

Different mutational processes are active in the fetal intestine than in the fetal liver.
Abstract
A developing human fetus needs to balance rapid cellular expansion with maintaining genomic stability. Here, we accurately quantified and characterized somatic mutation accumulation in fetal tissues by analyzing individual stem cells from human fetal liver and intestine. Fetal mutation rates were about fivefold higher than in tissue-matched adult stem cells. The mutational landscape of fetal intestinal stem cells resembled that of adult intestinal stem cells, while the mutation spectrum of fetal liver stem cells is distinct from stem cells of the fetal intestine and the adult liver. Our analyses indicate that variation in mutational mechanisms, including oxidative stress and spontaneous deamination of methylated cytosines, contributes to the observed divergence in mutation accumulation patterns and drives genetic mosaicism in humans.
INTRODUCTION
Mutations that arise during fetal development result in somatic mosaicism and can affect a large population of cells in the adult organism. Potential consequences for human health are congenital disorders and increased cancer risk (1–4). Understanding the processes that drive somatic mosaicism in development would therefore expand our knowledge on the origin and evolution of diseases. A powerful method for obtaining insight into active and past mutational processes is through the analysis of mutational profiles, which reflect balances between DNA damage and activity of DNA repair pathways. So far, 30 mutational signatures have been extracted from the mutational landscapes of cancer genomes and listed in the COSMIC database (5). Mutational signature analysis of human adult stem cells (SCs) has revealed that mutation accumulation in healthy cells is tissue specific and happens with a continuous rate throughout adult life (6). Adult intestinal SCs have a large contribution of COSMIC signature 1, which is characterized by C to T changes at CpG sites and has been attributed to spontaneous deamination of methylated cytosines. In contrast, adult liver SCs show minimal signature 1 contribution, while signature 5, characterized by T to C transitions, is prevalent (6). Signatures 1 and 5 were found to accumulate with age and act in a clock-like manner (7) and were also found to explain mutations that occur very early in development in two-cell–stage embryos (8). To date, it is unknown what the contribution of fetal development to cell-specific mutation accumulation is and when in life patterns of mutation accumulation start to diverge between tissues. Therefore, we accurately cataloged genome-wide somatic mutations in individual liver and intestinal SCs of human fetuses ranging in age from 13 to 20 weeks after conception.
RESULTS
Fetal mutation accumulation rates
Measuring extremely low mutation loads in individual cells is challenging because of the high noise rates that are associated with single-cell DNA sequencing techniques that typically involve error-prone amplification methods (9). To circumvent this hurdle, we studied mutation accumulation in individual SCs by expanding them in vitro as organoids before whole-genome sequencing (WGS) and bioinformatic filtering for clonal variants—a method that we previously applied successfully to identify somatic variants in adult SCs from different tissues and donors of different ages (6, 10). Using routine organoid culture conditions (11, 12), we successfully derived SC lines from the liver and intestine of four fetuses at weeks 15, 17, and 22 (n = 2) after gestation, which amounts to an estimated post-conceptional age of 13, 15, and 20 weeks, respectively. Individual organoids from the primary cultures were manually picked, expanded to obtain 14 clonal lines (6 of the intestine and 8 of the liver) (fig. S1), and whole genome–sequenced to a minimum average coverage of 30×. No chromosomal aberrations and aneuploidies were observed in the copy number profiles. At the base pair level (see Materials and Methods for details), we identified a total of 834 somatic base substitutions in 14 SCs from four independent fetuses (table S1). Independent validation using amplicon-based resequencing of 569 base substitutions confirmed 556 (98%) of the variants (table S2).
On average, each SC accumulated 67 base substitutions. Particularly for the liver, there was a high degree of variation (minimum = 20, maximum = 153), which is likely caused by the spread in fetal age, as there was little variation between fetuses of the same age (Fig. 1A). A linear mixed-effects random slope model analysis (in which the fetus is a random effect) confirmed a significant correlation (corrected P = 0.04) between the number of base substitutions in the liver SCs and fetal age (Fig. 1A), indicating accumulation of mutations over time. Because SC mutation accumulation rates have been measured previously for adult liver and intestine (6), we can now directly compare mutation accumulation rates between human adult and fetal SCs of the same organ. For 13 of 14 fetal SCs, the mutation rates fell outside the 95% confidence intervals of the slope estimates of the adult liver and small intestine SCs (linear mixed-effects model, table S1), with about fivefold more somatic variants per week of fetal life than per week of adult life (Fig. 1B).
