

Abstract
Background
Although cell lines are an essential resource for studying cancer biology, many are of unknown ancestral origin, and their use may not be optimal for evaluating the biology of all patient populations.
Methods
An admixture analysis was performed using genome‐wide chip data from the Catalogue of Somatic Mutations in Cancer (COSMIC) Cell Lines Project to calculate genetic ancestry estimates for 1018 cancer cell lines. After stratifying the analyses by tissue and histology types, linear models were used to evaluate the influence of ancestry on gene expression and somatic mutation frequency.
Results
For the 701 cell lines with unreported ancestry, 215 were of East Asian origin, 30 were of African or African American origin, and 453 were of European origin. Notable imbalances were observed in ancestral representation across tissue type, with the majority of analyzed tissue types having few cell lines of African American ancestral origin, and with Hispanic and South Asian ancestry being almost entirely absent across all cell lines. In evaluating gene expression across these cell lines, expression levels of the genes neurobeachin line 1 (NBEAL1), solute carrier family 6 member 19 (SLC6A19), HEAT repeat containing 6 (HEATR6), and epithelial cell transforming 2 like (ECT2L) were associated with ancestry. Significant differences were also observed in the proportions of somatic mutation types across cell lines with varying ancestral proportions.
Conclusions
By estimating genetic ancestry for 1018 cancer cell lines, the authors have produced a resource that cancer researchers can use to ensure that their cell lines are ancestrally representative of the populations they intend to affect. Furthermore, the novel ancestry‐specific signal identified underscores the importance of ancestral awareness when studying cancer.
Keywords: admixture, African ancestry, Asian ancestry, cancer genomics, clinical genetics, genomic ancestry, population genetics, precision medicine
Short abstract
Preclinical cancer cell line research is often conducted without an awareness of ancestral background, which results in incongruities between the genetic backgrounds of used cell lines and the patient populations they are intended to represent. By calculating genetic ancestry for 1018 common cancer cell lines and identifying ancestry‐specific expression and somatic mutation patterns, the importance of ancestral awareness is emphasized, and a resource is provided that can be used by cancer researchers to ensure that their cell lines are ancestrally representative of the populations they aim to impact.
Introduction
Cell lines function as an essential resource for studying cancer biology, and their use in the testing of initial hypotheses has resulted in seminal findings and important progress.1, 2 Therefore, the National Institutes of Health (NIH) and other experts have placed an emphasis on improving cell line characterization, including genetically, so that phenotypic heterogeneity between cell types can be better controlled and research reproducibility and generalizability can be improved.3, 4, 5, 6 One aspect of cancer cell line characterization that is notably lacking is the identification and annotation of ancestral composition.7 The Catalogue of Somatic Mutations in Cancer (COSMIC)8 (Wellcome Sanger Institute, Hinxton, UK) reports ancestry or race information for only approximately 30% of the >1000 cell lines they annotate and for which they provide genomic data. With the remaining approximately 70% of COSMIC cell lines reporting unknown ancestral status, and with preclinical studies often failing to report the ancestral characterization of the cell lines they use, it is easy to imagine scenarios in which researchers use cell lines and/or cell line data that are not ancestrally representative of the patient populations for whom their findings are intended to apply.
This is likely of important consequence, as studies have demonstrated significant interancestral differences in cancer cell phenotypes and in the types and frequencies of molecular alterations that drive oncologic disease.9, 10, 11, 12, 13, 14 For example, Bateman et al recently used proteomics and transcriptomics to identify ancestry‐specific molecular alterations in European and African ancestry individuals with endometrial cancer and then correlated a subset of these to ancestry‐specific progression‐free survival.15 The prostate cancer‐driving fusion gene TMPRSS2‐ERG (transmembrane serine protease 2–v‐ets erythroblastosis virus E26 oncogene homolog) has been identified at significantly different rates in ancestrally distinct populations, with twice the frequency of this cancer causing alteration reported in European populations compared with African and Asian populations.16 Thus, a lack of awareness about the ancestral makeup of cancer cell lines may lead to unaccounted for biologic differences, and, in turn, can reduce the ability to control for heterogeneity between ancestrally distinct cell lines and/or limit the replication of previous study results.
Therefore, it is essential that researchers have the information necessary to ensure that their cell lines and preclinical research studies are maximally reflective of the diseases and patient populations they are studying. Recent NIH and National Cancer Institute (NCI) initiatives emphasize this and have called for increased ancestry awareness and minority‐focused resource development.17 To this end, we use genetic estimates to ancestrally characterize the genomes of 1018 cell lines from the COSMIC database for which genotypic data are available. By utilizing chip data for these samples in combination with genome sequence data from ancestrally diverse samples from the 1000 genomes project and a separate Native American cohort, we employed admixture analysis to produce the first quantitative ancestral annotations of these cancer cell lines. We then stratify our ancestral annotations by primary tissue and histology type, and demonstrate distinct ancestral imbalances, including marked African and Hispanic under‐representation, in the majority of cell lines regardless of source. In providing further support of the biologic differences between cancer cell lines of differing ancestral origins, we observe significant differences in gene expression and single‐base mutation types across ancestrally distinct cell lines. These annotations of genetic ancestry can serve as a resource for preclinical scientists interested in knowing the ancestral compositions of the cell lines with which they work. It is our hope that this characterization will help control for phenotypic heterogeneity between cell lines, improve research reproducibility, aid in experimental design and clinical trial patient selection, and facilitate more appropriate and precise cancer research for patients of all ancestral backgrounds.
Materials and Methods
Data Source and Quality Control
We accessed the publicly available cell line panel Affymetrix 6.0 chip data (Affymetrix Inc, Santa Clara, CA) from COSMIC version 83 for 1018 cell lines. This contained certain annotation data, including tissue source and histology of the sample. The COSMIC group made both simple and complex calls of genotype, and we combined all cell lines according to the simple calls. Missing data were evaluated for each site and sample.
We then combined this call set with 2504 individuals from the 1000 genomes project phase 3 data18 and 88 Native American individuals from the study by Bigham et al,19 who also were genotyped using the Affymetrix 6.0 chip. We removed any variant that was not found at the intersection of the 3 data sources or that was missing in at least 1% of individuals. In addition, we removed all G‐C or A‐T variants for which the strand was ambiguous.
We then pruned single nucleotide polymorphisms (SNPs) for linkage and minor allele frequency (MAF), as all subsequent analyses assume independence among SNPs. To prune all SNPs, we used the plink20 linkage‐pruning algorithm command “–indep‐pairwise 50 5 0.1,” which uses a window of 50 with an r2 > 0.1 and a SNP step of 5. All SNPs with an MAF <1% were removed.
By using intermediate admixture analyses (described below), we filtered SNPs that were strongly associated with the unidentified cluster within the Cell Line Panel samples. We removed 7409 variants in the first iteration and 2847 variants in the second iteration. Although these were the variants that were most strongly associated with the unidentified cluster, we were unable to completely remove the signal, as it is genome‐wide. However, removal to this level did allow us to examine the ancestry signal, which is most pertinent to the scope of this article. With the removal of these variants, we were left with a data set of 94,593 SNPs.
