. Res. Commun 259, 483–488. [DOI] [PubMed] [Google Scholar]
- Woolstenhulme CJ, Guydosh NR, Green R, and Buskirk AR (2015). High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Burkhardt DH, Rouskin S, Li GW, Weissman JS, and Gross CA (2018). A stress response that monitors and regulates mRNA structure Is central to cold shock adaptation. Mol. Cell 70, 274–286 e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.