

Viral dynamics and host responses to vaginal infection of SIV in nonhuman primates may identify viral vulnerabilities for HIV.
Abstract
Understanding HIV transmission is critical to guide the development of prophylactic interventions to prevent infection. We used a nonhuman primate (NHP) model with a synthetic swarm of sequence-tagged variants of SIVmac239 (“SIVmac239X”) and scheduled necropsy during primary infection (days 3 to 14 after challenge) to study viral dynamics and host responses to the establishment and dissemination of infection following vaginal challenge. We demonstrate that local replication was initiated at multiple sites within the female genital tract (FGT), with each site having multiple viral variants. Local replication and spread in the FGT preceded lymphatic dissemination. Innate viral restriction factors were observed but appeared to follow viral replication and were ineffective at blocking initial viral establishment and dissemination. However, major delays were observed in time to dissemination in animals and among different viral variants within the same animal. It will be important to assess how phenotypic differences affect early viral dynamics.
INTRODUCTION
Nearly 40 million people are infected with HIV-1 worldwide, with 1.8 million new infections estimated to have occurred in 2016 (http://unaids.org). The vast majority of infections are still sexually transmitted, requiring the virus to successfully cross a mucosal barrier to establish infection. The estimated per-exposure infection rate for heterosexual transmission is highly variable and depends on multiple factors including the viral load of the transmitting partner (1), but outside of predisposing circumstances, successful transmission is generally considered a low-probability event (2). This low-average infectivity rate was confirmed molecularly when it was demonstrated that at least 80% of sexual transmissions originate from a single transmitted/founder (T/F) virus (3–6) despite great genetic diversity of virus present in semen (7, 8). Molecular clones of envelope and full-length viral genome sequences have been generated from many T/F viruses, demonstrating that viruses capable of crossing mucosal barriers retain specific features compared to clones typical of chronic infection. T/F viruses use CD4 and CCR5 as their dominant receptor and coreceptor, and they infect activated CD4+ T cells ~20-fold better than they infect monocyte-derived macrophages in vitro (5, 9–12). Transmission data combined with evidence of a single T/F variant in the vast majority of sexually infected individuals emphasize the stringent genetic bottleneck that occurs during mucosal transmission. While all the factors contributing to this bottleneck are not fully understood, they likely include features associated with both the virus (e.g., dose, phenotype, tropism, etc.) (12–16) and the susceptibility of the recipient (e.g., site of exposure, preexisting inflammation/coinfection, resident microbiota, mucus levels, mucosal damage, type of coital act, and target cell availability) (2, 4, 17–21).
While important advances in understanding HIV-1 transmission have been made by studying acutely infected humans, sampling relevant mucosal tissues at known, very early time points after exposure requires the use of an animal model. There are many similarities in the physiology and immunology of the female genital tract (FGT) between nonhuman primates (NHPs) and humans, including the relative paucity of available virus-susceptible target cells (20, 21). Vaginal transmission studies in rhesus macaques have demonstrated that virions can traverse the epithelial barrier from the vaginal lumen to establish infection (22). Recently, a novel dual reporter virus was used to show that many distinct regions of the FGT can act as sites of initial mucosal entry and infection (23). In samples collected within 24 hours of exposure, infecting virus that had penetrated the mucosal barrier was reported to be associated with dendritic cells or T cells (22). By 48 hours, virus was associated predominantly within T cells (70%) with evidence of only minor macrophage infection (23).
Other studies using replication-competent viruses required multiple days of viral replication, allowing for viral expansion, before unequivocal detection. Haase and colleagues (24, 25) reported that recruitment of CD4+ T cells to sites of initial mucosal infection was necessary to generate a sufficient critical mass of virus and infected cells to enable systemic dissemination. Although these studies demonstrated that some limited virus could be detected distally before extensive local replication, they conclude that these foci were unlikely to initiate a self-sustaining systemic infection (25). In contrast, Barouch et al. (26) provided evidence for early systemic viral dissemination after vaginal simian immunodeficiency virus (SIV) inoculation, without the apparent requirement for recruitment of CD4+ T cells or local amplification at initial sites of mucosal infection. In a monoclonal antibody–mediated passive immunoprophylaxis study involving intravaginal simian HIV (SHIV) challenge, Liu et al. (27) showed evidence for rare, early distal dissemination of replication-competent viruses. Administration of protective levels of antibody appeared to result in clearance of virus both at the vaginal portal of entry and at sites of viral dissemination. Hessell et al. (28) reported similar results in passive protection of infant macaques infected orally.
Notably, to understand vaginal infection, there are anatomic and cellular differences between potential sites of initial infection within the FGT, which present both opportunities and challenges for viral entry, replication, and dissemination (21). While the endocervical mucosal barrier consists of only a single columnar epithelial layer, it is protected by variable but often copious amounts of mucus (varying across the menstrual cycle) that can trap incoming virus and limit infection (25). The transformation zone between the ectocervix and endocervix has been identified as a likely site for infection since there are both more abundant targets and a single-cell epithelial barrier (21, 24). Furthermore, in the absence of local inflammation, CD4+ target cells are sparsely and heterogeneously distributed throughout the FGT (17). During subclinical infections, including bacterial vaginosis and trichomoniasis, susceptibility to HIV infection can increase (29) likely due to an increase in inflammation (30) or mucosal breaches.
To address key questions regarding vaginal transmission of HIV, we designed a scheduled serial sacrifice study in NHPs such that the entire FGT (vagina, cervix, uterus, and ovaries) could be sampled at defined times after inoculation to assess local parameters associated with anatomic sites of early replication. To allow the genetic discrimination and longitudinal tracking of individual viral transmission events while limiting the potential effects of viral phenotypic variability, we used a synthetic swarm of SIVmac239, designated “SIVmac239X” (31). The SIVmac239X model is based on infection with a pool of 10 clonal viruses that are isogenic except for two to three synonymous base substitutions in the integrase gene. These substitutions are all found in naturally occurring SIV, do not change the protein coding sequence, and were designed to not affect viral phenotype or fitness while still being easily distinguishable by sequence analysis using a real-time polymerase chain reaction (RT-PCR) approach for single-genome amplification (SGA), next-generation sequencing (NGS), and laser capture microdissection sequencing. In addition to sequence analysis, we used next-generation in situ hybridization (ISH; RNA/DNAScope) (32), immunohistochemistry (IHC), and quantitative PCR (qPCR) to quantify viral spread and antiviral host responses.
We applied these analytical approaches to tissue samples following necropsy between 3 and 14 days after challenge. The results established in this model system demonstrated that (i) local replication precedes viral dissemination, (ii) each focus of infection contained more than one variant, (iii) systemic dissemination occurs via the lymphatic system, (iv) time to dissemination and the relative proportion of each lineage varied substantially between animals, (v) CD4+ T cells are the primary targets during primary infection and were recruited to mucosal sites of infection, (vi) increased expression of viral restriction factors was ineffective at blocking initial viral establishment, and (vii) the relative proportions of different viral variants are established early and persist systemically. These data provide an unprecedented view into the dynamic and complex process of vaginal transmission.
RESULTS
Intravaginal infection with SIVmac239X
Fifteen adult rhesus macaques (Macaca mulatta) were intravaginally challenged during the early follicular phase of the menstrual cycle, a time associated with increased relative susceptibility to infection (33). Within 4 days following the end of menses, animals were challenged intravaginally with 2 ml (5 × 105 IU/ml) of a transfection-produced virus stock containing essentially equivalent proportions of each of the 10 variants of SIVmac239X (31). Animals were euthanized, and plasma viral load (pVL) measurements were determined at necropsy and before necropsy in some animals (Fig. 1). Only five animals (EZN, F74, D290, E052, and ELV) had detectable viremia with terminal pVL ranging from 2.0 × 103 copies/ml at day 6 to 1.4 × 107 copies/ml at day 14 after infection. The remaining 10 animals were below the level of detection (30 copies/ml). At days 6, 7, and 8 after challenge, only a fraction of animals (25% at days 6 and 7 and 50% at day 8) showed evidence of measurable plasma viremia with the remaining animals challenged on the same day below the limit of detection.
Fig. 1. Plasma viral load (pVL) data.

Longitudinal pVL determined by qRT-PCR from animals during ramp up and necropsy with day of necropsy indicated by different colors (red, d14; orange, d8; pink, d7; blue, d6; green, d5; cyan, d3). Open symbols represent animals with a plasma viremia of less than 30 copies/ml (limit of detection) at necropsy.
Viral dynamics in tissue compartments
At necropsy, the entire FGT (vagina, cervix, uterus, and ovaries) was excised en bloc and dissected into ~1-cm3 tissue pieces for bulk nucleic acid quantification and sequence analysis or for IHC and ISH. In addition to the FGT, the local draining lymph nodes (LNs; internal and common iliac and para-aortic LNs, which we had previously identified as draining the FGT) (34), distal LNs (axillary, inguinal, bronchial, cervical, mesenteric, and colonic), spleen, bone marrow, and various sections of the upper and lower gastrointestinal (GI) tract were also partitioned and analyzed. For each animal, overall tissue viral loads for a given tissue were calculated by averaging the normalized viral loads [copies per 106 cell equivalents (CE)] determined for each of the individual pieces of that tissue, providing a broad anatomic view on where the virus was at necropsy (Fig. 2). Within the FGT, mean tissue viral loads were determined for the cervix, vagina, and uterus (Fig. 2, green panel). For the lymphatic system, mean viral RNA (vRNA) and viral DNA (vDNA) quantities were determined for the draining, distal, and GI LNs, as well as individually extracted spleen sections (Fig. 2, yellow panel). Separate GI tract sections were also averaged for both the upper and lower GI tract (Fig. 2, purple panel). Bone marrow and peripheral blood mononuclear cells (PBMCs) were also extracted and quantified (Fig. 2, blue panel).
Fig. 2. Viral nucleic acid distribution during primary infection.
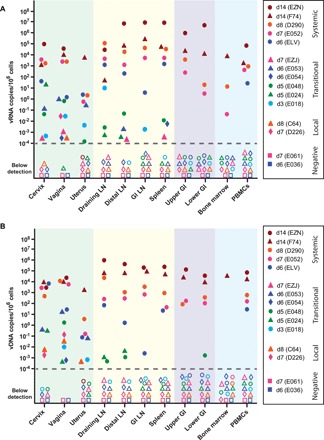
Viral RNA (vRNA) (A) and viral DNA (vDNA) (B) quantities were assessed by qRT-PCR or qPCR from nucleic acids obtained from ~1-cm3 pieces of tissue. Individual tissue viral loads were averaged and plotted for each tissue indicated. Background shading indicates different types of tissues [green, FGT; yellow, lymphatic system; purple, gastrointestinal (GI) tract; blue, bone marrow and peripheral blood mononuclear cells (PBMCs)]. Individual symbols are color-coded based on days after infection until necropsy (red, d14; orange, d8; pink, d7; blue, d6; green, d5; cyan, d3) with animals grouped based on phases of infection (negative, local, transitional, and systemic). Missing symbols indicate no data available. Open symbols indicate no detectable vRNA or vDNA. For simplicity, the limit of detection for each tissue type is not identified in this figure (see fig. S4 for limit of detection per sample).
We found vRNA and vDNA in various tissues from each animal except for two animals necropsied at day 6 (E036) and day 7 (E061) (Fig. 2 and fig. S1). In these two animals, there was no vRNA or vDNA signal detected by RT-PCR in any of the ≥57 tissue samples tested; however, in animal E061, there was evidence of rare vRNA-positive (vRNA+) and vDNA-positive cells in the draining LN identified by RNAscope and DNAscope (fig. S2). In two animals necropsied at day 7 (D226) and day 8 (C64), we found evidence of viral replication within the FGT only (Fig. 2, “local”). In six animals (E018, EZJ, E054, E024, E048, and E053) taken to necropsy from 3 to 7 days after challenge, we found evidence of both local replication and early dissemination (Fig. 2, “transitional”). In these animals, we found the most virus in the FGT, with evidence of spread into the lymphatic system and spleen but rarely to the GI tract, bone marrow, or blood. However, in viremic animals, there was widespread systemic dissemination with vRNA and vDNA found in all lymphoid tissues, in both upper and lower GI tract tissues, the bone marrow, and blood (Fig. 2, “systemic”). In three animals necropsied at day 6 (ELV), 7 (E052), or 8 (D290), vRNA and vDNA were detected in the same tissue compartments as the day 14 necropsied animals (EZN and F74), although lower in absolute magnitude. Overall, initial viral replication was demonstrable within the cervix, vagina, and uterus, with apparent lymphatic spread via draining LNs, which preceded systemic dissemination. vRNA or vDNA were only observed in the upper and lower GI tract and PBMCs in animals with measurable plasma viremia, suggesting virus seeding of these tissues via the circulatory system.
To better understand the complex spatial and temporal dynamics of local replication and dissemination, we next analyzed individual tissue pieces that were SIV positive (fig. S3). Individual foci of infection were detectable in only a small portion of the total tissues sampled with most tissue sections below the limit of detection, which depended on the variable amount of each tissue available for analysis, but often reached one viral template copy per 109 cells. For animals designated as local (D226 and C64), we found evidence of vRNA and vDNA only in a subset of all the distinct tissue pieces examined throughout the FGT with the highest levels of viral nucleic acid in the vagina (fig. S4, A and B). In the six transitional animals (E018, EZJ, E054, E024, E048, and E053), again, only a subset of tissue pieces examined contained measurable vRNA and vDNA, which was found in both the cervix and vagina, and evidence of lymphatic spread of virus to draining LNs and less often in distal LNs (fig. S4, C to F). For systemic animals (ELV, E052, and D290), we found robust vRNA and vDNA signal in nearly all FGT tissues analyzed, with even higher levels within the lymphatic tissues, consistent with the higher availability of CD4+ target cells in those tissues (fig. S4, G to I). We also detected some lower signal within the GI tract, bone marrow, and PBMCs, but virus was rarely detectable in the GI tract until animals had become measurably viremic, confirming that the earliest phase of viral replication after dissemination is dominated by lymphatic expansion followed by spread to and replication in the GI tract.
Viral dynamics of entry and local spread
Since we genetically labeled the virus before infection, we can analyze the sequences of these SIV-positive tissues to determine which lineages are present and in what proportion. The distribution of viral variants within the FGT can be used to assess several possible hypotheses. One is that if the FGT epithelium is relatively permeable, then individual lineages would cross at multiple sites initiating infection with single variants per site (represented schematically in Fig. 3A). Alternatively, if the epithelium were the major barrier to entry, then we might see evidence of one or all variants entering at a single susceptible site such as preexisting mucosal breach, area of inflammation, or increased numbers of potential target cells (Fig. 3B). These would lead to either a dispersed anatomical distribution of lineages or a highly clustered distribution, respectively. The final possibility is that there are multiple sites of vulnerability within the vaginal/cervical epithelium, which allowed for several individual lineages to enter together (Fig. 3C). Since foci of infection were often identified in individual pieces or adjacent sections as hotspots of viral replication surrounded by tissues without detectable viral nucleic acids, we sequenced each vRNA and vDNA tissue to assess which of the 10 genetically distinct variants were present. In all animals, there was genetic evidence for multiple variants per positive tissue (Fig. 3, D and E, and fig. S5). Therefore, using the distribution of variants within the FGT, we found that there was strong evidence for clustering; that is, not all sites in the vagina were infected, but where infection was observed, it was with more than one variant. This suggests that, overall, the vaginal epithelium prevents entry in most regions, but where entry occurred at “vulnerable sites,” multiple clones were able to enter simultaneously.
Fig. 3. Proportion and quantification of individual variants.
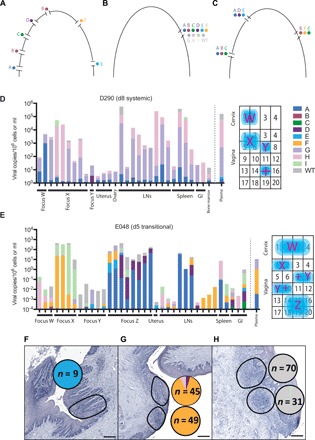
(A to C) Schematic representation of the three distinct possible mechanisms for multilineage infection; (A) multiple breaches in mucosal barrier allowing for a single variant each, (B) single breach where all detected lineages enter, and (C) a few breaches each containing more than one lineage. Nucleic acid–positive tissues are displayed as the height of the bar corresponding to the log scale on the left axis. The relative proportion of each viral variant is represented by color coding within each bar representing the linear proportion of variants A to wild type (WT). Individual foci of infection are indicated schematically within the FGT. In addition to PCR-positive tissue, RNAscope positive sites are indicated (+). Animal D290 (D) has detectable virus within the FGT and within some draining and distal LNs, GI tract, bone marrow, and plasma. Animal E048 (E) has detectable virus with multiple variants both locally and within some draining and distal LNs, spleen, GI tract, and plasma. Individual sections from the cervix of ELV were stained for vRNA, and highlighted regions were captured for DNA extraction and sequencing (F and G). The proportion of each variant is shown in the pie chart adjacent to the region isolated, with the number of sequences indicated within the pie chart. Cervix 1 tissues were analyzed in two separate sections containing one to three distinct variants. Cervix 2 contained WT virus found in two distinct aggregates (H). Scale bars, 100 μm.
In animals necropsied later after infection, the proportion of FGT sites that were positive for SIV increased. To investigate whether the increasing infection in the FGT occurred because of horizontal spread (i.e., to directly adjacent tissues) or more broadly (e.g., via lymphatic or blood circulation, potentially “reseeding” the FGT), we studied the relative ratios of different variants in adjacent tissue sections. A Mantel test was used to measure the association between the anatomic location of infected foci and the sharing of viral clonotype ratios. We observed a significant trend for local clustering within the FGT in six of the nine animals tested, suggesting lateral spread from the original site of entry (E018, P = 0.85; E024, P = 0.92; E048, P = 0.003; E053, P = 0.001; ELV, P = 0.04; EZJ, P = 0.84; D226, P = 0.03; E052, P = 0.047; D290, P = 0.003). Since the tissue sections are ~1 cm3 and clusters often appeared to span several sections, lateral spread can apparently occur over considerable anatomical distances.
Whereas we always observed clustered entry of multiple strains at the macroscopic level (analyzing viral strains in 1-cm3 tissue samples), sequence analysis at the microscopic level allows for the assessment of individual lineages within individual lymphoid aggregates. Laser capture microdissection sequencing was performed on vaginal and cervical tissues from viremic animals ELV, D290, and E052 (Fig. 3, F to H). Microscopic, localized sites of infection contain either a single variant or multiple lineages consistent with our previous data that multiple lineages can cross the mucosa in close proximity and establish infection within the same aggregate of CD4+ T cells. In all cases, the viral variants found within individual sections were found in other tissues and in plasma.
Local replication and the absence of bottleneck
Two key questions in transmission are amenable to testing with this study: (i) Do viral sequences bypass the FGT and move directly to the systemic compartment? (ii) Are there variants found within the FGT that do not progress out of the site of infection (dead-end infections)? We hypothesized that all lineages detectable systemically first replicated at the site of entry and that local replication is necessary before dissemination. To test this, we used animals with partially or fully disseminated infection to search for variants replicating in plasma that were not detected at point of entry. We used both real-time SGA (RT-SGA) (31) and NGS to improve sensitivity (35). A modified version of our NGS approach was adopted to amplify the molecular tag and was confirmed to provide an unbiased and proportional representation of the input virus that is a hallmark advantage of SGA (fig. S6). For animal D290, seven variants were detected in various tissue compartments and systemically (Fig. 4A). Using NGS, we were able to detect the FGT origins of variant E, which was only identified in plasma using RT-SGA (Fig. 4D). For EZN, we detected two lineages: a dominant lineage (H) and a minor lineage (I) approximately 3 logs less abundant but found throughout multiple tissues and in three separate plasma samples (Fig. 4B). In the remaining systemic animals, we found multiple variants at various proportions as much as 4 logs different (fig. S7). For all of the tissues examined, we found evidence of local replication of each variant to be systemically consistent with our hypothesis that initial replication at the mucosal portal of entry is important for successful systemic dissemination. Furthermore, the vast majority of variants found in the FGT were also detected in the blood plasma supporting the notion that, once across the epithelial barrier, dead-end infections are rare and dissemination is likely to occur. The exceptions to this were twofold: (i) variant A was found in the cervix of ELV without detection distally (fig. S7A) and (ii) variant D was found in several animals locally, but with limited or no spread systemically (Fig. 4A and fig. S7, A to C). Although the mechanism is unclear, we have previously shown that variant D has a lower replicative capacity than the other nine viral variants in SIVmac239X (31). While variant D can establish initial infection, its lower replicative capacity appears to markedly reduce its ability to successfully disseminate. We conclude that the vast majority of lineages detectable in the systemic compartment replicated at the site of entry and that local replication is most common before dissemination.
Fig. 4. Proportion of individual variants using NGS.
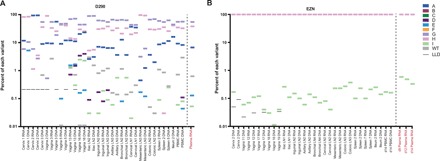
The proportion of each variant was determined in a subset of SIV-positive tissues, cells, and plasma using NGS and is plotted for animal D290 necropsied at day 8 (A) and animal EZN necropsied at day 14 (B). Rectangular symbols are used for each variant (A to WT; legend on the right). The lower limit of detection is indicated with a black line if above 0.01%. All other samples had a limit of detection less than 0.01%. Left of dashed line, tissue samples; right of dashed line, plasma samples.
A threshold replication requirement forsystemic dissemination
We next assessed the level of viral replication necessary in the FGT to allow for dissemination and systemic detection. We plotted the cumulative, normalized vRNA and vDNA levels from the entire FGT and all LN and spleen samples from all animals (Fig. 5A). Systemic animals had a higher level of vRNA and vDNA in the FGT compared to animals with only local or transitional infection, confirming a requirement of local viral amplification before dissemination. Furthermore, the differences in vRNA and vDNA levels were even greater when comparing the LN and spleen samples. We can estimate a threshold level of local viral replication in tissues that correlates with the appearance of measurable levels of virus in plasma. For example, the estimated threshold for sufficient tissue virus associated with detectable plasma viremia appears to be between 671 and 2820 vRNA copies per 106 cells and between 29 and 87,800 copies in the LN and spleen (indicated as shaded bars in Fig. 5A). Similarly, a threshold of 7770 to 13,000 vDNA copies per 106 cells in FGT and 1 to 2170 in the LN and spleen is consistent with the data. Given the viral loads measured here, it is unlikely that bypassing local replication is typical, supporting the hypothesis that early differences in viral replication within the FGT dictate the dynamics of dissemination.
Fig. 5. A tissue infection threshold for viral dissemination to plasma.
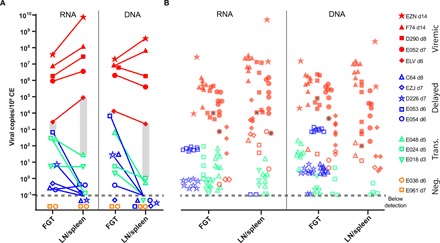
(A) The sum of vRNA and vDNA for all pieces of the FGT and for LNs/spleen is plotted for animals with different levels of viral dissemination. Viremic animals are indicated in red, delayed animals are in blue, transitional animals are in green, and negative animals are in orange. Open symbols indicate undetected in plasma. Gray shading indicates the approximate threshold level of tissue infection observed in animals that had detectable plasma virus. (B) The proportion of the total viral load for each individual lineage was plotted with the same symbol as the total but shaded. Where individual lineages were not found in plasma, this is displayed as an open symbol. Gray-filled symbols represent variant D. Similar threshold levels of infection appear to apply for individual lineages, since only a minority of lineages with concentrations below the threshold are observed in plasma. The limit of detection is indicated by a dashed line.
Since the overall viral load data can be partitioned into each individual lineage found in each animal, we can also estimate the necessary viral load required for each lineage to be observed in plasma in animals with systemic infection (Fig. 5B). In general, the threshold levels of local infection required for systemic spread of total viral load also seemed to apply to individual lineages. That is, almost all of the lineages with local infection levels above the threshold were observed in plasma. However, only a few lineages below the threshold in tissues were observed in plasma. This implies that the ability of any lineage to achieve systemic infection is reliant on its viral load within the FGT. Overall, we conclude that the delay to systemic viremia for individual animals and individual lineages within an animal is due to a delay in generating sufficient levels of total viral load within the FGT to disseminate.
Timing of dissemination
Since macaques are outbred with highly divergent genetics, we next sought to determine whether variability in exponential growth rates between animals could explain the delay to detectable viremia in some animals following vaginal infection. An average growth rate for SIVmac239 was calculated from 15 animals infected intravenously. The median exponential growth rate was 1.75 day−1 with a range of 1.5 to 2 day−1 (fig. S8). We compared the viral load of intravenously infected animals to intravaginal infection (Fig. 6A). We find that the five viremic animals fit well within the animal-to-animal variability following intravenous infection. However, assuming an equivalent growth rate for all animals, then the only thing that can account for being aviremic after day 6 is a prolonged eclipse phase. During this phase, the virus is either replicating at a slow rate or quiescent. We conclude that a delay in exponential replication within the first days following exposure can cause an important difference in the time to detectable plasma viremia.
Fig. 6. Using estimated growth rates to estimate the time to detectable viral load.
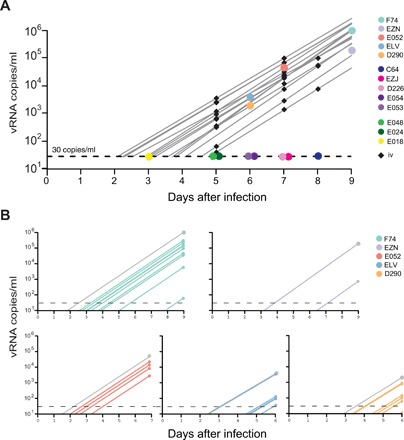
(A) Viral load data for viremic animals following vaginal infection (n = 5) were plotted with 15 intravenously (iv) infected animals. Viremic animals and animals sampled at days 3 and 5 fall within the animal-to-animal variability of exponential growth. Animals sampled after day 6 are outside of this range and are classified as delayed. (B) For individual viral lineages within each animal, the proportion of the total viral load is plotted with and the time each sample would reach 30 copies/ml are shown assuming a constant growth rate for each lineage. The differences in time to detectable viremia between individual lineages within the same animal can be as high as 5 days.
Again, our molecularly tagged virus model allows us not only to assess the overall growth rate per animal but also to study interlineage variability within the same animal. Here, we estimated the relative time delay for individual lineages using the calculated growth rate of 1.75 day−1 (Fig. 6B). Since the growth rates were assumed to be constant within each lineage, the several log difference in the relative proportion of each transmitted lineage corresponds to a 2- to 5-day delay in exponential replication for lineages with the lowest relative proportion. Since the proportion of each lineage is constant during exponential growth (Fig. 4B and fig. S7C), we conclude that the proportional differences of each variant are due to heterogeneity at the site of infection that can diminish individual lineage during the first few days following infection. Although this delay is unlikely to be viral in this model (outside of variant D), differences in viral phenotypes of HIV could add substantial variability in growth between distinct viral variants.
Correlation of CD4+ T cells and viral load
Previously published data suggested a requirement for recruitment of CD4+ T cells to sites of initial infection (24); we therefore quantified the target cells in the vagina and cervix following challenge. We found a positive correlation between the percentage of CD4+ T cells in a given tissue area and the average tissue viral load per million cells for both vRNA and vDNA (Spearman correlation P < 0.0001 and P < 0.05, respectively) (Fig. 7, A and B). We found CD4+ T cells variably distributed along the FGT mucosal barrier with T cell aggregates in the lamina propria evident sporadically throughout the entire FGT, and these aggregates were greater in size and number in animals with progressive infection (Fig. 7, C to E). The change in the density of CD4+ T cell targets was associated with a concomitant augmentation of local inflammation and immune activation (Ki67 expression). Using RNAscope to detect the vRNA+ cells, most of the productively infected cells were found within lymphoid aggregates with lymphocytic morphology consistent with CD4+ T cells being the dominant target for early viral replication (Fig. 7, F to H).
Fig. 7. Early recruitment of CD4+T cells and antiviral factors.
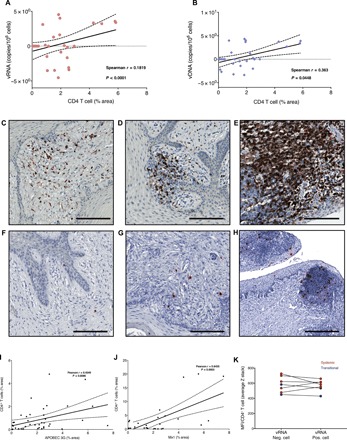
CD4+ T cell recruitment to FGT was monitored by quantitative IHC image analysis. CD4 recruitment was correlated with vRNA (A) and vDNA (B) within the FGT. Representative images of CD4+ T cell recruitment (brown) are shown during local (C), transitional (D), and systemic (E) infection with macrophages (red). RNAscope was used to localize and identify the cell morphology of vRNA+ cells in the FGT from animals displaying local (F), transitional (G), and systemic (H) infection. The cell morphology is consistent with CD4+ T cells being the dominant target cell following vaginal transmission. Scale bars, 100 μm. Quantitative image analysis was used to identify the increase in Mx1 (I) and APOBEC3G (J) expression during progressive infection, which correlated to CD4+ T cell recruitment. The relative expression levels of Mx2, APOBEC3G, Tetherin, and TRIM5α were quantified together in SIV-negative or SIV-infected CD4+ cells using fluorescent confocal microscopy and expressed as the median fluorescence intensity (MFI) per T cell. Neither transitional nor systemic animals exhibited any difference in the MFI between SIV-positive and SIV-negative cells (K).
Innate antiretroviral factors
Since the loss of initially present viral variants was rare, we used quantitative image analysis to assess whether the innate host responses to infection could diminish viral replication in individual animals or viral lineages. Since host innate pathways have been shown to exert antiviral functions that could affect viral replication, we quantified the magnitude of multiple key antiviral restriction factors [apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC-3G), Tetherin, and Tripartite Motif Containing 5 (TRIM5α)], as well as the type I interferon (IFN)–stimulated gene (ISG) products Mx1 and Mx2, the proinflammatory mediator tumor necrosis factor–α (TNFα), and cytolytic effector molecule granzyme B expressed by effector CD8+ T cells and natural killer cells within the FGT. Expression levels of each analyte were highly variable between animals and between tissues from the same animal. While there were trends for increased expression of innate antiviral factors (AVFs) as the infection progressed, there was no individual analyte that was significantly increased after correcting for multiple comparisons (fig. S9). There was a significant positive correlation between the magnitude of both Mx1 and APOBEC3G with CD4+ T cell density, supporting a role for early host inflammatory responses driving the recruitment of CD4+ T cells into the portal of entry (Pearson r = 0.64, P = 0.0003 and r = 0.40, P = 0.0086, respectively) (Fig. 7, I and J).
To assess the potential activity of AVFs within individual cells to limit infection, we used quantitative confocal microscopy and combined antibodies specific for Mx2, APOBEC3G, Tetherin, and TRIM5α into a single assay and measured the median integrated intensity [reported as median fluorescence intensity (MFI)] of AVF expression in the cellular volume from Z-stack images (representative image; fig. S10). To determine whether AVFs were differentially expressed within infected and uninfected cells, we compared the expression levels of AVFs in vRNA+ T cells compared to adjacent T cells that were vRNA-negative (vRNA−) (Fig. 7K). There was no significant difference in the AVF expression levels in productively infected and vRNA− cells, consistent with the inability of these AVFs to meaningfully suppress viral replication (Wilcoxon signed-rank test, P = 0.19).
We also assessed whether expression of ISG products were spatially or temporally associated with viral spread. Uninfected animals (Fig. 8A) exhibited low levels of Mx1 in both draining and distal LNs. SIV-infected animals with evidence of only local viral replication displayed increased Mx1 expression only in draining LNs, but this increase was heterogeneously spread across the LN in small punctate regions (Fig. 8B). In those animals in which infection progressed past local replication into the transitional phase, we observed increased Mx1 expression with a more homogeneous, diffuse pattern in local draining LNs with small, multifocal increases in expression in distal LNs (Fig. 8C). Once systemic infection occurred, we found high levels of homogeneous, diffuse Mx1 in both draining and distal LNs (Fig. 8D). Therefore, although there is a large and robust Mx1 response, it seemingly lacks sufficient activity to block viral dissemination, likely due to a delay in induction of expression. In support of this conclusion, we compared the growth rates in SIV-positive tissues within the FGT in eight animals necropsied between days 3 and 8 to the growth rate in plasma in three of these animals and found no significant difference (1.9 day−1 in FGT versus 1.8 day−1 in plasma, P = 0.98; fig. S8). We conclude that rapid viral replication within the FGT prevents host antiviral activity from effectively blocking viral spread—the virus replicates and spreads faster than a naïve host can arrest it. These data confirm that, in naïve animals, host responses are “too little and too late” to efficiently arrest the early spread of virus (36).
Fig. 8. Accumulation of Mx1 in draining and distal LNs during progressive infection.
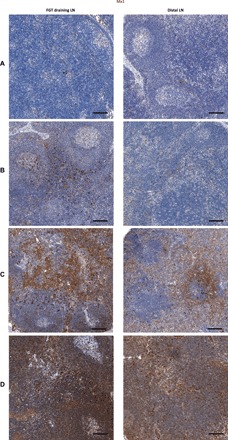
Mx1 staining (brown) in FGT draining and distal LNs from an SIV-negative animal (A), local infection animal D226 (B), transitional infection animal E024 (C), and systemic infection animal D290 (D). Mx1 was increased in the draining LNs as early as during the local infection phase, which progressed throughout the rest of the phases of infection. Distal LNs showed evidence of up-regulated Mx1 commencing only in animals in the transitional phase, which increased further as animals became systemically infected. Scale bars, 100 μm.
DISCUSSION
An improved understanding of the mechanisms involved in mucosal transmission may guide the development of prophylactic interventions to prevent HIV-1 infection. Here, we used an NHP model with a sequence-tagged synthetic swarm of isogenic viral variants and a sequential scheduled necropsy study to track initial viral infection though systemic dissemination. qPCR of tissue samples from multiple anatomic sites showed that local replication within the FGT precedes viral dissemination, and this dissemination occurs predominantly via the lymphatic system. Furthermore, in this model, which uses a large viral inoculum, in all cases, individual foci of initial infection contained two or more viral variants that contained varying proportions of the total viral populations, and these proportions are maintained even as the virus disseminates systemically. This is consistent with viral transmission occurring at sites of nonhomogeneously disturbed mucosal sites of increased susceptibility. We confirm that CD4+ T cells are the primary targets during primary infection and increase locally in the FGT during infection through recruitment and/or proliferation. Last, while several AVFs were increased at sites showing evidence of viral replication, expression of these factors appeared to follow, not precede, viral spread and seemed to be ineffective at blocking viral lineages once they were established within the FGT. The apparent requirement for local viral replication at the mucosal portal of entry suggests that strategies that can contain this process may be able to halt systemic dissemination of the infection.
Previously, a key obstacle to understanding viral transmission and the pathways and timing of viral dissemination, even in NHP studies, was the daunting technical challenges inherent in tracking the virus population from the portal of entry through systemic dissemination. Use of a clonal challenge virus precludes the ability to track distinct chains of viral transmission and spread events, while use of a mixed swarm challenge adds to the confounding influence of potential phenotypic differences between variants and limits the feasibility of deep sequence analysis. Here, the novel SIVmac239X molecularly tagged virus approach provided evidence of the number of variants within each focal site of infection and allowed tracking of the spread of distinct individual lineages systemically. With this virus model, we were able to use an RT-SGA strategy that allows for the rapid assessment of PCR-positive wells while retaining all the benefits of SGA (e.g., no Taq-induced error, no in vitro recombination, and proportional representation of templates). We also developed an NGS assay that allowed for the proportional representation of each variant while assessing hundreds of thousands of vRNA and vDNA templates. Overall, there was agreement between the two sequencing approaches, with NGS providing several logs more sensitivity to detect minor variants. Last, laser capture microdissection sequencing allowed for the microscopic discrimination of local sites of infection (made up of one or only a few variants) compared to the bulk tissue-based sequencing where many smaller foci were sampled together. Using laser capture sequencing, we found that multivariant infection in the FGT was common even within microscopic regions of actively replicating virus, suggesting that whatever mechanism allowed for infection at a particular site facilitated infection with more than one variant. Therefore, host factors, potentially including preexisting mucosal breaches, inflammation, or increased local concentrations of potential target cells, may increase susceptibility to viral transmission.
Our results also indicate the importance of viral characteristics in determining viral transmission. Although the goal of generating the tagged virus swarm was to create a genetically distinguishable but phenotypically identical population, we confirmed that variant D had a diminished replicative capacity compared to the other variants in the challenge mixture. Here, we were able to demonstrate that variant D was transmitted across the mucosa but lacked sufficient replicative capacity to disseminate much beyond the FGT. Despite the less than 1 log difference in viral replication in vitro between variant D and all the other variants (31), in vivo, this difference markedly affected successful dissemination from the site of exposure. This observation emphasizes the importance of viral phenotype on initiating a successful infection. Some of the known differences between viruses that establish transmission and those that do not include coreceptor usage, IFN resistance, and replication capacity (3–6, 9, 12, 37). These differences and possibly other as of yet unidentified characteristics highlight the potential disparity in the efficiency of different viruses in finding new targets, actively replicating and producing infectious progeny at a rate sufficient for successful dissemination.
Excluding variant D, there was only a single instance of identifying a viral lineage in the FGT that was also not found systemically (either in plasma or in distal lymphatic tissues). These data are consistent with a requirement for local replication before dissemination. Furthermore, we were able to test the alternative hypothesis that some variants may have bypassed local expansion in the FGT by looking for variants that were replicating systemically without evidence of local infection. Although this analysis is considerably more complicated because of potential viral reseeding of sites of initial infection following dissemination, we found no evidence of any lineage peripherally that also did not have a presence at the site of infection. Therefore, we conclude that local replication is important for eventual successful dissemination following mucosal exposure.
One of the most interesting and unexpected findings in this study was the heterogeneous patterns of dissemination in animals challenged during the same time of the menstrual cycle with identical inocula and with an essentially clonal virus population. Viral nucleic acid measurements from both plasma and tissue showed dramatic evidence of rapid replication in some animals and much slower dynamics in others. Explanations for the variable viral levels of different clones in different animals might include differences in inocula, the permissiveness for viral replication at the site of entry, or variability in growth rates in different animals. In our system, with equal levels of each variant in the inoculum, the initial population size of each variant should follow a binomial distribution, and thus, differences in initial copy number should be relatively limited. While it is often assumed that early growth in the FGT may be slower than that observed later in plasma, when we compared early growth rates in FGT compared to later growth rates in plasma, we found no significant difference. In a rectal mucosal infection model, it has been reported that all infected animals had rapid dissemination (38). The differences between these studies could be the lack of target cells in the FGT that are abundant in rectal mucosa. In some animals, and for some variants, perhaps additional targets in the FGT allowed for a more rapid dissemination compared to other animals or other lineages within the same animal.
One potential factor contributing to delay in local replication or spread in individual viral variants or in individual animals might be variability in local expression of innate AVFs, but no statistically significant difference in expression of AVFs was detected. Therefore, we conclude that these innate responses are unlikely to halt the spread of virus from the FGT to distal sites. Further support of this is found in the expression of Mx1 in draining and distal LNs, which occurred only after infection was present. Last, there was no difference in the AVF expression levels between infected and adjacent vRNA− cells, highlighting the challenges associated with the ability of the innate immune system to block infection. It will be important to determine whether the innate host immune response can be altered following interventions intended to boost innate immunity (30).
Human mucosal HIV infection likely parallels these results with two major differences. First, the virus inoculum used is different. The virus is suspended in culture medium, not in semen, and the amount of virus in the inoculum used in this NHP model study, intended to infect the majority of animals with a single challenge, is much greater than is typical for human HIV infection. The infection rate in this study was nearly 90%, while in humans, it is notably lower. While this model is not physiologically identical to clinically relevant HIV-1 vaginal exposure, it is nevertheless interesting to note that this model appeared to capture the spectrum of transmission from no infection to infection with several T/F lineages. Second, the virus used here was designed to be phenotypically homogeneous. In HIV infection, the virus population will be vastly more heterogeneous than this virus stock (even including variant D), which may affect the translatability of this study to HIV transmission (13). We hypothesize that, in humans, there may be additional viral lineages that cross the mucosal barrier but fail to disseminate due to poor replicative capacity. Alternatively, since infection rates are so much lower in humans, it is possible that only a single variant is actually transmitted in humans and that if a particular lineage cannot sustain a productive infection, then there is no lasting evidence of infection. When transmission of multiple founders is observed in humans, it seems to occur with more variants than would be expected from a simple Poisson process (3). These data are consistent with our observation that sites of infection with multiple variants per tissue are clustered together, potentially at sites of preexisting inflammation or target cell localization that may be permissive for infection. This may be a fundamental property of vaginal transmission.
Furthermore, some T/F clones and acute-phase virus isolates from acute infection were shown to be relatively resistant to IFN (13, 14); other cohorts reported no IFN effect of transmission (15, 39). Therefore, while variant D provides a notable example of selection between viral entry and dissemination of a less replicative clone, it will be important to assess other viral clones with different phenotypes (including variable sensitivity to IFN) to understand the viral requirements necessary for rapid local replication and successful dissemination.
This study provides insights into HIV transmission and dissemination dynamics at the early phase of transmission. Previous studies have relied on measuring the entire viral population rather than individual lineages. In high-dose challenge models, the overall population dynamics are driven by the more rapid lineage(s) and leave little room to identify important differences in local replication dynamics very early following infection. Here, we found that some lineages were quicker to expand and disseminate than other lineages, and even between animals, differences were measurable. We conclude that the relative proportions of different viral variants are established early with local replication, resulting in an apparent delay of up to 3 days before exponential amplification. We hypothesize that the activation state of the initial infected cells might contribute to these delays, a feature that cannot be directly assessed in this study. It will be important to determine whether these findings also apply for other mucosal routes of infection (oral, rectal, or penile) and whether phenotypic differences can alter the dynamics of early replication and dissemination.