Cryptic Kinship
With these data, we identified low levels of kinship within our combined data set. We estimated kinship coefficients using the program KING,21 which suggests a threshold of 0.0442 as the lower end of third‐degree (ie first cousin) relations. By using this threshold, we identified 73 pairs, 11 of which came from the Cell Line Panel. We then used a heuristic to retain the largest sample set by removing a sample identified in the most pairs, then updating the number of the remaining samples’ connections. After this procedure, we removed 55 individuals, including 5 from the Cell Line Panel and 7 from the Native American data. The remaining related individuals from the 1000 Genomes Project represent known relationships.18 This left us with a total of 3555 individuals for the analysis of ancestry.
Ancestry Analyses
We calculated the principal components from this unrelated data set with the program KING21 and then observed the output using R (R Foundation for Statistical Computing, Vienna, Austria).22 We also estimated ancestral components using the program ADMIXTURE,23 which uses an unsupervised learning algorithm to estimate the proportion of the genome in each sample that corresponds to a given number of clusters (K) and does this in a manner similar to that of a K‐means clustering algorithm. When used in combination with individuals of known ancestry, the output from this approach can be interpreted as genome‐wide ancestry proportions. To correctly optimize the estimates, we ran 20 replicates with random start seeds for each K tested, and we selected the replicate with the best log‐likelihood (ie, model fit) for that K. We ran K from 2 to 8 to estimate the proportions of continental admixture. The results of K = 6 are presented throughout, because this is most representative of continental divisions, and subsequent clustering divides the continents in ways that cannot be validated as thoroughly. After sorting based on population label and major admixture cluster (eg, African proportion in African populations and European proportion in European populations), as determined by individuals from known source populations (ie, 1000 Genomes Project and Bigham et al), we used R for visualization.
Accounting for Copy Number Changes Within Admixture Analysis
To evaluate any potential effects of large‐scale copy number changes on our admixture estimates, we repeated our admixture analysis after removing all genotypes that existed in regions with copy number changes that effected allelic balance. In other words, using COSMIC’s complex genotype calls, which take copy number into account by using calls generated by the predicting integral copy numbers in cancer (PICNIC) algorithm,24 we only retained genotypes for which no copy number changes existed (ie, complex genotype is identical to simple genotype) or for which the allelic ratio of the complex genotype was identical to that of the simple genotype (eg, AATT vs AT, AAA vs AA, etc). Because all simple genotypes are made up of only 2 alleles, this ensures that there are no SNPs remaining in the analysis that have allelic balances other than 0%, 50%, or 100%. We refer to the genotypes that remain in this analysis as being harmonized between complex and simple calls, and this approach should mitigate any effects from copy number changes.
NCI60 Cell Lines Analysis
The list of cell lines used in anticancer drug screens by the NCI’s Developmental Therapeutics Program was obtained from the NCI website.25 Considerations for which cell lines make up the original NCI60 cell lines and which have been added more recently also were determined from information available on these NCI web pages. Numerous NCI60 cell lines are duplicates, including the MDA‐MB‐435, MDA‐N, and M14 cell lines, which are all derived from the same individual; NCI/ADR‐RES, which derives from the same individual as OVCAR‐8; and SNB‐19, which is from the same individual as U251.26 Ultimately, we determined genetic estimates for 59 of the 70 nonredundant NCI60 cell lines that are used in anticancer drug screens.
Stratification by Tissue and Histology Type
We used cell line annotations from COSMIC to stratify cell lines according to the primary tissue site and histology type listed for each cell line. Tissue sites and histology types that were represented by fewer than 5 cell lines in our data set were excluded from our stratification analyses and visualizations. Of the 1013 unrelated cell lines for which we estimated genetic ancestry, all 1013 had tissue site and histology data.
Correlation of Gene Expression and Ancestry
Gene expression data were downloaded from COSMIC within the file named “CosmicCompleteGeneExpression.tsv.gz.” After parsing the data and combining them with our ancestral estimates, we had 959 cell lines remaining that had both ancestry and gene expression data. With these normalized expression data, we ran linear models on each of 16,681 genes to assess the relationship between expression level and ancestry after accounting for tissue site and histology type. These models can be represented as:
| (1) |
with βa (correlation coefficient) estimated separately for European, African, and East Asian ancestry, and all histology and tissue types included as covariates, with residual ε. When correcting for multiple testing in the maximally conservative fashion with a total number of 50,043 (16,681 × 3), our threshold for family‐wide significance at the .05 level is 9.99 × 10−7. At this level, the neurobeachin line 1 (NBEAL1) gene is associated significantly with ancestry. We also corrected for multiple testing in a slightly less conservative fashion by using the number of annotated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, which was 523, instead of the number of genes tested. This was based on the logic that gene expression between genes is not independent and is likely to associate for genes in similar biologic pathways. Applying this less conservative multiple testing correction required a P value of 9.56 × 10−5 to achieve significance at the .05 family‐wide level. At this level, we identified additional genes with expression levels that were associated significantly with ancestry.
Relation Between Mutation Types and Ancestry
Mutation data were downloaded from COSMIC and represented coding and noncoding point mutations from targeted and genome‐wide screens (downloaded files named “CosmicMutantExport.tsv.gz” and “CosmicCLP_NCVExport.tsv.gz”). Only single nucleotide mutations were included, and 1009 cell lines had both mutation and ancestry data. After counting the number of mutations of each of the 12 possible types for each cell line, we ran linear models for each mutation type to evaluate the contribution of ancestry to mutation counts and proportions. First, we ran separate analyses in which only cell lines of the same tissue type were tested for linear relations between mutation type and ancestry. Next, the relation between each mutation type and ancestry was tested across all cell lines within a linear framework that accounted statistically for tissue site and histology type (Eq. 2). Thus, these models can be understood as:
| (2) |
and we independently tested the influence of European, African, and East Asian ancestry on each of the 12 mutation types. Tissue and histology were controlled for as covariates with residual ε. To correct for multiple testing, we used a Bonferroni correction with a conservative number of 36 (3 ancestries × 12 mutation types). This is notably conservative, because each ancestry is not completely independent of another, and one‐half of the 12 mutations are related to 1 another by base pairing relations. Nonetheless, we produced significant results after conservatively correcting.
Results
By using the genetic resources developed by the COSMIC Cell Lines Project, we were able to characterize the genetic ancestry of 1018 commonly used cancer cell lines. After identifying and removing cryptic relatedness between different cell lines, we used admixture analysis to estimate the ancestral proportions of each cell line.23 We then demonstrated imbalances in the ancestral composition of cell lines across a variety of primary tissue and primary histology types. Finally, we used these genetic estimates to test genome‐wide for differences in gene expression and somatic mutation type across cell lines of differing ancestral proportions, and we report significant differences.
