

Summary
Fungi represent a significant proportion of the gut microbiota. Aberrant immune responses to fungi are frequently observed in inflammatory bowel diseases (IBD) and colorectal cancer (CRC), and mutations in the fungal sensing pathways are associated with the pathogenesis of IBD. Fungal recognition receptors trigger downstream signaling via the common adaptor protein CARD9 and the kinase SYK. Here we found that commensal gut fungi promoted inflammasome activation during AOM-DSS induced colitis. Myeloid cell -specific deletion of Card9 or Syk reduced inflammasome activation and interleukin (IL)-18 maturation, and increased susceptibility to colitis and CRC. IL-18 promoted epithelial barrier restitution and interferon-γ production by intestinal CD8+ T cells. Supplementation of IL-18 or transfer of wild-type myeloid cells reduced tumor burden in AOM-DSS treated Card9−/− and Sykfl/flLysMCre/+ mice, whereas treatment with anti-fungal agents exacerbated colitis and CRC. CARD9 deletion changes the gut microbial landscape, suggesting that SYK-CARD9 signaling maintains a microbial ecology that promotes inflammasome activation and thereby restrains colitis and colon tumorigenesis.
Keywords: Inflammatory Bowel Diseases, colorectal cancer, microbiota, CARD9, SYK
Graphical Abstract
ETOC
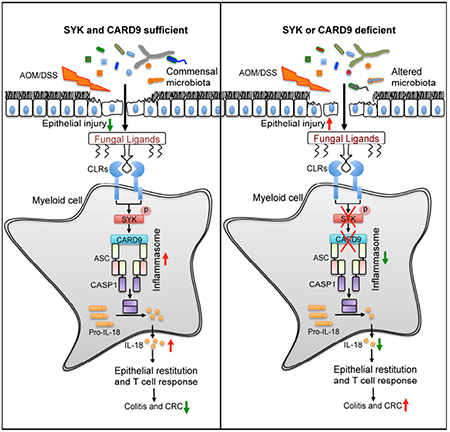
Fungi represent a significant proportion of the gut microbiota, but how anti-fungal immunity contributes to tumor-promoting inflammatory responses is unclear. Malik et al. find that recognition of commensal gut fungi, sensed via the Card9-Syk signaling axis, is protective in the context of inflammation-associated cancer. IL-18 maturation downstream from inflammasome activation promotes epithelial barrier restitution and IFN-γ production by intestinal CD8+ T cells.
INTRODUCTION
Crohn’s disease (CD) and Ulcerative Colitis (UC) are common Inflammatory Bowel Diseases (IBD) that together affect over 3 million individuals in the United States of America, constituting a major health crisis (Dahlhamer, 2016). IBD is characterized by chronic inflammation in the intestine with episodic abdominal pain, diarrhea, rectal bleeding and body weight loss (Kappelman et al., 2013). Mucus and epithelium form a physical barrier between the commensal microbes and the underlying host immune system, and deterioration of this barrier is prominent in IBD patients (Podolsky and Isselbacher, 1984; Rhodes, 1996). IBD also increases the risk of developing colorectal cancer (CRC) (Grivennikov et al., 2010). CRC is a leading cause of cancer-related deaths in adults with 160,000 cases being diagnosed annually in the USA, and inflammation is associated with approximately 90% of these cases (Howlader et al., 2015).
We and others have shown that gut bacteria modulates IBD and CRC pathogenesis (Elinav et al., 2011a; Malik et al., 2016; Man et al., 2015). Fungi are also a significant component of the gut microbiota and IBD patients develop increased titers of anti-fungal antibodies, suggesting a role for fungi in the pathogenesis of these diseases (McKenzie et al., 1990). Fungal diversity in stool samples from UC patients is significantly when compared with healthy controls (Sokol et al., 2017). In line with decreased fungal diversity during IBD in humans, fungal diversity also narrows during DSS-induced colitis in mice (Qiu et al., 2015). Further, deficiency in the fungal recognition C-Type Lectin Receptors (CLRs) Dectin-1, Dectin-3 or a common downstream signaling adaptor Caspase recruitment domain 9 (CARD9) are reported to increase severity of DSS-induced colitis in mice (Iliev et al., 2012; Lamas et al., 2016; Sokol et al., 2013). In contrast to these studies, CARD9 was shown to promote colitis-associated cancer by regulating IL-22 production from group 3 innate lymphoid cells (Bergmann et al., 2017a). Whereas anti-fungal treatment ameliorates colitis in Dectin-1-deficient mice, it exacerbates colitis in wild-type mice (Wheeler et al., 2016). Mutations in CARD9 are highly associated with increased susceptibility to both fungal infections and IBD in humans (Glocker et al., 2009; Zhernakova et al., 2008). These reports suggest that interaction of commensal gut fungi with the mucosal immune system can modulate IBD. Moreover, mycobial diversity during colon tumorigenesis in humans is also decreased (Luan et al., 2015). However, the role of gut fungi or associated immune sensing pathways in tumorigenesis has not been explored.
Inflammasomes are multimeric protein complexes that form upon sensing of a diverse range of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). The inflammasome forms a platform to activate the cysteine protease caspase-1, which triggers the proteolytic processing of cytokines, interleukin (IL)-1β and IL-18, and induces pyroptosis, a form of cell death (Sharma and Kanneganti, 2016). Inflammasome-forming receptors are cytosolic sensors that include nucleotide-binding oligomerization domain-like receptors (NLRs) and absent in melanoma (AIM2)-like receptors (ALRs). Mutations in genes that encode components of the inflammasome are frequently associated with auto-inflammatory diseases and cancer (Karki et al., 2017; Zhang et al., 2015). NLRP1 (Williams et al., 2015) NLRP3 (Allen et al., 2010; Zaki et al., 2010a) NLRC4 (Carvalho et al., 2012; Hu et al., 2010), Pyrin (Sharma et al., 2017a; Sharma et al., 2017b) NLRP6 (Chen et al., 2011; Elinav et al., 2011a; Elinav et al., 2011b) and IL-18 have been shown to modulate colitis and colon tumorigenesis (Zaki et al., 2010b). We and others have shown that fungi are potent inducers of the inflammasome (Karki et al., 2015; Lamkanfi et al., 2009). However, the relative contribution of the components of fungal sensing pathways including CARD9 and its upstream activator spleen tyrosine kinase (SYK) in modulating inflammasome activation and gut homeostasis has not been explored in detail.
Here we found that commensal gut fungi signal via the SYK-CARD9 pathway to restrain colitis and colorectal cancer. Commensal gut fungi contributed to inflammasome activation and IL-18 maturation during colitis. Early IL-18 maturation by inflammasome promoted epithelial barrier restitution and interferon gamma (IFN-γ) production from the CD8+ T cells. Mice lacking CARD9 and SYK were similarly defective in inflammasome activation and susceptible to colitis and colorectal cancer. Consequently, colitis and tumor-promoting inflammation in these mice was reduced by supplementation with IL-18 or by transfer of myeloid cells capable of inflammasome activation after fungal recognition. These findings reveal a role for commensal fungi and the associated signaling cascade in restraining colitis and colon cancer by promoting inflammasome activation.
RESULTS
CARD9 restrains colitis and colorectal cancer
Because mycobial diversity and mutations in CARD9 are associated IBD and colon tumorigenesis in humans (Glocker et al., 2009; Zhernakova et al., 2008), we sought to further explore the function of CARD9 in colitis and CRC. Expression of CARD9 was detectable in colon tissues from healthy humans and was increased in colon tumor biopsies in the GENT database (Shin et al., 2011) (Figure 1A). Expression of CARD9 mRNA and protein were also increased in tumor-bearing colons from mice subjected to axozymethane (AOM)–dextran sodium sulfate (DSS) model of colon cancer (Figures 1B–D) (Tanaka et al., 2003). Expression of Card9 was increased in immune cells (CD45+EpCAM−) during both acute colitis and tumorigenesis and was below detection limit in the epithelial cells (CD45−EpCAM+)(Figures S1A–B). In contrast, expression of Il18 was detectable in both immune cells and epithelial cells (Figures S1A–B). Immunofluorescence analysis showed increased staining for CARD9 at day 14, which was distinct from EpCAM+ cells (Figures S1C). Immunohistochemical staining of CARD9 in human colon biopsies cataloged in the human protein atlas (Uhlen et al.) also demonstrated positively stained cells to be restricted to the lamina propria (Figures S1D). Increased expression of CARD9 in human colon tumor tissue, compared with the normal control tissue, also correlated with the increased expression of the myeloid cell marker, ITGAM (CD11b) (Figures S1E). Therefore, the expression of CARD9 in the colon is increased in the immune cells during colon tumorigenesis.
Figure 1. CARD9 decreases susceptibility to colitis-associated cancer.