Fig. 1. Mutation accumulation in SCs of the human fetal liver and intestine.
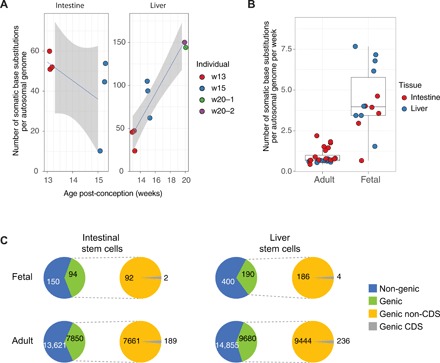
(A) Left panel: Number of somatic base substitutions in each intestinal fetal SC (extrapolated to the whole autosomal genome). Right panel: Number of somatic base substitutions in each liver fetal SC (extrapolated to the whole autosomal genome) as a function of the estimated fetal age in weeks after conception. Colors indicate the different fetuses. (B) Number of somatic base substitutions that accumulated per week during life in SCs of adult and fetal liver and intestine. Each SC is represented by a data point. (C) Genomic location of somatic base substitutions for adults and fetuses per indicated cell type. CDS, coding sequence.
The proportion of base substitutions in intergenic, genic, and protein-coding sequences is found to be similar for SCs from both fetal tissues and comparable to the proportions found in adult SCs (Fig. 1C). Most of the identified base substitutions are located outside genes. In fetal SCs, five nonsynonymous and one nonsense mutations were detected in protein-coding sequences of six different genes (Fig. 1C and table S3). None of these genes are involved in cell proliferation or have been causally implicated in cancer according to the COSMIC cancer gene census (13), suggesting that there was no positive selection for cells with functional somatic mutations that confer a selective advantage.
Mutational patterns of fetal somatic mutations are tissue specific
The mutation spectrum of the fetal intestinal SCs was characterized by a large fraction of C to T changes, particularly at CpG sites and thus likely resulting from deamination of methylated cytosines (Fig. 2A). The fetal intestinal SC mutation spectrum closely resembled that of adult intestinal SCs, with a cosine similarity of 0.94 between the 96-type mutational profiles (Fig. 2B). This similarity suggests that the balance in activity of DNA damage and repair processes is similar in adult and fetal intestinal SCs. In contrast, the spectrum of the fetal liver SCs was different between individuals of different ages (P < 0.001, Pearson’s χ2 test; Fig. 2A) largely caused by increasing number of C to A changes with age. The fetal liver spectrum was also distinct from that of adult liver SCs (P < 2.2 × 10−16, Pearson’s χ2 test; Fig. 2, A and B), characterized by fewer T to G changes and more C to A in the fetal than in the adult liver SCs. Notably, the spectrum of the fetal liver was also significantly different from that of the fetal intestine (P < 1.2 × 10−12, Pearson’s χ2 test; Fig. 2, A and B), with more C to A changes in the liver and more C to T changes at methylated cytosines in the intestine. These results demonstrate that the liver and the intestine accumulate different types of mutations during fetal development.
Fig. 2. The fetal liver and fetal intestine have distinct mutational patterns.
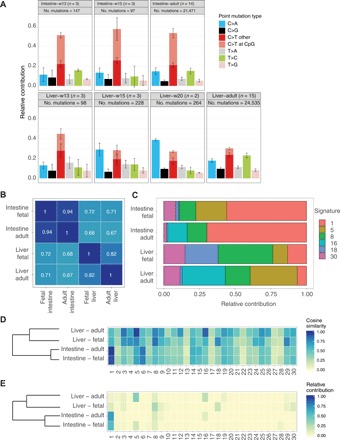
(A) Mutation spectra for all tissues and ages. Error bars represent SDs. The total number of identified somatic base substitutions per spectrum is indicated. (B) Cosine similarities between the average 96-type mutational profiles of liver and intestinal SCs from fetal and adult origin. (C) Relative contribution of the COSMIC signatures to the different SC types that have been analyzed in the current study. (D) Cosine similarity heat map between the COSMIC signatures and the mutational profiles of the adult and fetal SCs. Samples are grouped by unsupervised hierarchical clustering. (E) Relative contribution heat map of the COSMIC signatures to the mutational profiles of the adult and fetal SCs. Samples are grouped by unsupervised hierarchical clustering.
Fetal mutational signature analysis
We reconstructed the mutational profiles of the adult and fetal SCs with the pan-cancer–derived COSMIC signatures (14, 15). The cosine similarities of the reconstructed profiles with the fetal SC profiles were 0.95 to 0.99 (fig. S2), from which we conclude that the mutational spectrum can be largely explained by the known COSMIC signatures. However, to avoid biases originating from the existing COSMIC signatures, we also performed a de novo extraction of mutational signatures using nonnegative matrix factorization, using the mutational landscapes of all adult and fetal samples as input (14). We identified three signatures, of which signature A is highly similar to COSMIC signatures 5 and 16, signature B resembled COSMIC signature 1, and signature C showed resemblance to COSMIC signatures 8 and 18 (fig. S3).
The mutation profile of fetal as well as adult intestinal SCs is most similar to COSMIC mutational signature 1 (Fig. 2, C to E). In contrast, there was only a minimal contribution of signature 1 to the mutational profile of the fetal liver (Fig. 2, C to E). Instead, the fetal liver mutation profiles show resemblance to COSMIC signatures 8 and 18, both of which are characterized by C to A changes, while adult liver SCs are highly similar to COSMIC signatures 5 and 16, the etiology of which is unknown (Fig. 2, C to E). The etiology of signatures 8 and 18 is also unknown but has been linked to oxidative stress–related mechanisms (16). These findings demonstrate that in fetal liver SCs, other mutational mechanisms are dominant than in adult liver SCs.
Because the fetal intestine and liver do not yet fulfill their adult tissue functions, the observed divergence between these tissues in mutation accumulation is probably not caused by differences in exposure to exogenous mutagens but is more likely the result of cell- or tissue-intrinsic processes that are regulated differently between the developing organs. Principal components analysis of RNA sequencing data revealed similar overall transcriptome profiles for fetal and adult intestinal SCs, whereas the fetal liver SCs are distinct from the intestinal SCs and the adult liver SCs (Fig. 3A). Unsupervised hierarchical clustering of the top 100 differentially expressed genes yielded similar results (Fig. 3B).
Fig. 3. RNA expression analysis.
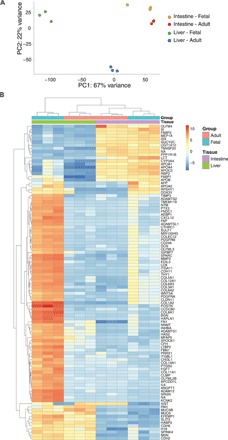
(A) Principal components analysis of the gene expression profiles. The individual samples are projected onto the first two principal components. PC1, principal component 1; PC2, principal component 2. (B) Gene expression heat map of the top 100 differentially expressed genes between adult and fetal liver and intestinal SCs. Genes are ordered by nonsupervised hierarchical clustering.
To gain more insight into the putative mechanisms leading to fetal tissue–specific mutation accumulation, we focused on the dominant observed substitution types, i.e., C to A changes in the fetal liver and C to T changes at CpG sites in the fetal intestine. C to A transversions can result from DNA damage by oxidation, resulting in 8-oxoguanine pairing aberrantly with adenine (17). MUTYH and OGG1 are the major glycosylases that identify oxidized bases and initiate base excision repair (BER) (18). Inactivating mutations in these genes lead to more C to A substitutions (19, 20). The expression of MUTYH and OGG1 is lower in the fetal liver than in the fetal intestine (fig. S4), which could result in increased C to A transversions in the fetal liver.
The predominant base changes in fetal and adult intestinal SCs (C to T changes at CpG sites) are also frequent in early embryogenesis (8, 21). The underlying cause, spontaneous deamination of methylated cytosines, therefore appears to be a dominant mutagenic factor from fertilization until adulthood in intestinal SCs and their progenitors. Spontaneous deamination of methylated cytosines at CpG dinucleotides results in T:G mismatches (5). These mismatches can be effectively repaired by BER (22) and potentially noncanonical mismatch repair (23), but a mutation becomes fixed if the cell divides before the mismatch is resolved, resulting in a C to T change (7). Methyl-CpG binding domain 4 (MBD4) and thymine DNA glycosylase (TDG) are the two major glycosylases that specifically catalyze the removal of thymines opposite of guanines at CpG sites (22, 24). Both glycosylases are expressed at higher levels in SCs of the fetal liver than in SCs of the fetal intestine (fig. S4). This may lead to tissue-specific rates of thymine excision at the T:G mismatches by BER, resulting in differences in C to T substitution rates at CpG sites. These results suggest that tissue-specific mutation accumulation in fetal development may be the result of cell-intrinsic processes such as differential expression of BER glycosylases, although the distinction between cause and consequences remains difficult and would require induction and measurement of DNA damage and the activity of DNA repair in fetal tissues.