Cryptic Relatedness Among the COSMIC Cell Line Panel
We identified 11 pairs of cell lines that exhibited relatedness at the same level as first cousins (Supporting Table 1). According to the COSMIC website, all samples sent for genotyping were prescreened using a barcode of 94 SNPs and 16 short tandem repeats, and all identical samples were removed.27, 28 Consistent with this, we identified no sample pairs with relatedness levels of siblings to monozygotic twins, and the relations we did identify likely were the result of both real first‐cousin relationships as well as relationships that appear close because of similar types and levels of contamination. It is noteworthy that 9 of 11 related cell line pairs derive from different primary tumor sources, but may share substantial genetic background. Furthermore, we observed multiple cell lines that belong to 2 or more closely related pairs and likely represent contamination sources or biases in sample acquisition. By using a heuristic approach to identify the maximum number of unrelated cell lines (see Materials and Methods, above), we removed 5 related cell lines before proceeding to downstream analyses.
Quantitative Estimates of Ancestry for Cancer Cell Lines
Across the remaining unrelated 1013 cell lines for which we estimate genetic ancestry (Supporting Table 2), we observe significant ancestral heterogeneity, with European, East Asian, and African ancestries most dominantly represented. Among the 312 cell lines for which ancestry is reported by the COSMIC database, we observed that the genetic ancestral estimates failed to match the reported ancestry in 7 cases (Fig. 1, Supporting Fig. 1, Supporting Table 3). Part of this is because of the mislabeling of individual cell lines between different data repositories. For example, the popular cell line FaDu, which represents 1 of only 3 cell lines in our data set with >40% South Asian ancestry, is listed as Caucasian or white within COSMIC and by at least 1 vendor.29 However, at least 1 other vendor lists FaDu cells as Indian in origin,30 and the original publication concurs with our assessment of a South Asian (Indian) origin.31
Figure 1.
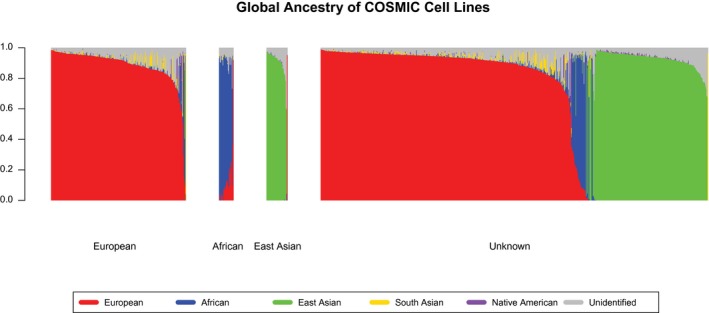
Genomic Ancestry estimates for 1009 cell lines from the Catalogue of Somatic Mutations in Cancer (COSMIC) database are illustrated. Each vertical bar represents a different cell line, and the height of the bar (y‐axis) represents the total genomic ancestry proportion. The height of each color represents the proportion of the ancestry represented by that color. The group labeled “European” on the x‐axis represents cells lines for which the ancestry was reported by COSMIC as European (n = 244). The vast majority of these cell lines are comprised of European ancestry (red), with some cell lines exhibiting predominantly African ancestry (blue) and some exhibiting small amounts of South Asian ancestry (gold). Cell lines reported as African (n = 26) exhibit predominantly African ancestry (blue) as well as a gradation of European ancestry proportion (red). Cell lines reported as East Asian (n = 38) are almost exclusively of East Asian ancestry, except for 1 inaccurately reported cell line that exhibits exclusively European ancestry (red). Among the 701 cell lines for which ancestry was reported as “unknown,” 453 were of predominantly European ancestry (red), 30 were of predominantly African ancestry (blue), and 215 were of predominantly East Asian ancestry. Within this group of cell lines with previously unknown ancestry, the predominantly African cell lines were admixed the most, followed by the predominantly European cell lines, and then the mostly nonadmixed cell lines of East Asian origin.
Within the 701 cell lines for which ancestry was reported as unknown or not listed in the COSMIC database, we identify additional ancestral heterogeneity, including 453 cell lines with predominantly (ie, >50%) European ancestry, 215 with predominantly East Asian ancestry, 30 with predominantly African ancestry, and 1 with exclusively South Asian ancestry (CAL‐85‐1) (Fig. 1, Supporting Figs. 1 and 2,18 Supporting Table 2). The predominantly African ancestry cell lines exhibit significantly higher levels of admixture than the predominantly European or predominantly East Asian cell lines, and the various degrees of European and African admixture observed in these cell lines fits with the demographic histories of the African Americans from whom these cell lines likely originate.32, 33 The predominantly European ancestry cell lines exhibit some Native American ancestry (Fig. 1, Supporting Fig. 1, purple), as well as notable levels of South Asian ancestry (Fig. 1, Supporting Fig. 1, gold). The latter is likely reflective of noise that results from the long‐term relatedness of Europeans and South Asians deriving from out‐of‐Africa migrations, rather than from recent admixture between modern‐day European and South Asian individuals.34 The cell lines identified as predominantly East Asian exhibit the lowest levels of admixture, as most feature exclusively East Asian ancestry or trace amounts of Native American ancestry (with whom Asians are more closely related than are Europeans or Africans35).
Native American ancestry, which is generally present at significant levels in Latino individuals,18 is notably underrepresented across the entire cancer cell line data set. Only 1 cell line had predominantly Native American ancestry (UACC‐812 cells; 49.1%); and only 14 individuals, or 1.38% of our data set, had Native American ancestry proportions ≥8% (Supporting Table 2). This underrepresentation of Native American ancestry is similar to that observed across The Cancer Genome Atlas36 and indicates the need to increase ancestral representation across cancer genome resources.6, 37
To evaluate any potential effects of large‐scale copy number changes on our admixture estimates, we repeated our admixture analysis after removing genotypes that exist in regions with copy number changes that effect allelic balance. In other words, we only retained genotypes for which no copy number changes exist or for which the allelic ratio of the genotype is not affected by copy number changes. This should enable an admixture analysis that is unaffected by copy number changes. The resultant admixture analysis produces ancestry estimates that are correlated nearly perfectly with the original estimates (Supporting Fig. 3), which support the robustness of the admixture algorithm and our use of it to the copy number changes that exist across the cancer cell lines we analyzed.