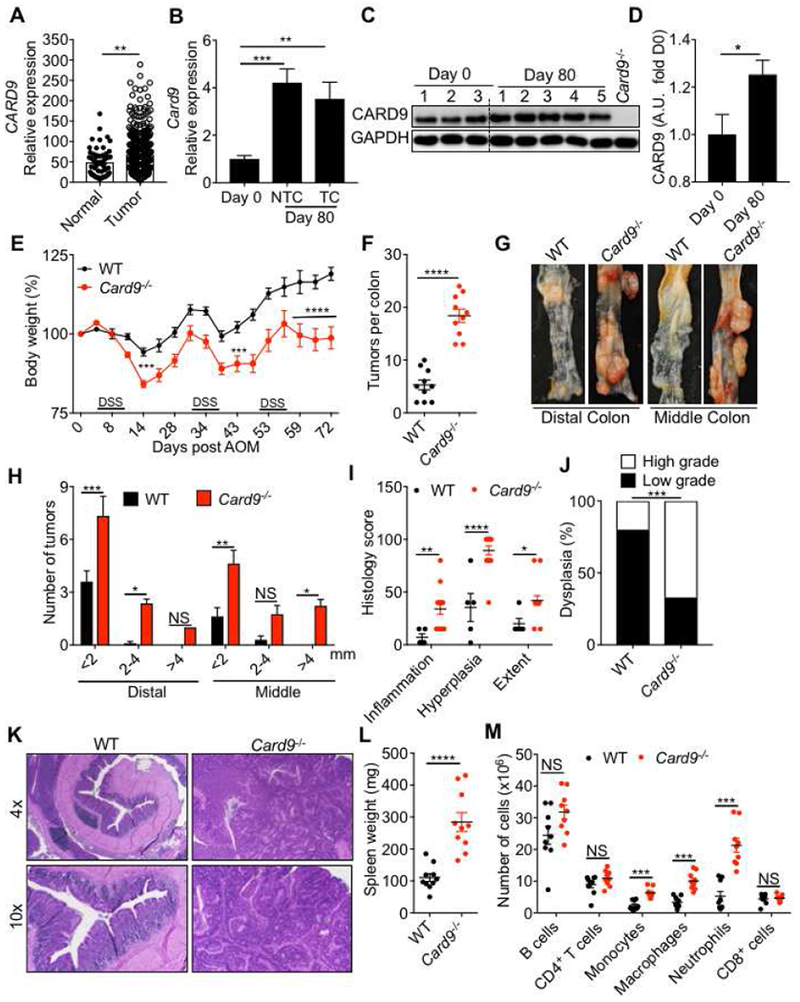
(A) CARD9 expression in normal human colon and tumor tissue from GENT database (B) Card9 expression in tumor and non-tumor colon tissue (NTC) relative to expression in the basal colon tissue (Day 0) of WT mice. (C) Immunoblot for CARD9 in colon tissue of WT mice at indicated times after AOM–DSS treatment and (D) the densitometric analysis for the CARD9 immunoblot. The last lane represents the lysate from the colon of a Card9−/− mouse on day 0. (E) Change in body weight in WT and Card9−/− mice injected with AOM on day 0 and administered with three rounds of 2% DSS in drinking water. (F) Number of tumors, (G) representative pictures of colons and (H) tumor distribution in the colons at day 80 post-AOM. (I) Colon histology scores and (J) proportion of mice with low- or high-grade dysplastic adenomas in the colons at day 80 post-AOM. (K) Representative H&E stained images at 4× and 10× magnifications of colon sections at day 80. (L) Spleen weight and (M) flow cytometric analysis of the indicated cellular populations in the spleens of WT and Card9−/− mice at day 80. Each symbol represents an individual mouse. NS, not significant; *P < 0.05; **P < 0.01, ***P < 0.001, ****P < 0.0001; Mann–Whitney U test (A, D, F and L), two-way ANOVA (E, H, I, M), one-way ANOVA followed by the Holm–Sidak post-hoc test (B) or Fisher’s exact test (J). Data are from one experiment representative of two independent experiments with n = 5-10 mice per group (A-M; mean and SEM [A, B, D, E, F, H, I, L and M]). See also Figure S1.
To evaluate the role of CARD9 in colitis and colon cancer, we subjected WT and Card9−/− mice that were separated upon weaning (3-4 weeks old) to AOM–DSS treatment (Figure S1F). Compared to WT mice, Card9−/− mice lost more body weight during DSS treatment (Figure 1E). At day 80, colons of Card9−/− mice exhibited increased number of tumors in the distal (bottom 1/3rd) and middle (central 1/3rd) region that were also bigger in size when compared to the WT mice (Figure 1F–H). Histological analysis revealed greater extent of inflammation and epithelial hyperplasia in Card9−/− colons (Figures 1I–K). Splenomegaly, an indicator of systemic inflammation, was also significantly enhanced in Card9−/− mice (Figure 1L). Whereas the numbers of B cells, CD4+ T and CD8+ T cells were similar between the spleens of WT and Card9−/− mice, the numbers of macrophages, neutrophils and monocytes was significantly increased (Figure 1M).
Colitis is a predisposing factor for tumorigenesis (Grivennikov et al., 2010). Similar to the observations during tumorigenesis, expression of both CARD9 mRNA and protein were increased in the colon tissue during acute colitis (day 14) (Figures 2A–C). Day 14 was considered a time point for acute colitis because on day 14 mice typically display significant body weight loss, colon shortening and histological manifestations of colitis, without the presence of macroscopic tumors (Figures 2D–G). To determine the role of CARD9 in acute colitis, WT and Card9−/− mice were injected with AOM and subjected to a single round of DSS in drinking water. Consistent with the previous data (Figure 1E), Card9−/− mice lost significantly more body weight when compared to WT mice (Figure 2D). At day 14, Card9−/− mice also had significantly shorter colons (Figures 2E and 2F), a characteristic of acute colitis (Gaudio et al., 1999). Histological analysis revealed significant increase in inflammation, ulceration and extent of lesions in the colons of the Card9−/− mice when compared to WT mice (Figure 2G and 2H). Increased colitis in Card9−/− mice was also associated with decreased IFN-γ and TNF production from CD4+ T and CD8+ T cells (Figure 2I). Thus, signaling via CARD9 limits inflammation and inflammation-associated cancer in the AOM-DSS model.
Figure 2. CARD9 decreases susceptibility to DSS-induced colitis.
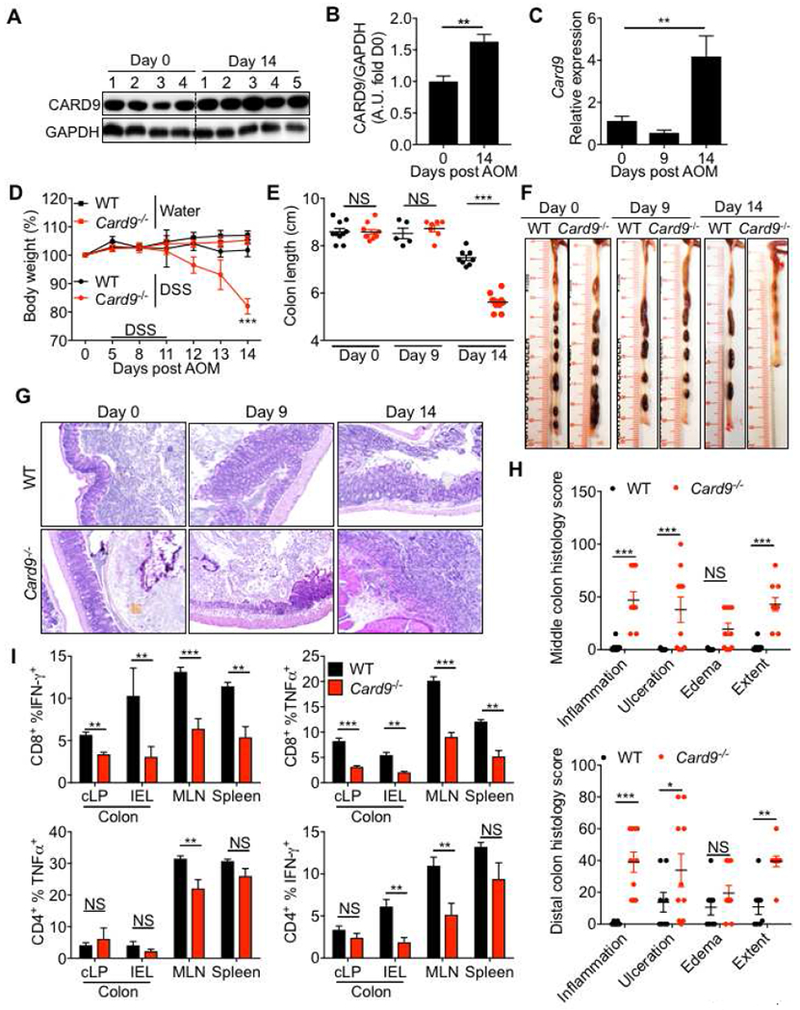
(A) Immunoblot for CARD9 in the colon and (B) densitometric analysis of the CARD9 immunoloblot. (C) RT-PCR analysis for Card9 expression in whole colon. (D) Percent body weight (E) colon length and (F) representative photographs of colons from WT and Card9−/− mice at the indicated days post-AOM treatment. (G) Representative pictures of H&E stained colon sections and (H) histological analysis of colons at day 14. (I) Flow-cytometric analysis for TNF-α and IFN-γ production from CD8+ and CD4+ T cells from colonic epithelial, lamina propria fractions and mesenteric lymph nodes and spleens of WT and Card9−/− mice. Each symbol represents an individual mouse. NS, not significant; *P < 0.05; **P < 0.01, ***P < 0.001; Mann–Whitney U test (B and I), two-way ANOVA followed by the Holm–Sidak post-hoc test (D, E and H) or one-way ANOVA followed by the Holm–Sidak post-hoc test (C). Data are from one experiment representative of two independent experiments with n = 5-8 mice per group (A-H; mean and SEM [B, C, D, E, H and I]).
CARD9 promotes inflammasome activation in the colon
To ascertain the immunological basis of increased colitis and colon cancer in Card9−/− mice, the immune response was assessed at days 9 and 14 after AOM injection. IL-18 secretion was significantly decreased in the Card9−/− colon explants, both on day 9 and day 14, compared to the WT explants (Figure 3A). While, there was no difference in the release of G-CSF, KC or IL-6 at day 9, these cytokines were increased in the colons and sera of Card9−/− mice at day 14 (Figure 3A). Concentration of MCP-1 was also increased in the sera on day 14 (Figure 3A). Expression of Il22 and its associated anti-microbial peptides (AMPs) Reg3g and Reg3b were also unperturbed at day 9 post-AOM. (Figure 3B). There were no significant differences in the expression of goblet cell differentiation factors Klf4 and Spdef or the major colon mucin Muc2 at basal or pre-clinical time between WT and Card9−/− colons (Figure S2A). Consistently, activation of ERK1, ERK2 and NF-kB were similar at day 9, but increased at day 14 in the Card9−/− colons, while phosphorylation of MAPKs p38 and JNK was comparable (Figure 3C and S2B). Activation of STAT3, a major driver of colon tumorigenesis (Grivennikov et al., 2009), was also increased in the colons of the Card9−/− mice (Figure 3C and S2B). Therefore, reduced IL-18 secretion precedes the enhanced inflammatory response observed in colons of Card9−/− mice.
Figure 3. CARD9 promotes inflammasome activation in the colon.
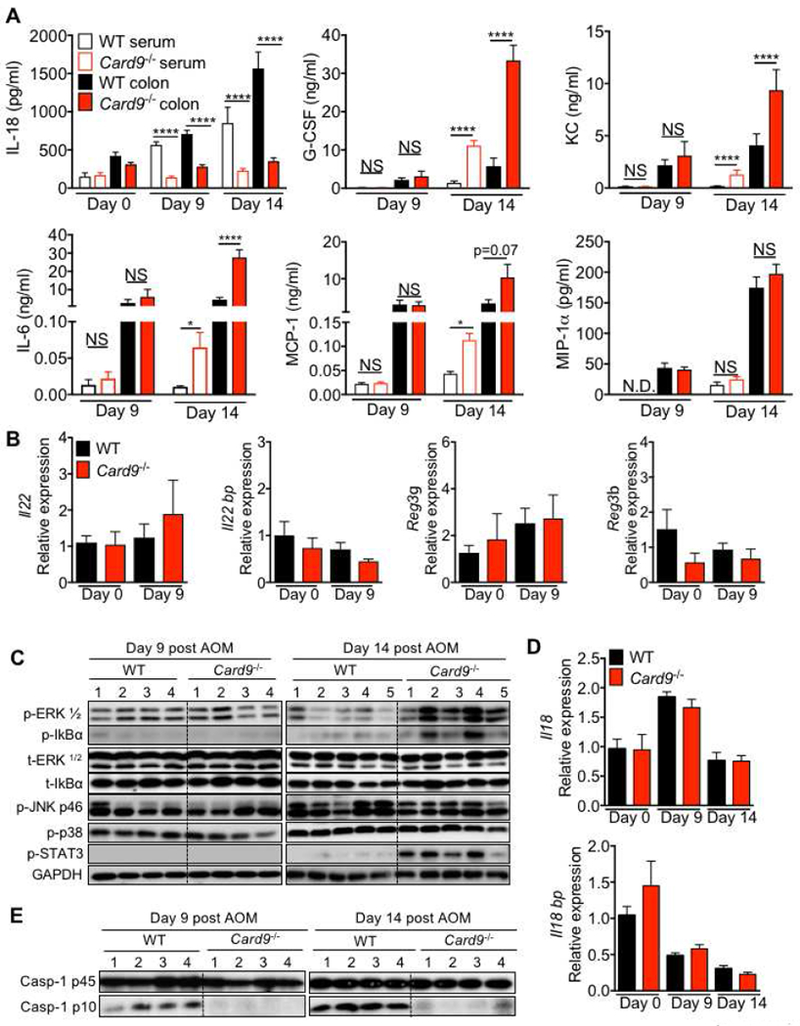
(A) ELISA in colon explants and sera of WT or Card9−/− mice at indicated days post-infection. (B and D) qPCR analysis for expression of indicated genes in colon tissue from WT and Card9−/− mice at indicated days after AOM treatment. (C and E) immunoblotting for indicated proteins in homogenates from colon tissue. NS, not significant; ****P < 0.0001; Two-way ANOVA followed by Holm-Sidak post-test (A). Data are from one experiment representative of two independent experiments with n =4-8 mice per group for each time point (A-E; mean and SEM [A, B and D]). See also Figure S2.
The decrease in IL-18 release was independent of transcriptional regulation because expression of Il18 and the decoy receptor Il18bp, were similar in colons of Card9−/− and WT mice (Figure 3D). The proportion of CD11b+ cells (Figure S2C), and the expression of Il18 in the CD11b+ cells (Figure S2D) of the colon, MLN and spleen were also similar between WT and Card9−/− mice. The decrease in IL-18 secretion was due to lower activation of caspase-1, as evidenced by reduced caspase-1 activation in the colon tissue of Card9−/− mice (Figure 3E and S2B) at both day 9 and day 14. These findings demonstrate that CARD9 is required to promote inflammasome activation and optimum IL-18 maturation in the colon during colitis and colon cancer.
IL-18 production by myeloid cells restricts colitis in Card9−/− mice
To directly test if pro-tumorigenic inflammation observed in Card9−/− mice during AOM-DSS treatment was due to the deficiency in IL-18 maturation, we exogenously supplemented IL-18 during AOM-DSS administration. IL-18 supplementation significantly ameliorated weight loss, disease parameters and colon shortening in Card9−/− mice (Figures 4A–D). IL-18 supplementation also protected the colon against histological perturbations of inflammation, ulceration, edema, epithelial hyperplasia and extent of lesions (Figures 4E–G). Consistent with a role for IL-18 in promoting IFN-γ production (Kohno et al., 1997), IL-18 supplementation significantly increased the frequencies of IFN-γ+ CD8+ and CD4+ T cells in the mesenteric lymph nodes and spleen (Figures 4H).
Figure 4. Supplementation with IL-18 or myeloid cells protects Card9−/− mice from colitis and epithelial hyperplasia.
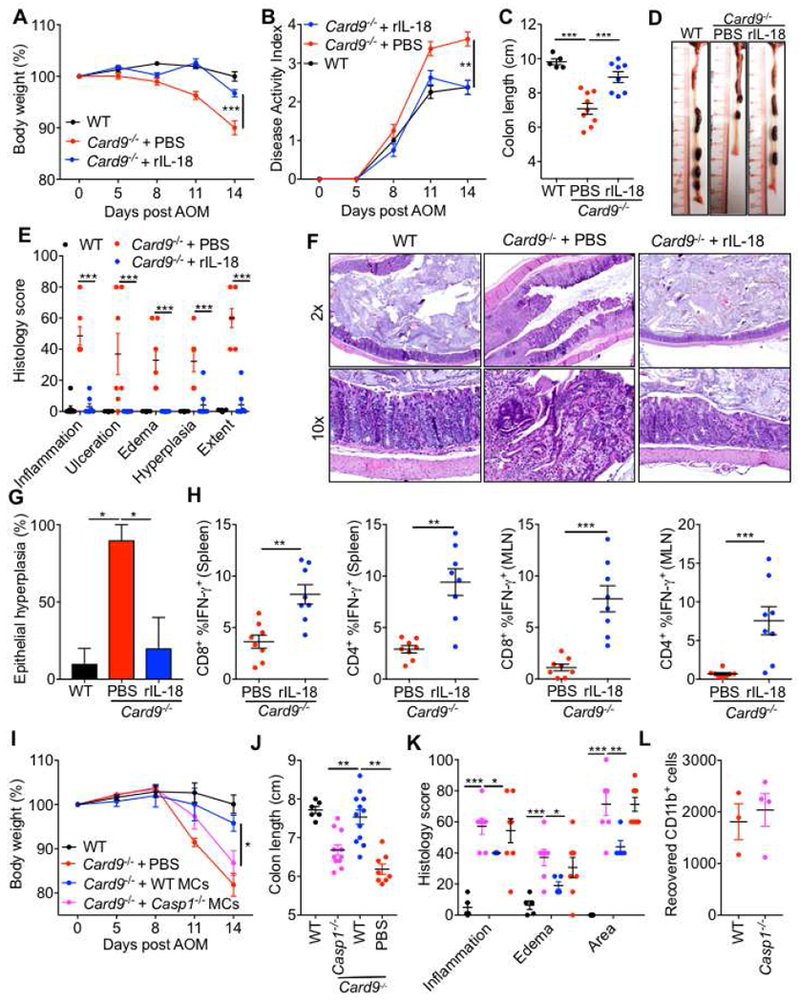
(A) Percent body weight (B) disease activity index (C) colon length and (D) representative colon photographs from WT and Card9−/− mice given PBS or IL-18 supplementation during AOM-DSS treatment. (E) Histological analysis of colons at day 14 and (F) representative pictures of H&E stained sections. (G) Percentage of mice with epithelial hyperplasia in colons at day 14. (H) Flow-cytometric analysis of TNF-α and IFN-γ production from CD8+ and CD4+ T cells from mesenteric lymph nodes and spleens of WT and Card9−/− mice at day 14. (I) Percent body weight, (J) colon length and (K) histological analysis of the colons from WT and Card9−/− mice at day 14 given WT or Casp1−/− myeloid cells (MCs). (L) Number of CD45.2+ CD11b+ cells recovered at day 14 from the colons of CD45.1 WT mice given WT or Casp1−/− MCs. Each symbol represents an individual mouse. NS, not significant; *P < 0.05; **P < 0.01, ***P < 0.001; Two-way ANOVA (A, B, E, I and K), one-way ANOVA followed by the Holm–Sidak post-hoc test (C, G and J) or Mann-Whitney U test (H). Data are from one experiment representative of two independent experiments with n = 5-10 mice per group (A-J) or n = 3-4 mice per group (L; mean and SEM [A, B, C, E, G, H, I, J, K and L]). See also Figures S3 and S4.
To directly test if CARD9 promotes inflammasome activation in myeloid cells to restrain colitis, we adoptively transferred WT or Casp1−/− bone marrow-derived myeloid cells (MCs) into Card9−/− mice during AOM-DSS administration. WT cells significantly ameliorated body weight loss and colon shortening in Card9−/− mice when compared to Casp1−/− cells (Figures 4I–J), demonstrating that CARD9 exerts its protective effect at least in part via Caspase-1 activation in myeloid cells. Consistent with the protection from body weight loss and colon shortening, Card9−/− mice given WT MCs had decreased inflammation, edema and extent of lesions when compared with Card9−/− mice given Casp1−/− MCs (Figure 4K). To determine the transfer potential and survival of Casp1−/− cells compared to WT cells, we adoptively transferred CD45.2 WT or Casp1−/− MCs into CD45.1 recipient mice and enumerated the donor cells in the colons at day 14 post. The number of recovered Casp1−/− cells were comparable to WT (Figure 4L). Therefore, deficiency of Caspase-1 does not hinder the survival or migration of myeloid cells during adoptive transfer. Taken together, these findings demonstrate that CARD9 promotes inflammasome activation and subsequent IL-18 maturation in the colon, and that this alleviates colitis and associated epithelial hyperplasia in the AOM-DSS model.
Card9−/− mice exhibit altered microbial landscape
CARD9 regulates the gut fungal and bacterial microbiota (Lamas et al., 2016). IL-18 promotes a symbiotic gut microbiota such that WT mice that are cohoused with the Il18−/− mice during AOM-DSS administration have more severe colitis (Levy et al., 2015). To test if the CARD9-IL-18 axis similarly affected the gut microbiota and the susceptibility to colitis and colorectal cancer, we cohoused the WT and Card9−/− mice during AOM-DSS treatment. WT mice housed with Card9−/− mice throughout the AOM-DSS treatment displayed increased weight loss and colon shortening at day 14, and a higher colonic tumor burden on day 80, as compared to WT mice housed separately (Figure S3A–F). The cohoused WT and Card9−/− mice showed similar extent of body weight loss, colon shortening and overall number of tumors in the colon. However, the cohoused WT mice developed significantly more number of tumors in the distal colon as compared to the Card9−/− mice (Figure S3A–F), in line with recent observations (Bergmann et al., 2017b).
To directly test if cohousing led to normalization of the gut microbiota between the WT and Card9−/− mice, we characterized th fungal and bacterial diversity in WT, Card9−/− and the cohoused WT (CH) and Card9−/− (CH) mice by 18s Internal transcribed spacer (ITS) and 16s rRNA sequencing. The mycobiota of WT mice was dominated by the members of the Leotiomycetes (Ascomycota) and the Tremellomycetes (Basidiomycota). Principal coordinate (PCoA) analysis based on the Bray-Curtis dissimilarity of the genus composition revealed a distinct mycobial landscape in the WT and Card9−/− mice (Figure S3G). Compared to the WT mice, Card9−/− mice harbored decreased occurrence of several members of Ascomycota, including Claussenomyces of Leotiomycetes, Pseudocercospora cordiana of Dothideomycetes and Agaricomycetes, along with increased representation of Saccharomycetes, particularly Diutina catenulata and Cladosporium of Dothideomycetes (Figure S3H). Upon cohousing, the overall fungal landscape in the WT and Card9−/− mice became similar, and the PCA plots indicated cohoused mice to be more similar to Card9−/− mice than the WT mice (Figure S3G). Similar analyses were performed to identify the bacterial taxa that show difference in abundance amongst these mice. The bacterial microbiota was dominated by bacteriodetes in all mice. Similar to the fungal landscape, the bacterial composition of Card9−/− mice formed a distinct group from the WT mice (Figure S3H). Compared to the WT mice, the relative abundance of members of Mycoplasmataceae, Bacteroidaceae, Anaeroplasmataceae, Rikenellaceae and Odoribacteraecae was increased in the Card9−/− mice, while the relative abundance of alphaproteobacteria RF32, Helicobacteraceae, Prevotellaceae, Ruminococcaceae, Paraprevotellaceae, Deferribacteraceae, Acetobacteraceae and Phormidiaceae was decreased in the Card9−/− mice (Figure S4). While cohousing normalized the composition of the bacteria, the abundance of Phormidiaceae and alphaproteobacteria RF32 was lower while Mycoplasmataceae was higher in the Card9−/− mice housed with the WT mice. Taken together, these data show that CARD9 controls the gut mycobial and bacterial landscape, and the deficiency of CARD9 can transfer the increased susceptibility to the cohoused WT mice.
CARD9 interacting partner SYK is required for protection from colitis and colon cancer
We next explored the upstream pathways involved in CARD9 mediated inflammasome activation during colitis and CRC. Activation of SYK is a common link between CLRs and downstream CARD9 activation (Gross et al., 2006; Ruland, 2008). Further, SYK activation (phosphorylation) was detectable in mouse colons under homeostatic conditions (day 0), and substantially increased in colons from inflamed (day 14) and tumor-bearing mice (day 80) (Figure 5A).
Figure 5. SYK activity in myeloid cells decreases susceptibility to colitis-associated cancer.
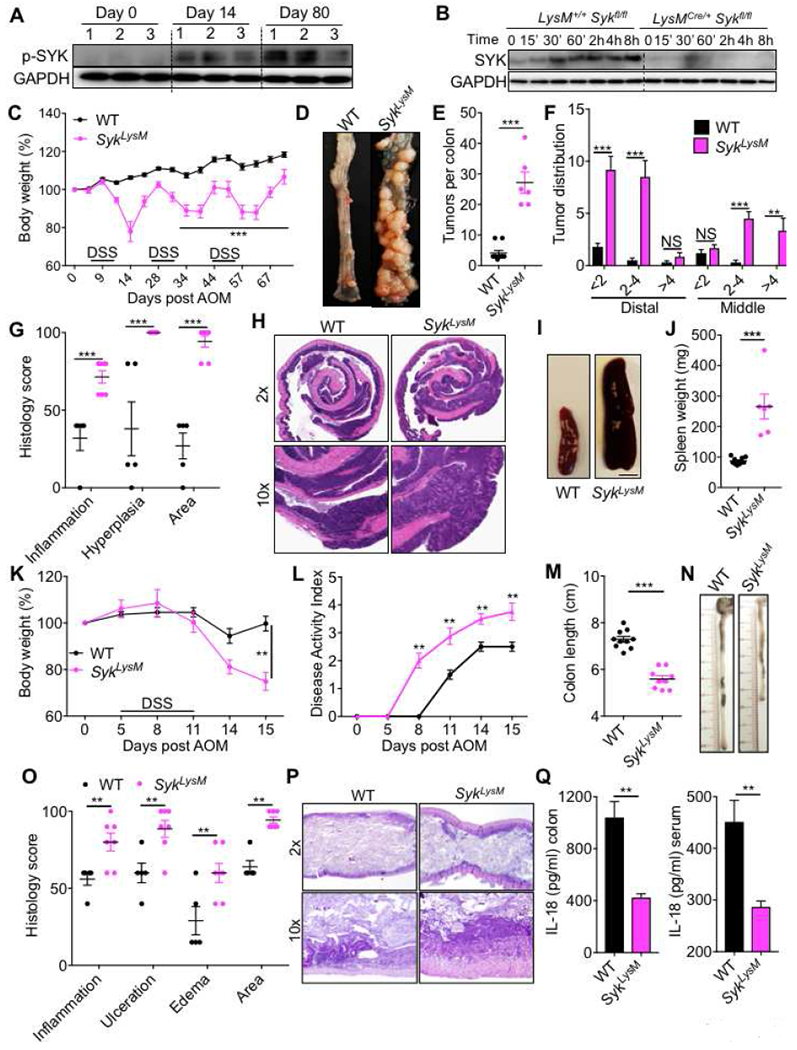
(A) Immunoblot for p-SYK (Tyr 519/520) in colon tissue of WT mice at indicated times after AOM–DSS treatment. (B) Immunoblot for SYK in bone marrow-derived myeloid cells from LysM+/+ Sykfl/fl (WT) and LysMCre+/− Sykfl/f (SykLysM) mice stimulated with 100 ng/ml of LPS for indicated times. (C) Change in body weight in WT and SykLysM mice injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. (D) Representative pictures of colons (E) number of tumors, (F) tumor distribution and (G) histological analysis in the colons of WT and SykLysM mice at day 80 post-AOM. (H) Representative H&E stained images from 2× and 10× magnification of colon sections at day 80 post-AOM injection. (I) Representative pictures of spleens of WT and SykLysM mice (scale bar = 4mm) and (J) spleen weights at day 80. (K) Body weight changes (L) disease activity index in WT and SykLysM mice during AOM-DSS treatment. (M) Colon length, (N) representative photographs of colons, (O) histological analysis of colons and (P) representative pictures of H&E stained sections from colons of WT and SykLysM mice at day 15. (Q) IL-18 ELISA from colon explants and serum of WT and SykLysM mice. Each symbol represents an individual mouse. NS, not significant; **P < 0.01, ***P < 0.001; two-way ANOVA followed by the Holm–Sidak post-hoc test (C, F, G, K, L and O) or Mann–Whitney U test (E, J, M and Q). Data are from one experiment representative of two independent experiments with n = 6-10 mice per group (A-Q; mean and SEM [C, E, F, G, J, K, L, M, O and Q]). See also Figure S4
Mice with global deficiency in SYK die in the perinatal stage (Cheng et al., 1995). Since CARD9 functions in myeloid cells to regulate disease, we evaluated the role of SYK in myeloid cells by generating myeloid cell-specific Syk-deficient mice (Sykfl/flLysMCre/+ - referred to as SykLysM). The efficiency of SYK deletion was confirmed by western blotting for SYK in the bone marrow-derived myeloid cells during LPS stimulation (Figure 5B). Upon AOM-DSS administration SykLysM mice lost significantly more body weight during each round of DSS administration, compared with the SYK-sufficient littermates (Sykfl/flLysM+/+ - referred to as WT) that were separated into respective cages upon weaning at 3-4 weeks of age (Figure 5C). At day 80, SykLysM mice exhibited an increased number of tumors that were also bigger in size when compared to WT mice (Figure 5D–F). Histological analysis revealed greater extent of inflammation and epithelial hyperplasia in colons (Figures 5G and 5H) and splenomegaly (Figure 5I and 5J). These findings are in line with Card9−/− mice and demonstrate that SYK-CARD9 function in myeloid cells protects from colon cancer.
Further, SykLysM mice also displayed characteristics of acute colitis - increased weight loss, disease activity and colon shortening at day 15 when compared to WT mice (Figures 5K–N). Histological analysis of the colons at day 15 also demonstrated increased inflammation, ulceration, edema and extent of lesions in SykLysM mice (Figures 5O and 5P). IL-18 was significantly decreased in the colon and serum at preclinical time (day 9) demonstrating a critical role of SYK-CARD9 axis in promoting inflammasome activation early during disease pathogenesis (Figure 5Q). These findings demonstrate that both SYK and CARD9 are required to promote inflammasome activation and optimum IL-18 maturation in the colon during colitis and colon cancer.
We also confirmed a significant role for SYK and CARD9 in caspase-1 activation and IL-18 maturation in bone marrow-derived myeloid cells after stimulation with fungi ex vivo. SYK and CARD9 were required for caspase-1 activation and IL-18 release in response to stimulation with Candida albicans (Figures S5A and S5B). Canonical and non-canonical NLRP3 activation triggered by LPS+ATP and intracellular LPS respectively was unaffected by loss of SYK or CARD9 (Figures S5C and S5D). SYK and CARD9 also did not affect AIM2 or NLRC4 inflammasome activation after DNA transfection or Salmonella infection respectively (Figures S5E and S5F). Therefore, CARD9 and SYK are specifically required for inflammasome activation in myeloid cells in response to fungal stimuli.
Commensal fungi are major inducers of inflammasome activation in the colon
Fungi and anaerobic bacteria are major components of the gut microbiota, especially in the distal colon where inflammation and tumors are most evident with AOM-DSS treatment (Donaldson et al., 2016). To determine the relative contribution of fungi in provoking inflammasome activation during AOM-DSS treatment, mice were depleted of commensal fungi by treatment with amphotericin B (AmpB) or Itraconazole (Itra) orally for 4 weeks prior to AOM-DSS administration. Treatment with AmpB led to a significant decrease in the total fungal loads (Figure 6A and S6). We further characterized the fungal and bacterial diversity in mice treated with AmpB or Itra by 18s ITS and 16s rRNA sequencing. Principal coordinate analysis based on the Bray-Curtis dissimilarity of the genus composition revealed that amphotericin B or itraconazole treatment led to a similar shift in the mycobial landscape, when compared to the water treated group (Figure S6A). Treatment of mice with AmpB or Itra led to a significant decrease in several members of Ascomycota particularly the Leotiomycetes, Helotiales and Pseudocercospora (Dothideomycetes) (Figure S6B). 16s rRNA analysis however revealed a distinct bacterial landscape in mice treated with AmpB or Itra (Figure S6C). In the AmpB-treated mice, the members of Bacteroidetes including Prevotellaceae, Rikenellaceae, and Bacteroidaceae were increased, while Opitutae (Verrucomicrobia), Phormidiaceae (Cyanobacteria) and Bifidobacteriaceae (Actinobacteria) were decreased (Table S2). For the Itra-treated mice, significantly increased Prevotellaceae and RF16 of Bacteroidales were observed, while the occurrence of Alcaligenaceae (Proteobacteria), Ruminococcus (Firmicutes), Helicobacteraceae (Proteobacteria) and several other members of Firmicutes were decreased (Table S3). Therefore, treatment of mice with AmpB or Itra leads to similar depletion of commensal fungi, but distinct changes in the bacterial landscape.
