

Abstract
Replication fork stability during DNA replication is vital for maintenance of genomic stability and suppression of cancer development in mammals. ATR is a master regulatory kinase that activates the replication stress response to overcome replication barriers. While many downstream effectors of ATR have been established, the upstream regulators of ATR and the impact of such regulation on liver cancer remain unclear. The ubiquitin conjugase BRUCE is a guardian of chromosome integrity and activator of ATM signaling which promotes DNA double-strand break repair via homologous recombination. Here we demonstrate new functions for BRUCE in ATR activation in vitro and liver tumor suppression in vivo. BRUCE is recruited to induced DNA damage sites. Depletion of BRUCE inhibited multiple ATR-dependent signaling events during replication stress, including activation of ATR itself, phosphorylation of its downstream targets CHK1 and RPA, and the mono-ubiquitination of FANCD2. Consequently, BRUCE deficiency resulted in stalled DNA replication forks and increased firing of new replication origins. The in vivo impact of BRUCE loss on liver tumorigenesis was determined using the hepatocellular carcinoma model induced by genotoxin Diethylnitrosamine. Liver-specific knockout of murine Bruce impaired ATR activation and exacerbated inflammation, fibrosis and hepatocellular carcinoma, which exhibited a trabecular architecture, closely resembling human HCC. In humans, the clinical relevance of BRUCE downregulation in liver disease was found in hepatitis, cirrhosis and hepatocellular carcinoma specimens and deleterious somatic mutations of the Bruce gene was found in human hepatocellular carcinoma in TCGA database. Conclusion: These findings establish a new BRUCE-ATR signaling axis in accurate DNA replication and suppression of liver cancer in mice and humans and provides a clinically relevant HCC mouse model.
Keywords: Liver cancer, DEN, DNA repair, Liver injury
Introduction
Genome integrity is crucial for cell survival and tumor suppression. To protect the genome, cells have evolved a DNA damage response (DDR) program that coordinates DNA replication, DNA repair and cell cycle progression (1). The ATM (ataxia-telangiectasia mutated) and ATR (ATM and RAD3-related) protein kinases act as master regulators of the DDR program. While they share many functional similarities, ATM primarily functions in response to DNA double-strand breaks (DSBs), whereas ATR mainly regulates damaged replication forks and is essential for the viability of replicating cells (2).
BIR repeat containing ubiquitin-conjugating enzyme (BRUCE) is a large protein (530kD) that was initially identified as an inhibitor of apoptosis (3, 4). BRUCE is a ubiquitin (Ub) conjugase and ligase, which catalyzes post-translational protein ubiquitination to regulate apoptosis (5–8), promotes cell division and mouse embryogenesis (6, 8). Importantly, we have discovered BRUCE promotes ATM activation and permits efficient access of multiple DDR factors to repair DNA DSBs via homologous recombination (HR) repair. Additionally, we published that BRUCE-deficient cells display spontaneous chromosomal breaks and gaps (9, 10).
Replication fork stability is also vital for genome stability (11). The slow progression or stalling of replication forks is known as replication stress, which arise from replication barriers of nucleotide depletion, DNA damage, RNA-DNA hybrids, and oncogene activation. As stalled forks are vulnerable to breakage and collapse, cells have developed genome surveillance mechanisms to resolve replication barriers which are orchestrated by the ATR kinase. ATR is activated during every S-phase of the cell cycle by the presence of single-stranded DNA (ssDNA) coated by the RPA protein complex at replication forks (12). ATR phosphorylates a number of downstream proteins to stabilize stalled forks and delay cell cycle progression. Among the downstream proteins is the checkpoint kinase, CHK1, which diffuses to the cell nucleus to prohibit new origin firing. Meanwhile, ATR orchestrates multiple DNA repair pathways at stalled forks, including HR to repair DNA breaks and the Fanconi Anemia (FA) pathway to fix DNA interstrand cross links (ICLs).
DNA ICLs are highly toxic replication stressors that prevent separation of DNA strands and block replication and transcription (13). DNA ICLs can be generated by endogenous metabolite products with DNA crosslinking capabilities or exogenously by exposure to chemotherapeutic drugs, including Mitomycin C (MMC) and Cisplatin (14). Upon DNA crosslink formation, FANCD2, a key molecule required for ICL repair, is mono-ubiquitinated in an ATR-dependent manner (15) by the FA core complex which is an E3 ligase complex composed of eight FA proteins (13, 14). Mono-ubiquitinated FANCD2 along with FANCI form the I-D2 complex which is subsequently loaded onto ICL lesions where the complex serves as a platform for recruitment and activation of multiple DNA repair factors required to resolve DNA ICLs (16–18).
Genome instability can be induced by DNA DSBs and replication stress both of which can generate the spontaneous chromosomal abnormalities we observed in BRUCE-deficient cells (9, 10). In this study, we utilized biochemistry, cell biology, and proteomic approaches to assess the function of BRUCE in ATR signaling under replication stress conditions. We further investigated the in vivo role of this BRUCE-ATR signaling axis in the suppression of liver tumorigenesis by using a mouse liver cancer model. We then examined the clinical relevance of BRUCE deficiency in hepatocellular carcinoma using a human liver disease array, as well as the somatic gene mutation data available from The Cancer Genome Atlas (TCGA). Collectively, this study provides the first critical view on the functional importance of the BRUCE-ATR axis in replication fork stability and liver tumor suppression in mice and humans.
Materials and Methods
Generation of Genetic Conditional LKO Mice
The Bruce floxed conditional mouse strain was generated by inGenious Targeting Lab; https://www.genetargeting.com). Loxp sequences were inserted into the endogenous Bruce locus flanking exon 2. Bruce loxp/+ embryonic stem cells were microinjected into C57BL/6 blastocysts. Resulting chimeras with a high percentage agouti coat color were mated to C57BL/6 FLP mice to remove the Neo cassette. Germline transmission of the Bruce loxp/+ allele was confirmed by PCR genotyping.