Unidentified Cluster Among Cell Lines
We also identified an additional genetic cluster within the data that is present at consistent proportions throughout all cell lines (Fig. 1, Supporting Fig. 1, gray). Although this signal may represent a type of batch effect or contamination, which is known to exist in cell lines,38 its persistence after multiple rounds of filtering on variants that drive the signal, and its nonclustered and genome‐wide nature, suggest that contamination is an unlikely source. Although HeLa cells have been reported as contaminants of cell banks,39 the proportion of the HeLa cell line’s genome that is comprised by this signal is low (estimated at 6% after variant removal) and does not seem to be a major driver of this cluster signal. The absence of this cluster in the reference samples we analyzed, some of which were genotyped using the same technology, suggests it is not an artifact of our workflow. Somatic mutations almost certainly are not responsible for this cluster, because we restrict our analyses to SNPs identified in all reference samples, which should leave virtually no somatic mutations across our analysis. In addition, our analysis controlling for copy number changes demonstrates that these also do not significantly influence this cluster (Supporting Fig. 3). We also performed an admixture analysis that used supervised learning to cluster the genome according to reference samples reflecting 5 continental ancestries (European, African, East Asian, South Asian, and Native American). Although such an approach is highly dependent on the genomic variation represented by the used reference samples, and it is not as effective as our original unsupervised approach at identifying inherent genomic clustering, it helps to demonstrate the stability of our ancestral estimates despite the existence of the unidentified sixth cluster. Our original estimates are highly concordant with these supervised admixture estimates, despite the supervised approach increasing the proportion of European, African, and East Asian admixture to accommodate the portions of the genome no longer being attributed to the 6th cluster (Supporting Fig. 4). Therefore, because this signal seems to represent some kind of database or laboratory artifact, and because we were able to selectively filter the variants that drive this signal to unambiguously estimate genetic ancestry, we report this signal but do not address it further (for additional filtering descriptions, see Materials and Methods, above).
Ancestry of the NCI60 Cell Lines
Next, we examined ancestry estimates for the subset of cell lines that belong to the NCI60 cell lines, which are curated by the NIH and used regularly for anticancer drug assessments and screens.2, 40 Of the 1013 COSMIC cell lines for which we estimated genetic ancestry, 59 belonged to the expanded NCI60 list, which now consists of 70 nonredundant cell lines (Supporting Table 4). We observed a strong dominance of European ancestry within this group of cell lines, with all but 3 (94.9%) exhibiting almost exclusively European ancestry (Fig. 2). While these other 3 cell lines were of predominantly African ancestry, our analysis suggests that anticancer drug screens that use the NCI60 cell line resource are being done almost exclusively on European genetic backgrounds. This poses serious questions about whether the results of these screens are applicable to populations of predominantly non‐European ancestry. Regarding the unidentified cluster described above, the significantly lower level (P = 1.964 × 10−5; Welch 2‐sample t test) of this signal estimated within NCI60 cell lines (Supporting Fig. 5) supports the possibility that this signal reflects contamination, as the NCI60 cell lines are known to have reduced contamination levels.26
Figure 2.
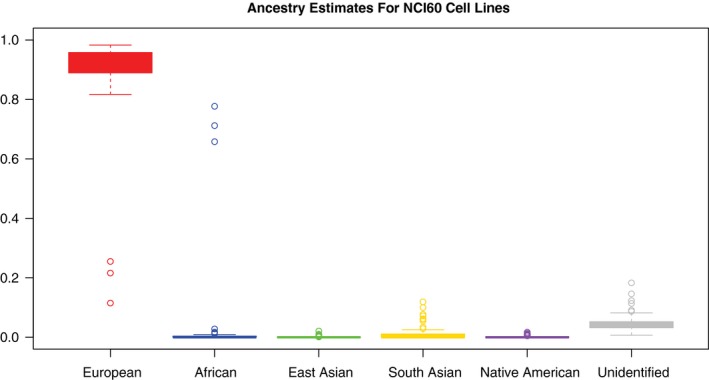
Ancestry estimates for NCI60 cell lines are illustrated. Boxplots represent the distribution of ancestry proportion estimates across 59 of the 70 nonredundant cell lines belonging to the expanded NCI60 anticancer drug screening resource. Nearly all of the cell lines are of almost entirely European ancestry: only 3 cell lines exhibit predominantly African ancestry, and none of the cell lines exhibit notable proportions of East Asian, South Asian, or Native American ancestry.
Tissue‐Specific Imbalances in Ancestral Sampling
To determine how well represented each ancestry is across the cell lines of different cancers, we grouped cell lines by their derived tissue and histology types and evaluated the resultant distributions (Fig. 3, Supporting Fig. 6). We observed that the ancestral representation between tissue types was unbalanced, which is particularly informative when considered in the context of the incidence and mortality rates between races.41 Cell lines from stomach, liver, prostate, pancreas, and kidney cancers, which have significantly increased incidence rates in individuals of African ancestry,41 have relatively little representation of African ancestry. Other than 1 stomach cell line and 1 kidney cell line with significant African ancestry, the remaining cell lines across these cancers are dominated by either East Asian cell lines (stomach and liver cancer cell lines), European cell lines (prostate and kidney cancer cell lines), or both (pancreatic cancer cell lines). Prostate cancer cell lines are notable for having entirely European ancestry in our data set, despite prostate cancer being the most common cancer among all men42 and having an estimated incidence rate that is 70% higher among men of African ancestry than it is among men of European ancestry.41 This is concordant with recent results from Woods‐Burnham and colleagues, who identified substantial African ancestry in a commercially available prostate cancer cell line with previously unknown ancestry, and noted the lack of racial diversity in commercially available prostate cancer cell lines.7 In esophageal cancer, which is less common among males of African ancestry than among those of European ancestry but significantly more common among females of African ancestry than among those of European ancestry,41 cell lines demonstrate a similar dearth of African genomic ancestry. Although endometrial cancer is more common and more severe among individuals of African ancestry, and has distinct molecular alterations in these individuals,9, 10, 11, 13, 14, 15 no endometrial cancer cell lines from our data set have any African ancestry. In contrast to these patterns, while breast and lung cancers have disparate incidence rates between individuals of African and European ancestry that invert across sex (higher in males of African ancestry and lower in females of African ancestry),41 both organ site malignancies have significant numbers of cell lines with predominantly African ancestry. However, although there are significant numbers of lung cancer cell lines with predominantly European, African, and East Asian ancestry, <5% of breast cancer cell lines in our data set represent East Asian genomes. Finally, hematopoietic and lymphoid cancers, which have varying degrees of ancestral disparity in incidence depending on the specific cancer subtype, appear to be the best balanced with regard to ancestral composition, and have significant numbers of cell lines with predominantly European, African, and East Asian ancestry. In fact, one of only 2 cancer genomes in our data set with >40% Native American ancestry is of hematopoietic and lymphoid origin.
Figure 3.