Figure 6. Commensal gut fungi promote inflammasome activation.
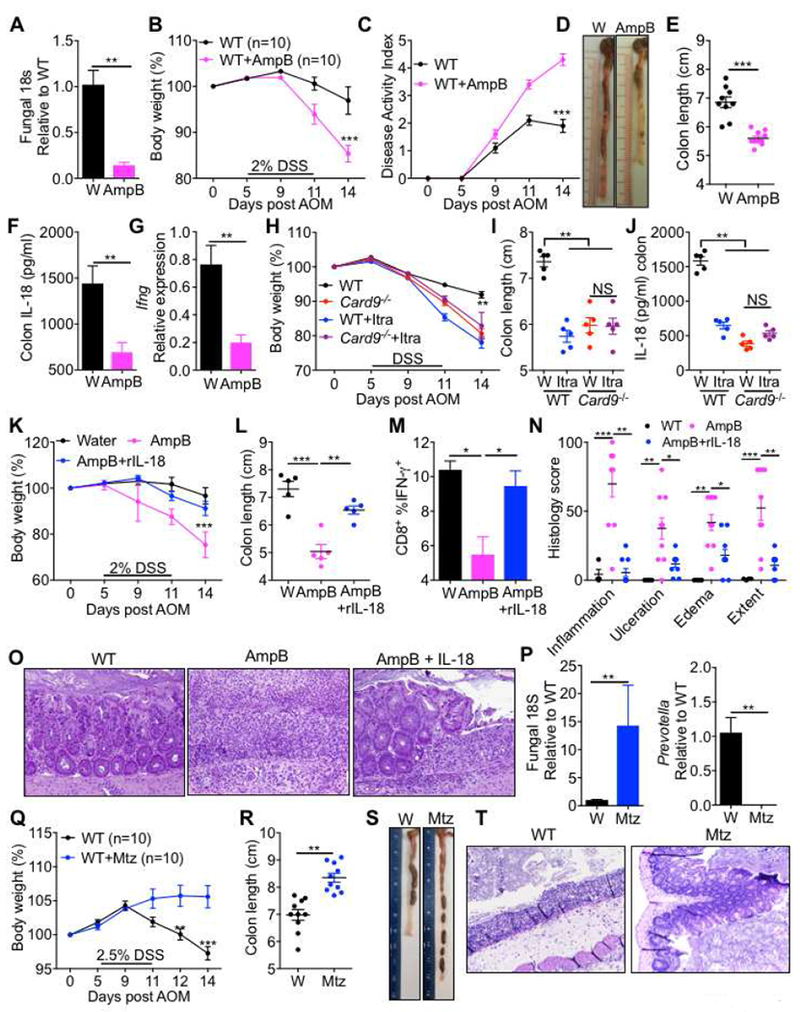
(A) qPCR analysis for fungi in fecal samples of mice treated with amphotericin B (AmpB) for 4 weeks. (B) Body weight change (C) disease activity index (D) representative colon photographs and (E) colon length at day 14. (F) Analysis of IL-18 in the explants and (G) Ifng expression in colon tissue at day 9 in WT mice treated with AmpB or drinking water for 4 weeks prior to AOM-DSS treatment. (H) Body weight change (I) colon length at day 14 and (J) Secretion of IL-18 in the colon explants at day 9 of WT and Card9−/− mice treated with Itraconazole for 4 weeks prior to AOM-DSS administration. (K) Body weight change in WT mice pretreated with AmpB, along with IL-18 or PBS supplementation during AOM-DSS treatment. (L) Colon length (M) IFN-γ production in CD8+ T cells of the MLN (N) histological analysis of colons and (O) representative pictures of H&E stained sections from colons at day 14. (P) qPCR analysis of indicated organisms in fecal samples of mice pre-treated with Metronidazole (Mtz) or control solution (1% sucrose in drinking water) for 5 days. (Q) Body weight change in WT mice treated with Mtz or control solution before AOM-DSS administration. (R) Colon length (S) representative colon photographs and (T) representative pictures of H&E stained sections from colons at day 14. Each symbol represents an individual mouse. *P < 0.05; **P < 0.01, ***P < 0.001; Mann–Whitney U test (A, E, F, G, P and R), two-way ANOVA (B, C, H, K, N and Q), one-way ANOVA followed by the Holm–Sidak post-hoc test (I, J, L and M). Data are from one experiment representative of two independent experiments with n = 5-10 mice per group (A-T; mean and SEM [A, B, C, E, F, G, H, I, J, K, L, M, N, P, Q, R and T]). See also Figures S5–S8.
Upon treatment with a low dose (2%) of DSS, mice treated with AmpB lost significantly more body weight, displayed increased signs of disease, and had shorter colons on day 14 (Figure 6B–6E), indicating that commensal fungi are protective against acute colitis. Consistent with the role of molecules involved in fungal sensing, depletion of fungi also led to decreased release of inflammasome-associated cytokine IL-18 (Figures 6F) and decreased transcription of Ifng early during AOM-DSS treatment (Figures 6G). Similar results were obtained when commensal fungi were depleted in WT mice by Itraconazole (Figure 6H–6J). Further, depletion of commensal fungi in the Card9−/− mice by treatment with Itraconazole did not affect the susceptibility to AOM-DSS induced colitis, or secretion of IL-18 from the colon explants (Figure 6H–6J). Therefore, commensal fungi in the gut promote inflammasome activation to protect from colitis in a CARD9-dependent manner.
To directly test if increased inflammation in mice depleted of fungi was due to decreased IL-18 release, we supplemented AmpB pretreated mice with exogenous IL-18 during AOM-DSS treatment. Supplementation with IL-18 was able to significantly protect the AmpB treated mice from exacerbated weight loss and colon shortening (Figures 6K and 6L). IL-18 supplementation also restored the IFN-γ production from CD8+ T cells in the mesenteric lymph nodes (Figures 6M). Histological analysis of the colon further revealed significant increases in inflammation, ulceration, edema and the extent of lesions in AmpB-treated mice, which were ameliorated by supplementation with IL-18 (Figures 6N and 6O).
In sharp contrast, depletion of anaerobic bacteria by Mtz treatment led to a significant increase in the overall fungal loads while depleting anaerobic bacteria, including Prevotella (Figure 6P and S9A-B). We further characterized the bacterial and fungal microbiota by 16s rRNA and 18s ITS sequencing. PCoA analysis of the bacterial and fungal composition at the genus level revealed that mice given Mtz formed an independent group (Figure S7A–B). 16s analyses of the bacterial taxa demonstrated significant decrease in the occurrence of Rikenellaceae and S24_7 of Bacteroidales, Deferribacteraceae and Alphaproteobacteria and increase in Lactobacillaceae, Enterobacteriaceae, Mycoplasmataceae, Aerococcaceae and Porphyromonadaceae (Figure S7C) while the 18s analysis revealed an increase in an unassigned group. Upon AOM-DSS treatment, Mtz-treated mice were significantly protected from signs of acute colitis including body weight loss, colon shortening, and associated histological perturbations (Figures 6Q–6T). Therefore, AmpB or Itra-sensitive commensal fungi promote inflammasome activation and protect from colitis, while Mtz-sensitive commensal anaerobes promote colitis.
Commensal fungi-SYK-CARD9-IL-18 axis is protective from colon cancer
To directly test if the loss of IL-18 is also responsible for the increased tumorigenesis in the colons of Card9−/− and SykLysM we supplemented these mice with exogenous IL-18 during each round of the DSS treatment after AOM injection and evaluated the tumor burden in the colon at day 80. Consistent with amelioration of colitis (Figures 4 and 6), exogenous IL-18 treatment also significantly decreased the body weight loss, as well as the tumor burden in the Card9−/− mice during the AOM-DSS treatment, when compared to the Card9−/− or Il18−/− mice given control injections during AOM-DSS treatment (Figures 7A–C). Exogenous IL-18 treatment in WT mice also led to a decrease in the number of tumors in the colon (Figures 7A–C). Similar to the observations in the Card9−/− mice, exogenous IL-18 supplementation during AOM-DSS treatment also protected the SykLysM mice from the exacerbated weight loss and colon tumorigenesis by (Figures 7D–E).
Figure 7. IL-18 supplementation or metronidazole treatment protects Card9−/−, SykLysM and AmpB-treated mice from colon tumorigenesis.