Liver-specific Mouse Experiments
All animal experiments were performed in accordance with guidelines approved by our Institutional Animal Care and Use Committee. To generate the Albumin (Alb)-Cre Bruce liver-specific KO mice: Bruceflox/flox mice (C57BL/6) were crossed with Alb-Cre mice (Jackson Labortories; stock number 018961). Genotypes were confirmed by PCR. The Bruce Alb-Cre KO mice were analyzed with the WT mice (Bruceflox/flox; Cre-) as control. For initiating acute hepatic stress responses, 2- to 3-month-old male mice were intraperitoneally (i.p.) injected with DEN (Sigma, #N0756) at 100mg/kg of body weight. WT and KO mice were randomly grouped and sacrificed after 1, 3, 5, and 10 days of DEN exposure. For HCC induction, DEN was delivered into 14-day old male mice by i.p. at 25mg/kg of body weight. WT and KO mice were sacrificed upon 14 months of exposure to DEN and livers collected for further studies.
DNA Fiber Analysis of DNA Replication
DOX-BRUCE U2OS cells were pre-labeled with IdU for 30 min, treated with HU (2 mM) for 120 min, and then post-labeled with CldU for 60 min. Cells were then fixed, immunostained with IdU (green) and CldU (red) antibodies, and counterstained with DAPI (blue). DNA fiber labeling and quantification of percentages of cells with three major types of labeled DNA tracts (stalled forks after HU treatment; new origin firing and fork stalling) were conducted using a published method [3]. Statistics were conducted by Paired Student’s t test.
Clonogenic Assay
Cells grown in 60 mm culture dish with a confluency at ~ 300 cells per dish in the medium containing MMC or HU were harvested after 7–10 days of continuous culture, fixed and stained in crystal violet solution (Sigma, HT901). Cell clones with more than 50 cells were counted and survival percentage was normalized to untreated cells which calculated as 100%.
Liver RNA and cDNA Isolation
Liver samples were placed in RNAlater® Solution (Ambion, #AM7020) and kept at 4°C. Liver RNA isolation was performed using the mirVana™ miRNA Isolation Kit (Ambion, #AM1560) according to the manufacturer’s protocol. For liver cDNA isolation: cDNA was isolated from RNA templates using the reaction setup according to instructions from the BioRad iScript™ cDNA Synthesis Kit (BioRad, #1708890). Reaction protocol includes: 5 mins at 25°C, 30 mins at 42°C, 5 mins at 85°C and (optional) hold at 4°C. For semi-quantitative and RT-PCR, a 1/10 cDNA solution was made using RNAse free water.
ImageJ Analysis of IHC data
For pATR, pRPA32, and PRP19, nuclear staining positivity was analyzed using the “Fiji” version of ImageJ software. Image was opened. Color Deconvolution was selected for images stained specifically in the nuclei. To decrease the interference of cytoplasmic staining, images that had nuclear and cytoplasmic staining, under the image pull down, RGB stack was selected under type. To decrease cytoplasmic signal, go to Image>Type>RGB stack. Once the RGB window appears, select Image>Stacks>Make Montagne then perform color deconvolution. For both nuclear-specific and other images, select the Vectors pull-down> “H DAB”. The “Colour_2” image window was selected and measured. The units of intensity derived in the Results window were transferred to an Excel spreadsheet. The optimal density (O.D.) was calculated using the formula, O.D. = log (max intensity/mean intensity), where the max intensity should be 255. The average optimal density and standard deviations were calculated and graphed.
Results
BRUCE is required for cell clonogenic survival and activation of the ATR-CHK1-RPA pathway during DNA replication stress
MMC generates DNA ICLs between complimentary DNA strands that block replication forks, while hydroxyurea (HU) induces replication stress by depleting the nucleotide pool required for DNA synthesis. To determine the impact of BRUCE on replication stress response, wild type and Bruce KO mouse embryonic fibroblasts (MEFs) were exposed to MMC and HU at a range of doses and clonogenic survival was measured. BRUCE KO cells were more sensitive to both MMC and HC exposure as compared to WT cells (Fig. 1 A and 1B), suggesting a role for BRUCE in protecting cells against replication stressors.
Fig. 1. BRUCE is required for the replication stress response and ATR activation.
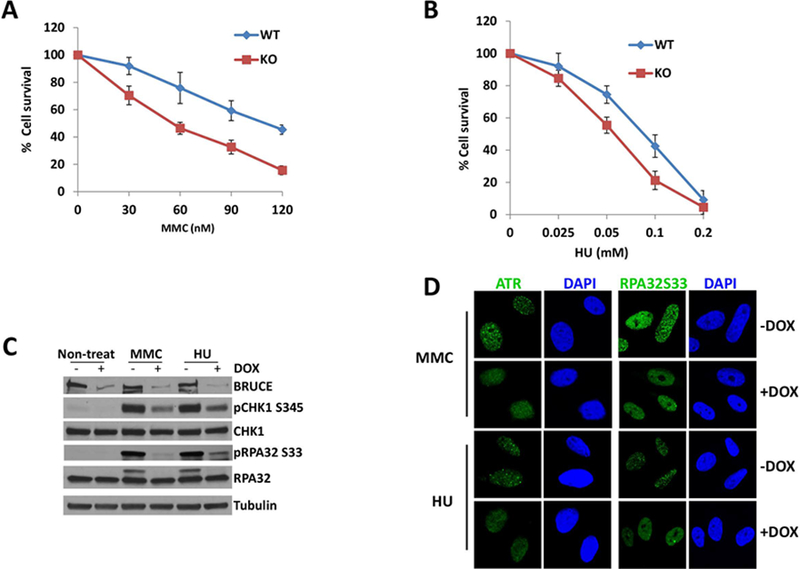
(A) Clonogenic survival assay shows that cells are sensitive to MMC treatment in the absence of BRUCE.
(B) Clonogenic survival assay shows that cells are more sensitive to HU treatment in the absence of BRUCE.
(C) U2OS shBRUCE cells were treated with DOX to deplete BRUCE. MMC or HU was added to medium at day 3 with a final concentration of 1 μm and 2 mM respectively. After another 24 hrs in culture, cells were collected and subjected to immunoblotting against antibodies as indicated.