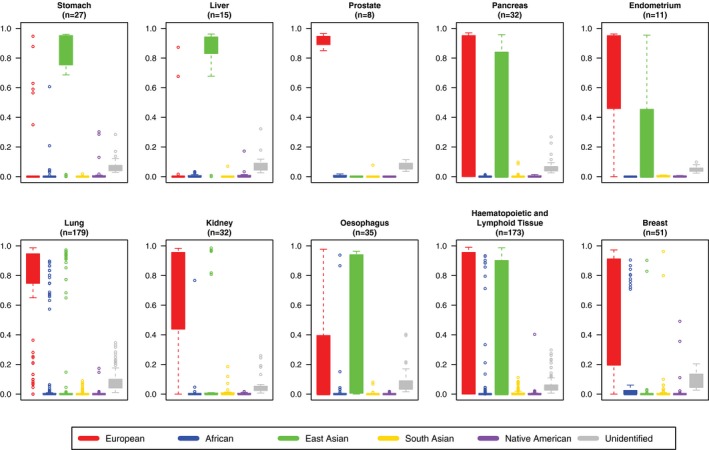
Distributions of genetic ancestry estimates across 10 racially disparate cancers are illustrated. Boxplots represent the distributions of ancestry proportion estimates for cell lines belonging to each of 10 cancer types. These 10 representative cancers, which are indicated above each chart (with the numbers of cell lines shown below the name of each tumor type), exhibit different incidence and/or mortality rates across ancestrally distinct populations (for all analyzed cancer types, see Supporting Fig. 2). Most cancer cell lines are of predominantly European (red), East Asian (green), or European and East Asian ancestry. Exceptions to this are lung, hematopoietic and lymphoid, and breast tumors, which have significant numbers of cells lines with predominantly African ancestry (blue). Cell lines from prostate cancer, which is 1 of the most common cancers in all men, have only European ancestry. Few or no cell lines from cancers that have significantly high incidence rates among individuals of African ancestry, like cancers of the stomach, liver, pancreas, and kidney, have African ancestry (blue).
Similarly, we observe unbalanced ancestral representation across 13 histologic types, with a predominance of European and East Asian ancestry cell lines, and an absence of African and Native American ancestry cell lines (Supporting Fig. 7). One exception to these patterns is within cell lines from rhabdomyosarcomas, which have significant African ancestry and lack East Asian ancestry. Although carcinoma cell lines are the most ancestrally diverse of all of our groupings (including tissue type) and have significant ancestral representation from Europeans, Africans, East Asians, and even South Asians and Native Americans, this seems to be a feature of large sample size, which is able to overcome the study selection bias that shapes which human populations are represented in cancer cell line research.
Gene Expression Differences by Ancestry: NBEAL1 and Other Candidates
For each of the 16,681 genes for which expression data were available from COSMIC, we tested the association between European, African, and East Asian ancestry proportions and normalized gene expression across 959 cell lines for which all data were available. After accounting for tissue and histology type and correcting for multiple testing, we observed evidence of a significant correlation between African ancestry proportion and the expression level of the NBEAL1 gene. NBEAL1 has been associated with brain function and cancers,43, 44, 45, 46 and we observe that cancer cells with higher proportions of African ancestry have lower NBEAL1 expression levels (Fig. 4, Supporting Table 5). If we relax the stringency of the multiple testing correction and correct for the number of genome‐wide pathways (as denoted by KEGG47) rather than the number of genes (with the logic that gene expression between genes is not independent), expression levels of the solute carrier family 6 member 19 (SLC6A19), HEAT repeat containing 6 (HEATR6), and epithelial cell transforming 2 like (ECT2L) genes also become significantly associated with ancestry proportion (Fig. 4, Supporting Table 5). Overall, numerous other genes demonstrate suggestive associations with ancestry proportion, and although they are not significant after correcting for multiple testing, these genes are likely to represent good follow‐up candidates for research into the effects of ancestry on cancer biology.
Figure 4.
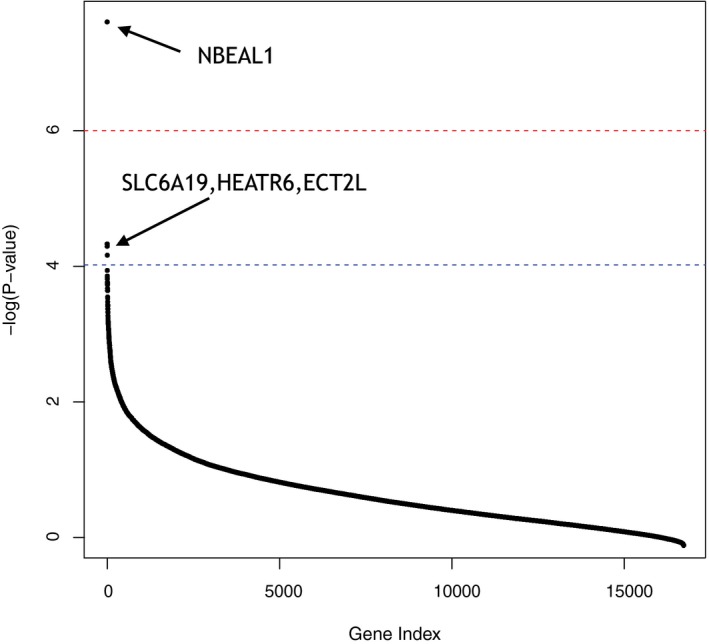
Associations between ancestry proportion and gene expression levels are illustrated. Results shown are from analyses estimating the relation between ancestry proportions and gene expression levels for 16,681 genes across 959 cancer cell lines. After the most conservative multiple testing correction, the NBEAL1 gene (arrow) is associated significantly with gene expression and African ancestry (Supporting Table 5). Three additional genes, SLC6A19, HEATR6, and ECT2L (arrow) are associated significantly with European ancestry at a threshold set by a more moderate multiple testing correction.
Differences in Mutation Type by Ancestry
To directly assess how differences in ancestry relate to differences in cancer genome architecture, we used our ancestral estimates to determine the correlation between single‐base somatic mutation proportions (eg, A>T, G>A) and ancestral proportions. Across the 1009 COSMIC cell lines that had mutation data, tissue‐of‐origin metadata, and ancestry estimates, we tested whether European ancestry, African ancestry, or East Asian ancestry proportions were correlated with mutation type for each of the 12 possible single‐base mutations. First, we performed individual analyses among cell lines that had the same tissue type (ie, intratissue‐type analyses), which demonstrated significant relations between mutation type and ancestry in some tissues (eg, lung, central nervous system, autonomic ganglia, skin, pancreas) and almost no relations in other tissues (eg, stomach, ovary, kidney) (Supporting Table 6). Interestingly, hematopoietic and lymphoid tissues exhibited a significant increase in overall mutation burden in cell lines that had increased proportions of African ancestry.
To get a better sense of the association between ancestry and somatic mutation in cancer overall, we performed additional analysis of the relationship between ancestry and mutation after accounting for tissue and histology type. This was done using linear models that account for variation in mutation due to cancer type by featuring tissue and histology type as covariates. After correcting for multiple testing, we observed significant associations across all cancer cell lines between both European and East Asian ancestry and the proportion of A>G, C>A, G>T, and T>C mutations (P < 4 × 10−4) (Table 1, Supporting Table 7, Supporting Figs. 8 and 9). These relationships persist in both coding and noncoding regions across the genome, and represent the most robust associations observed in our previous intratumor‐type analyses. Of these 4 mutations, there were 2 complimentary pairs (eg, A>G is the same as T>C on the opposite strand) that had similar correlation coefficients (βa) and P values, and that acted as an internal corroboration of this result.
Table 1.
