

Abstract
Objective
To perform a retrospective analysis examining the incidence and prognosis of glioma patients with leptomeningeal disease (LMD) at Memorial Sloan Kettering Cancer Center over a 15-year period and correlate these findings with clinicopathologic characteristics.
Methods
We conducted a retrospective review of glioma patients with LMD at Memorial Sloan Kettering Cancer Center diagnosed from 2001 to 2016. Patients were identified through a keyword search of their electronic medical record and by ICD-9 codes.
Results
One hundred three patients were identified with disseminated LMD and 85 patients with subependymal spread of disease, 4.7% of all patients with glioma. These cohorts were analyzed separately for time to development of disseminated LMD/subependymal LMD, median overall survival, and survival from LMD diagnosis. Patients were pooled for subsequent analyses (n = 188) because of comparable clinical behavior. LMD was present at glioma diagnosis in 10% of patients. In the remaining 90% of patients diagnosed at recurrence, time to LMD diagnosis, survival after LMD diagnosis, and overall survival varied by original histology. Patients with oligodendroglioma had a median survival of 10.8 (range 1.8–67.7) months, astrocytoma 6.5 (0.1–28.5) months, and glioblastoma 3.8 (0.1–32.6) months after LMD diagnosis. In addition, we found that treatment of LMD was associated with superior performance status and increased survival.
Conclusion
Patients with LMD diagnosed at relapse may not have decreased overall survival as compared to historical controls with parenchymal relapse and may benefit from treatment.
Glioma cell invasion into the CSF or leptomeninges, known as leptomeningeal spread of disease (LMD), occurs in 4% of patients with glioma.1–4 LMD is often ascribed a worse prognosis than progression of parenchymal disease, with a median survival in oligodendroglioma of 16 to 22 months,1,2 astrocytoma of 4 months,1 and glioblastoma, 3.5 to 3.9 months.3–5 Recent studies indicate that the incidence of LMD is increasing, possibly because of prolonged survival, more frequent testing, and antiangiogenic therapy.6–9
Suspicion of LMD arises when a patient develops cranial neuropathies, cerebellar signs, conus medullaris/cauda equina symptoms, or signs of hydrocephalus.10 Evaluation involves MRI of the brain and spine, and often lumbar puncture. LMD diagnosis can be made radiographically or through positive CSF cytology, but this is rare in glioma.11 In addition, disease can spread through the CSF along the ventricles, leading to subependymal or ependymal enhancement on MRI. Controversy exists regarding the prognostic significance of subependymal spread and whether it should be considered a harbinger for CSF dissemination.12–14 However, no studies have compared outcomes between glioma patients with subependymal and disseminated disease.
Several series report on treatment of LMD in glioma. Systemic and intrathecal chemotherapy, radiation, and antiangiogenic therapies have been tested, with some evidence that combination therapy is more effective.1–3,5,15 A recent case report demonstrated efficacy for intratumoral and intrathecal chimeric antigen receptor T cells.16
We collected data retrospectively from glioma patients with LMD diagnosed over a 15-year period at our institution. We investigated the incidence of LMD over time, clinicopathologic correlates of LMD, treatment, and the effect of LMD on survival.
Methods
Standard protocol approvals, registrations, and patient consents
The institutional review board at Memorial Sloan Kettering Cancer Center approved the study, granting a waiver for informed consent because of its retrospective nature.
Case ascertainment
This is a retrospective study of patients with leptomeningeal spread of glioma diagnosed between January 1, 2001, and June 16, 2016. After institutional review board approval, all potential patients were identified through our institutional database at Memorial Sloan Kettering Cancer Center. We identified glioma patients with at least one office visit at Memorial Sloan Kettering Cancer Center using the following criteria: (1) clinical documentation of LMD in chart notes including the terms “leptomeningeal,” “CSF dissemination,” or “meningeal seeding”; (2) CSF cytology reports, for the terms “malignant” or “positive”; and (3) ICD-9 codes 198.4 (spinal cord/meninges metastases), C79.32 (secondary malignant neoplasm of cerebral meninges), 198.3 (brain/spine metastases), 192.2 (spinal cord cancer), C79.31 (secondary malignant neoplasm of the brain), and C72.0 (malignant neoplasm of the spinal cord).