(D) U2OS cells were depleted of BRUCE in the presence of DOX and treated with 1 μm MMC or 2 mM HU for 24 hours. Cells were fixed and immunostained for ATR and phospho-RPA32; representative images are shown.
ATR plays an essential role in replication stress response; therefore, we investigated whether the increased cell sensitivity to the replication stressors is due to impaired ATR signaling. Active ATR kinase phosphorylates CHK1 at S345, RPA32 at S33 (the small subunit of the RPA complex) and a variety of other proteins through which ATR activation counters replication stress. BRUCE levels were depleted in U2OS cells by DOX-inducible shBRUCE as we previously described (9, 10). Subsequent Western blot analysis indicated that BRUCE depletion attenuated ATR-dependent phosphorylation of CHK1 at S345 (pCHK1-S345) and RPA32 at S33 (pRPA32-S33) upon induction of replication stress by either MMC or HU treatment (Fig. 1C). Additionally, BRUCE depletion impaired ATR recruitment to damaged chromatin and prevented the formation of ATR repair foci in MMC and HU treated cells (Fig. 1D). This impairment of ATR activity is further supported by the lack of recruitment of ATR-dependent RPA32-pSer33 to DNA repair foci (Fig. 1D; the bright diffuse nuclear staining in +DOX samples does not reflect the punctate pattern associated with repair foci). Similar results were obtained from WT and BRUCE KO mouse tail fibroblasts (not shown). Together these results suggest a requirement for BRUCE in activating the ATR-CHK1-RPA pathway in response to replicative stress.
BRUCE accumulates at DNA damage sites and protects against replication fork stalling and new origin firing
One major function of the ATR-CHK1-RPA signaling is to prohibit fork collapse and suppress unscheduled firing of new origins (19). Due to the compromised ATR replication stress signaling in BRUCE KD cells, we investigated whether BRUCE deficiency also affects replication forks. We conducted DNA fiber analysis following previously published methods (20) which is depicted in the schemes shown in Fig. 2A. We found distinct DNA labeling patterns between WT and KD samples (Fig. 2B and 2C) which indicated that BRUCE KD resulted in significantly elevated stalling of replication forks (Fig. 2D) and an increase in new origin firing (Fig. 2E), two well-established defects observed in ATR-deficient cells (19). ChIP-qPCR analysis found that BRUCE is recuited to DNA damage sites induced by the endounclease I-SecI in a human fibroblast DR95 cell line, which expresses a pDR-GFP plasmid containing I-SceI cleavage sequence (21) (Fig. 2F). These data support a critical function of BRUCE in permitting continuous DNA replication at local replication forks and suppressing distal replication origins in the genome from aberrant firing.
Fig. 2. BRUCE localizes to DNA breaks induced by I-SceI endonuclease and suppresses replication fork stalling and new replication origin firing upon replication stress.
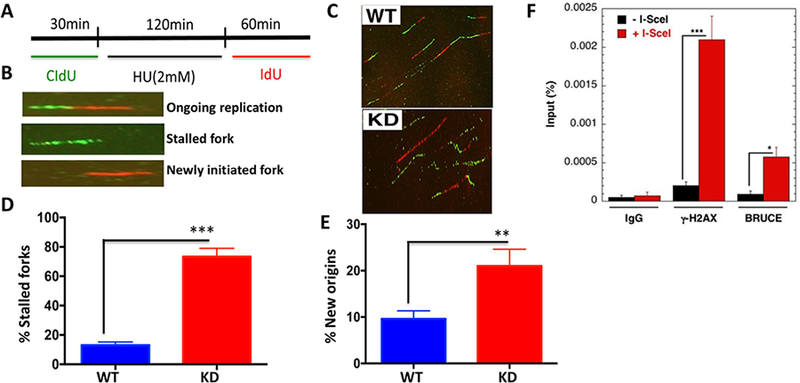
(A) DNA fibers in HU-treated shBRUCE U2OS cells were labeled following the scheme and analyzed as outlined (B). (C) Representative DNA fiber images were taken and the frequency of stalled forks (D) and new origin firing
(E) was quantified. **p < 0.01; ***p<0.001; Paired Student’s t test. (F) Chromatin IP/qPCR of BRUCE and γH2AX (control) with human fibroblast DR-95 cell line showing recruitment of BRUCE to sites of DNA breaks induced by I-SecI endounuclease.
BRUCE is required for ATR-dependent FANCD2 mono-ubiquitination and localization to DNA damage sites in response to replication stress
The more pronounced sensitivity of BRUCE depleted cells to MMC exposure (Fig. 1A) suggested that BRUCE is critical for DNA ICL resolution which requires activation of the canonical FA DNA repair pathway (14, 22). While HU does not induce ICLs, HU induces fork stalling (23) and activates the non-canonical function of the FA pathway (16). The recently defined non-canonical activity of the FA pathway involves stabilizing stalled replication forks, preventing fork collapse (16, 23), suppressing new origin firing and controlling the proper rate of fork progression (24–27). Due to the observed sensitivity to MMC and HU and the impaired replication forks in BRUCE depleted cells (Fig. 2), we examined the impact of BRUCE on the activation of the FA pathway.