Associations Between Ancestry Proportion and Single Nucleotide Mutation Proportions
| Mutation | Ancestry | |||||||
|---|---|---|---|---|---|---|---|---|
| European | African | East Asian | Unidentified | |||||
| βa | P | βa | P | βa | P | βa | P | |
| A>C | 0.001 | .287 | 0.001 | .703 | −0.002 | .088 | .009 | .330 |
| A>G b | −0.013 | 1.675 × 10−8 c | 0.010 | .041 | 0.012 | 1.267 × 10−6 c | −0.009 | .573 |
| A>T | 0.002 | .123 | −0.002 | .509 | −0.002 | .182 | 0.008 | .417 |
| C>A b | 0.009 | 1.688 × 10−3 c | 0.008 | .156 | −0.012 | 6.9 × 10−5 c | −0.015 | .426 |
| C>G | 0.002 | .188 | −0.005 | .232 | −0.002 | .448 | 0.038 | .004 |
| C>T | −0.000 | .935 | −0.020 | .027 | 0.006 | .173 | −0.008 | .796 |
| G>A | −0.006 | .168 | −0.009 | .286 | 0.008 | .057 | −0.020 | .502 |
| G>C | 0.003 | .103 | −0.005 | .169 | −0.001 | .505 | 0.022 | .103 |
| G>T b | 0.012 | 1.814 × 10−4 c | 0.007 | .221 | −0.015 | 6.984 × 10−7 c | −0.022 | .276 |
| T>A | 0.002 | .181 | 0.002 | .389 | −0.003 | .048 | 0.010 | .304 |
| T>C b | −0.013 | 2.78 × 10−8 c | 0.011 | .024 | 0.011 | 7.625 ×10−6 c | −0.016 | .352 |
| T>G | 0.000 | .805 | 0.001 | .734 | −0.001 | .458 | 0.004 | .651 |
Abbreviations: A, adenosine; βa, correlation coefficient; C, cytosine; G, guanine; T, thymidine.
Results are from association analyses estimating the effect of ancestry on the proportion of single nucleotide mutations made up by each of 12 possible mutation types. The proportions of 4 mutation types differ significantly by ancestry across the 1009 cancer cell lines for which mutation and other data were available.
bThese 4 mutation types represent 2 independent mutation classes after accounting for reverse complementation (ie, A>G implies T>C), with significance levels and βa values representing concordant associations between complimentary mutation types.
cThese P values were significant after corrections for multiple testing.
Discussion
The objective of the current study was to investigate the ancestral origins of >1000 cancer cell lines commonly used in preclinical cancer research. By using admixture analysis to generate the first quantitative estimates of ancestry proportion across these cell lines, we provide a resource that can be used by scientists and clinicians to select cancer cell lines in an ancestry‐aware fashion for various experimental purposes. Given the recent focus by the cancer research community on the limited quality, reproducibility, and translatability of cancer cell line research1, 3, 4, 48 and on the nonrepresentation by cancer resources of minority populations,6, 17 having this knowledge is timely and important. Our findings of ancestral imbalances across cancer cell lines, and of novel ancestry‐specific biologic differences, underscore the need to increase the diversity of available cell lines and improve the equitability of cancer research.
The ancestral heterogeneity we identified across COSMIC cell lines is important and likely results primarily from the ways that cancer data are accumulated globally. Factors such as which countries are most heavily involved in preclinical cancer research, which cancers are predominantly studied in which countries, and how heavily integrated a country’s research initiatives are into the global science community are likely to shape the ancestral makeup of available cancer cell lines. For example, the hundreds of East Asian genomes we identified are almost certainly from Japan and China, where medical research, and cancer research in particular, is very robust,49 and where study populations are comprised almost exclusively of East Asian individuals. Nonetheless, regardless of the multitude of factors that shape the ancestral imbalances we report across cancer cell lines, the underrepresentation of African, Native American, and South Asian ancestry cell lines, as well as cell lines from other non‐European and non‐East Asian individuals, represents a problem in the preclinical cancer setting governed by traditional cancer cell lines. Currently, many cancers can be studied only as specific genetic backgrounds. Therefore, it is imperative that we establish new and ancestrally diverse cell lines that more accurately represent diverse populations.
By using our genetic ancestral estimates to identify multiple genes with expression differences across ancestry, we produced novel evidence of biologic differences between ancestrally distinct cancer cell lines. These differences persist after accounting for tissue type and histology, and further support the need to be aware of ancestry, and potentially to account for it, when working preclinically with cancer cells. The NBEAL1 gene has significantly lower gene expression levels in more African cancer genomes (Fig. 4, Supporting Table 5). Although the precise role of NBEAL1 has not been clearly defined, it is linked to vesicle trafficking, signal transduction, and neuronal proliferation and development.43, 44, 46 It is noteworthy that NBEAL1 overexpression has been implicated in brain cancer,44, 45 so this reduced NBEAL1 expression could play a role in the significantly lower incidence of brain cancer in individuals of African ancestry.41 The SLC6A19 gene exhibits lower expression in increasingly European cells lines (Fig. 4, Supporting Table 5), and represents a transporter family that reportedly is up‐regulated in and associated with cancers.50, 51 The HEATR6 gene, which is part of a highly expressed breast cancer amplicon52 and has also been associated with disease through interactome studies,53 has higher expression in more European cell lines and lower expression in more East Asian cell lines. Furthermore, recent efforts have identified elevated expression of HEATR6 in endometrial tumors from individuals of African ancestry, compared with those of European ancestry, and have associated HEATR6 protein levels with disease outcome only in individuals of African ancestry.15 Although these findings represent different analyses and separate results, they are all complementary in their support of an ancestry‐specific role for HEATR6 in cancer biology. The ECT2L gene has lower expression levels in more European cell lines and reportedly has recurrent mutations in leukemia.54 These genes represent promising targets for follow‐up research into the relation between cancer biology, gene expression, and ancestry. Genes that do not reach significance after multiple testing correction but still have a suggestive correlation with ancestry may represent reasonable candidates for follow‐up research that has increased power (see Supporting Table 5). Although we did not observe a signal of ancestry‐specific expression among consensus cancer genes (as annotated by COSMIC), these genes have large oncogenic effect sizes and may exert similar influences across various ancestral backgrounds. Furthermore, many of these genes may shape oncologic processes through nontranscriptional mechanisms. Nonetheless, despite our conservative assessment of the correlation between expression and ancestry (with limited statistical power), we still observed novel ancestry‐specific gene expression differences that underscore the importance of ancestry awareness in cancer research.