DISCUSSION
We have previously demonstrated that adult SCs of different organs show tissue-specific mutation accumulation (6). Here, we extend on these findings by demonstrating divergence between liver and intestinal SCs in patterns of mutation accumulation at the second trimester of fetal development. Moreover, we show that mutation accumulation rates in fetal SCs are higher than in adult SCs of the same organ. The observed mutation accumulation rates in SCs of the fetal liver and intestine are well below the ~36 base substitutions per week that have been described for neural progenitors of similarly aged human fetuses (25). Thus, in contrast to what has recently been hypothesized (25), mutation rates in prenatal development appear not to be equal between tissues, although the effects of technical differences in measurement methods cannot be excluded. Nevertheless, elevated mutation accumulation rates appear to be general for fetal cells of different tissues, suggesting that the rapid cellular expansion in development comes at the cost of increased mutation accumulation. Unlike neural progenitors, SCs of the liver and the intestine fulfill important roles throughout adult life in tissue self-renewal and regeneration upon damage, thereby running a lifelong risk that somatic mutations are propagated to daughter SCs and contribute to tumor development. It may therefore be expected that these cells maintain higher levels of genomic integrity in early development.
The mutation spectrum of the fetal liver is characterized by many C to A changes. We have previously described that in vitro culture also leads to more C to A substitutions (11). To discriminate somatic mutations from mutations that arise during the culture, the variant allele frequency filter (VAF) was set at ≥0.3. Nevertheless, in vitro–acquired mutations may escape the VAF filter and cause the high number of C to A changes in the fetal liver (Fig. 2A). To escape the VAF filter, in vitro–induced mutations would have to arise at the clonal step at the onset of the culture (fig. S1). Although we cannot formally exclude this possibility, we consider this unlikely, because we do not observe a high contribution of C to A changes in the fetal intestine and the week 15 and 17 liver clones. Therefore, the relative high contribution of C to A mutations probably has an in vivo origin, as is the case for the adult liver (6). The mutation spectrum of fetal intestinal SCs is dominated by C to T mutations at CpG sites, most likely as a result of spontaneous deamination of methylated cytosines. This process does not have a strong contribution in fetal liver SCs. Thus, irrespective of the number of C to A substitutions, the low (absolute) contribution of C to T mutations at CpG sites in the liver also supports our conclusion that patterns of mutation accumulation diverge early between tissues.
Although liver cells divide rapidly during development (26), we observed little contribution of signature 1 to the fetal liver, which conflicts with the suggestion that cell types with a high cell division rate exhibit more signature 1 mutations and that this signature could serve as a clock that registers the number of mitoses that a cell has experienced since fertilization (7). Our results indicate that a high cell turnover rate is, at least in fetal development, not necessarily associated with a pronounced contribution of signature 1–like mutations.
In conclusion, our results show that fetal growth comes at the cost of elevated mutation rates. Furthermore, distinct mutational mechanisms shape the mutational landscapes of the fetal liver and the fetal intestine. Spontaneous deamination of methylated cytosines is the main driver of mutation accumulation in fetal intestinal SCs, while oxidative mechanisms appear to be important for mutation accumulation in fetal liver SCs.
MATERIALS AND METHODS
Collection of human material
The Medical Ethical Committee of the Leiden University Medical Center approved this study (P08.087). Informed consent was obtained on the basis of the Declaration of Helsinki (World Medical Association). Human fetal tissues (intestine, liver, and skin) at gestational age weeks 15 to 22 (weeks 13 to 20 after conception) were collected from elective abortion material (vacuum aspiration) without medical indication. In this study, weeks of gestation, which is equivalent to the last menstrual period, was determined by ultrasonography. The age in weeks after conception was determined by subtracting 2 weeks from the weeks of gestation. After collection, the material was washed with 0.9% NaCl (Fresenius Kabi, France) and stored on ice until further processing.
Fetal SC isolation and clonal organoid culture
Organoid cultures from liver and intestinal tissue material were derived as previously described (12, 27). In short, liver biopsies were minced and subsequently dissociated into single-cell solutions using human liver digestion solution [Earle's balanced salt solution supplemented with collagenase type 1A (1 mg/ml) and deoxyribonuclease I (0.1 mg/ml)]. Cells were plated at limiting dilution, and organoid cultures were initiated by culturing these cells in basement membrane extract (BME) overlaid with human liver isolation medium [60% advanced Dulbecco’s modified Eagle’s medium (DMEM)/F-12 (supplemented with 1% penicillin/streptomycin, 1% GlutaMAX, and 10 mM Hepes), 30% WNT3A conditioned medium (produced in house), 10% R-spondin 1 conditioned medium (produced in house), 1× B27 supplement without retinoic acid, 1× N2 supplement, 1× Primocin, 1:1000 hES Cell Cloning & Recovery Supplement, 10 mM nicotinamide, 1.25 mM N-acetylcysteine, 5 μM A83-01, 10 μM forskolin, 10 μM Rho kinase inhibitor, 10 nM gastrin I, Noggin (100 ng/ml), FGF10 (100 ng/ml), human EGF (50 ng/ml), and HGF (25 ng/ml)]. For the intestine, villi were removed from intestinal biopsies by applying mechanical strength. Subsequently, the biopsies were minced and dissociated into single-cell solutions by incubating these tissue pieces with TrypLE at 37°C for 10 to 20 min. Cells were plated at limiting dilution, and organoid cultures were initiated by culturing these cells in Matrigel overlaid with human intestinal isolation medium [30% advanced DMEM/F-12 (supplemented with 1% penicillin/streptomycin, 1% GlutaMAX, and 10 mM Hepes), 50% WNT3A conditioned medium (produced in house), 20% RSPOI conditioned medium (produced in house), 1× B27 supplement, 1× Primocin, 1:1000 hES Cell Cloning & Recovery Supplement,10 μM SB 202190, 10 mM nicotinamide, 1.25 mM N-acetylcysteine, 0.5 μM A83-01, 10 μM Rho kinase inhibitor, Noggin (100 ng/ml), and hEGF (50 ng/ml)]. After 2 to 3 days, organoids started to appear, and the medium was changed to human liver expansion medium and human intestinal expansion medium, respectively (recipe as described above, without Rho kinase inhibitor and hES Cell Cloning & Recovery Supplement). Clonal cultures were derived by picking single organoids and expanding them until there was sufficient material to perform WGS.
WGS and variant identification
DNA was isolated from the organoid cultures and skin and liver tissue using the QIAsymphony DNA Mini Kit (Qiagen). DNA libraries for Illumina sequencing were generated from 200 ng of genomic DNA using standard protocols (Illumina) and sequenced 2 × 100 base pair (bp) paired-end to 30× base coverage with the Illumina HiSeq Xten at the Hartwig Medical Foundation. WGS data were processed, and copy number analyses and base substitutions were called using GATK (28) and filtered as described previously (10). Skin was used as a control in the filtering pipeline. For one fetus (week 13), not enough DNA was retrieved from the skin and a piece of bulk liver tissue was used as reference instead. Because this tissue contains cells from multiple germ layers (i.e., mesoderm-derived endothelial cells, nucleated red blood cells, and mesenchyme, and endoderm-derived hepatoblasts and biliary epithelium), only very early mutational events will be shared by all these different cell types. The mutation catalogs will not contain these very early mutational events, and therefore, the actual fetal SC somatic mutation loads could be slightly higher for this particular fetus.
In addition to the filtering described in (10), we filtered on genotype quality (GQ): GQ ≥ 99 in the clonal organoid culture and GQ ≥ 10 in the polyclonal control sample, as these positions commonly represent false positive somatic variants. To generate a set of high-confidence variants, we visually inspected all base substitutions using the Integrative Genomics Viewer (IGV) (29) and excluded 44% of all base substitutions. Base substitutions in repetitive sequences (e.g., poly-G stretches), nearby an indel (within ~20 bp of base substitution), in regions with a very high coverage (as these are most likely mismapped reads), which were found in most of the other clones (as these are most likely missed germline positions), and within a deletion or duplication were flagged as false positives. For adult SCs, we used data from previously established human liver and intestinal SC lines (6, 11).
For 569 of the variants that were identified in fetal SCs, 1138 custom capture probes of 120 bp were designed (one for the variant and one for the reference allele) and ordered. DNA sequencing libraries were prepared according to the manufacturer’s (Twist Bioscience) protocol. In short, DNA was enzymatically fragmented followed by end repair and dAMP tailing. Subsequently, index adapters were ligated and a pre-capture polymerase chain reaction (PCR) amplification was performed. Libraries were pooled, and a bead-based size selection was performed. The size-selected libraries were hybridized to the capture probes. After binding to streptavidin beads, the enriched libraries were PCR-amplified, purified, and sequenced 2 × 150 bp on the NextSeq500 platform. All samples were thus analyzed for all target loci, thereby functioning as each other’s controls. Variants were called using GATK (28) and considered positive if unique to the sample in which the sample was originally called. All other variants were manually inspected in IGV. This validation resulted in the confirmation of 98% of all variants (555 after initial automated filtering, 1 after manual inspection in IGV; table S2). For the 13 remaining variants, the results were inconclusive, because the custom capture failed at these positions.
Base substitution load and type
True-positive base substitutions were extracted from the variant call format (VCF) files. The base substitution load was computed by extrapolating the number of mutations to the autosomal genome, as described previously (10). To allow comparison between the accumulation during fetal development and adult life, we downloaded VCF files and surveyed bed files of healthy liver and small intestine adult SCs from https://wgs11.op.umcutrecht.nl/mutational_patterns_ASCs/. In addition, mutation catalogs of five clonal liver organoid cultures derived from four different healthy liver donors were included in this study (data available upon request).
Using the Linear and Nonlinear Mixed Effects Models R package version 3.1-137, the slope and 95% confidence intervals for the mutation rates of the adult liver and small intestine were determined. Next, we tested whether the mutation rates per week after conception fell within the 95% confidence interval.
Mutational pattern analysis was performed as described previously, using the MutationalPatterns R package (15). Seven-type mutation spectra were extracted from the VCF files. Ninety-six–type mutational profiles were subsequently generated, and the average profile (centroid) was determined for the assessed SC types. Centroids were reconstructed with the 30 COSMIC mutational signatures. Subsequently, signatures that contributed at least 5% to one or more centroids were selected, and the centroids were reconstructed again using this subset of the mutational signatures. Cosine similarities between samples and signatures and relative contributions of signatures were calculated using MutationalPatterns (15).
Gene definitions for hg19 were retrieved from the University of California, Santa Cruz (UCSC) Genome Browser (30) as a TxDb annotation package from Bioconductor. Functional consequences of coding mutations were identified using the VariantAnnotation R package (31).
RNA sequencing
Early passage bulk organoid cultures were lysed with TRIzol (Life Technologies), and RNA was isolated using the QIAsymphony RNA Kit (Qiagen). For the liver, two fetal lines were from week 20 fetuses (after conception) and were also used to study mutation accumulation. One liver SC line was from a week 14 fetus. Because subsequent DNA sequencing revealed a trisomy 21, this sample was excluded from the mutation accumulation study. This sample was included in the RNA sequencing analysis, because it clusters well with the two other lines and the examined genes are on unaffected chromosomes. For the intestine, RNA was sequenced from one line of the week 13 fetus and two lines of the week 15 fetus that were derived separately from distal origins. mRNA was isolated from 50 ng of total RNA, and libraries were subsequently generated using the Illumina NeoPrep TruSeq Stranded mRNA Library Prep Kit (Illumina, NP-202-1001). mRNA libraries were sequenced 1 × 75 bp on the NextSeq500 platform.
RNA sequencing reads were mapped with STAR v.2.4.2a to the human genome assembly GRCh37. The BAM files were sorted with Sambamba v0.5.8, and reads were counted with HTSeq-count version 0.6.1p1 (default settings) to exons. DESeq2 v_1.16.1 was used to normalize counts and for rlog transformation, followed by nonsupervised hierarchical clustering of the top 100 differentially expressed genes (32). Principal components analysis was performed on normalized and rlog-transformed counts. Gene ontology enrichment analysis was performed using the clusterProfiler package version 3.8.1 (33).
Supplementary Material
Acknowledgments
We thank the Centre for Contraception, Abortion and Sexuality (CASA) in Leiden and the Hague for their efforts to obtain the human fetal material and the USEQ for their support with next-generation sequencing experiments. We would also like to thank L.J.W. van der Laan of the Erasmus MC for providing adult liver samples and S. Middendorp for providing adult intestinal samples, for which previously published data have been used in the current study. Funding: This work was financially supported by the NWO Gravitation Program Cancer Genomics.nl, NWO/ZonMW (Zenith project 93512003), and NWO VICI grant (865.13.004) to E.C. Author contributions: E.K., M.J., and N.B. performed wet-lab experiments. E.K., F.B., M.J., and S.B. performed bioinformatic analyses. S.M.C.d.S.L. obtained and isolated fetal tissues. E.K., F.B., M.J., R.v.B., and E.C. were involved in the conceptual design of the study. E.K., F.B., M.J., and E.C. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The RNA and DNA sequencing data have been deposited at the European Genome-Phenome Archive (EGA) under accession numbers EGAS00001002886, EGAS00001001682, and EGAS00001002983. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw1271/DC1
Fig. S1. Experimental setup.
Fig. S2. Reconstruction of mutational profiles using known mutational signatures.
Fig. S3. De novo identification of mutational signatures.
Fig. S4. Gene expression of important DNA repair components.
Table S1. Summary of all samples and the number of mutations identified per sample.
Table S2. Results for the validation of 569 base substitutions by capture using custom-designed probes, followed by resequencing.
Table S3. All identified base pair substitutions that affect protein-coding regions of the genome.
REFERENCES AND NOTES
- 1.Frank S. A., Somatic evolutionary genomics: Mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 107 ( suppl. 1), 1725–1730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank S. A., Nowak M. A., Cell biology: Developmental predisposition to cancer. Nature 422, 494 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Lupski J. R., Genetics. Genome mosaicism—One human, multiple genomes. Science 341, 358–359 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Fernández L. C., Torres M., Real F. X., Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 16, 43–55 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Aparicio S. A. J. R., Behjati S., Biankin A. V., Bignell G. R., Bolli N., Borg A., Børresen-Dale A.-L., Boyault S., Burkhardt B., Butler A. P., Caldas C., Davies H. R., Desmedt C., Eils R., Eyfjörd J. E., Foekens J. A., Greaves M., Hosoda F., Hutter B., Ilicic T., Imbeaud S., Imielinski M., Jäger N., Jones D. T. W., Jones D., Knappskog S., Kool M., Lakhani S. R., López-Otín C., Martin S., Munshi N. C., Nakamura H., Northcott P. A., Pajic M., Papaemmanuil E., Paradiso A., Pearson J. V., Puente X. S., Raine K., Ramakrishna M., Richardson A. L., Richter J., Rosenstiel P., Schlesner M., Schumacher T. N., Span P. N., Teague J. W., Totoki Y., Tutt A. N. J., Valdés-Mas R., van Buuren M. M., van ‘t veer L., Vincent-Salomon A., Waddell N., Yates L. R.; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain, Zucman-Rossi J., Futreal P. A., McDermott U., Lichter P., Meyerson M., Grimmond S. M., Siebert R., Campo E., Shibata T., Pfister S. M., Campbell P. J., Stratton M. R., Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blokzijl F., de Ligt J., Jager M., Sasselli V., Roerink S., Sasaki N., Huch M., Boymans S., Kuijk E., Prins P., Nijman I. J., Martincorena I., Mokry M., Wiegerinck C. L., Middendorp S., Sato T., Schwank G., Nieuwenhuis E. E. S., Verstegen M. M. A., van der Laan L. J. W., de Jonge J., IJzermans J. N. M., Vries R. G., van de Wetering M., Stratton M. R., Clevers H., Cuppen E., van Boxtel R., Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexandrov L. B., Jones P. H., Wedge D. C., Sale J. E., Campbell P. J., Nik-Zainal S., Stratton M. R., Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ju Y. S., Martincorena I., Gerstung M., Petljak M., Alexandrov L. B., Rahbari R., Wedge D. C., Davies H. R., Ramakrishna M., Fullam A., Martin S., Alder C., Patel N., Gamble S., O’Meara S., Giri D. D., Sauer T., Pinder S. E., Purdie C. A., Borg Å., Stunnenberg H., van de Vijver M., Tan B. K. T., Caldas C., Tutt A., Ueno N. T., van ’t Veer L. J., Martens J. W. M., Sotiriou C., Knappskog S., Span P. N., Lakhani S. R., Eyfjörd J. E., Børresen-Dale A.-L., Richardson A., Thompson A. M., Viari A., Hurles M. E., Nik-Zainal S., Campbell P. J., Stratton M. R., Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543, 714–718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gawad C., Koh W., Quake S. R., Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 17, 175–188 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Jager M., Blokzijl F., Sasselli V., Boymans S., Janssen R., Besselink N., Clevers H., van Boxtel R., Cuppen E., Measuring mutation accumulation in single human adult stem cells by whole-genome sequencing of organoid cultures. Nat. Protoc. 13, 59–78 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Huch M., Gehart H., van Boxtel R., Hamer K., Blokzijl F., Verstegen M. M. A., Ellis E., van Wenum M., Fuchs S. A., de Ligt J., van de Wetering M., Sasaki N., Boers S. J., Kemperman H., de Jonge J., Ijzermans J. N. M., Nieuwenhuis E. E. S., Hoekstra R., Strom S., Vries R. R. G., van der Laan L. J. W., Cuppen E., Clevers H., Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato T., Stange D. E., Ferrante M., Vries R. G. J., van Es J. H., van den Brink S., van Houdt W. J., Pronk A., van Gorp J., Siersema P. D., Clevers H., Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Forbes S. A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C. G., Ward S., Dawson E., Ponting L., Stefancsik R., Harsha B., Kok C. Y., Jia M., Jubb H., Sondka Z., Thompson S., de T., Campbell P. J., COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Campbell P. J., Stratton M. R., Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 3, 246–259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blokzijl F., Janssen R., van Boxtel R., Cuppen E., MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 10, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viel A., Bruselles A., Meccia E., Fornasarig M., Quaia M., Canzonieri V., Policicchio E., Urso E. D., Agostini M., Genuardi M., Lucci-Cordisco E., Venesio T., Martayan A., Diodoro M. G., Sanchez-Mete L., Stigliano V., Mazzei F., Grasso F., Giuliani A., Baiocchi M., Maestro R., Giannini G., Tartaglia M., Alexandrov L. B., Bignami M., A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine 20, 39–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacolla A., Cooper D. N., Vasquez K. M., Mechanisms of base substitution mutagenesis in cancer genomes. Genes 5, 108–146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ide H., Kotera M., Human DNA glycosylases involved in the repair of oxidatively damaged DNA. Biol. Pharm. Bull. 27, 480–485 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Al-Tassan N., Chmiel N. H., Maynard J., Fleming N., Livingston A. L., Williams G. T., Hodges A. K., Davies D. R., David S. S., Sampson J. R., Cheadle J. P., Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Minowa O., Arai T., Hirano M., Monden Y., Nakai S., Fukuda M., Itoh M., Takano H., Hippou Y., Aburatani H., Masumura K.-i., Nohmi T., Nishimura S., Noda T., Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 4156–4161 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang A. Y., Yang X., Wang S., Zheng X., Wu Q., Ye A. Y., Wei L., Distinctive types of postzygotic single-nucleotide mosaicisms in healthy individuals revealed by genome-wide profiling of multiple organs. PLOS Genet. 14, e1007395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellacosa A., Drohat A. C., Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 32, 33–42 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J., Miller B. F., Furano A. V., Repair of naturally occurring mismatches can induce mutations in flanking DNA. eLife 3, e02001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sjolund A. B., Senejani A. G., Sweasy J. B., MBD4 and TDG: Multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 743-744, 12–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae T., Tomasini L., Mariani J., Zhou B., Roychowdhury T., Franjic D., Pletikos M., Pattni R., Chen B.-J., Venturini E., Riley-Gillis B., Sestan N., Urban A. E., Abyzov A., Vaccarino F. M., Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 359, 550–555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Archie J. G., Collins J. S., Lebel R. R., Quantitative standards for fetal and neonatal autopsy. Am. J. Clin. Pathol. 126, 256–265 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Broutier L., Andersson-Rolf A., Hindley C. J., Boj S. F., Clevers H., Koo B.-K., Huch M., Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 11, 1724–1743 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Van der Auwera G. A., Carneiro M. O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., Banks E., Garimella K. V., Altshuler D., Gabriel S., DePristo M. A., From FastQ data to high-confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorvaldsdottir H., Robinson J. T., Mesirov J. P., Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obenchain V., Lawrence M., Carey V., Gogarten S., Shannon P., Morgan M., VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu G., Wang L.-G., Han Y., He Q.-Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw1271/DC1
Fig. S1. Experimental setup.
Fig. S2. Reconstruction of mutational profiles using known mutational signatures.
Fig. S3. De novo identification of mutational signatures.
Fig. S4. Gene expression of important DNA repair components.
Table S1. Summary of all samples and the number of mutations identified per sample.
Table S2. Results for the validation of 569 base substitutions by capture using custom-designed probes, followed by resequencing.
Table S3. All identified base pair substitutions that affect protein-coding regions of the genome.