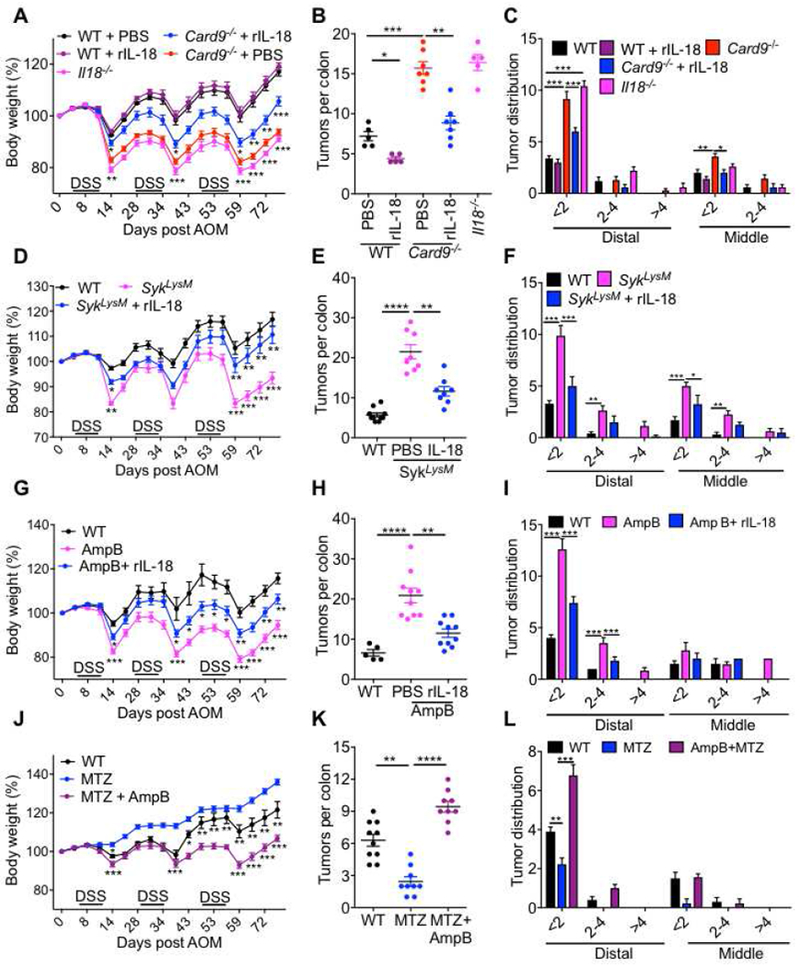
(A) Change in body weight in WT, Card9−/− and Il18−/− mice injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. A group of WT and Card9−/− mice were also given IL-18 supplementation during AOM-DSS treatment. (B) number of tumors and (C) tumor distribution in the colons. (D) Change in body weight in WT and SykLysM mice injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. A group of SykLysM mice were also given IL-18 supplementation during AOM-DSS treatment. (E) number of tumors and (F) tumor distribution in the colons. (G) Change in body weight in WT mice treated with drinking water or AmpB for 4 weeks before being injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. A group of AmpB-treated WT mice were also given IL-18 supplementation during AOM-DSS treatment. (H) number of tumors and (I) tumor distribution in the colons. (J) Change in body weight in WT mice treated with d MTZ or both MTZ and AmpB for 4 weeks before being injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. (K) number of tumors and (L) tumor distribution in the colons. Each symbol represents an individual mouse. *P < 0.05; **P < 0.01, ***P < 0.001; Two-way ANOVA (A, C, D, F, G, I, J and L), one-way ANOVA followed by the Holm–Sidak post-hoc test (B, E, H and K). Data are from one experiment representative of two independent experiments with n = 8-10 mice per group (A-L; mean and SEM [A-L]).
To test if commensal fungi play an active role in the pathogenesis of colitis-associated colon cancer, we depleted WT mice of commensal fungi (with AmpB treatment) or anaerobic bacteria (with Mtz treatment) or both (simultaneous AmpB and Mtz treatment) before AOM-DSS treatment. Similar to the observations with the Card9−/− and SykLysM mice, AmpB treatment increased the extent of body weight loss and colon tumorigenesis upon AOM-DSS administration, which was also ameliorated significantly by exogenous IL-18 supplementation (Figures 7 G–I). On the other hand, Mtz treatment ameliorated the body weight loss and colon tumor burden (Figures 7 J–L). To test if the protection conferred by Mtz was due to the increase in fungal loads, we simultaneously depleted both commensal fungi and anaerobic bacteria by simultaneous Mtz and AmpB administration. Mice receiving both Mtz and AmpB had exacerbated body weight loss and colon tumorigenesis when compared to mice receiving Mtz alone, demonstrating an active role for commensal fungi in suppressing colitis-associated colon cancer (Figure 7 J–L). Therefore, commensal fungi-SYK-CARD9 axis is protective from colitis-associated colon cancer, at least in part, by prompting inflammasome activation and IL-18 maturation in the colon.
Discussion
Currently available anti-TNF biologics provide sustained disease remission in only 25% of IBD patients, while another 30% require total colectomy (Nielsen, 2014). IBD is also a strong predisposing factor for CRC, a leading cause of cancer-related death in adults (Howlader et al., 2015). While the etiology of IBD is unknown, aberrant immune responses to commensal microbes are widely thought to underlie it. Therefore, the role of specific components of the microbiota, and associated signaling pathways that regulate disease susceptibility are avenues of active research. Inflammasome activation is one such critical aspect of innate immunity that has been shown to modulate colitis and CRC pathogenesis (Zaki et al., 2010b). In this study, we demonstrate that commensal gut fungi signaling via SYK-CARD9 axis is essential for IL-18 maturation and protection from experimental colitis and CRC.
SYK-CARD9 signaling axis is activated downstream of CLRs and has been shown to induce NF-κB, ERK, p38 MAPK and JNK signaling and cytokine production during fungal infection (Gross et al., 2006; Ruland, 2008). SYK and CARD9 are also required for inflammasome activation by Candida and Aspergillus (Gringhuis et al., 2012). We found that colons of Card9−/− mice did not exhibit any defects in expression of cytokines or chemokines, or NF-κB, ERK, p38 MAPK and JNK signaling early during experimental colitis. Consistent with increased inflammation and epithelial hyperplasia during acute disease ERK, IκBα and STAT-3 phosphorylation were increased during acute disease. In contrast, inflammasome activation and IL-18 secretion were significantly lower in colons of Card9−/− and SykLysM mice, and mice depleted of commensal gut fungi prior to clinical disease. IL-18 is known to be a limiting factor in IFN-γ production in the colon (Salcedo et al., 2010a) and in line with our observations of defective IL-18 secretion in colons from Card9−/− mice, they also have lower production of IFN-γ from T cells during experimental colitis. Further, IL-18 supplementation was able to significantly ameliorate colitis and CRC and promote IFN-γ production from T cells. Therefore, commensal fungi are a critical contributor to inflammasome activation via the SYK-CARD9 pathway, and IL-18 production downstream of the inflammasome is required for maximal IFN-γ production from T cells. Further studies will be needed to determine if the IFN-γ producing CD8+ T cells are specific to the antigens presented by the colon tumors and directly mediate anti-tumor immunity, and the cell-type specific contribution of CARD9 in promoting inflammasome activation in the colon. Further, treatment with two different anti-fungal agents led to a similar shift in the mycobial landscape but distinct changes in the bacterial landscape. This suggests that increased susceptibility observed with these anti-fungal agents is likely attributable to depletion of fungi, and not the changes in the bacterial microbiome. Nevertheless, it is possible that fungal depletion leads to an overall ecological shift that favors lower inflammasome activation and increased colitis and colon cancer. Therefore, commensal fungi may have an indirect role in protecting from colitis and CRC as well.
IL-18 and inflammasomes play multifaceted roles in colitis and colon cancer. Expression of IL-18 and the accessory protein IL-18RAP are lower in human colon cancer biopsies, and polymorphisms in the components of the IL-18 signaling pathway are associated with increased susceptibility to IBD in humans (Barrett et al., 2008; Hedl et al., 2014). Mice lacking IL-18, or the inflammasome components such as Nlrp3−/−, Asc−/− and Caspase1−/− also have increased susceptibility to AOM-DSS induced colitis and colon cancer, which can be ameliorated by IL-18 supplementation (Allen et al., 2010; Salcedo et al., 2010b; Zaki et al., 2010a; Zaki et al., 2010c). However, excessive inflammasome activation and loss of the IL-18 decoy receptor increase the susceptibility to experimental colitis (Neudecker et al., 2017; Nowarski et al., 2015). Therefore, both the lack and excess of inflammasome activation and IL-18 can promote inflammation in the colon (Mager et al., 2016; Sharma and Kanneganti, 2017). Mucosal IL-18 responses have also been implicated in governing a transmissible gut microbiota (Levy et al., 2015). In line with this observation, we showed here that co-housed WT and Card9−/− mice show similar disease susceptibility to acute colitis and colon cancer. However, in a recent study (Bergmann et al., 2017b), the authors found a slight reduction in tumor size in co-housed Card9−/− mice at a later time point. They demonstrated the impaired production of IL-1β in Card9−/− mice and mechanistically reasoned that IL-1β supports tumor-promoting activity of group 3 innate lymphoid cells through IL-22 expression. Though these studies demonstrated pathogenic effects of CARD9–mediated IL-1 β production, the beneficial effects of the other major inflammasome-dependent cytokine IL-18 was apparently normalized by prolonged co-housing of Card9−/− mice. In summary, our study demonstrates that commensal gut fungi are critical for protection from colitis and CRC, while anaerobic bacteria promote disease pathogenesis. Fungi signal via SYK-CARD9 axis to promote inflammasome assembly, and subsequent IL-18 maturation protects against colitis and colon cancer and promotes anti-tumorigenic T cell responses. Therefore, promoting healthy gut mycobiota could provide protection from IBD and CRC.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thirumala-Devi Kanneganti (thirumala-devi.kanneganti@stjude.org). The mouse lines obtained from other laboratories are described below and may require a Material Transfer Agreement (MTA) with the providing scientists.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
Card9−/− mice have been described previously (Gross et al., 2006) were rederived in C57BL/6J mice and backcrossed for 10 generations in the colony. SykLysM mice were generated by crossing Sykfl/fl mice (Saijo et al., 2003) with LysMCre/+ mice (Lys2tm(cre)/fo), and deletion of SYK was confirmed in bone marrow-derived myeloid cells. Mice were maintained in a specific pathogen-free facility at St. Jude Children’s Research, used at 6-12 weeks of age, assigned randomly to experimental groups and males and females were used at approximately equal ratios. The St. Jude Children’s Research Hospital Animal Care and Use Committee approved all animal study protocols.
Induction of Colitis and Tumors
The AOM–DSS murine model for colorectal tumorigenesis has been previously described (Zaki et al., 2010b). For the DSS model of colitis, mice were administered the indicated dose of DSS (mol wt 36–40 kDa; Affymetrix) for 6 days, followed by regular water. For IL-18 supplementation, mice were injected with 0.5μg of recombinant mouse IL-18 (MBL) in 100μl sterile PBS on days 1, 3 and 0.1 μg on days 5 and 7 via retro-orbital route during DSS administration. Mice were administered with 2.5mg/ml metronidazole (Teva pharmaceuticals) with 1% sucrose in drinking water for 5 days for depletion of anaerobic bacteria, and 0.25mg/ml of Amphotericin B (X-gen pharmaceuticals) or 1mg/ml of Itraconazole (Patriot pharmaceuticals) for three weeks for depletion of fungi, prior to DSS administration. For co-housing experiments, equal numbers of female WT and female Card9−/− littermates were housed in the same cage for 4 weeks. Mice were then remained co-housed and subjected to AOM and DSS treatment.
METHODS DETAILS
Histology and Microscopy Analysis
Colons were fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Histological analysis for inflammation, epithelial hyperplasia, and tumorigenesis was performed by a board-certified pathologist (PV) as previously described (Karki et al., 2016). The histology scoring criteria is as follows: 0 = normal, 2 = minimal, 15 = mild, 40 = moderate, 80 = marked and 100 = severe.