In testing how single‐base mutation types correlate with ancestry proportions, we identified mutational differences that correlated with ancestry within the cell lines of numerous cancer types. The proportions of 4 single‐base mutation types that represent 2 distinct mutational classes (ie, A>G/T>C and C>A/G>T) (Table 1) are associated most robustly with ancestry after accounting for tissue and histology type across all cancer cell lines. Although the differences we observed in mutation type proportions are on the order of 1% or 2% and may seem small, they are actually quite relevant when compared with the mutation differences and heterogeneity often observed within and between tumor types.55 When not accounting for ancestry, the mutational differences identified across cancers might be ascribed exclusively to differences in tumor type (for example, kidney cancer vs ovarian cancer), whereas our results demonstrate that these differences may be significantly influenced by ancestry (ie, European vs East Asian). Furthermore, these differences agree with recent findings of mutational differences across ancestry,56, 57 and extend such mutational work into a cancer genome context. However, it is important to note that calling somatic mutations is notoriously difficult, and that even though COSMIC somatic calls represent one of the best curated sets to date, somatic mutation call sets are often plagued by high false‐positive rates.58, 59, 60 Therefore, it is best to interpret these mutational results conservatively, and to focus on what these patterns suggest about considering ancestry when studying the genomic variation of cancers. Ultimately, these mutational patterns represent ancestrally correlated genomic differences, and accounting for them can help preclinical and translational studies become more representative of one another.
Our findings of differential gene expression and distinct mutational profiles across ancestry require additional research and add to the increasingly robust evidence of biologic differences across ancestrally distinct cancers.9, 10, 11, 13, 14, 15 If the cancer community wants to be able to effectively study various forms of oncologic disease and to capture and adequately model the biologic distinctness that different ancestral backgrounds represent, then it is imperative that we rectify the ancestral imbalances identified here and widen the scope of our preclinical studies. Such an effort will likely increase our knowledge of cancer biology while also increasing the number of patients who can benefit from advances in cancer research.
Funding Support
Michael D. Kessler is supported by a grant from the National Institutes of Health (NIH) (T32CA154274). Michael D. Kessler and Timothy D. O’Connor were supported by funding from the Center for Health Related Informatics and Bioimaging at the University of Maryland School of Medicine, institutional support for the Institute for Genome Sciences and Program in Personalized Genomic Medicine at the University of Maryland School of Medicine, an NIH Genomic Commons award (OT3 OD025459‐01), and a National Heart, Lung, and Blood Institute Trans‐Omics for Precision Medicine Program high‐performance grant (U01 HL137181‐01). This work was supported in part by an NIH Transformative Research Award (1‐R01‐CA 206,188 to Julie C. Dunning Hotopp), the Inova Schar Cancer Institute (Thomas P. Conrads), the Congressionally Directed Medical Research Program (W81XWH‐16‐2‐0038 to Thomas P. Conrads), the Murtha Cancer Center (HU0001‐16‐2‐0014 to Thomas P. Conrads), and the Uniformed Services University of the Health Sciences from the Defense Health Program (HU0001‐16‐2‐0006 to Nicholas W. Bateman, Thomas P. Conrads, and George L. Maxwell).
Conflict of Interest Disclosures
The author made no disclosures.
Author Contributions
All authors conceived of the project. Michael D. Kessler and Timothy D. O’Connor designed and performed experiments, analyzed data, and wrote the manuscript. Nicholas W. Bateman, Julie C. Dunning Hotopp, Thomas P. Conrads, and George L. Maxwell assisted with the editing and review of the manuscript.
Supporting information
We thank Stuart S. Martin, Toni M. Antalis, Curt I. Civin, Andrew Berchuck, and Aksinija A. Kogan for helpful comments and discussions.
Genomic data that support the findings from this study are available from the Catalogue of Somatic Mutations in Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cell_lines/download). Other supporting data, including genetic ancestry estimates and source data for figures, are available within the paper and its Supporting Information files.
Contributor Information
Michael D. Kessler, Email: michael.kessler@som.umaryland.edu.
Timothy D. O’Connor, Email: timothydoconnor@gmail.com.
References
- 1. Masters JR. HeLa cells 50 years on: the good, the bad and the ugly. Nat Rev Cancer. 2002;2:315‐319. [DOI] [PubMed] [Google Scholar]
- 2. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813‐823. [DOI] [PubMed] [Google Scholar]
- 3. Begley CG, Ellis LM. Drug development: raise standards for preclinical cancer research. Nature. 2012;483:531‐533. [DOI] [PubMed] [Google Scholar]
- 4. Lorsch JR, Collins FS, Lippincott‐Schwartz J. Fixing problems with cell lines. Science. 2014;346:1452‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. National Institutes of Health (NIH) . Implementing Rigor and Transparency in NIH & AHRQ research grant applications NOT‐OD‐16‐011. Bethesda, MD: NIH; 2015. Available at: https://grants.nih.gov/grants/guide/notice-files/not-od-16-011.html. Accessed October 17, 2018. [Google Scholar]
- 6. Polite BN, Adams‐Campbell LL, Brawley OW, et al. Charting the future of cancer health disparities research: a position statement from the American Association for Cancer Research, the American Cancer Society, the American Society of Clinical Oncology, and the National Cancer Institute. CA Cancer J Clin. 2017;67:353‐361. [DOI] [PubMed] [Google Scholar]
- 7. Woods‐Burnham L, Basu A, Cajigas‐Du Ross CK, et al. The 22Rv1 prostate cancer cell line carries mixed genetic ancestry: implications for prostate cancer health disparities research using pre‐clinical models. Prostate. 2017;77:1601‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high‐resolution. Nucleic Acids Res. 2016;45(D1):D777‐D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clifford SL, Kaminetsky CP, Cirisano FD, et al. Racial disparity in overexpression of the p53 tumor suppressor gene in stage I endometrial cancer. Am J Obstet Gynecol. 1997;176:S229‐S232. [DOI] [PubMed] [Google Scholar]
- 10. Maxwell GL, Risinger JI, Hayes KA, et al. Racial disparity in the frequency of PTEN mutations, but not microsatellite instability, in advanced endometrial cancers. Clin Cancer Res. 2000;6:2999‐3005. [PubMed] [Google Scholar]
- 11. Santin AD, Bellone S, Siegel ER, et al. Racial differences in the overexpression of epidermal growth factor type II receptor (HER2/neu): a major prognostic indicator in uterine serous papillary cancer. Am J Obstet Gynecol. 2005;192:813‐818. [DOI] [PubMed] [Google Scholar]
- 12. Malone KE, Daling JR, Doody DR, et al. Prevalence and predictors of BRCA1 and BRCA2 mutations in a population‐based study of breast cancer in white and black American women ages 35 to 64 years. Cancer Res. 2006;66:8297‐8308. [DOI] [PubMed] [Google Scholar]
- 13. Oliver KE, Enewold LR, Zhu K, et al. Racial disparities in histopathologic characteristics of uterine cancer are present in older, not younger blacks in an equal‐access environment. Gynecol Oncol. 2011;123:76‐81. [DOI] [PubMed] [Google Scholar]
- 14. Maxwell GL, Shoji Y, Darcy K, et al. MicroRNAs in endometrial cancers from black and white patients. Am J Obstet Gynecol. 2015;212:191.e1‐191.e10. [DOI] [PubMed] [Google Scholar]
- 15. Bateman NW, Dubil EA, Wang G, et al. Race‐specific molecular alterations correlate with differential outcomes for black and white endometrioid endometrial cancer patients. Cancer. 2017;123:4004‐4012. [DOI] [PubMed] [Google Scholar]
- 16. Zhou CK, Young D, Yehoah ED, et al. TMPRSS2: ERG gene fusions in prostate cancer of West African men and a meta‐analysis of racial differences. Am J Epidemiol. 2017;186:1352‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. National Cancer Institute, National Institutes of Health, Department of Health and Human Services . Minority Patient‐Derived Xenograft (PDX) Development and Trial Centers (M‐PDTCs) (U54). Vol 2018. Available at: https://grants.nih.gov/grants/guide/rfa-files/rfa-ca-17-032.html. Accessed October 17, 2018.