MATERIALS AND METHODS
Animals
All Institutional Animal Care and Use Committee (IACUC) guidelines were followed. Fifteen Indian-origin rhesus macaques (M. mulatta) were housed at the National Institutes of Health (NIH) in accordance with the Association for the Assessment and Accreditation of Laboratory Animal Care standards, and all procedures were performed according to protocols approved by the IACUC of the National Cancer Institute (Assurance no. A4149-01). Animals were female nulliparous sexually mature Indian-origin rhesus between 5 and 9 kg at the time of study initiation. Fifteen additional Indian-origin rhesus macaques (M. mulatta) used as control animals were housed at the Oregon National Primate Center with the approval of the Oregon National Primate Research Center’s Animal Care and Use Committee (Assurance no. A3304-01), under the standards of the U.S. NIH Guide for the Care and Use of Laboratory Animals.
Viral challenge
Fifteen control animals were infected intravenously with 200 IU (TZM-bl) of SIVmac239M. Blood was drawn frequently during primary infection, and viral load data were used to estimate viral growth rates. Fifteen additional macaques were challenged intravaginally with 2 ml of transfection-derived SIVmac239X (TZM-bl titer of 5 × 105 IU/ml). Animals were placed at a ~20° down angle in an inverted Trendelenburg position, and the challenge was performed using a 3-ml slip tip syringe with a small amount of nonbacteriostatic, single-use, sterile lubricant. Menstrual cycles were assessed by visual inspection of menstrual blood following vaginal swab. Animals were challenged in the early follicular phase of menstrual cycle (within 4 days following the last positive swab).
Plasma viral loads
Plasma SIV RNA loads were quantified, essentially as previously described, with an assay sensitivity of 30 SIV gag RNA copies per milliliter (40).
Tissue nucleic acid extraction and quantification
vRNA and vDNA were extracted from various tissues using a phenol extraction protocol after tissue pieces were homogenized in 1 ml of TRI Reagent (Molecular Research Center) in 2-ml extraction tubes of Lysing Matrix D (MP Biomedicals) using Precellys 24 (Bertin Instruments). Total RNA and DNA were prepared from the homogenates following the manufacturer’s recommendations but specifically following the alternative, back-extraction method for DNA extraction. Recovered RNA and DNA were dissolved in minimal volumes of 10 mM tris-Cl at pH 8.0 and 10 mM tris-Cl at pH 9.0, respectively, for qRT-PCR and qPCR, as well as for use in various sequencing approaches, as described below. Quantification for vRNA and vDNA was performed using a real-time assay designed to encompass the molecularly tagged portion of the integrase gene, as previously described (31). To quantify the number of cells assayed, a qPCR assay for the single-copy rhesus CCR5 gene was used with the following primers: RHR5F01, 5′-CCAGAAGAGCTGCGACATCC-3′; RHR5R01, 5′-CTAATAGGCCAAGCAGCTGAGG-3′; and probe RHR5P01, 5′-(Texas Red) TTCCCCTACAAGAAACTCTCCCCGGTAAGTA (BHQ2)-3′. CEs were calculated based on diploid genome equivalents of the CCR5 gene.
SGA and MiSeq sequencing
For samples with limited vRNA and vDNA, virus-positive tissues were sequenced using an RT-SGA and Sanger sequencing approach we described previously (31). For samples with high levels of viral nucleic acid, representative samples were sequenced using both RT-SGA and an Illumina-based sequencing approach implemented on the MiSeq instrument, as previously described (35) with primers SIV.INT.P5 (5′-GAAGGGGAGGAATAGGGGATATG-3′) and SIV.INT.P7 (5′-CCTCCATGTGGGAACTGCTATCC-3′).
Immunohistochemistry
IHC and quantitative image analysis were performed on 5-μm tissue sections mounted on glass slides, as previously described (41). Antibodies used in this study were mouse anti-CD68 (1:400; clone KP1, Dako), mouse anti-CD163 (1:400; clone 10D6, Novocastra/Leica), rabbit monoclonal anti-CD4 (1:200; clone EPR6855, Abcam Inc.), rabbit anti-APOBEC3G (1:500; HPA001812, Sigma), rabbit anti–granzyme B (1:200; HPA003418, Sigma), mouse anti-Mx1 (1:2000; clone M143; a gift from G. Kochs and the Department of Virology of the University of Freiburg), rabbit anti-Mx2 (1:500; HPA030235, Sigma), rabbit anti-Tetherin/BST2 (1:1000; HPA017060, Sigma), mouse anti-TNFα (1:1000; clone P/T2, ab9579, Abcam), and rabbit anti-TRIM5α (1:1000; HPA023422, Sigma).
ISH and laser capture microdissection sequencing
SIV ISH was performed on serial sections using next-generation ISH RNAscope, as previously described (32). The slides were scanned at high magnification (×400) using the Aperio AT2 digital Whole Slide Scanning System (Leica Biosystems), yielding high-resolution data from the entire tissue section. After regions of interest (ROIs) were determined and mapped on the digital Aperio scans of each tissue, laser capture microdissection was performed on the subjacent serial sections aligned to the specified ROIs using an ArcturusXT, Nikon TE2000 Microscope laser capture microdissection system. The targeted areas were captured on different caps (CapSure Macro LCM Caps, Applied Biosystems), as previously described (42). Each tube was inverted to allow total immersion of the tissue in the protease K solution and incubated at 65°C for 20 hours. The samples were then used for viral sequencing, as described above.
RNAscope, DNAscope, and quantitative immunofluorescence analysis
Chromogenic RNAscope and DNAscope and fluorescent RNAscope with protein immunofluorescence detection were performed as previously described (32). Quantification of antiviral restriction factors (APOBEC3G, Mx1, Mx2, Tetherin, and TRIM5α multiplexed to detect signal in a single fluorescent channel) in T cells (CD3+) and myeloid cells (CD68/CD163+), which were productively infected cells (vRNA+) or not (vRNA−), was performed on high-magnification confocal image volumetric Z-stacks, using an Olympus FV10i (×60 objective). The Nyquist sampling method was used from ROIs along the vaginal and cervical mucosa including lymphoid aggregates when present. Laser settings (gains and power) were set to ensure that there was no pixel saturation in the antiviral restriction factor channel and were kept constant for all sample collections for each animal imaged. Images were analyzed using Olympus FV10-ASW software (v3.1) and individual cells were identified and outlined with well-defined cellular regions that were manually drawn; the median integrated intensity of AVF expression in the cellular volume from Z-stack images for each cell within these defined cellular ROIs was measured and reported as MFI.
Statistics
To quantify the spatial relationship in the viral spread, we used Mantel test to find the correlation between genetic distance and geographical distance. We used R (v3.1.2) function mantel from library vegan to perform this mantel test. Function mantel accepts two arguments, x-dis and y-dis. We used Euclidian distance between sites in the vagina as x-dis and the Morisita-Horn index of similarity as y-dis. The Morisita-Horn similarity index accounts for both the number of common variants and the distribution of variants. This index ranges between 0 and 1, representing minimal and maximal similarity, respectively.
Viral growth rate was measured by linear regression of the log-transformed values (of viral copies) against time (days). In plasma, this was simply the increase in concentration of vRNA measurements. For FGT virus, this was the sum of vRNA copies per million cells (in each FGT tissue portion) multiplied by the estimated total cells in each tissue sample (assumed to be 109).
Supplementary Material
Acknowledgments
We thank the NHP care staff in the Laboratory Animal Sciences Program, Leidos Biomedical Research Inc., Frederick National Laboratory for Cancer Research for expert animal care and the Nonhuman Primate Research Support Core, AIDS and Cancer Virus Program, Leidos Biomedical Research Inc., Frederick National Laboratory for Cancer Research for specimen processing and animal support. We thank A. Okoye and L. Picker for helpful discussion and intravenously infected viral load data. Funding: This project has been funded in whole or in part with Federal funds from the National Cancer Institute, NIH, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. M.P.D. and A.R. were supported by NHMRC grants 1080001 and 1052979. J.M.C. was supported by NSF grant no. DMS-1714654. J.D.E. was supported in part by the Oregon National Primate Research Center NIH grant P51OD011092. Author contributions: C.D., C.M.F., C.R., L.N., L.L., C.C., S.O., G.Q.D.P., J.D.L., J.D.E., and B.F.K. generated and analyzed the primary data. T.T.I., A.R., J.M.C., M.P.D., and B.F.K. conducted the statistical analysis and modeling. J.S., G.Q.D.P., and J.D.L. provided animal support. In addition, C.D., J.D.E., and B.F.K. designed the study and wrote the paper. All authors contributed to the final draft of the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav7116/DC1
Fig. S1. No viral nucleic acid signal detected in two exposed animals.
Fig. S2. Detection of rare vRNA- and vDNA-positive cells in draining LN before systemic dissemination.
Fig. S3. Schematic representation of FGT tissue collection schema.
Fig. S4. Viral nucleic acid distribution within individual tissue pieces.
Fig. S5. Proportion and quantification of each variant in virus-positive tissues.
Fig. S6. Proportional representation of sequences following NGS.
Fig. S7. Proportion of individual variants using NGS.
Fig. S8. Growth rates during FGT and systemic viral replication.
Fig. S9. No significant difference in the local expression of many proinflammatory and AVFs.
Fig. S10. AVFs measured in SIV-positive and SIV-negative cells.
REFERENCES AND NOTES
- 1.Cohen M. S., Chen Y. Q., McCauley M., Gamble T., Hosseinipour M. C., Kumarasamy N., Hakim J. G., Kumwenda J., Grinsztejn B., Pilotto J. H., Godbole S. V., Mehendale S., Chariyalertsak S., Santos B. R., Mayer K. H., Hoffman I. F., Eshleman S. H., Piwowar-Manning E., Wang L., Makhema J., Mills L. A., de Bruyn G., Sanne I., Eron J., Gallant J., Havlir D., Swindells S., Ribaudo H., Elharrar V., Burns D., Taha T. E., Nielsen-Saines K., Celentano D., Essex M., Fleming T. R.; HPTN 052 Study Team , Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 365, 493–505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray R. H., Wawer M. J., Brookmeyer R., Sewankambo N. K., Serwadda D., Wabwire-Mangen F., Lutalo T., Li X., vanCott T., Quinn T. C.; Rakai Project Team , Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet 357, 1149–1153 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Abrahams M. R., Anderson J. A., Giorgi E. E., Seoighe C., Mlisana K., Ping L. H., Athreya G. S., Treurnicht F. K., Keele B. F., Wood N., Salazar-Gonzalez J. F., Bhattacharya T., Chu H., Hoffman I., Galvin S., Mapanje C., Kazembe P., Thebus R., Fiscus S., Hide W., Cohen M. S., Karim S. A., Haynes B. F., Shaw G. M., Hahn B. H., Korber B. T., Swanstrom R., Williamson C.; CAPRISA Acute Infection Study Team; Center for HIV-AIDS Vaccine Immunology Consortium , Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J. Virol. 83, 3556–3567 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haaland R. E., Hawkins P. A., Salazar-Gonzalez J., Johnson A., Tichacek A., Karita E., Manigart O., Mulenga J., Keele B. F., Shaw G. M., Hahn B. H., Allen S. A., Derdeyn C. A., Hunter E., Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLOS Pathog. 5, e1000274 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keele B. F., Giorgi E. E., Salazar-Gonzalez J. F., Decker J. M., Pham K. T., Salazar M. G., Sun C., Grayson T., Wang S., Li H., Wei X., Jiang C., Kirchherr J. L., Gao F., Anderson J. A., Ping L. H., Swanstrom R., Tomaras G. D., Blattner W. A., Goepfert P. A., Kilby J. M., Saag M. S., Delwart E. L., Busch M. P., Cohen M. S., Montefiori D. C., Haynes B. F., Gaschen B., Athreya G. S., Lee H. Y., Wood N., Seoighe C., Perelson A. S., Bhattacharya T., Korber B. T., Hahn B. H., Shaw G. M., Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U.S.A. 105, 7552–7557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Bar K. J., Wang S., Decker J. M., Chen Y., Sun C., Salazar-Gonzalez J. F., Salazar M. G., Learn G. H., Morgan C. J., Schumacher J. E., Hraber P., Giorgi E. E., Bhattacharya T., Korber B. T., Perelson A. S., Eron J. J., Cohen M. S., Hicks C. B., Haynes B. F., Markowitz M., Keele B. F., Hahn B. H., Shaw G. M., High multiplicity infection by HIV-1 in men who have sex with men. PLOS Pathog. 6, e1000890 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delwart E. L., Mullins J. I., Gupta P., Learn G. H. Jr., Holodniy M., Katzenstein D., Walker B. D., Singh M. K., Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 72, 617–623 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta P., Leroux C., Patterson B. K., Kingsley L., Rinaldo C., Ding M., Chen Y., Kulka K., Buchanan W., McKeon B., Montelaro R., Human immunodeficiency virus type 1 shedding pattern in semen correlates with the compartmentalization of viral Quasi species between blood and semen. J. Infect. Dis. 182, 79–87 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Ochsenbauer C., Edmonds T. G., Ding H., Keele B. F., Decker J., Salazar M. G., Salazar-Gonzalez J. F., Shattock R., Haynes B. F., Shaw G. M., Hahn B. H., Kappes J. C., Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 86, 2715–2728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ping L.-H., Joseph S. B., Anderson J. A., Abrahams M. R., Salazar-Gonzalez J. F., Kincer L. P., Treurnicht F. K., Arney L., Ojeda S., Zhang M., Keys J., Potter E. L., Chu H., Moore P., Salazar M. G., Iyer S., Jabara C., Kirchherr J., Mapanje C., Ngandu N., Seoighe C., Hoffman I., Gao F., Tang Y., Labranche C., Lee B., Saville A., Vermeulen M., Fiscus S., Morris L., Karim S. A., Haynes B. F., Shaw G. M., Korber B. T., Hahn B. H., Cohen M. S., Montefiori D., Williamson C., Swanstrom R.; CAPRISA Acute Infection Study and the Center for HIV-AIDS Vaccine Immunology Consortium , Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J. Virol. 87, 7218–7233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilen C. B., Parrish N. F., Pfaff J. M., Decker J. M., Henning E. A., Haim H., Petersen J. E., Wojcechowskyj J. A., Sodroski J., Haynes B. F., Montefiori D. C., Tilton J. C., Shaw G. M., Hahn B. H., Doms R. W., Phenotypic and immunologic comparison of clade B transmitted/founder and chronic HIV-1 envelope glycoproteins. J. Virol. 85, 8514–8527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parrish N. F., Gao F., Li H., Giorgi E. E., Barbian H. J., Parrish E. H., Zajic L., Iyer S. S., Decker J. M., Kumar A., Hora B., Berg A., Cai F., Hopper J., Denny T. N., Ding H., Ochsenbauer C., Kappes J. C., Galimidi R. P., West A. P. Jr., Bjorkman P. J., Wilen C. B., Doms R. W., O’Brien M., Bhardwaj N., Borrow P., Haynes B. F., Muldoon M., Theiler J. P., Korber B., Shaw G. M., Hahn B. H., Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. U.S.A. 110, 6626–6633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyer S. S., Bibollet-Ruche F., Sherrill-Mix S., Learn G. H., Plenderleith L., Smith A. G., Barbian H. J., Russell R. M., Gondim M. V. P., Bahari C. Y., Shaw C. M., Li Y., Decker T., Haynes B. F., Shaw G. M., Sharp P. M., Borrow P., Hahn B. H., Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc. Natl. Acad. Sci. U.S.A. 114, E590–E599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenton-May A. E., Dibben O., Emmerich T., Ding H., Pfafferott K., Aasa-Chapman M. M., Pellegrino P., Williams I., Cohen M. S., Gao F., Shaw G. M., Hahn B. H., Ochsenbauer C., Kappes J. C., Borrow P., Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 10, 146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deymier M. J., Ende Z., Fenton-May A. E., Dilernia D. A., Kilembe W., Allen S. A., Borrow P., Hunter E., Heterosexual transmission of subtype C HIV-1 selects consensus-like variants without increased replicative capacity or interferon-α resistance. PLOS Pathog. 11, e1005154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson J. M., Schaefer M., Monaco D. C., Batorsky R., Claiborne D. T., Prince J., Deymier M. J., Ende Z. S., Klatt N. R., DeZiel C. E., Lin T.-H., Peng J., Seese A. M., Shapiro R., Frater J., Ndung’u T., Tang J., Goepfert P., Gilmour J., Price M. A., Kilembe W., Heckerman D., Goulder P. J. R., Allen T. M., Allen S., Hunter E., Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 345, 1254031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galvin S. R., Cohen M. S., The role of sexually transmitted diseases in HIV transmission. Nat. Rev. Microbiol. 2, 33–42 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Powers K. A., Poole C., Pettifor A. E., Cohen M. S., Rethinking the heterosexual infectivity of HIV-1: A systematic review and meta-analysis. Lancet Infect. Dis. 8, 553–563 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chehoud C., Stieh D. J., Bailey A. G., Laughlin A. L., Allen S. A., McCotter K. L., Sherrill-Mix S., Hope T., Bushman F., Associations of the vaginal microbiota with HIV infection, bacterial vaginosis, and demographic factors. AIDS (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller C. J., Shattock R. J., Target cells in vaginal HIV transmission. Microbes Infect. 5, 59–67 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Keele B. F., Estes J. D., Barriers to mucosal transmission of immunodeficiency viruses. Blood 118, 839–846 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu J., Gardner M. B., Miller C. J., Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 74, 6087–6095 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stieh D. J., Maric D., Kelley Z. L., Anderson M. R., Hattaway H. Z., Beilfuss B. A., Rothwangl K. B., Veazey R. S., Hope T. J., Vaginal challenge with an SIV-based dual reporter system reveals that infection can occur throughout the upper and lower female reproductive tract. PLOS Pathog. 10, e1004440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q., Estes J. D., Schlievert P. M., Duan L., Brosnahan A. J., Southern P. J., Reilly C. S., Peterson M. L., Schultz-Darken N., Brunner K. G., Nephew K. R., Pambuccian S., Lifson J. D., Carlis J. V., Haase A. T., Glycerol monolaurate prevents mucosal SIV transmission. Nature 458, 1034–1038 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller C. J., Li Q., Abel K., Kim E.-Y., Ma Z.-M., Wietgrefe S., la Franco-Scheuch L., Compton L., Duan L., Shore M. D., Zupancic M., Busch M., Carlis J., Wolinksy S., Haase A. T., Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 79, 9217–9227 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barouch D. H., Ghneim K., Bosche W. J., Li Y., Berkemeier B., Hull M., Bhattacharyya S., Cameron M., Liu J., Smith K., Borducchi E., Cabral C., Peter L., Brinkman A., Shetty M., Li H., Gittens C., Baker C., Wagner W., Lewis M. G., Colantonio A., Kang H. J., Li W., Lifson J. D., Piatak M. Jr., Sekaly R. P., Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell 165, 656–667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J., Ghneim K., Sok D., Bosche W. J., Li Y., Chipriano E., Berkemeier B., Oswald K., Borducchi E., Cabral C., Peter L., Brinkman A., Shetty M., Jimenez J., Mondesir J., Lee B., Giglio P., Chandrashekar A., Abbink P., Colantonio A., Gittens C., Baker C., Wagner W., Lewis M. G., Li W., Sekaly R.-P., Lifson J. D., Burton D. R., Barouch D. H., Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science 353, 1045–1049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hessell A. J., Jaworski J. P., Epson E., Matsuda K., Pandey S., Kahl C., Reed J., Sutton W. F., Hammond K. B., Cheever T. A., Barnette P. T., Legasse A. W., Planer S., Stanton J. J., Pegu A., Chen X., Wang K., Siess D., Burke D., Park B. S., Axthelm M. K., Lewis A., Hirsch V. M., Graham B. S., Mascola J. R., Sacha J. B., Haigwood N. L., Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat. Med. 22, 362–368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mavedzenge S. N., Pol B. V., Cheng H., Montgomery E. T., Blanchard K., de Bruyn G., Ramjee G., van der Straten A., Epidemiological synergy of Trichomonas vaginalis and HIV in Zimbabwean and South African women. Sex. Transm. Dis. 37, 460–466 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Sandler N. G., Bosinger S. E., Estes J. D., Zhu R. T. R., Tharp G. K., Boritz E., Levin D., Wijeyesinghe S., Makamdop K. N., del Prete G. Q., Hill B. J., Timmer J. K., Reiss E., Yarden G., Darko S., Contijoch E., Todd J. P., Silvestri G., Nason M., Norgren R. B. Jr., Keele B. F., Rao S., Langer J. A., Lifson J. D., Schreiber G., Douek D. C., Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Prete G. Q., Park H., Fennessey C. M., Reid C., Lipkey L., Newman L., Oswald K., Kahl C., Piatak M. Jr., Quiñones O. A., Alvord W. G., Smedley J., Estes J. D., Lifson J. D., Picker L. J., Keele B. F., Molecularly tagged simian immunodeficiency virus SIVmac239 synthetic swarm for tracking independent infection events. J. Virol. 88, 8077–8090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deleage C., Wietgrefe S. W., Del Prete G., Morcock D. R., Hao X.-P., Piatak M. Jr., Bess J., Anderson J. L., Perkey K. E., Reilly C., McCune J. M., Haase A. T., Lifson J. D., Schacker T. W., Estes J. D., Defining HIV and SIV reservoirs in lymphoid tissues. Pathog. Immun. 1, 68–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kersh E. N., Henning T., Vishwanathan S. A., Morris M., Butler K., Adams D. R., Guenthner P., Srinivasan P., Smith J., Radzio J., Garcia-Lerma J. G., Dobard C., Heneine W., McNicholl J., SHIV susceptibility changes during the menstrual cycle of pigtail macaques. J. Med. Primatol. 43, 310–316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smedley J., Turkbey B., Bernardo M. L., Del Prete G. Q., Estes J. D., Griffiths G. L., Kobayashi H., Choyke P. L., Lifson J. D., Keele B. F., Tracking the luminal exposure and lymphatic drainage pathways of intravaginal and intrarectal inocula used in nonhuman primate models of HIV transmission. PLOS ONE 9, e92830 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fennessey C. M., Pinkevych M., Immonen T. T., Reynaldi A., Venturi V., Nadella P., Reid C., Newman L., Lipkey L., Oswald K., Bosche W. J., Trivett M. T., Ohlen C., Ott D. E., Estes J. D., del Prete G. Q., Lifson J. D., Davenport M. P., Keele B. F., Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLOS Pathog. 13, e1006359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davenport M. P., Ribeiro R. M., Perelson A. S., Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection. J. Virol. 78, 10096–10103 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salazar-Gonzalez J. F., Bailes E., Pham K. T., Salazar M. G., Guffey M. B., Keele B. F., Derdeyn C. A., Farmer P., Hunter E., Allen S., Manigart O., Mulenga J., Anderson J. A., Swanstrom R., Haynes B. F., Athreya G. S., Korber B. T. M., Sharp P. M., Shaw G. M., Hahn B. H., Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82, 3952–3970 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ribeiro Dos Santos P., Rancez M., Prétet J.-L., Michel-Salzat A., Messent V., Bogdanova A., Couëdel-Courteille A., Souil E., Cheynier R., Butor C., Rapid dissemination of SIV follows multisite entry after rectal inoculation. PLOS ONE 6, e19493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oberle C. S., Joos B., Rusert P., Campbell N. K., Beauparlant D., Kuster H., Weber J., Schenkel C. D., Scherrer A. U., Magnus C., Kouyos R., Rieder P., Niederöst B., Braun D. L., Pavlovic J., Böni J., Yerly S., Klimkait T., Aubert V., Trkola A., Metzner K. J., Günthard H. F.; The Swiss HIV Cohort Study (SHCS) , Tracing HIV-1 transmission: Envelope traits of HIV-1 transmitter and recipient pairs. Retrovirology 13, 62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Wang S., Kong R., Ding W., Lee F.-H., Parker Z., Kim E., Learn G. H., Hahn P., Policicchio B., Brocca-Cofano E., Deleage C., Hao X., Chuang G.-Y., Gorman J., Gardner M., Lewis M. G., Hatziioannou T., Santra S., Apetrei C., Pandrea I., Alam S. M., Liao H.-X., Shen X., Tomaras G. D., Farzan M., Chertova E., Keele B. F., Estes J. D., Lifson J. D., Doms R. W., Montefiori D. C., Haynes B. F., Sodroski J. G., Kwong P. D., Hahn B. H., Shaw G. M., Envelope residue 375 substitutions in simian–human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl. Acad. Sci. U.S.A. 113, E3413–E3422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deleage C., Schuetz A., Alvord W. G., Johnston L., Hao X. P., Morcock D. R., Rerknimitr R., Fletcher J. L. K., Puttamaswin S., Phanuphak N., Dewar R., McCune J. M., Sereti I., Robb M., Kim J. H., Schacker T. W., Hunt P., Lifson J. D., Ananworanich J., Estes J. D.; RV254/SEARCH 010 and RV304/SEARCH 013 Study Groups , Impact of early cART in the gut during acute HIV infection. JCI Insight 1, e87065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klase Z., Ortiz A., Deleage C., Mudd J. C., Quinones M., Schwartzman E., Klatt N. R., Canary L., Estes J. D., Brenchley J. M., Dysbiotic bacteria translocate in progressive SIV infection. Mucosal Immunol. 8, 1009–1020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav7116/DC1
Fig. S1. No viral nucleic acid signal detected in two exposed animals.
Fig. S2. Detection of rare vRNA- and vDNA-positive cells in draining LN before systemic dissemination.
Fig. S3. Schematic representation of FGT tissue collection schema.
Fig. S4. Viral nucleic acid distribution within individual tissue pieces.
Fig. S5. Proportion and quantification of each variant in virus-positive tissues.
Fig. S6. Proportional representation of sequences following NGS.
Fig. S7. Proportion of individual variants using NGS.
Fig. S8. Growth rates during FGT and systemic viral replication.
Fig. S9. No significant difference in the local expression of many proinflammatory and AVFs.
Fig. S10. AVFs measured in SIV-positive and SIV-negative cells.