All charts were manually reviewed. Patients were included in the study if clinical radiology reports specifically mentioned leptomeningeal dissemination of disease (figure 1, A and B) on either the brain or spine MRI or subependymal or ependymal enhancement on the brain MRI (which differ by one cell layer and are radiographically indistinguishable; figure 1, C and D). Patients with positive CSF cytology per the official pathology report were also included in the study.
Figure 1. MRIs of leptomeningeal tumor spread in patients with glioma.
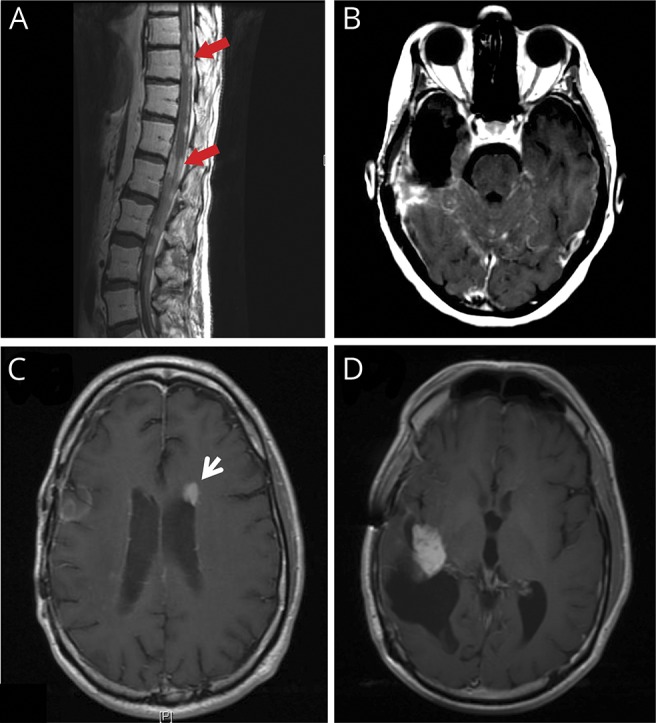
(A) Sagittal T1-post contrast of the lower spine demonstrating superficial enhancement (red arrows) along the distal cord, conus medullaris, and cauda equina nerve roots secondary to LMD in a patient with anaplastic astrocytoma. (B) Axial T1 postcontrast brain MRI showing superficial leptomeningeal enhancement along the cerebellar folia and surface of the pons in a patient with glioblastoma. (C) Axial T1 postcontrast brain MRI showing subependymal LMD (white arrow) in a patient with anaplastic oligodendroglioma. The primary tumor site (D) was contralateral to and distant from the new subependymal disease. LMD = leptomeningeal disease.
Further eligibility criteria included age 18 years or older at original tumor diagnosis, and a confirmed pathologic diagnosis of a grade 2 to 4 glioma. Specifically, patients with grade 1 tumors, ependymoma, or primary brainstem or spinal cord gliomas were excluded. The date of LMD diagnosis was defined as the earliest date of imaging on which LMD was noted or positive CSF cytology. When radiographic evidence of LMD preceded the surgery that diagnosed the glioma, the date of LMD diagnosis used was the date of surgery and glioma diagnosis.
Original search hits outnumbered true cases approximately 4 to 1. We manually confirmed all search hits, rather than reviewing all patients with glioma in the study period, because of practical limitation. Cases of glioma within the radiation field that did not show clear evidence of tumor growth over time were excluded, as radiation-induced pseudoprogression could not be excluded. Cases in which subependymal disease preceded disseminated LMD (dLMD) were assigned to the dLMD cohort and date of LMD diagnosis was the time dLMD developed.
Data collection
Data collected for each patient included date of birth, date of initial glioma diagnosis, date of LMD diagnosis, Karnofsky Performance Status (KPS) score at LMD diagnosis (estimated by the investigator based on chart review if not recorded), histologic and molecular pathologic analysis, initial and subsequent treatment of LMD, MRI results, CSF results, and date of death or last follow-up.
Original histologic diagnosis was made in accordance with the 2016 World Health Organization classification system when possible. Grade 2 and 3 tumors that were negative for 1p19q codeletion or in which 1p19q analysis was not done were considered astrocytoma, while 1p19q codeletion was a requirement for diagnosis of an oligodendroglioma. Isocitrate dehydrogenase (IDH) mutations were considered present if patients tested positive for IDH1 R132H by immunohistochemistry, PCR for alternate R132 mutations, or if IDH1 or 2 was mutated on next-generation sequencing. Patients without a documented date of death were censored at date of last follow-up; there were 5 patients with oligodendroglioma, 3 patients with astrocytoma, and 4 patients with glioblastoma censored from survival analysis.
Statistical analysis
Overall survival was defined as the time from glioma diagnosis to the time of death or last follow-up. Survival rates were determined using the Kaplan-Meier method, and survival curves were compared using the log-rank test. A p value <0.05 was considered significant for all analyses. All statistical analyses were performed in GraphPad Prism version 6.04 (GraphPad Software, La Jolla, CA).
Data sharing
Any data not available within this article will be anonymized and made available on request from any qualified investigator.
Results
Patient characteristics
A total of 4,082 unique patients with glioma with at least one office visit at Memorial Sloan Kettering Cancer Center over a 15-year period (2001–2016) were identified. Cases detected in the initial database inquiry were manually confirmed by chart review, including clinical notes, radiology reports, pathology reports, and molecular data when available. One hundred three patients had dLMD as identified on the clinical radiology reports for the brain or spine MRI, or positive CSF cytology. Upon manual review of charts, there were also 85 patients who exhibited clinical signs concerning for leptomeningeal spread of disease who were noted on official radiology reports to have ependymal or subependymal enhancement and not dLMD. There were 9 patients who developed subependymal spread of disease, then later developed dLMD, who were included in the dLMD cohort with date of LMD diagnosis being the time of disseminated disease.
We initially conducted a separate analysis on these 2 groups of patients—dLMD and subependymal LMD (sLMD)—to assess whether the clinical outcomes were comparable between these 2 groups. Patients with sLMD or dLMD diagnosed at glioma recurrence were analyzed by original histology (n = 168). We found that the time to development of sLMD or dLMD did not differ (glioblastoma, 14.7 vs 11.4 months, respectively, p = 0.32; astrocytoma, 18.4 vs 23.5 months, p = 0.51; and oligodendroglioma, 76.4 vs 61.7 months, p = 0.84). In addition, survival from LMD diagnosis (glioblastoma, 3.8 for sLMD vs 3.2 months for dLMD, p = 0.31; astrocytoma, 6.6 vs 5.2 months, p = 0.75; oligodendroglioma, 11.7 vs 10.66 months, p = 0.77) and overall survival for the groups with sLMD and dLMD were also equivalent (glioblastoma, 21.7 [range 3.1–99.1] vs 16.3 [5.3–83.7] months, p = 0.17; figure 2A; astrocytoma, 28.7 vs 42.2 months, p = 0.35; oligodendroglioma, 191.2 vs 132 months, p = 0.64). Moreover, of 17 patients with glioblastoma found to have LMD at glioma diagnosis, there were 9 with sLMD and 8 with dLMD (median survival 5.0 vs 9.0 months, p = 0.49). We also found that symptoms of increased intracranial pressure were equal between sLMD and dLMD (14% vs 17%, p = 0.63; table 1), and the number of patients who required ventriculoperitoneal (VP) shunting was equal for those with sLMD and dLMD (n = 18; 23% vs 19%; p = 0.98). Based on the comparable clinical outcomes between these cohorts, they were pooled for all further analyses.
Figure 2. Median survival times.

(A) Median survival in GBM was 21.7 (range 3.1–99.1) months for subependymal LMD and 16.3 (5.3–83.7) months for disseminated LMD (p = 0.21). (B) Median survival was 8.3 (range 1.5–25.9) months for patients whose LMD was present at GBM diagnosis and 17.1 (3.1–82) months for those with LMD diagnosed at recurrence (p < 0.0001). GBM = glioblastoma; LMD = leptomeningeal disease.
Table 1.
Symptoms at time of diagnosis of leptomeningeal spread of disease

In total, our study included 188 patients (n = 188, 4.6%) with dLMD and sLMD. Incidence at our institution appears to be increasing: 1.8% of patients diagnosed with gliomas between 2001 and 2005 developed LMD, 4.4% in 2006–2010, and 6.9% in 2011–2016. Of these 188 patients, 31 had astrocytoma (n = 7 World Health Organization grade 2, n = 24 grade 3), 29 had oligodendroglioma (n = 13 grade 2, N = 16 grade 3), and 128 had glioblastoma. Only the glioblastoma cohort was sufficiently large for most within-group comparisons. While almost all patients with LMD became symptomatic from LMD, nearly half of patients were diagnosed on surveillance imaging and were asymptomatic (n = 90, 47%; table 1). Fifty patients (26%) were diagnosed with LMD after they developed cranial symptoms, such as cranial neuropathies and ataxia, and 31 patients (16%) developed symptoms of increased intracranial pressure. Twenty-two patients (12%) developed spinal symptoms such as radicular pain, urinary or bowel dysfunction, or meningismus. Twenty-one patients (11%) presented with symptoms due to a combination of cranial, spinal, or intracranial hypertension symptoms.
Within our cohort, 20 patients were diagnosed with LMD at glioma diagnosis (10% of patients with LMD and 0.5% of patients with glioma); 17 with glioblastoma, 2 with astrocytoma (one initially a grade 2 and one initially a grade 3 tumor), and one with an anaplastic oligodendroglioma. The 17 patients with LMD at presentation of glioblastoma survived a median of 8.3 (range 1.5–25.9) months after diagnosis, approximately half the time of the patients with glioblastoma who developed LMD at recurrence, who had an overall median survival of 17.1 (3.1–82) months (p < 0.0001; figure 2B).
There were 168 patients diagnosed with LMD at recurrence, 29 with astrocytoma, 28 with oligodendroglioma, and 111 with glioblastoma. The median age was 44 (range 20–88) years and median KPS score was 70 (range 30–100) at LMD diagnosis for all groups except oligodendroglioma, in which it was 80 (50–100; table 2). There were more men (61%) than women. One hundred fifty-nine of 168 patients (95%) were diagnosed with radiographic LMD on brain MRI and 42 of 75 patients (56%) were found to have LMD on MRI of the spine. Thirty-three patients met radiographic criteria in both brain and spine MRI, and 9 patients met criteria only on spine MRI (table 1). Forty-four patients also underwent lumbar puncture in order to diagnose LMD or to predict responsiveness to CSF diversion; CSF cytology was positive in 11 (25%). Nine of 11 patients with positive CSF cytology were also found to have radiographic LMD. Of the 9 patients with positive CSF cytology and radiographic evidence of LMD, 8 were found to have radiographic LMD on both the brain and spine MRI.
Table 2.
Patient characteristics

Within our cohort, the median lines of treatment prior to LMD was 2 in the patients with glioblastoma and astrocytoma, indicating LMD was most often associated with advanced disease (second recurrence). Of 28 patients originally diagnosed with astrocytoma, 8 (28%) had histologically confirmed progression to glioblastoma at LMD diagnosis. Eleven of 28 patients (39%) with oligodendroglioma were originally diagnosed with grade 2 tumors, and all had progressed to grade 3 by the time of LMD diagnosis; moreover, patients with oligodendroglioma received a median of 4 lines of therapy before LMD diagnosis (table 2), reflecting their longer disease course. Among all 168 patients with glioma, 156 (93%) had received prior radiation, 152 (90%) received cytotoxic chemotherapy, 40 (24%) received antiangiogenic therapy, 51 (30%) had been in a clinical trial, and 25 (15%) received targeted therapy (table 2).
Histopathology among glioblastoma patients with LMD at presentation was compared with those diagnosed with LMD at recurrence; 2 patients with LMD at diagnosis (11.7%; 2/17) had gliosarcoma, while there were 8 patients with gliosarcoma (7.1%; 8/112) who developed LMD at recurrence, giving a total frequency of gliosarcoma of 7.8% (10/129) within our cohort. None of the patients with LMD at disease presentation had primitive neuroectodermal tumor (PNET) features, while 6 (5.3%; 6/112) with LMD at recurrence had PNET features, giving an overall frequency of 4.7% (6/129) for glioblastoma with PNET features. Three patients with an original diagnosis of astrocytoma, progression to glioblastoma, and LMD at recurrence had histologic PNET features (11%; 3/28 for astrocytoma).
Time to leptomeningeal spread of disease
Time from glioma diagnosis to development of LMD varied by histology; patients with oligodendroglioma developed LMD at a median of 61.7 (range 3.9–274.8) months, astrocytoma at a median of 19.1 (1.4–202.8) months, and glioblastoma at a median of 11.9 (0.9–92.6) months (p < 0.0001; figure 3A).
Figure 3. Time to LMD, overall survival, and survival from LMD in patients with glioblastoma diagnosed with LMD at recurrence.

(A) Median time from glioma diagnosis was 11.9 (range 0.9–92.6) months for glioblastoma, 19.1 (1.4–202.8) months for astrocytoma, and 61.7 (3.9–274.8) months for oligodendroglioma (p < 0.0001). Median survival (B) from glioma diagnosis was 17.1 (range 3.1–99) months for glioblastoma, 33.5 (1.5–207) months for astrocytoma, and 132 (8.5–279) months for oligodendroglioma (p < 0.0001). (C) Median survival after LMD diagnosis was 3.8 (range 0.1–32.6) months for glioblastoma, 6.5 (0.1–28.5) months for astrocytoma, and 10.8 (1.8–67.7) months for oligodendroglioma (p = 0.0004). LMD = leptomeningeal disease.
Survival
Of the 168 patients with LMD at recurrence, 159 (93%) have died. Median overall survival from initial tumor diagnosis for patients with LMD and oligodendroglioma was 132 (range 8.5–279) months, for astrocytoma 33.5 (1.5–207) months, and for glioblastoma 17.1 (3.1–99) months (p < 0.0001; figure 3B). Survival from the time of LMD diagnosis also varied by histology: patients with oligodendroglioma survived a median of 10.8 (range 1.8–67.7) months, while those with astrocytoma and glioblastoma survived a median of 6.5 (0.1–28.5) and 3.8 (0.1–32.6) months, respectively (p < 0.0004; figure 3C).
Treatment of leptomeningeal spread of disease
A median of one line of treatment was given following diagnosis of LMD in all 3 histologic groups (table 3). Of the 168 patients with LMD at recurrence, 118 (69%) received some form of therapy, whereas 52 (31%) did not receive further tumor-directed therapy. Sixteen patients (9%) received radiotherapy, which included intensity-modulated radiotherapy to the brain or spine, whole-brain radiotherapy, or whole-spine radiotherapy. Cytotoxic chemotherapy was used in 59 patients (35%) and antiangiogenic therapy was administered to 49 patients (29%). Four patients were enrolled in a clinical trial immediately following a diagnosis of LMD (2%), and 9 (5%) were treated with targeted therapy off-label. The development of hydrocephalus is a common complication of leptomeningeal spread of cancer, and symptoms may be alleviated by VP shunt placement. Thirty-six patients (23%) underwent a VP shunt for CSF diversion, of whom 11 (7%) had shunting immediately after diagnosis of LMD (table 3). One patient underwent Ommaya reservoir placement and treatment with intrathecal cytarabine.
Table 3.
Initial treatment of leptomeningeal disease in patients with glioma diagnosed at recurrence

After diagnosis of LMD, the 36 patients with glioblastoma who ceased tumor-directed therapy had a median KPS score of 60 and survived a median of 1.6 months. The 75 patients with glioblastoma undergoing some form of treatment had a median KPS score of 80 and survived a median of 5.0 months after LMD diagnosis. Time to LMD did not differ between these 2 groups (median 11.5 months in treated LMD vs 12.3 months in untreated LMD; p = 0.88).
Furthermore, patients with glioblastoma who received treatment were divided into those who received any radiotherapy, antiangiogenic therapy with or without chemotherapy, cytotoxic chemotherapy alone, or targeted therapy/clinical trial therapy. Patients with LMD who initially received some form of radiotherapy, with or without other treatments, survived a median of 5.0 months. Patients initially receiving antiangiogenic therapy survived a median of 7.6 months, patients who received chemotherapy alone survived a median of 4.8 months, and those treated with targeted or clinical trial therapies survived 7.6 months; median survival did not differ among these treatment groups (p = 0.33; data not shown).
Discussion
This retrospective series examines the clinical disease course and survival outcomes of 188 patients with glioma who had LMD over the past 15 years. The estimated overall incidence of LMD in patients with glioma was 4.7% at our institution, but over time the incidence increased. Incidence may have increased because of more frequent testing for LMD, the improvement in MRI resolution at our center, improved overall survival, and/or changing referrals to our tertiary care center. While rigorously matched comparisons are not available, median overall survival from time of glioma diagnosis in our cohort was roughly equal to historic cohorts, except for slightly prolonged survival in anaplastic astrocytoma and glioblastoma.17,18 Patients given standard of care treatment for glioblastoma in recent prospective trials, although receiving the same regimens as in original publications, are living several months longer, an improvement thought to be attributable to optimization of standard of care.18–20 This incremental improvement in overall survival in a large number of patients may have contributed to increased incidence of LMD. Overall, the increasing incidence in our study echoes that of other studies and discussions in the neuro-oncology community that LMD is an increasingly prominent clinical problem.
Time to development of LMD, survival from LMD, and overall survival in patients with LMD are highly dependent on original histology. LMD in patients with oligodendroglioma appears at a median time of the fourth recurrence. The relatively slower course of oligodendroglioma is consistent with previous reports on LMD in oligodendroglioma21 and is likely reflective of the underlying growth rate and rate of clonal evolution of these tumors.
We searched for factors that predisposed to the development of LMD, but none were definitively identified. Our limited molecular data could not establish whether unfavorable molecular features such as IDH wild type in grade 2 and 3 tumors or other genetic mutations predispose to LMD development. Many but not all cases of LMD involved concomitant parenchymal progression of disease, with all cases of grade 2 oligodendroglioma progressing to grade 3, and 8 of 29 cases of astrocytoma progressing from grade 2 or 3 to glioblastoma. There were also cases of LMD, disseminated and subependymal, in which LMD appeared in the absence of parenchymal progression. This variability in spread of glioma is in keeping with knowledge that specific gene mutations are implicated and required for LMD in animal models in systemic solid malignancies.22 Larger tumor burden may enable enough genomic diversity to increase the probability of a CSF-adaptable mutation, but such a mutation may occasionally develop without clear evidence of parenchymal tumor growth.
It has also been hypothesized that antiangiogenic therapies may induce invasive cellular programs and increase the risk of LMD in glioma. In our cohort, 24% of 168 patients had received bevacizumab prior to development of LMD, compared to 49% of patients with recurrent anaplastic astrocytoma and glioblastoma at our institution from the same time period.23 Thus, our study provides no evidence that antiangiogenic therapies predispose to LMD. More research is required to better understand whether a relationship exists between anti-angiogenic therapy and the development of LMD in patients with glioma.
We also searched for histopathologic PNET features, which are more frequently associated with LMD. PNET features were overrepresented in our glioblastoma population (4.7% in our cohort vs 0.5% of cases in the general glioblastoma population); the relative prevalence of PNET features in patients originally diagnosed with astrocytoma who progressed to glioblastoma is consistent with previous literature, which noted that glioblastomas with PNET features appear to have a tropism for CSF and more frequently arise from previously lower grade astrocytomas.24
Subependymal spread of glioma may indicate high risk of CSF dissemination, but it has not historically been equated with LMD in glioma. Our data demonstrate that both groups have equivalent clinical behavior regarding the following: (1) time to development of LMD/subependymal disease; (2) survival from LMD/subependymal diagnosis; (3) overall survival; (4) rates of hydrocephalus and VP shunt placement; and (5) frequency of symptoms of increased intracranial pressure. Together, these results support the fact that subependymal spread is clinically equivalent to dLMD.
Ten percent of our patients had LMD at glioma diagnosis, but most patients developed LMD approximate to the second recurrence. LMD at glioblastoma diagnosis carries a worse overall prognosis, with median overall survival of only 8.3 months despite full treatment, which is substantially shorter than the median of 16 months reported in most series. It is unclear whether progression of glioblastoma in the form of LMD carries a worse prognosis than progression of parenchymal disease, as proper comparisons are lacking.
Limitations of our study include its single-center, retrospective nature and the extended time period of the study (with inevitable heterogeneity in diagnosis and treatment over this period). Our calculated incidence is probably lower than the true incidence; the terms “subependymal” and “ependymal” were not part of the initial database search, probably resulting in failure to identify all cases. Identification of LMD through chart documentation, such as “leptomeningeal,” “CSF dissemination,” or “meningeal seeding,” or ICD codes, is admittedly prone to bias from the clinician, who uses these terms in notes and enters billing information. Determination of treatment efficacy in patients with LMD was not possible because of strong selection bias between untreated patients with poor performance and treated patients with adequate performance. Our study also did not permit an assessment of morbidity or quality of life in those with LMD. Determination of these critical clinical features would require a prospective study.
Despite these limitations, our cohort demonstrates that glioblastoma patients survive a median of 5 months after LMD diagnosis if the KPS score is ≥80. Overall, this retrospective series of glioma patients with LMD suggests that patients who develop subependymal or dLMD at the time of recurrence should be evaluated clinically, and their LMD could be treated if their performance status is adequate and aligned with the patient's/family's wishes.
Glossary
- dLMD
disseminated leptomeningeal disease
- ICD-9
International Classification of Diseases, Ninth Revision
- IDH
isocitrate dehydrogenase
- KPS
Karnofsky Performance Status
- LMD
leptomeningeal disease
- PNET
primitive neuroectodermal tumor
- sLMD
subependymal leptomeningeal disease
- VP
ventriculoperitoneal
Appendix. Authors
Study funding
Supported through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Disclosure
B. Andersen, C. Miranda, and V. Hatzoglou report no disclosures relevant to the manuscript. L. DeAngelis received scientific advisory board honoraria from Sapience Therapeutics, Tocagen, BTG International, Roche, and Syndax. A. Miller reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Dardis C, Milton K, Ashby L, Shapiro W. Leptomeningeal metastases in high-grade adult glioma: development, diagnosis, management, and outcomes in a series of 34 patients. Front Neurol 2014;5:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roldan G, Scott J, George D, et al. Leptomeningeal disease from oligodendroglioma: clinical and molecular analysis. Can J Neurol Sci 2008;35:204–209. [DOI] [PubMed] [Google Scholar]
- 3.Lawton CD, Nagasawa DT, Yang I, Fessler RG, Smith ZA. Leptomeningeal spinal metastases from glioblastoma multiforme: treatment and management of an uncommon manifestation of disease. J Neurosurg Spine 2012;17:438–448. [DOI] [PubMed] [Google Scholar]
- 4.Mandel JJ, Yust-Katz S, Cachia D, et al. Leptomeningeal dissemination in glioblastoma: an inspection of risk factors, treatment, and outcomes at a single institution. J Neurooncol 2014;120:597–605. [DOI] [PubMed] [Google Scholar]
- 5.Chamberlain MC. Combined-modality treatment of leptomeningeal gliomatosis. Neurosurgery 2003;52:324–329; discussion 330. [DOI] [PubMed] [Google Scholar]
- 6.de Groot JF, Fuller G, Kumar AJ, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 2010;12:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayana A, Golfinos JG, Fischer I, et al. Feasibility of using bevacizumab with radiation therapy and temozolomide in newly diagnosed high-grade glioma. Int J Radiat Oncol Biol Phys 2008;72:383–389. [DOI] [PubMed] [Google Scholar]
- 8.Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 2008;70:779–787. [DOI] [PubMed] [Google Scholar]
- 9.Vertosick FT Jr, Selker RG. Brain stem and spinal metastases of supratentorial glioblastoma multiforme: a clinical series. Neurosurgery 1990;27:516–521; discussion 521–522. [DOI] [PubMed] [Google Scholar]
- 10.DeAngelis LM, Posner JB. Neurologic Complications of Cancer, 2nd ed. New York: Oxford University Press; 2009. [Google Scholar]
- 11.Pentsova EI, Shah RH, Tang J, et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol 2016;34:2404–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaidar-Person O, Darawshe F, Tzuk-Shina T, Eran A. The clinical significance of ependymal enhancement at presentation in patients with malignant glioma. Rambam Maimonides Med J 2015;6:e0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsa AT, Wachhorst S, Lamborn KR, et al. Prognostic significance of intracranial dissemination of glioblastoma multiforme in adults. J Neurosurg 2005;102:622–628. [DOI] [PubMed] [Google Scholar]
- 14.Witham TF, Fukui MB, Meltzer CC, Burns R, Kondziolka D, Bozik ME. Survival of patients with high grade glioma treated with intrathecal thiotriethylenephosphoramide for ependymal or leptomeningeal gliomatosis. Cancer 1999;86:1347–1353. [DOI] [PubMed] [Google Scholar]
- 15.Burger MC, Zeiner PS, Jahnke K, Wagner M, Mittelbronn M, Steinbach JP. Addition of anti-angiogenetic therapy with bevacizumab to chemo- and radiotherapy for leptomeningeal metastases in primary brain tumors. PLoS One 2016;11:e0155315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 2005;64:479–489. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014;370:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017;18:1373–1385. [DOI] [PubMed] [Google Scholar]
- 20.Stupp R, Taillibert S, Kanner A, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 2017;318:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roldan G, Chan J, Eliasziw M, Cairncross JG, Forsyth PA. Leptomeningeal disease in oligodendroglial tumors: a population-based study. J Neurooncol 2011;104:811–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massague J. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell 2017;168:1101–1113.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omuro A, Chan TA, Abrey LE, et al. Phase II trial of continuous low-dose temozolomide for patients with recurrent malignant glioma. Neuro Oncol 2013;15:242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry A, Miller CR, Gujrati M, et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol 2009;19:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]