- 18. 1000 Genomes Project Consortium , Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bigham A, Bauchet M, Pinto D, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data [serial online]. PLoS Genet. 2010;6:e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome‐wide association studies. Bioinformatics. 2010;26:2867‐2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 23. Alexander DH, Novembre J, Lange K. Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655‐1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Greenman CD, Bignell G, Butler A, et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Developmental Therapeutics Program, Division of Cancer Treatment & Diagnosis, National Cancer Institute, National Institutes of Health . Cell Lines in the In Vitro Screen. Frederick, MD: National Cancer Institute; 2015. Available at: https://dtp.cancer.gov/discovery_development/nci-60/cell_list.htm. Accessed October 17, 2018. [Google Scholar]
- 26. Lorenzi PL, Reinhold WC, Varma S, et al. DNA fingerprinting of the NCI‐60 cell line panel. Mol Cancer Ther. 2009;8:713‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wellcome Sanger Institute . COSMIC Catalogue of Somatic Mutations in Cancer. Available at: https://cancer.sanger.ac.uk/cosmic. Accessed October 17, 2018.
- 28. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43(database issue):D805‐D811, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. American Type Culture Collection (ATCC) . FaDu cells (ATCC HTB‐43). Available at: https://www.atcc.org/en/Products/All/HTB-43.aspx. Accessed October 17, 2018.
- 30. German Collection of Microorganisms and Cell Cultures GmbH . FaDu cells. Available at: https://www.dsmz.de/catalogues/details/culture/ACC-784.html?tx_dsmzresources_pi5%5BreturnPid%5D=192. Accessed October 17, 2018.
- 31. Rangan S. A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer. 1972;29:117‐121. [DOI] [PubMed] [Google Scholar]
- 32. Bryc K, Auton A, Nelson MR, et al. Genome‐wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci U S A. 2010;107:786‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kessler MD, Yerges‐Armstrong L, Taub MA, et al. Challenges and disparities in the application of personalized genomic medicine to populations with African ancestry [serial online]. Nat Commun. 2016;7:12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461:489‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nielsen R, Akey JM, Jakobsson M, Pritchard JK, Tishkoff S, Willerslev E. Tracing the peopling of the world through genomics. Nature. 2017;541:302‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19:A68‐A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spratt DE, Chan T, Waldron L, et al. Racial/ethnic disparities in genomic sequencing. JAMA Oncol. 2016;2:1070‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. MacLeod RA, Dirks WG, Matsuo Y, Kaufmann M, Milch H, Drexler HG. Widespread intraspecies cross‐contamination of human tumor cell lines arising at source. Int J Cancer. 1999;83:555‐563. [DOI] [PubMed] [Google Scholar]
- 39. Gartler SM. Apparent HeLa cell contamination of human heteroploid cell lines. Nature. 1968;217:750‐751. [DOI] [PubMed] [Google Scholar]
- 40. Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev Res. 1995;34:91‐109. [Google Scholar]
- 41. American Cancer Society . Cancer Facts & Figures for African Americans 2016‐2018. Atlanta, GA: American Cancer Society; 2016. [Google Scholar]
- 42. National Cancer Institute, Centers for Disease Control and Prevention . US Cancer Statistics: Data Visualizations. Leading Cancer Cases and Deaths, Male, 2015. Atlanta, GA: CDC; 2017. Available at: https://gis.cdc.gov/cancer/USCS/DataViz.html. Accessed February 3, 2019. [Google Scholar]
- 43. Verhaaren BF, Debette S, Bis JC. Multi‐ethnic genome‐wide association study of cerebral white matter hyperintensities on MRI. Circ Cardiovasc Genet. 2015;8:398‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen J, Lu Y, Xu J, et al. Identification and characterization of NBEAL1, a novel human neurobeachin‐like 1 protein gene from fetal brain, which is up regulated in glioma. Mol Brain Res. 2004;125:147‐155. [DOI] [PubMed] [Google Scholar]
- 45. Bao C, Yang H, Li N, et al. Cloning, expression and purification of novel gene NBEAL1 and its relationship with pathological grades of glioma. Chinese J Cancer Biother. 2010;17:77‐81. [Google Scholar]
- 46. Karimzadhagh J, Salehgargari S, Omrani M. Characterization of a de novo constitutional balanced translocation t(2;11)(q33.2;q23.2) with break point on the human NBEAL1‐GeneHo. Iran J Child Neurol. 2018;12:94‐100. [PMC free article] [PubMed] [Google Scholar]
- 47. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016;45:D353‐D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gillet JP, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst. 2013;105:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elsevier BV . Cancer Res.: Current Trends & Future Directions. New York: Elsevier BV; 2016. Available at: https://www.elsevier.com/?a=230374. Accessed October 17, 2018. [Google Scholar]
- 50. Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. Gain at chromosomal region 5p15. 33, containing TERT, is the most frequent genetic event in early stages of non‐small cell lung cancer. Cancer Genet Cytogenet. 2008;182:1‐11. [DOI] [PubMed] [Google Scholar]
- 51. Bhutia YD, Ganapathy V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta. 2016;1863:2531‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sinclair CS, Rowley M, Naderi A, Couch FJ. The 17q23 amplicon and breast cancer. Breast Cancer Res Treat. 2003;78:313‐322. [DOI] [PubMed] [Google Scholar]
- 53. Stebbing J, Zhang H, Xu Y, et al. Reprogramming of the tyrosine kinase‐regulated proteome in breast cancer by combined use of RNA interference (RNAi) and stable isotope labelling with amino acids in cell culture (SILAC) quantitative proteomics. Mol Cell Proteomics. 2015;14:2479‐2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T‐cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kandoth C, McLelland MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harris K. Evidence for recent, population‐specific evolution of the human mutation rate. Proc Natl Acad Sci U S A. 2015;112:3439‐3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Harris K, Pritchard JK. Rapid evolution of the human mutation spectrum [serial online]. Elife. 2017;6:e24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation [serial online]. Sci Transl Med. 2015;7:283ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Teer JK, Zhang Y, Chen L, et al. Evaluating somatic tumor mutation detection without matched normal samples [serial online]. Hum Genomics. 2017;11:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mannakee BK, Balaji U, Witkiewicz AK, Gutenkunst RN, Knudsen ES. Sensitive and specific post‐call filtering of genetic variants in xenograft and primary tumors. Bioinformatics. 2018;34:1713‐1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials