

Abstract
Objective
To report results of intrathecal nusinersen in children with later-onset spinal muscular atrophy (SMA).
Methods
Analyses included children from a phase 1b/2a study (ISIS-396443-CS2; NCT01703988) who first received nusinersen during that study and were eligible to continue treatment in the extension study (ISIS-396443-CS12; NCT02052791). The phase 1b/2a study was a 253-day, ascending dose (3, 6, 9, 12 mg), multiple-dose, open-label, multicenter study that enrolled children with SMA aged 2–15 years. The extension study was a 715-day, single-dose level (12 mg) study. Time between studies varied by participant (196–413 days). Assessments included the Hammersmith Functional Motor Scale–Expanded (HFMSE), Upper Limb Module (ULM), 6-Minute Walk Test (6MWT), compound muscle action potential (CMAP), and quantitative multipoint incremental motor unit number estimation. Safety also was assessed.
Results
Twenty-eight children were included (SMA type II, n = 11; SMA type III, n = 17). Mean HFMSE scores, ULM scores, and 6MWT distances improved by the day 1,150 visit (HFMSE: SMA type II, +10.8 points; SMA type III, +1.8 points; ULM: SMA type II, +4.0 points; 6MWT: SMA type III, +92.0 meters). Mean CMAP values remained relatively stable. No children discontinued treatment due to adverse events.
Conclusions
Nusinersen treatment over ∼3 years resulted in motor function improvements and disease activity stabilization not observed in natural history cohorts. These results document the long-term benefit of nusinersen in later-onset SMA, including SMA type III.
Clinicaltrials.gov identifier
NCT01703988 (ISIS-396443-CS2); NCT02052791 (ISIS-396443-CS12).
Classification of evidence
This study provides Class IV evidence that nusinersen improves motor function in children with later-onset SMA.
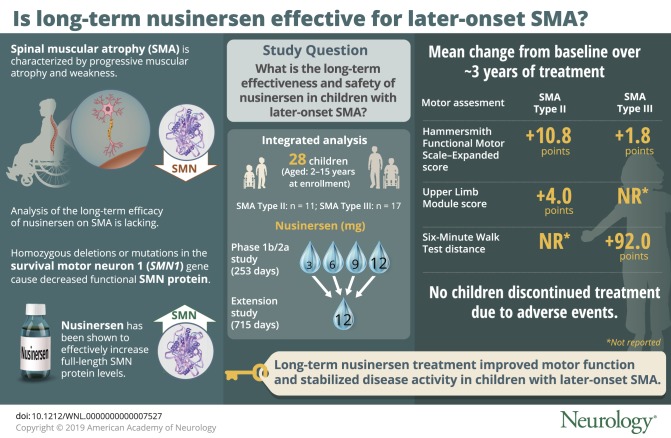
Spinal muscular atrophy (SMA) is a neuromuscular disorder characterized by progressive muscular atrophy and weakness.1 Infants with SMA type I (symptom onset ≤6 months) are unable to sit independently. Children with SMA type II (symptom onset 7–18 months) can sit unsupported, though some may lose this ability over time, and some may stand but are unable to walk independently. Children with SMA type III (symptom onset >18 months) can walk independently, but some may lose this ability over time.
SMA is caused by homozygous deletions or mutations in the survival motor neuron 1 (SMN1) gene, resulting in decreased full-length functional SMN protein expression and spinal cord and brainstem motor neuron degeneration.2 The paralogous SMN2 gene also codes for the SMN protein but produces very little functional SMN protein due to aberrant splicing.1
Nusinersen is an antisense oligonucleotide drug that modifies SMN2 premessenger RNA splicing, resulting in increased full-length SMN protein levels.3 In previous clinical studies, nusinersen showed efficacy over placebo or the expected clinical course of SMA in individuals with infantile-onset SMA (most likely to develop SMA type I or II) and later-onset SMA (most likely to develop SMA type II or III) as well as a favorable benefit–risk profile.4–7
We report effectiveness and safety results in children with later-onset SMA (have or are most likely to develop SMA type II or III) who received their first nusinersen dose in the phase 1b/2a ISIS-396443-CS2 (CS2) study and who were eligible to continue treatment in the ISIS-396443-CS12 (CS12) extension study.
Methods
Standard protocol approvals, registrations, and patient consents
The CS2 and CS12 study protocols and informed consent/assent forms were approved by institutional review boards at each study center before participant recruitment. Parent or legal guardian written informed consent and, when guidelines and participant age required, informed assent were obtained for each participant. The World Medical Association Declaration of Helsinki and Good Clinical Practice guidelines were strictly followed throughout the study. An independent data and safety monitoring board (DSMB) provided oversight of the studies in collaboration with the sponsors. CS2 and CS12 were registered with ClinicalTrials.gov (NCT01703988 and NCT02052791, respectively).
Participants
In CS2, eligible children were aged 2–15 years, had genetic documentation of 5q SMA, had clinical symptoms attributable to SMA, and met all additional eligibility criteria. Children eligible for CS12 completed dosing in either the CS2 or ISIS-396443-CS10 (CS10; NCT01780246) studies between 6 and 13 months before screening and met all additional eligibility criteria. Only those children who enrolled in CS2 are included in the current integrated analysis because the loading dose schedule in CS10 differed from the intended treatment regimen in CS12. Full listings of CS2 and CS12 eligibility criteria are available from Dryad (e-Methods, doi.org/10.5061/dryad.n37q406).
CS2 enrolled a total of 34 children; 28 were treatment-naive and received their first dose of nusinersen in CS2. Of the children who first received nusinersen in CS2, 24 continued onto CS12 (figure 1B). For the integrated analyses reported in this article, all children in CS2 who received their first nusinersen dose while in CS2 and were eligible to continue treatment in CS12 are included (n = 28).
Figure 1. CS2 (phase 1b/2a open-label) and CS12 (extension) study designs and patient disposition.

(A) Study designs: aOverall enrollment. bPatients received treatment on days 1 and 85 only. (B) Patient disposition: The CS2-CS12 integrated efficacy analysis included 28 children from CS2 who received their first dose of nusinersen in CS2 and could have been treated in CS12. The total number of nusinersen doses administered in CS2 was 2 (cohort 3) or 3 (cohorts 1, 2, and 4), and the total number of nusinersen doses administered in CS12 was 4. D = day; SMA = spinal muscular atrophy.
Study designs, treatments, and procedures
CS2 was an open-label, phase 1/2a, multicenter, multiple-dose, dose-escalation study in which participants received 2–3 doses of intrathecally administered nusinersen over 85 days. Following a screening period of ≤28 days, 4 dose levels were evaluated sequentially in 4 separate cohorts: cohort 1 received nusinersen 3 mg on days 1, 29, and 85 (n = 8); cohort 2 received nusinersen 6 mg on days 1, 29, and 85 (n = 8); cohort 3 received nusinersen 9 mg on days 1 and 85 (n = 9); and cohort 4 received nusinersen 12 mg on days 1, 29, and 85 (n = 9). During the study, progression from the first dosing cohort to each subsequent dosing cohort was based on lack of occurrence of dose-limiting toxicity as determined by the sponsors and the independent DSMB. Nusinersen intrathecal lumbar puncture (LP) injection volume was 5 mL. Anesthesia/sedation may have been used for the LP procedure per each participating institution's guidelines. Within each cohort, ≤4 children aged >7 years at screening were enrolled. A posttreatment follow-up period of ∼6 months followed the day 85 dose. Safety monitoring visits occurred on days 8, 36, 92, 169, and 253 (≤24 weeks after the last dose of nusinersen).
For children who continued onto the CS12 extension study, the time interval between the completion of CS2 and enrollment in CS12 was generally longer in those who received lower doses in CS2 and shorter in those who received higher doses due to the sequential nature of enrollment of ascending dose cohorts in CS2. The time between studies was ∼13 months for those in CS2 cohort 1 (3 mg; range, 386–413 days), ∼11 months for those in CS2 cohort 2 (6 mg; range, 288–348 days), ∼10 months for those in CS2 cohort 3 (9 mg; range, 255–361 days), and ∼7 months for those in CS2 cohort 4 (12 mg; range, 196–271 days).
CS12 was a multiple-dose, open-label extension study in which participants received 4 doses of 12 mg intrathecally administered nusinersen at 6-month intervals on days 1, 169, 351, and 533. Nusinersen intrathecal LP injection volume was 5 mL. Anesthesia/sedation may have been used for the LP procedure per each participating institution's guidelines. A screening period of ≤28 days preceded the first dose of nusinersen on day 1, and a posttreatment follow-up period of ∼6 months followed the fourth dose on day 533. Safety monitoring visits occurred on days 8, 85, 260, 442, 624, and 715 (≤26 weeks after the last dose of nusinersen). The date of the last CS12 participant's last visit was January 24, 2017. Children who completed treatment in CS12 could transition into the open-label extension study SHINE (NCT02594124).
Study assessments and outcomes
All physical therapists involved in CS2 attended a comprehensive outcomes training course before study commencement and a refresher training session annually thereafter. Investigators, subinvestigators, and site staff were trained during the site initiation visit. Efficacy assessments conducted during the course of both studies included the Hammersmith Functional Motor Scale–Expanded (HFMSE), Upper Limb Module (ULM) test, 6-Minute Walk Test (6MWT), compound muscle action potential (CMAP), and quantitative multipoint incremental motor unit number estimation (MUNE). The HFMSE is a valid and reliable measure of motor function in SMA.8–10 The ULM assesses upper limb function in nonambulant individuals with SMA.11 The 6MWT is a valid and reliable measure of functional capacity in ambulatory individuals with SMA.12,13 CMAP and MUNE are electrophysiologic measures that can be used to estimate the degree of innervation of a muscle.14 For more details, see the e-Methods (doi.org/10.5061/dryad.n37q406). Analyses of clinically meaningful change in efficacy endpoints utilized the following thresholds of improvement identified in previous studies as clinically meaningful: ≥3-point change on HFMSE,20 ≥2-point change on the ULM,17 and ≥30 meter change in 6MWT walking distance.14 Of note, while some children underwent efficacy assessments at day 1,150 (n = 4–6), assessments of clinically meaningful improvement for all efficacy endpoints are presented among those assessed at day 1,050 because the larger numbers of children assessed at this time point reflect results more representative of the entire cohort.
In CS2, efficacy endpoints were assessed at baseline (last nonmissing assessment before the first dose of nusinersen) and days 92, 169, and 253. In CS12, HFMSE, ULM, and 6MWT endpoints were assessed at baseline and days 85, 169, 260, 351, 442, 533, 624, and 715. CMAP and MUNE endpoints were assessed at baseline and days 85, 260, 442, 624, and 715.
Safety and tolerability assessments included adverse event (AE) monitoring, neurologic examinations, vital signs, physical examinations and weight, clinical laboratory tests, CSF laboratory tests, EKGs, and use of concomitant medications.
Statistical analysis
To allow for an integrated analysis over time while taking into account the differing times between the end of CS2 and the beginning of CS12 between participants, a windowing approach was utilized. Study days were derived from the first day of dosing on CS2 for all study visits in CS2 and CS12. Study visits were then assigned specific study days as follows: study visits that took place >50 to ≤131 days from the first day of dosing on CS2 were labeled study day 92, study visits >131 to ≤211 days were labeled study day 169, study visits >211 to ≤302 days were labeled study day 253, study visits >302 to ≤400 days were labeled study day 350, and study visits >X − 50 to ≤X + 50 days were labeled study day X (beginning at day 450 and increasing by 100 to day 1,150). When >1 study visit for a participant fit into a specific study day interval, the corresponding scores were averaged and the mean was assigned to the visit label and used for analysis purposes. In addition, due to the gap between studies and windowing of visits, some windowed visits do not include all participants.
Efficacy assessments were summarized using descriptive statistics. Correlations between changes in HFMSE and ULM scores were calculated.
Classification of evidence
This study provides Class IV evidence that children with later-onset SMA treated with multiple doses of intrathecal nusinersen (≤12 mg per dose) over ∼3 years demonstrated clinically meaningful improvements in measures of motor function, including the HFMSE (SMA type II), ULM (SMA type II), and 6MWT (SMA type III), and stabilization of disease activity (HFMSE, SMA type III) that were not seen in comparable natural history cohorts.
Data availability
Requests for data supporting this article should be submitted to the Biogen Clinical Data Request Portal (biogenclinicaldatarequest.com).
Results
Patients
Of the 28 children included in the integrated analysis, 11 (39%) had SMA type II and 17 (61%) had SMA type III (table). In accordance with SMA subtypes classifications based on natural history,2,8 none of the children with SMA type II were reported as ambulatory (defined as having the ability to walk ≥15 feet independently without support or braces) at their baseline assessment. All 17 children with SMA type III had previously walked, but 4 lost that ability before the CS2 baseline assessment; thus, 13 were ambulatory at CS2 baseline. Two of 11 (18%) children with SMA type II and 15 of 17 (88%) children with SMA type III were able to walk with support at CS2 baseline. None of the children with SMA type II and 12 of the 17 (71%) children with SMA type III were able to stand without support. All participants were able to sit without support. Children included in these analyses were enrolled in CS2 and CS12 for an average of 965.1 (range, 31–1,219) days and received a median of 7 (range, 1–7) nusinersen doses. Baseline functional scores were similar to those observed in prior SMA natural history studies.14–18
Table.
Baseline demographics and characteristicsa,b

Efficacy results
Hammersmith Functional Motor Scale–Expanded
Children with SMA type II demonstrated progressive improvement in mean HFMSE score over time, with mean (standard error [SE]) scores improving by 10.8 (4.3) points from baseline to day 1,150 (figure 2). Nine of 11 (82%) children demonstrated clinically meaningful improvements (previously defined as an increase of ≥3 points from baseline)19 by day 253 and 7 of 9 (78%) children demonstrated clinically meaningful improvements by day 1,050.
Figure 2. Mean change from baseline in Hammersmith Functional Motor Scale–Expanded (HFMSE) score.

Due to the gap between CS2 and CS12 and windowing of visit days, some windowed visits do not contain all children. SE = standard error; SMA = spinal muscular atrophy.
Overall, children with SMA type III demonstrated a relatively modest change in mean HFMSE score over time, with mean (SE) scores improving by 1.8 (0.9) points from baseline by day 1,150 (figure 2). Three of 16 (19%) children demonstrated clinically meaningful improvements by day 253, including 1 nonambulant child, and 4 of 11 (36%) children demonstrated clinically meaningful improvements by day 1,050.
Among the 13 ambulant children with SMA type III, mean (SE) HFMSE scores improved by 2.6 (0.8) points from baseline by day 1,150, and clinically meaningful improvements were demonstrated by 2 of 12 (17%) children by day 253 and 4 of 9 (44%) children by day 1,050.
Upper Limb Module
Progressive improvements in mean ULM score were demonstrated over time; mean (SE) scores improved by 4.0 (2.4) points from baseline by day 1,150 (figure 3A). Five of 11 (45%) children demonstrated clinically meaningful improvements (previously defined as ≥2-point increase from baseline)17 by day 253 and 5 of 9 (56%) children demonstrated clinically meaningful improvements by day 1,050.
Figure 3. Mean change from baseline in Upper Limb Module (ULM) score and 6-Minute Walk Test (6MWT) distance.

(A) ULM: Assessed only in nonambulant participants. (B) 6MWT: Assessed only in ambulant participants. Due to the gap between CS2 and CS12 and windowing of visit days, some windowed visits do not contain all children. SE = standard error.
All nonambulant children with SMA type III assessed at the day 350 visit had reached the maximum score of 18 points and maintained this score through day 1,150 assessments.
Correlation between HFMSE and ULM
In children with SMA type II, change from baseline in HFMSE score was strongly correlated with change from baseline in ULM score at every visit day, with a correlation coefficient of r = 0.87 demonstrated at day 1,150.
Six-Minute Walk Test
One of the 11 children with SMA type II (age 2.1 years at first dose in CS2) gained the ability to walk independently during the course of the studies. The child first completed the 6MWT at the day 650 visit (total distance walked was 25.5 meters) and demonstrated continued improvements over time; by the day 1,150 visit, the child's 6MWT distance was 180 meters.
Children with SMA type III demonstrated progressive improvements from baseline in 6MWT distance over time, with mean (SE) distances improving by 92.0 (21.5) meters by day 1,150 (figure 3B). Two of the 4 children who were previously able to walk but had lost that ability before the baseline assessment regained the ability to walk independently during the course of the studies. In addition, 6 of the 12 (50%) children assessed at the day 253 visit demonstrated clinically meaningful improvements in 6MWT distance (previously defined as ≥30-meter increase from baseline)13 and 8 of 8 (100%) demonstrated clinically meaningful improvements by day 1,050.
CMAP and MUNE
Mean CMAP amplitudes, CMAP areas, and MUNE values demonstrated small fluctuations over time in children with SMA type II (figure 4, A–C). By day 1,150, mean (SE) CMAP amplitude increased by 0.4 (0.8) mV, mean (SE) CMAP area increased by 3.0 (2.4) mV/ms, and mean (SE) MUNE value increased by 2.0 (14.5) from baseline.
Figure 4. Mean change from baseline in compound muscle action potential (CMAP) amplitude, CMAP area, and motor unit number estimation (MUNE) value.
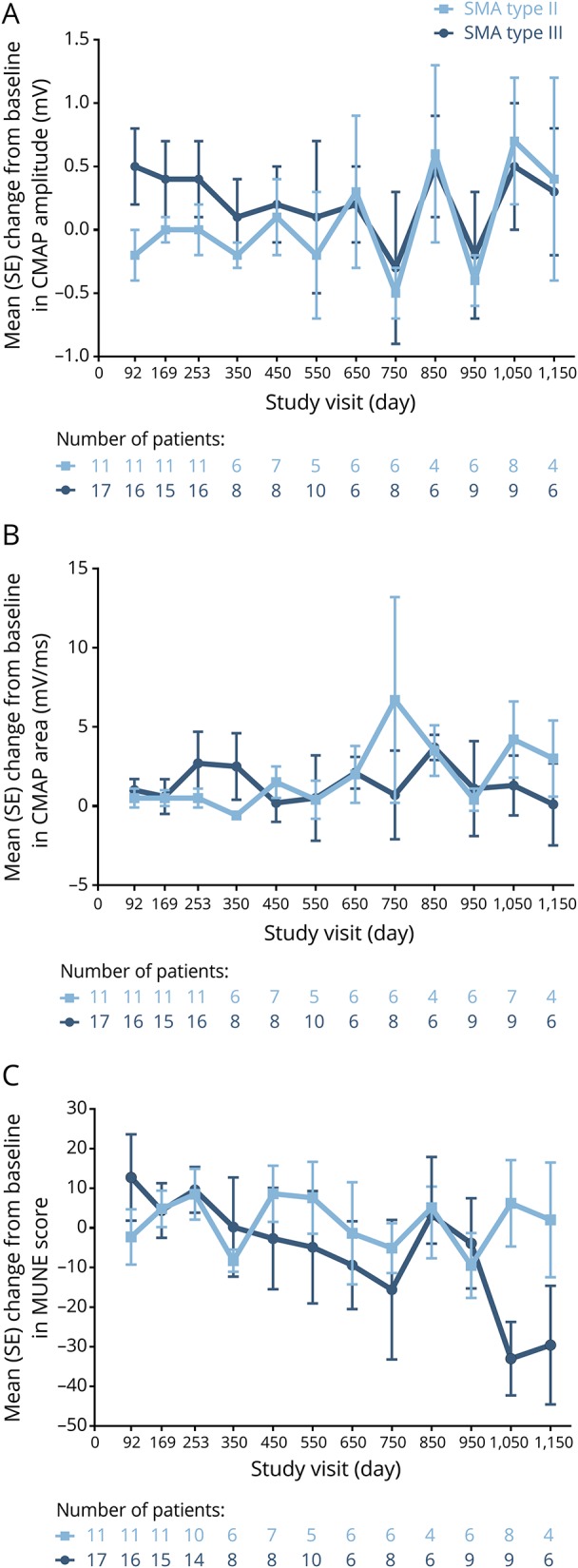
(A) CMAP amplitude, (B) CMAP area, and (C) MUNE value. Due to the gap between CS2 and CS12 and windowing of visit days, some windowed visits do not contain all children. SE = standard error; SMA = spinal muscular atrophy.
In children with SMA type III, CMAP amplitude and area also demonstrated small fluctuations over time; mean (SE) CMAP amplitude increased by 0.3 (0.5) mV (figure 4A) and mean (SE) CMAP area increased by 0.1 (2.6) mV/ms from baseline by day 1,150 (figure 4B). Mean MUNE motor unit numbers progressively decreased over time in children with SMA type III, with mean (SE) MUNE values decreasing by 29.6 (15.0) by day 1,150 (figure 4C).
Safety results
All children experienced ≥1 AE while on study (n = 28; 100%). The most common AEs (occurring in ≥20% of children) were post-LP syndrome (n = 16; 57%), headache (n = 13; 46%), nasopharyngitis (n = 12; 43%), upper respiratory tract infection (n = 12; 43%), puncture site pain (n = 11; 39%), back pain (n = 9; 32%), scoliosis (n = 8; 29%), pyrexia (n = 7; 25%), joint contracture (n = 6; 21%), rhinorrhea (n = 6; 21%), and vomiting (n = 6; 21%). Most AEs were mild or moderate in severity and were considered by investigators as unrelated to the study drug. Serious AEs (SAEs) were reported by 5 (18%) children and included post-LP syndrome (n = 2); lower respiratory tract infection, respiratory distress, and viral pneumonia (n = 1); acute respiratory failure and respiratory syncytial viral pneumonia (n = 1); and vesicoureteral reflux and pyelonephritis (n = 1). No SAEs were considered related to study drug, and no children discontinued treatment due to AEs. No clinically relevant changes in laboratory values or neurologic examinations considered related to nusinersen treatment were observed.
Discussion
This integrated analysis of the CS2 and CS12 studies provides evidence-based data that extend the age range and length of time of nusinersen treatment in children with later-onset SMA over what has been observed in previous clinical studies.7,20 A previously published phase 1 study reported positive safety and preliminary efficacy results in children with SMA type II or type III aged 2–14 years over the course of 9–14 months.20 In a previously published phase 3 study of nusinersen in children with later-onset SMA (most likely to develop SMA type II or type III), statistically significant and clinically meaningful improvements in motor function were seen in children age 2–9 years at treatment initiation over the course of 15 months.7 In the current study, clinically meaningful improvements in motor function were observed in children with SMA type II or type III age ≤15 years at treatment initiation over the course of ∼3 years of follow-up. Thus, the current results extend the age range over which motor function improvements have been observed following nusinersen treatment and more than double the length of treatment time over which progressive improvements in motor function have been observed in children with SMA as well as the time over which nusinersen has demonstrated a favorable safety profile.
Mean HFMSE and ULM scores in children with SMA type II and mean 6MWT distances in children with SMA type III increased over time in a relatively linear trajectory without plateau, suggesting that the positive effects of nusinersen in these populations are time-dependent. These results represent a vast improvement over the documented natural history of children with SMA classified as type II or type III, a progressive neuromuscular condition where patients may gain function or remain stable initially but eventually lose function over time.7,15 These results also suggest that treatment with nusinersen may not just prevent motor function deterioration, but could also allow for continued motor function improvement and even reversal of motor function loss. The biological basis for this time-dependent effect remains to be studied, but may be related to continued axonal growth and reinnervation of muscle fibers, improved neuronal/axonal function, maturation of abnormal synapses, and/or transneuromuscular junction effects on muscles, all potentially induced by increased SMN protein production related to continued treatment with nusinersen. Further studies would be necessary to elucidate the mechanism of action responsible for the nusinersen-induced improvements in motor function. In addition, the measures of motor function used in this study assess motor abilities directly relevant to activities of daily living10,11,13; thus, the improvements observed in this study could represent an increased standard of living for these children.
In the current study, HFMSE scores improved by a mean of 10.8 points in children with SMA type II and a mean of 1.8 points in children with SMA type III at day 1,150, respectively, compared with declines in score of 1.7 points at 36 months (1,095 days) in a natural history study of children with SMA type II or III.15 Mean improvements in HFMSE score were generally larger in children with SMA type II compared with those with SMA type III at each time point, possibly due to the lower mean HFMSE score observed in this group at baseline. Indeed, clinically meaningful increases were observed in the majority of children (78%) with SMA type II by day 1,050, a result rarely seen in this SMA population,21 while clinically meaningful increases were only observed in 36% of children with SMA type III over the same time frame. The relatively modest increase in mean HFMSE score in children with SMA type III could be due to the fact that some of the more difficult items on the HFMSE (i.e., squatting, jumping, stair climbing) are simply harder to achieve regardless of SMA type. This explanation is supported by our results; only 6 of the 17 (35%) children with SMA type III in this study achieved a highest score of >60 points despite 11 (65%) having baseline scores >50 points. Regardless, of the children with SMA type III with HFMSE score increases <3 points at day 1,050 (indicating no apparent clinically meaningful improvement), 5 of the 5 (100%) who had 6MWT distances recorded at baseline and day 1,050 demonstrated clinically meaningful improvements on the 6MWT. It should also be noted that any increase or stabilization of HFMSE score is a distinct and evident improvement over comparable natural history cohorts that have demonstrated progressive decreases in score over similar time frames.15,21 Among individuals with SMA type II or III and their caregivers, slowing of disease progression and stabilization of disease course were considered clinically meaningful.10,22
Children with SMA type II demonstrated greater mean improvements in ULM scores compared with mean changes observed in a comparable natural history cohort (+2.8 points at day 350 vs +0.04 over 12 months, respectively).17 Mean ULM scores generally demonstrated consistent improvement over the course of the study period, similar to the progressive improvement in mean HFMSE score demonstrated by children with SMA type II over time. Indeed, changes from baseline in HFMSE and ULM scores were strongly correlated at each visit day.
Children with SMA type III demonstrated greater mean improvements in 6MWT distances compared with mean changes observed in a natural history cohort of patients with SMA type III (+36.3 meters at day 350 vs −1.5 meters over 12 months, respectively).16 In addition, 1 previously nonambulant child with SMA type II achieved independent walking over the course of the studies, an achievement not congruent with the definition of SMA type II. Moreover, 2 of the 4 children with SMA type III who had previously achieved independent walking but had lost that ability before the baseline assessment of CS2 regained the ability to walk independently during the course of the studies. These results show that nusinersen can alter SMA disease progression and that reversal of motor function loss is possible in individuals with later-onset SMA.
Mean CMAP amplitudes, CMAP areas, and MUNE values, with the exception of MUNE values in children with SMA type III, fluctuated but remained generally stable in contrast to the increases observed in HFMSE score, ULM scores, and 6MWT distances over time. While CMAP and MUNE values have been shown to correlate with disease progression in neuromuscular disorders,14,23 increased MUNE and stable CMAP values have been observed in some natural history populations despite motor function declines.24 CMAP amplitude and area were stable in healthy control infants (age ≤6 months) over 2 years of follow-up in the NEURONEXT study25 in contrast to the increase expected in normally developing children.26 Because the NEURONEXT study also used a single G1 electrode placement, this result suggests that the use of 1 G1 placement may result in decreased sensitivity compared with previously published techniques in which a minimum of 3–5 G1 placements were used to ensure measurement of the maximum motor response.14,18 These results suggest that the benefit of nusinersen treatment on distal innervation capacity may be less robust than its overall benefit on motor function and is likely influenced by factors including age, severity of denervation at the time of enrollment, duration of exposure, and nerve muscle group tested. As distal peripheral reinnervation appears to be more challenging to rescue in a growing child whose limbs are lengthening over time and whose distal motor units, neuromuscular junctions, and muscles continually require adequate SMN levels at distal terminals, the decline in MUNE values observed in children with SMA type III over time is not surprising in this cohort of children with later-onset SMA, despite the gains in motor function demonstrated by other assessments. Furthermore, it reinforces the potential benefit of using an incremental multipoint MUNE technique as a complementary endpoint in future clinical trials.
No new safety concerns beyond those previously reported were identified in these studies.4,5,7,20 Nusinersen demonstrated a favorable benefit–risk profile in this population, with most AEs reported being mild or moderate in severity and no children discontinuing treatment due to AEs. In addition, no clinically relevant changes in laboratory values considered related to nusinersen were observed.
This integrated analysis had some limitations. CS2 and CS12 were open-label studies with no internal control groups. Because of the small sample size, additional analyses assessing the effects of covariates on the endpoints tested were not possible. Similarly, because of the small number of children with efficacy assessments available at day 1,150, measurements of clinically meaningful improvement are presented out of those assessed at day 1,050. Due to the windowing approach utilized for study visit categorization, data incorporated into a single study visit could have been collected ≤100 days apart for each individual and, due to the gap between studies and visit windowing, some windowed visits do not include all participants. In addition, children in CS2 cohorts 1–3 received lower doses of nusinersen compared with those in CS2 cohort 4; these children may have demonstrated even greater improvements had they received the higher dose from the beginning of CS2. Moreover, due to the ascending dose level study design of CS2 and resultant larger gap between CS2 and CS12 in children enrolled in the lower-dose cohorts, it may have taken longer for children in cohorts 1–3 to reach steady-state levels of nusinersen. Finally, single G1 electrode placement for CMAP measurement may lead to decreased CMAP sensitivity, and MUNE and CMAP values could potentially be influenced by the reading neurologist's expertise, which may lead to result variability, especially in multicenter studies such as CS2 and CS12. These issues could be ameliorated by additional training or recording of these studies, or with enhanced opportunities for real-time centralized review and feedback.
The results of the CS2-CS12 integrated analysis support previous results demonstrating the statistically significant and clinically meaningful effects of nusinersen in infants and children with infantile- and later-onset SMA,4,7 and further extend the age range and length of treatment over which the benefits of nusinersen in children with later-onset SMA who have or are most likely to develop SMA type II or III have been observed.
Acknowledgment
The authors thank the patients who participated in CS2 and CS12 and their parents/guardians and family members; and the CS2 and CS12 study investigators and all the contributors to CS2 and CS12, including the clinical monitors, Data Safety and Monitoring Board members, study coordinators, physical therapists, pharmacists and laboratory technicians, and patient advocacy groups, who assisted in promoting awareness of these studies. This integrated analysis was funded by Biogen and Ionis Pharmaceuticals, Inc. Biogen provided funding for medical writing support in the development of this paper; Allison Green, PhD, from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors; and Kristen DeYoung from Excel Scientific Solutions copyedited and styled the manuscript per the journal requirements. Biogen and Ionis Pharmaceuticals, Inc. reviewed and provided feedback on the manuscript to the authors. The authors had full editorial control of the manuscript and provided their final approval of all content.
Glossary
- 6MWT
6-Minute Walk Test
- AE
adverse event
- CMAP
compound muscle action potential
- DSMB
data and safety monitoring board
- HFMSE
Hammersmith Functional Motor Scale–Expanded
- LP
lumbar puncture
- MUNE
motor unit number estimation
- SAE
serious adverse event
- SE
standard error
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
- ULM
Upper Limb Module
Appendix 1. Authors
Appendix 2. Coinvestigators
Footnotes
Class of Evidence: NPub.org/coe
See page 985
Study funding
Biogen and Ionis Pharmaceuticals, Inc.
Disclosure
B. Darras has been a member of advisory boards for AveXis, Biogen, Bristol-Myers Squibb, Cytokinetics, Marathon, PTC, Roche, Santhera, and Sarepta; has received research support from the NIH/National Institute of Neurologic Disorders and Stroke, the Slaney Family Fund for SMA, the SMA Foundation, and the Working on Walking Fund; has received grants from Biogen, CureSMA, and Ionis Pharmaceuticals, Inc. during the ENDEAR, CHERISH, CS2, CS12, and CS11 studies, and from AveXis, Cytokinetics, Fibrogen, PTC, Roche, Santhera, Sarepta, and Summit; and reports no personal financial interests in these companies. C. Chiriboga has been a member of advisory boards for SMA studies for AveXis, Biogen, Ionis Pharmaceuticals, Inc., Genentech, and Roche; and reports no financial interest in these companies. She has received grants from Biogen, Ionis Pharmaceuticals, Inc., the NIH, and the SMA Foundation. S. Iannaccone has been a consultant for Ionis Pharmaceuticals, Inc.; has been a member of advisory boards for AveXis, Biogen, Santhera, and Sarepta; and has received research grants from the Muscular Dystrophy Association and the NIH. K. Swoboda has received support from the Eunice Kennedy Shriver National Institute for Child Health and Human Development (R01-HD69045); has been a member of advisory boards for AveXis and Biogen; and has received clinical trial funding from Biogen and Ionis Pharmaceuticals, Inc. for the CS1, CS2, CS12, and NURTURE studies. J. Montes has been a member of advisory boards for Biogen, Cytokinetics, Roche, Scholar Rock, and the SMA Foundation; has been a consultant for Biogen and Ionis Pharmaceuticals, Inc.; has received research support from the Eunice Kennedy Shriver National Institute for Child Health and Human Development (1K01HD084690-01A1); and has received a conference grant from the Muscular Dystrophy Association. L. Mignon is an employee of and holds stock/stock options in Ionis Pharmaceuticals, Inc. S. Xia is an employee of and holds stock/stock options in Ionis Pharmaceuticals, Inc. C. Bennett is an employee of and holds stock/stock options in Ionis Pharmaceuticals, Inc. K. Bishop was an employee of Ionis Pharmaceuticals, Inc. at the time these studies were conducted; is currently an employee of Otonomy; and has acted in an advisory capacity for the following nonprofit organizations: the Myotonic Dystrophy Foundation and the SMA Foundation. J. Shefner has been an advisor/consultant for AveXis, Biogen, Cytokinetics, and Ionis Pharmaceuticals, Inc.; and has received research support from the ALS Association, ALS Finding a Cure, Biogen, Cytokinetics, and Neuraltus. A. Green is an employee of Excel Scientific Solutions. Biogen provided funding for medical writing support in the development of this report to Excel Scientific Solutions. P. Sun is an employee of and holds stock/stock options in Biogen. I. Bhan is an employee of and holds stock/stock options in Biogen. S. Gheuens is an employee of and holds stock/stock options in Biogen. E. Schneider is an employee of and holds stock/stock options in Ionis Pharmaceuticals, Inc. W. Farwell is an employee of and holds stock/stock options in Biogen. D. De Vivo has been an advisor/consultant for AveXis, Biogen, Cytokinetics, Ionis Pharmaceuticals, Inc., Metafora, Roche, Sanofi, Sarepta, and the SMA Foundation, with no financial interests in these companies; has received grants from the Department of Defense, Hope for Children Research Foundation, the NIH, and the SMA Foundation; and has received clinical trial funding from Biogen, Mallinckrodt, PTC, Sarepta, and Ultragenyx. Go to Neurology.org/N for full disclosures.
References
- 1.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008;371:2120–2133. [DOI] [PubMed] [Google Scholar]
- 2.Darras BT, Markowitz JA, Monani UR, De Vivo DC. Spinal muscular atrophies. In: Darras BT, Royden Jones H Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician's Approach, 2nd ed. San Diego: Academic Press; 2015:117–145. [Google Scholar]
- 3.Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010;24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017;377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 5.Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016;388:3017–3026. [DOI] [PubMed] [Google Scholar]
- 6.De Vivo DC, Hwu W-L, Reyna SP, et al. Interim efficacy and safety results from the phase 2 NURTURE study evaluating nusinersen in presymptomatic infants with spinal muscular atrophy. In: 69th Annual Meeting of the American Academy of Neurology; April 22–28, 2017; Boston.
- 7.Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 2018;378:625–635. [DOI] [PubMed] [Google Scholar]
- 8.Finkel R, Bertini E, Muntoni F, Mercuri E. 209th ENMC International Workshop: outcome measures and clinical trial readiness in spinal muscular atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord 2015;25:593–602. [DOI] [PubMed] [Google Scholar]
- 9.Glanzman AM, O'Hagen JM, McDermott MP, et al. Validation of the expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J Child Neurol 2011;26:1499–1507. [DOI] [PubMed] [Google Scholar]
- 10.Pera MC, Coratti G, Forcina N, et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol 2017;17:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzone E, Bianco F, Martinelli D, et al. Assessing upper limb function in nonambulant SMA patients: development of a new module. Neuromuscul Disord 2011;21:406–412. [DOI] [PubMed] [Google Scholar]
- 12.Montes J, McDermott MP, Martens WB, et al. Six-Minute Walk Test demonstrates motor fatigue in spinal muscular atrophy. Neurology 2010;74:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunaway Young S, Montes J, Kramer SS, et al. Six-Minute Walk Test is reliable and valid in spinal muscular atrophy. Muscle Nerve 2016;54:836–842. [DOI] [PubMed] [Google Scholar]
- 14.Lewelt A, Krosschell KJ, Scott C, et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve 2010;42:703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufmann P, McDermott MP, Darras BT, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012;79:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazzone E, Bianco F, Main M, et al. Six Minute Walk Test in type III spinal muscular atrophy: a 12 month longitudinal study. Neuromuscul Disord 2013;23:624–628. [DOI] [PubMed] [Google Scholar]
- 17.Sivo S, Mazzone E, Antonaci L, et al. Upper limb module in non-ambulant patients with spinal muscular atrophy: 12 month changes. Neuromuscul Disord 2015;25:212–215. [DOI] [PubMed] [Google Scholar]
- 18.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol 2005;57:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swoboda KJ, Scott CB, Crawford TO, et al. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 2010;5:e12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016;86:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercuri E, Finkel R, Montes J, et al. Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul Disord 2016;26:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouault F, Christie-Brown V, Broekgaarden R, et al. Disease impact on general well-being and therapeutic expectations of European type II and type III spinal muscular atrophy patients. Neuromuscul Disord 2017;27:428–438. [DOI] [PubMed] [Google Scholar]
- 23.Shefner JM, Watson ML, Simionescu L, et al. Multipoint incremental motor unit number estimation as an outcome measure in ALS. Neurology 2011;77:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang PB, Gooch CL, McDermott MP, et al. The motor neuron response to SMN1 deficiency in spinal muscular atrophy. Muscle Nerve 2014;49:636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolb SJ, Coffey CS, Yankey JW, et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol 2017;82:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang PB. Pediatric nerve conduction studies and EMG. In: Blum AS, Rutkove SB, eds. The Clinical Neurophysiology Primer. Totowa: Humana Press; 2007:369–389. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Requests for data supporting this article should be submitted to the Biogen Clinical Data Request Portal (biogenclinicaldatarequest.com).