For Inflammation, normal - small numbers of lymphocytes and plasma cells in lamina propria only; minimal - mixed infiltration (including macrophages and neutrophils) small, focal, or widely separated, limited to lamina propria; mild - multifocal mixed infiltration, often extending into submucosa; moderate - large multifocal lesions with mixed infiltration involving mucosa and submucosa; marked - marked extensive mixed infiltration with edema with or without small erosions or ulcers and small areas of transmural inflammation; severe – diffuse transmural inflammation with or without multiple ulcers.
For Ulceration, normal – none, minimal - only one small focus of ulceration involving fewer than 5 crypts; mild - a few small ulcers up to 5 crypts; moderate - multifocal ulcers up to 10 crypts; marked - multifocal to coalescing ulcers involving more than 10 crypts each; severe - extensive to diffuse with multiple ulcers covering >20 crypts each.
For edema, normal - edema absent; minimal - small areas of mucosal edema only; mild - plus restricted mild submucosal edema; moderate - moderate plus multiple foci of moderate submucosal edema; marked - marked submucosal edema with foci of architectural distortion with or without subserosal edema; severe - edema with extensive architectural distortion of colon mucosa.
For hyperplasia, normal - typical; minimal - some areas with crypts elongated and increased mitoses; mild - multifocal areas with crypts elongated up to 2 times normal thickness; normal goblet cells still present; moderate - extensive areas with crypts up to 2 times normal thickness, reduced goblet cells; marked - mucosa over 2 times normal thickness, hyperchromatic epithelium, reduced or rare goblet cells, possibly foci of arborization; severe - mucosa over 2 times normal thickness, marked hyperchromasia, crowding/stacking, absent goblet cells, high mitotic index, arborization.
For extent, normal - rare or inconspicuous lesions (0% involvement); minimal - less than 5%; mild - multifocal or small, focal, or widely separated, but conspicuous lesions (5 to 10% involvement); moderate - multifocal, prominent lesions (10 to 50% involvement), marked - coalescing to extensive lesions or areas of inflammation with some loss of structure (50% to 90% involvement); severe - diffuse lesion with effacement of normal structure (over 90% involvement).
Cytokine Analysis
Cytokines in colon explants, homogenates and sera were measured by ELISA. Culture of colon explants was described previously (Malik et al., 2013). Briefly, for explant culture, 0.5 cm of the distal colon was incubated in IMDM media supplemented with 10%FBS and penicillin, streptomycin and gentamycin for 48 hours. Cytokine quantities are presented as pg or ng/ml of the clarified supernatant media. Explants were also weighed before incubation, and equivalent data is obtained if the cytokine quantities are normalized to their wet weight. The IL-18 ELISA was from eBioscience; multiplex ELISAs were used for all other cytokines (Millipore) according to manufacturers’ instructions. IL-18 ELISA (BMS618/3TEN, Affymetrix eBioscience) and multiplex ELISAs (MCYTOMAG-70K, Millipore) were performed according to manufacturers’ instructions.
Cytokine expression in colon homogenates, and sera were measured by the enzyme-linked immunosorbent assay (ELISA). Protein from colon tissue was extracted as previously described (Sharma et al., 2015). Cytokine quantitiesare presented as picogram or nanogram per milliliter (pg or ng/mL) of the clarified supernatant medium.
Flow Cytometry Analysis
Single-cell suspensions were prepared from the colon as previously described (Malik et al., 2013). Briefly, to remove epithelial cells, the colon was washed, cut into small pieces, and pieces were incubated with calcium- and magnesium-free Hank’s balanced salt solution supplemented with 5% FBS and 5 mM EDTA (Sigma-Aldrich) and centrifuged at 140 rpm at 25°C for 30 min. Tissues were then incubated with RPMI 1640 containing 10% FBS and 0.5 mg/mL collagenase type IV for 1 h at 37°C with shaking at 150 rpm. Liberated cells were collected by passage through a 70-μm nylon mesh. Isolated cells from the EDTA-treated (epithelial) and collagenase-treated (lamina propria) fractions were separated on a 40%/80% discontinuous Percoll gradient (GE Bioscience). The following monoclonal antibodies were used in appropriate combinations: anti-EpCAM (clone G8.8) and anti-CD45.2 (clone 104).
Colon, spleen and MLN cells were processed and stimulated as previously described (Malik et al., 2013). Briefly, single-cell suspensions were prepared and stimulated with phorbol-12-myristate-13-acetate and ionomycin for 4 h, with brefeldin A (00-4506, Affymetrix eBioscience) added for the last 2 h. For intracellular cytokine staining, cells were fixed and permeabilized by using a fixation and permeabilization solution (00-5523, eBioscience). Antibodies used for staining were anti-CD3 (clone 145 – 2C11), anti-CD4 (clone RM4-5), anti-CD19 (clone 6D5), anti-IFNg (50–7311, Tonbo), and anti-TNF (506322, Biolegend). Flow cytometry data were acquired on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Western Blotting
Proteins were extracted from colon tissues using RIPA lysis buffer supplemented with proteinase and phosphatase inhibitors (Roche). Samples were resolved by 8%–15% sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE) electrophoresis and transferred onto polyvinylidene difluoride membranes. Blocking was performed in 3% BSA in TBST for 1 h, and membranes were incubated with primary antibodies overnight at 4°C. Membranes were incubated with horseradish peroxidase–conjugated secondary antibody for 1 h, and proteins were visualized by using ECL substrate (ThermoScientific). Primary antibodies used were anti-CARD9 (1:1000, 12283, Cell Signaling Technology), anti-p-Syk (1:1000, 2710, Cell Signaling Technology), anti-Syk (1:1000, 2712, Cell Signaling Technology), anti-p-ERK (1:1,000 dilution, #9101, Cell Signaling Technology), anti-ERK (1:1,000 dilution, #9102, Cell Signaling Technology), anti-p-IκBα (1:1,000 dilution, #9241, Cell Signaling Technology), anti-IκBα (1:1,000 dilution, #9242, Cell Signaling Technology), anti-p-p38 MAPK (1:1,000 dilution, #9211, Cell Signaling Technology), anti-p-JNK (1:1,000 dilution, #9251, Cell Signaling Technology), anti-caspase-1 (1:500 dilution, sc-515, Santa Cruz Biotechnology), anti-p-STAT3 Tyr705 (1:1,000, 9131, Cell Signaling Technology), and anti-GAPDH (1:10,000; 5174, Cell Signaling Technology).
Cell Culture and Stimulations
Primary mouse bone marrow–derived myeloid cells (BMMCs) were cultured as previously described (Lupfer et al., 2013). Cells were stimulated with 500 ng/mL ultra-pure LPS from Salmonella minnesota R595 (tlrl-smlps, InvivoGen) for 4 hours before stimulation with 5 mM ATP (10127531001, Roche). For DNA transfection, each reaction consisted of 1 μg of poly(dA:dT) (tlrl-patn, InvivoGen) resuspended in PBS and mixed with 0.3 μl of Xfect polymer in Xfect reaction buffer (631318, Clontech Laboratories). After 10 min, DNA complexes were added to BMDMs in Opti-MEM (31985-070, ThermoFisher Scientific) and incubated for 4 hr. For NLRC4 activation, cells were stimulated with S. Typhimurium at MOI 1 for 2 h. C. albicans ATCC MYA-3573 (UC 820), was grown overnight in Sabouraud broth at 37°C and used at MOI of 5.
Quantitative Real-Time PCR
RNA was isolated by using RNeasy kit (Sigma-Aldrich) as per manufacturer’s instructions and converted into cDNA as previously described (Sharma et al., 2015). Gene expression was assessed by using the 2× SYBR Green master mix as per the manufacturer’s instructions (Applied Biosystems). Sequences for quantitative RT-PCR (qRT-PCR) primers are listed in Table S1. qRT-PCR data were analyzed by the 2−ΔΔCT method, with β-actin as the housekeeping gene.
Microbiota sequencing
Genomic DNA from feces was extracted by bead beating followed by QiAMP DNA stool Mini Kit (Qiagen, Valencia, CA). Sequencing libraries were generated from 1-50 ng genomic DNA by using the Bioo Scientific NEXTflex 16s V1-V3 amplicon protocol. This method prepares amplicon libraries that span the hypervariable domains (V1–V3) of microbial 16s ribosomal RNA genes. The protocol uses a two-step amplification protocol. The first PCR step targets the V1-V3 domains, then the second PCR integrates adaptor and index sequences that are compatible with paired-end sequencing on the Illumina MiSeq platform. Sequencing libraries generated from microbial 16s ribosomal DNA were quality assessed for insert size distribution using an Agilent Bioanalyzer and quantified by fluorescence assay (picogreen) before pooling in equimolar ratios. Samples were pooled and sequenced using an Illumina MiSeq system using paired-end 300 base sequencing chemistries. Between 200,000 and 1 million total reads (~120 – 600 Mb per sample) were generated. The Illumina pair-ended reads with low-quality bases were trimmed to reduce sequencing or PCR errors at this step. The sequences were subsequently assembled into contigs. The contigs with ambiguous bases, chimeric sequences or incorrect assembly size were excluded from downstream analysis. The open reference OTU picking strategy as implemented in QNME(Caporaso et al., 2010) was used to identify the OTUs from the assembled contigs. Briefly, the contigs were clustered against the Greengene 16S rRNA database(DeSantis et al., 2006). The contigs that failed to hit in the reference sequence were excluded from downstream analysis. Taxonomy of each OTU was assigned by comparing with the Greengene reference OTUs. The final result contained an OTU matrix detailing the information about the number of sequences in each OTUs and their taxonomy assignment. Similar work flow was utilized to characterize fungal diversity with NEXTflex 18s ITS Amplicon-Seq Kit and UNITE fungal 18S ITS database(Kõljalg et al., 2013). Kruskal-Wallis non-parametric tests was used to test the significance of the difference of diversity. The linear discriminant analysis (LDA) effect size (LEfSe) method was used to test the significant difference of relative abundance of taxa among groups(Segata et al., 2011).
Quantification and Statistical Analysis
Data on changes in body weight were analyzed by two-way ANOVA, followed by the Holm–Sidak post-hoc test. Statistical significance for other datasets was determined by parametric or non-parametric tests when appropriate and is indicated in the respective figure legends. The number of repeats for each experiment is also indicated in the respective figure legends. N in the figure legends indicates the number of mice used in the experiments. P<0.05 was considered significant, and is represented as *P<0.05, **P<0.01, ***P<0.001, and NS: not significant.
Supplementary Material
HIGHLIGHTS.
Commensal gut fungi contribute to inflammasome activation during experimental colitis
Microbial landscape is altered in Card9−/− mice
Defective inflammasome activation, IL-18 maturation in myeloid cells lacking Card9 or Syk
Metronidazole treatment protects Card9−/− and SykLysM mice from colon tumorigenesis
Acknowledgments
Supported by the US National Institutes of Health (AI101935, AI124346, AR056296 and CA163507 to T.D.K.), the American Lebanese Syrian Associated Charities (to T.-D.K.). We thank Amanda Burton and Vivekanudeep Karri for their generous help and would like to apologize to our colleagues whose work could not be cited due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors have declared that no conflict of interest exists.
Declaration of Interests
The authors declare no competing interests.
References
- Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, and Ting JP (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207, 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. (2008). Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature genetics 40, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, Diefenbach A, Greten FR, and Ruland J (2017a). Card9-dependent IL-1beta regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur J Immunol 47, 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwälder M, Diefenbach A, Greten FR, and Ruland J (2017b). Card9-dependent IL-1β regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. European Journal of Immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, and Gordon JI (2010). QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, Doho GH, Vijay-Kumar M, and Gewirtz AT (2012). Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol 5, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Liu M, Wang F, Bertin J, and Nunez G (2011). A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. Journal of immunology (Baltimore, Md. : 1950) 186, 7187–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, and Pawson T (1995). Syk tyrosine kinase required for mouse viability and B-cell development. Nature 378, 303–306. [DOI] [PubMed] [Google Scholar]
- Dahlhamer JM (2016). Prevalence of inflammatory bowel disease among adults aged≥ 18 years—United States, 2015. MMWR. Morbidity and mortality weekly report 65. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson GP, Lee SM, and Mazmanian SK (2016). Gut biogeography of the bacterial microbiota. Nature Reviews Microbiology 14, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, and Flavell RA (2011a). NLRP6 inflammasome is a regulator of colonic microbial ecology and risk for colitis. Cell 145, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, and Flavell RA (2011b). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudio E, Taddei G, Vetuschi A, Sferra R, Frieri G, Ricciardi G, and Caprilli R (1999). Dextran sulfate sodium (DSS) colitis in rats: clinical, structural, and ultrastructural aspects. Digestive diseases and sciences 44, 1458–1475. [DOI] [PubMed] [Google Scholar]
- Glocker E-O, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, and Tahami F (2009). A homozygous CARD9 mutation in a family with susceptibility to fungal infections. New England Journal of Medicine 361, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, Van Der Vlist M, Boekhout T, and Geijtenbeek TB (2012). Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1 [beta] via a noncanonical caspase-8 inflammasome. Nature immunology 13, 246–254. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, and Eckmann L (2009). IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell 15, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, and Karin M (2010). Immunity, Inflammation, and Cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, Förster I, and Ruland J (2006). Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656. [DOI] [PubMed] [Google Scholar]
- Hedl M, Zheng S, and Abraham C (2014). The IL 18RAP region disease polymorphism decreases IL-18RAP/IL-18R1/IL-1R1 expression and signaling through innate receptor-initiated pathways. Journal of immunology (Baltimore, Md. : 1950) 192, 5924–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader N, Noone A, Krapcho M, Miller D, Bishop K, Altekruse S, Kosary C, Yu M, Ruhl J, and Tatalovich Z (2015). SEER Cancer Statistics Review, 1975–2013, National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/csr/1975_2013/. based on November. [Google Scholar]
- Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, and Flavell RA (2010). Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proceedings of the National Academy of Sciences of the United States of America 107, 21635–21640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, and Dubinsky M (2012). Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappelman MD, Moore KR, Allen JK, and Cook SF (2013). Recent Trends in the Prevalence of Crohn’s Disease and Ulcerative Colitis in a Commercially Insured US Population. Digestive Diseases and Sciences 58, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R, Man SM, and Kanneganti TD (2017). Inflammasomes and Cancer. Cancer immunology research 5, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R, Man SM, Malireddi RKS, Kesavardhana S, Zhu Q, Burton AR, Sharma AR, Qi X, Pelletier S, Vogel P, et al. (2016). NLRC3 is an inhibitory sensor of PI3K–mTOR pathways in cancer. Nature 540, 583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R, Man SM, Malireddi RS, Gurung P, Vogel P, Lamkanfi M, and Kanneganti T-D (2015). Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection against Aspergillus Infection. Cell host & microbe 17, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno K, Kataoka J, Ohtsuki T, Suemoto Y, Okamoto I, Usui M, Ikeda M, and Kurimoto M (1997). IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. The Journal of Immunology 158, 1541–1550. [PubMed] [Google Scholar]
- Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AF, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, and Callaghan TM (2013). Towards a unified paradigm for sequence-based identification of fungi. Molecular ecology 22, 5271–5277. [DOI] [PubMed] [Google Scholar]
- Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G, Bridonneau A, Jegou S, Hoffmann TW, and Natividad JM (2016). CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nature medicine 22, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Malireddi RS, and Kanneganti T-D (2009). Fungal zymosan and mannan activate the cryopyrin inflammasome. Journal of Biological Chemistry 284, 20574–20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Thaiss Christoph A., Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi Jemal A., David E, Savidor A, Korem T, Herzig Y, et al. (2015). Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan C, Xie L, Yang X, Miao H, Lv N, Zhang R, Xiao X, Hu Y, Liu Y, and Wu N (2015). Dysbiosis of fungal microbiota in the intestinal mucosa of patients with colorectal adenomas. Scientific reports 5, 7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al. (2013). Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager LF, Wasmer M-H, Rau TT, and Krebs P (2016). Cytokine-induced modulation of colorectal cancer. Frontiers in oncology 6, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Sharma D, St Charles J, Dybas L, and Mansfield L (2013). Contrasting immune responses mediate Campylobacter jejuni-induced colitis and autoimmunity. Mucosal immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Sharma D, Zhu Q, Karki R, Guy CS, Vogel P, and Kanneganti T-D (2016). IL-33 regulates the IgA-microbiota axis to restrain IL-1α-dependent colitis and tumorigenesis. The Journal of Clinical Investigation 126, 4469–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, Sharma D, Li L, Malireddi RS, and Gurung P (2015). Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 162, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie H, Main J, Pennington C, and Parratt D (1990). Antibody to selected strains of Saccharomyces cerevisiae (baker’s and brewer’s yeast) and Candida albicans in Crohn’s disease. Gut 31, 536–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H, Biette K, Jedlicka P, Brodsky KS, and Gerich ME (2017). Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. Journal of Experimental Medicine, jem. 20160462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen OH (2014). New Strategies for Treatment of Inflammatory Bowel Disease. Frontiers in Medicine 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, Low JS, Harman CC, Graham M, Elinav E, and Flavell RA (2015). Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 163, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolsky DK, and Isselbacher KJ (1984). Glycoprotein composition of colonic mucosa. Specific alterations in ulcerative colitis. Gastroenterology 87, 991–998. [PubMed] [Google Scholar]
- Qiu X, Zhang F, Yang X, Wu N, Jiang W, Li X, Li X, and Liu Y (2015). Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Scientific reports 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JM (1996). Unifying hypothesis for inflammatory bowel disease and associated colon cancer: sticking the pieces together with sugar. The Lancet 347, 40–44. [DOI] [PubMed] [Google Scholar]
- Ruland J (2008). CARD9 signaling in the innate immune response. Annals of the New York Academy of Sciences 1143, 35–44. [DOI] [PubMed] [Google Scholar]
- Saijo K, Schmedt C, Su I. h., Karasuyama H, Lowell CA, Reth M, Adachi T, Patke A, Santana A, and Tarakhovsky A (2003). Essential role of Src-family protein tyrosine kinases in NF-kB activation during B cell development. Nature immunology 4, 274–279. [DOI] [PubMed] [Google Scholar]
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai R-M, Wang E, Ma W, Haines D, O’hUigin C, et al. (2010a). MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. The Journal of Experimental Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O’HUigin C, et al. (2010b). MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med 207, 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome biology 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, and Kanneganti TD (2016). The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. The Journal of cell biology 213, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, and Kanneganti TD (2017). Inflammatory cell death in intestinal pathologies. Immunological reviews 280, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Malik A, Guy CS, Karki R, Vogel P, and Kanneganti T-D (2017a). Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Malik A, Steury MD, Lucas PC, and Parameswaran N (2015). Protective Role of β-arrestin2 in Colitis Through Modulation of T-cell Activation. Inflammatory bowel diseases 21, 2766–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Sharma BR, Vogel P, and Kanneganti T-D (2017b). IL-1β and Caspase-1 Drive Autoinflammatory Disease Independently of IL-1α or Caspase-8 in a Mouse Model of Familial Mediterranean Fever. The American Journal of Pathology 187, 236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin G, Kang T-W, Yang S, Baek S-J, Jeong Y-S, and Kim S-Y (2011). GENT: gene expression database of normal and tumor tissues. Cancer informatics 10, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Conway KL, Zhang M, Choi M, Morin B, Cao Z, Villablanca EJ, Li C, Wijmenga C, and Yun SH (2013). Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology 145, 591–601. e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Leducq V, Aschard H, Pham H-P, Jegou S, Landman C, Cohen D, Liguori G, Bourrier A, Nion-Larmurier I, et al. (2017). Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, and Mori H (2003). A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer science 94, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström B, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, and Asplund A Tissue-based map of the human proteome,(2015). Science 347, 6220. [DOI] [PubMed] [Google Scholar]
- Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, Brown J, Funari VA, Wang HL, and Crother TR (2016). Immunological consequences of intestinal fungal dysbiosis. Cell host & microbe 19, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, Heid B, Cecere TE, and Allen IC (2015). The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. Journal of immunology (Baltimore, Md. : 1950) 194, 3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, and Kanneganti TD (2010a). The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, and Kanneganti T-D (2010b). IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. The Journal of Immunology 185, 4912–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, and Kanneganti TD (2010c). IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. Journal of immunology (Baltimore, Md. : 1950) 185, 4912–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Fan HW, Zhang JZ, Wang YM, and Xing HJ (2015). NLRP3 rs35829419 polymorphism is associated with increased susceptibility to multiple diseases in humans. Genetics and molecular research : GMR 14, 13968–13980. [DOI] [PubMed] [Google Scholar]
- Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC, Monsuur AJ, Bevova M, Nijmeijer RM, van’t Slot R, and Heijmans R (2008). Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CArD9 and IL 18RAP. The American Journal of Human Genetics 82, 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.