FANCD2 mono-ubiquitination and localization to DNA damage foci are two ATR-dependent steps which are hallmarks of FANCD2 activation at ICL lesions and stalled replication forks (15). Mono-ubiquitination of FANCD2 then facilitates the canonical ICL repair by coordinating multiple DNA repair activities required for the resolution of ICLs and protection of replication forks (16–18, 28). The ratio of mono-ubiquitinated to non-ubiquitinated FANCD2 (L/S ratio) is a quantitative indication for FANCD2 activation. Upon MMC and HU exposure, FANCD2 was present mainly in the non-ubiquitinated form in normally growing cells (Fig 3A, FANCD2-S, lane 1) and treatment with HU or MMC activated FANCD2 ubiquitination (FANCD2-L, lanes 3&5). Depletion of BRUCE (+DOX), however, strongly inhibited FANCD2-Ub induced by HU and MMC (lanes 4&6). The L/S ratio also supports an inhibitory effect of BRUCE depletion on HU- and MMC-induced FANCD2 activation (bar graph depicted below each lane; Fig. 3A). To rule out potential off-target effects by the shBRUCE, two BRUCE siRNAs, distinct from shBRUCE and each other, were used and demonstrated similar results (Fig. 3B). BRUCE depletion significantly reduced the amounts of MMC- and HU-inducible FANCD2 nuclear foci (Fig. 3C), which was verified to be BRUCE-specific using a reconstitution experiment involving our published cell lines (Fig. 3D–E) (9, 10). Together these results demonstrate that BRUCE-dependent activation of ATR is required for signal transmission to its downstream target FANCD2. Loss of this BRUCE-ATR-FANCD2 signaling transduction can explain the replication fork instability (Fig. 2) and the enhanced cellular sensitivity to MMC and HU treatment (Fig. 1A and 1B).
Fig. 3. BRUCE is required for activation of the ATR-FA pathway.
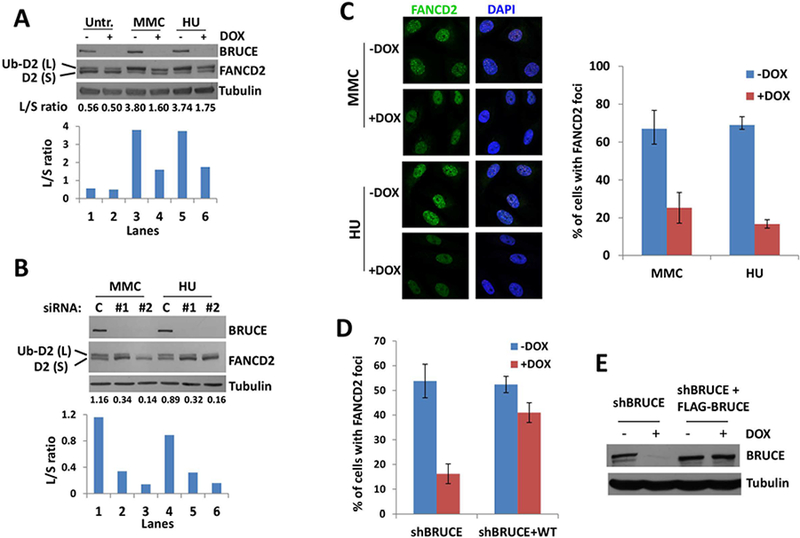
(A) U2OS shBRUCE cells were treated with DOX to deplete BRUCE. MMC or HU were added to medium at day 3 with final concentrations of 1 μm and 2 mM, respectively. After another 24 hrs culture, cells were collected and subject to immunoblot against BRUCE, FANCD2 and Tubulin. Lower bar graph represents the intensity ratio between FANCD2 large band (Ub-D2) and small band (D2); quantification was carried out with ImageJ software.
(B) U2OS cells were depleted of BRUCE by two distinct siRNAs and treated (or mock treated) with MMC or HU for 24 hrs with a final concentration of 1 μm and 2 mM, respectively. Cells were collected and subject to immunoblot against BRUCE, FANCD2 and Tubulin. The lower bar graph represents the intensity ratio of the FANCD2 large band (Ub-D2) and the small band (D2), quantification was performed with ImageJ software.
(C) U2OS shBRUCE cells were depleted of BRUCE by DOX and treated with 1 μm MMC or 2 mM HU for 24 hours. Cells were fixed and stained for FANCD2 foci. Representative images are shown and quantification of FANCD2 foci is displayed on the right, bars represent SD from a triplicate experiment.
(D) Quantification of FANCD2 foci in U2OS shBRUCE cells and shBRUCE cells reconstituted with FLAG-BRUCE (shBRUCE+WT). Error bars represent SD from a triplicate experiment.
(E) Western blot showing abolished BRUCE expression in shBRUCE cells treated with DOX and recovery of BRUCE in shBRUCE cells reconstituted with FLAG-BRUCE (shBRUCE+WT).
BRUCE acts as a scaffold that interacts with PRP19/PRPF19, promoting ATR signaling
BRUCE is a large scaffold protein that interacts with other proteins to orchestrate cellular functions (9, 10). To gain mechanistic insight into how BRUCE regulates ATR signaling, we took an unbiased approach to search for BRUCE interacting proteins. Mass spectrometric analysis of BRUCE immunoprecipitations (IPs) identified the PRP19 (Prpf19) protein as a major BRUCE-interacting partner (Fig. S1). All peptides identified are included in the Supplemental spreadsheet. Their interaction was validated by reciprocal IP showing the presence of PRP19 in BRUCE-IP products (Fig. 4A) and vice versa (Fig. 4B).
Fig. 4. BRUCE interacts with PRP19 which is required for FANCD2 mono-ubiquitination and foci formation.
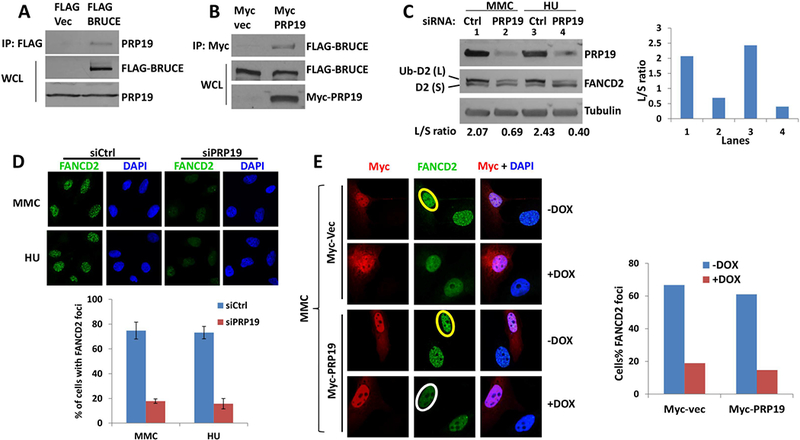
(A) U2OS cells stably expressing of FLAG-BRUCE were lysed and immunoprecipitation was carried out with anti-FLAG antibody. Elution was subjected to SDS-PAGE and immunoblotted for PRP19.
(B) U2OS cells that stably expressed FLAG-BRUCE were transfected with Myc-vector or Myc-PRP19 expression vector, and immunoprecipitation was carried out with anti-MYC antibody. The eluate was subjected to SDS-PAGE and immunoblotting against FLAG (BRUCE).
(C) U2OS cells were depleted of PRP19 by siRNA and treated with MMC or HU for 24 hrs with a final concentration at 1 μm and 2 mM respectively. Cells were collected and subject to immunoblot against BRUCE, FANCD2 and Tubulin, with the intensity of the ratio between FANCD2 large band (Ub-D2) and small band (D2) quantified with ImageJ software and shown to the right.
(D) U2OS cells were depleted of PRP19 by siRNA and treated with 1 μm MMC or 2 mM HU for 24 hours. Cells were fixed and stained for FANCD2 foci, representative images are present with the quantification of FANCD2 foci shown below. Bars represent SD from a triplicate experiment.
(E) U2OS cells were treated with DOX and transfected with MYC vector or MYC-PRP19 expression vector. After treatment with 1 μm MMC for 24 hours, cells were fixed and immunostained for MYC (Red) and FANCD2 (Green). Quantification of FANCD2 foci was shown to the right. Circle in yellow and white showing positive and negative nuclear FANCD2 foci, respectively.
PRP19 (Pre-mRNA-processing factor 19) is an E3 ubiquitin ligase originally found to be involved in pre-mRNA splicing from which its name was derived (29). PRP19 has been shown to regulate DNA repair (29, 30). Specifically, PRP19’s E3 ligase activity catalyzes the non-degradative ubiquitination of RPA thereby promoting RPA-Ub-mediated ATR activation (31, 32). Additionally, PRP19 is implicated in cellular resistance to DNA ICLs and the protection of replication forks during the replication stress response (30). However, it remains unclear whether PRP19 functions at the earlier steps of FANCD2 mono-ubiquitination and its foci formation or during later steps. To test whether BRUCE could promote PRP19-ATR-dependent activation of FANCD2, we measured MMC- and HU-inducible ATR-dependent FANCD2 activation in PRP19 depleted cells and found that FANCD2 mono-ubiquitination and foci formation were both significantly attenuated (Fig. 4C and 4D), implicating PRP19 at the step of ATR-dependent FANCD2 mono-ubiquitination and localization to ICL sites. Although the MMC-inducible FANCD2 foci were formed in BRUCE-proficient cells (Fig. 4E; yellow circles), ectopic overexpression of PRP19 in BRUCE-depleted cells could not rescue FANCD2 foci (Fig. 4E; white circle), demonstrating that BRUCE and PRP19 are new co-regulators required for the ATR-mediated FANCD2 activation.
Studies show that PRP19 mediates ATR activation by promoting non-degradative ubiquitination of RPA (31, 32). Considering BRUCE is a ubiquitin conjugase and is required for RPA foci formation (Fig. 1D), we examined whether BRUCE facilitates RPA ubiquitination to promote ATR signaling. Our results indicated that BRUCE did not impact RPA ubiqutination (Fig. S2). Altogether these data mechanistically establishes that BRUCE acts as a scaffold to bring PRP19 in close proximity to ATR to activate ATR signaling.
Generation of liver-specific Albumin (Alb)-Cre Bruce KO mice
Hepatocytes provide the major detoxification function of the liver and as a result, hepatocytes undergo cell death when damaged. Compensatory hepatocyte proliferation will replenish the liver with new hepatocytes. It is believed that DDR suppresses liver genotoxicity and hepatocarcinogenesis (33, 34); however, the underlying mechanisms are largely unclear. Therefore, we investigated whether Bruce-dependent DDR signaling is physiologically significant for protection of the liver.
A Bruce conditional knockout (cKO) mouse strain was generated by floxing the Bruce gene exon 2 (Bruceloxp/loxp; Fig. 5A) to bypass the embryonic lethality (5, 6). The Bruce liver-specific KO (LKO) mice were generated by crossing cKO with Alb-Cre transgenic mice, termed Bruce LKO (Bruceloxp/loxp;Alb-Cre+) and WT mice (Bruceloxp/loxp;Alb-Cre−) (Fig. 5A). Despite the loss of hepatic BRUCE expression (Fig. 5B), the adult Bruce LKO mice were viable without obvious phenotypic abnormalities under normal conditions (Fig. 5C).
Fig. 5. Bruce liver-specific KO mice are more prone to acute liver injury by DEN.
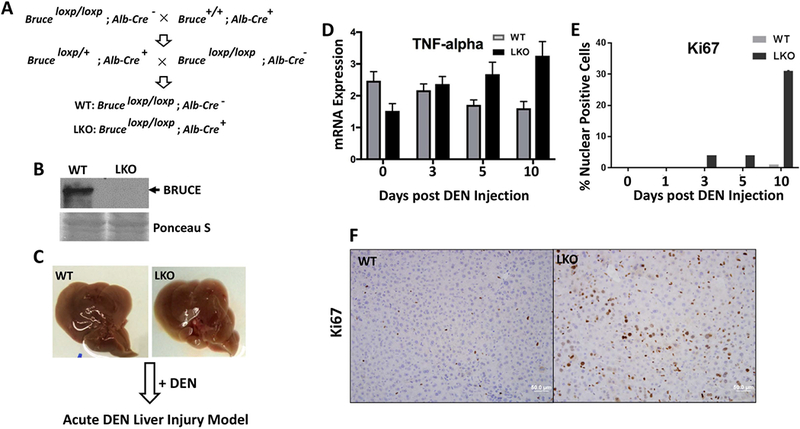
(A) Mouse breeding schematic demonstrates the cross of Bruceloxp/loxp mice with Albumin-Cre positive mice for generation of Bruce liver-specific KO (LKO) mice, genotypes confirmed.
(B) Western blot analysis shows absent of BRUCE protein expression in LKO mice.
(C) No detectible phenotypic differences among WT and LKO mouse livers (2-month age).
(D) Significantly increased mRNA transcription levels of inflammation marker the tumor necrosis factor alpha (TNFα) in LKO livers treated with DEN.
(E) Quantification of Ki67 IHC staining showed a significant increase in compensatory proliferation in LKO livers as compared to WT.
(F) Representative photos of Ki67 IHC staining 10 days post DEN treatment.
The genesis of liver injury and cancer relies not only on genes but also environment (35). The environmental hepatocarcinogen diethylnitrosamine (DEN) is a robust DNA alkylating agent in mice and humans (36). After being bioactivated in the hepatocytes, the metabolites of DEN generate DNA adducts, where the removal of which generates DNA SSBs, DSBs, and ICLs (36). DEN induces an acute liver injury response and when the injury prolongs over months, hepatocellular carcinoma (HCC) deveolops (37).
BRUCE suppresses DEN-induced acute liver injury in mice
Both WT and Bruce LKO male mice (8 weeks old) were given a single dose of DEN (100 mg/kg by i.p.; Fig. 5C). Using a time course study, we found that inflammation levels were increased in Bruce LKO livers as measured by tumor necrosis factor alpha (TNFα) gene expression on days 5–10 (Fig. 5D). Additional inflammation markers were increased including macrophage inflammation proteins MIP1-α (Fig. S3A), IL1β (Fig. S3B), F4/80 (Fig. S3C) and inducible nitric oxide synthase (iNOS) (Fig. S3D). Furthermore, compensatory hepatocyte proliferation (Ki67 staining) increased on days 3–10 in the LKO liver (Fig. 5E–F). These results demonstrate that Bruce LKO accelerates DEN induced acute liver injuries.
BRUCE promotes the ATR DDR signaling during repair of DEN-induced acute DNA damage in mice
Immunohistochemistry (IHC) analysis of the DNA damage marker γH2AX on paraffin embedded liver tissue revealed elevated γH2AX expression in both WT and LKO samples on day 1 (Fig. 6A; quantification Fig. 6B). Hepatic blood circulates from the portal vein (PV) to the midzone before draining into the central vein (CV). Hepatocytes surrounding the PV receive oxygenated blood whereas those in the CV zone are nutrient deficient and relatively hypoxic with low oxygenated blood (38).
Fig. 6. Bruce LKO liver has increased level of DEN-induced DNA damage and impaired ATR-PRP19 activity.
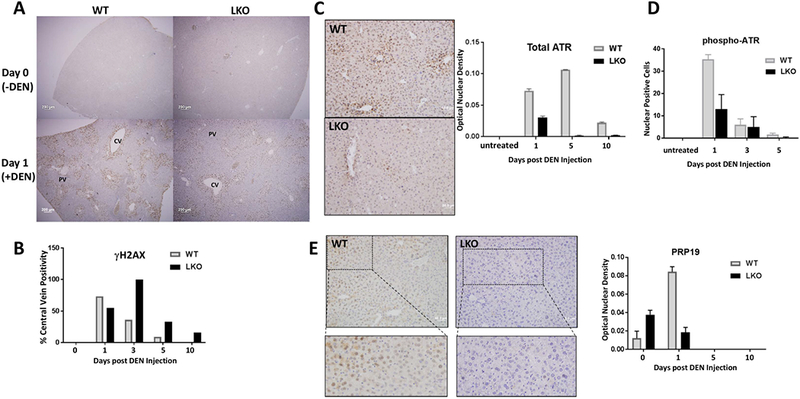
(A) IHC staining of γH2AX reveals DNA damage in both WT and LKO mouse livers one day post DEN induction.
(B) IHC quantification of γH2AX showing persistent DNA damage in LKO livers.
(C) Representative photos of the total ATR IHC five days post DEN treatment; IHC quantification of nuclear total ATR staining to the right.
(D) IHC quantification of pATR-Thr1989 showing a decrease in ATR activity in LKO livers upon DEN treatment in a time course study with days post DEN exposure indicated.
(E) Representative photos with zooms below for PRP19 IHC of the liver, one day post DEN treatment. To the right is IHC quantification of nuclear PRP19 staining showing a decrease in nuclear localization of PRP19 in LKO livers upon DEN treatment in a time course study.
We found γH2AX staining was negative in the nuclei of hepatocytes surrounding the PV and positive in hepatocytes in the CV and midzone areas (between PV and CV), suggesting that hepatocytes under hypoxic conditions are more vulnerable to DEN-induced DNA damage independent of BRUCE expression. However, a significant difference in DNA repair capacity was observed when the mice were given time to repair the DNA damage. The WT livers displayed a reduction for γH2AX staining on day 3 as compared to day 1, but a further increase of γH2AX staining was found in the LKO liver (Fig. S4A–B), demonstrating DNA repair has taken place in WT but not in LKO livers, which was further confirmed in days 5–10 (Fig. 6B). Hepatic oxidative stress also increased as demonstranted by enhanced lipid peroxidation end product 4-hydroxynonenal (4-HNE) (Fig. S4C).
To determine whether ATR and ATM signaling is impared as they are both reliant on BRUCE expression in vitro (Figs. 1–4 for ATR and our published work for ATM (9, 10)), we evaluated total ATR and its activation via autophosphorylation at Thr1989 (39) by IHC analysis and found both were reduced in LKO hepatocytes (Fig. 6C–D). As ATR activation in vitro requires the expression of both PRP19 (31, 32) and BRUCE (Fig. 4), an overall reduction of PRP19 levels in LKO was also observed (Fig. 6E). In contrast to the in vitro requirement for BRUCE in ATM activation (9, 10), we did not detect ATM activation in either WT or BRUCE LKO livers throughout the DEN study (not shown), which is consistent with a previous study that found no ATM kinase activation in DEN-induced pre-neoplastic hepatic lesions in the rat (40). Our earlier study showed that Bruce whole body KO mice are embryonic leathal because of increased apoptosis and activated p53 (6). However they are not the cause of the liver phenotypes, because hepatic p53 and p21 are not activated nor is caspase-3 (Fig. S5). Therefore, BRUCE has a new function in the liver for maximizing DEN-induced activation of PRP19-ATR DDR signaling for efficient repair of hepatic DNA damage.
BRUCE suppresses DEN-induced hepatocarcinogenesis in mice
The impaired DNA repair capacity in Bruce LKO livers implies a vulnerability of the liver to chronic DNA damage, which can increase the risk of HCC development in the DEN induced HCC model. To test this possibility, we generated a DEN-induced HCC model (36), which includes a single injection of DEN (25 mg/kg; i.p.) to WT and Bruce LKO male pups at 14-days of age, a stage where hepatocytes are naturally proliferating and vulnerable to DEN-induced DNA damage and inflammation (41).
Analysis of HCCs developed in 14-month old mice showed a higher incidence in Bruce LKO mice (100%, 17/17) and a lower incidence in WT littermates (80%; 8/10) (Fig. 7A). HCCs in Bruce LKO mice displayed larger tumor nodules (Fig. 7B and 7C) with a trabecular architecture that closely resembles HCCs commonly found in human biopsies (Fig. 7D). Furthermore, Bruce LKO HCCs displayed a significant increase in higher levels of lymphocyte infiltration (Fig. 7D). Liver fibrosis, a common risk factor associated with 80% of human HCCs (42), was exacerbated as demonstrated by increased collagen staining by sirrius red (Fig. 7E), α-smooth muscle actin (α-SMA) (Fig. 7F), and hydroxyproline (Fig. S6).
Fig. 7. Exacerbated fibrosis and HCC in the Bruce LKO liver.
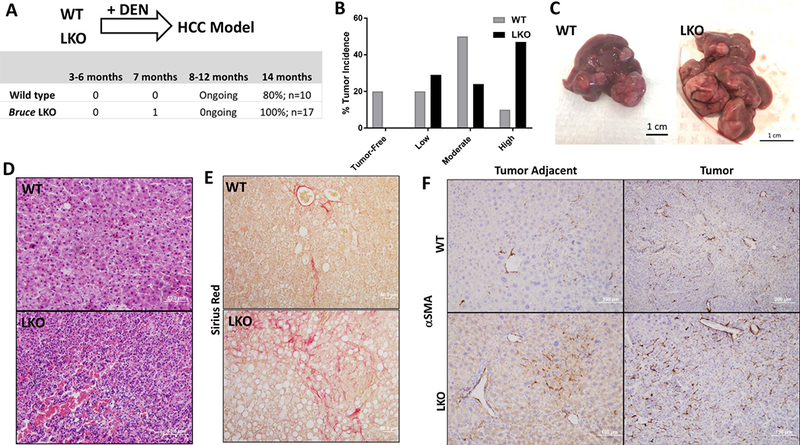
(A) DEN induced HCC model (Diagram) was established with WT and Bruce LKO mice by treatment with 25mg/kg of DEN and sacrificed after 14 months. BRUCE LKO mice developed HCC at a 100% incidence as compared to the lower 80% incidence in WT mice.
(B) HCC severity was categorized into: low-HCC, medium-HCC, and high-HCC. Low-HCC describes a liver with all of the liver lobes are distinguishable with a few smaller tumors. Medium-HCC livers possessed larger tumors that engulfed at least 1–2 of the lobes of the liver. Finally, high-HCC described livers that contained many large tumors that engulfed all lobes of the liver. The most common HCC severity found in the WT mice was the medium-HCC whereas that in LKO mice was the high-HCC category.
(C) Representation of a WT liver with a medium-HCC phenotype and a LKO liver with a high-HCC phenotype.
(D) Hematoxylin & Eosin staining of WT and LKO tumors showing the LKO liver with increased lymphocyte infiltration and a trabecular pattern, a common feature found in human HCC.
(E) Sirius Red staining (red staining) of WT and LKO tumors showing increased fibrosis in LKO livers.
(F) α-Smooth muscle actin IHC of WT and LKO tumors and tumor adjacent tissue demonstrating increased fibrosis in LKO livers.
BRUCE protein expression is down-regulated in a large subset of clinical HCC specimens
Since DEN-induced HCCs share similar gene expression profile signatures with HCC in humans (43), we examined the clinical relevance of BRUCE downregulation in human HCCs. HCC often progresses from hepatitis, fibrosis to cirrhosis in humans; therefore, we assessed the correlation of BRUCE expression levels with these different stages of liver disease in 91 liver specimens (both male and female patients) by IHC using a validated BRUCE antibody (10). BRUCE protein was readily detected in the normal liver, but its levels were reduced in both hepatitis and cirrhotic tissues, and further decreased or completely undetectable in HCC samples, regardless of patient gender (Fig. 8A). Using well-established clinical scoring procedures, we further validated the downregulation of BRUCE expression (Fig. 8B). Specifically, compared to normal tissue (n=14), BRUCE levels were decreased in 54.5% of hepatitis (n=22), 46.7% of cirrhosis (n=30), and 84% of HCC tissues (n=25) (Fig. 8B). The significant incidence of BRUCE downregulation in human HCCs coupled with the observed tumor promoting role of BRUCE-deficiency in mouse HCC development (Fig. 7) suggests that loss of hepatic BRUCE function may contributed to the development of HCC in human patients.
Fig. 8. BRUCE protein downregulation and Bruce gene somatic mutations in human HCC.
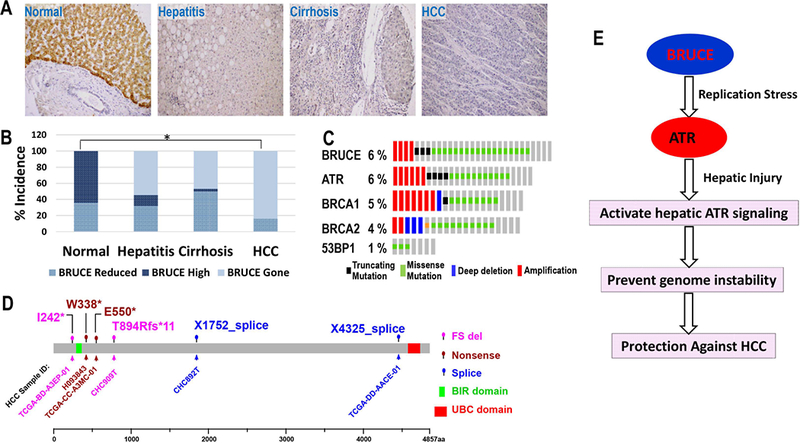
(A) Human normal liver section stained positive for BRUCE protein in hepatocytes (upper area, brown signals) by IHC; lower area bile duct; Steatohepatitic liver section stained for BRUCE showing both reduced BRUCE protein expression compared to normal liver tissue and significant lymphocyte infiltration and accumulated fat; Cirrhotic liver section showing both reduced BRUCE protein expression compared to normal liver tissue and one cirrhotic nodule in the right area of the photo; HCC section showing nearly negative BRUCE protein expression compared to normal liver tissue.
(B) The IHC results in each category in panel A were semi-quantified by clinical scoring and converted to the incidence of BRUCE expression alteration into high, reduced and being gone (absent). The percentage of “BRUCE being gone”: steatohepatitis (54.5%), cirrhosis (46.7%) and HCC (84%). *p = 2.0 × 10−7; ONE-SIDED Fisher’s exact test.
(C) TCGA HCC data portal showing a rate of 6% BRUCE somatic mutation, same or higher than that of ATR and BRCA1/2 genes with which BRUCE works together in the same DNA damage response pathway.
(D) The Amino acid residues with somatic mutations are shown in the BRUCE protein diagram (4857 amino acids with its BIR and UBC domains indicated). The TCGA HCC sample identification number (ID) that reported each mutation site are shown. Three major types of mutations are found: FS (frame shift), nonsense mutation and splice alterations.
(E) Diagram showing our working model: BRUCE promotes the ATR-replication stress response to assure accurate DNA replication and prevent genome instability. This new BRUCE-ATR axis is present in the liver and the in vivo significance of this pathway is to protect the liver against genotixin injury and suppress the HCC development.
Bruce gene mutations are found in human HCC specimens at The Cancer Genome Atlas (TCGA)
Multiple mechanisms can cause loss of BRUCE protein in HCCs, among which somatic mutations can inactivate or truncate BRUCE protein. We found a rate of 6% deleterious Bruce mutations in HCC patients documented at TCGA, a rate similar to the ATR, BRCA1, and BRCA2 genes (Fig. 8C). They are frameshifts and nonsense mutations (Fig. 8D) predicted to inactivate BRUCE function in DDR because they are large deletions leading to elimination of the UBC domain which is essential for BRUCE function in DDR (9, 10). For instance, two nonsense point mutations at W338* and E550* and one frameshift deletion at I242* would encode nonfunctional truncated BRUCE lacking ~90% of the amino acid residues, including the essential UBC domain (Fig. 8D). The remaining mutations are missense mutations that distribute throughout the entire gene which could destabilize the overall BRUCE structure and impair its DDR function (Fig. 8D). These mutations are less likely sequencing errors, because the “variant allele frequency” is relatively high for the X4326_splice mutation whose frequency is 0.39 and has 105 variant reads out of 269. I242* mutation has a frequency of 0.18 and 30 variant reads out of 171. They both are Shallow Deletions, meaning their expression level is reduced. Nonetheless, these potential deleterious mutations suggest that Bruce DDR function could be inactivated, which in our studies exacerbates HCC development (Fig. 7).
Discussion
As the 2nd leading cause of cancer deaths worldwide (44), HCC is closely associated with genome instability (34, 35). Major HCC risk factors including DEN (found in foods and drinks), aflatoxin and other diet contaminates, hepatitis B and C viral infection, alcohol consumption, and obesity directly or indirectly damage DNA, which can generate mutations and promote genome instability. In contrast to the tumorigenic outcome of DNA damage, the critical DDR proteins and signaling pathways that resolve DNA damage and thereby suppress HCC development and progression are largely unclear.
In this study we investigated the impact of BRUCE on DNA replication stress response and on the development of HCC in mice and clinical specimens. The study has demonstrated three aspects of significance. First, the previously unrecognized role of the BRUCE-ATR replication stress axis critical for stabilization of replication forks and suppression of unscheduled origin firing provides new insight into how ATR replication stress response is regulated by BRUCE. Secondly, we have demonstrated a novel tumor suppressing function of the BRUCE-ATR replication stress axis in protection of the mouse liver against genotoxic injury and hepatocarcinogenesis and provided translational relevance for both BRUCE downregulation and deleterious gene mutations in human HCC. Third, the genetically modified Bruce LKO mouse model is a clinically relevant model for further studies of pathogenic mechasnims underlying HCCs. Unlike other liver tumor models, the Bruce LKO mouse model under DEN treatement shares three clinical stages similar to human HCC development: inflammation, fibrosis, and tumorigenesis with the trabecular architecture resembling human HCC.
Based on these findings, we propose a working model as illustrated in Figure 8E. The key mechanism is that LKO of Bruce increases the accumulation of DNA damage which resulted from impaired BRUCE-ATR replication stress signaling. The impairment of DNA replication accelerates hepatocyte death and subsequent compensatory proliferation, promoting fibrosis and inflammation and exacerbating HCC development in Bruce LKO livers. Therefore, BRUCE deficiency predisposes the liver to earlier and more severe hepatocarcinogenic injury and HCC development (Fig. 7).
Although our previous studies have demonstrated that BRUCE regulates the activation of the ATM kinase in DNA DSB repair (9, 10), ATM did not appear to be activated in our DEN-treated WT or Bruce LKO mouse models which is consistent with a previous study (40). This finding establishes a tissue-specific requirement of BRUCE-ATR signaling in DEN-induced replication stress. Therefore, impaired DNA replication can be a major source of DNA damage and genome instability and has a profound effect on accelerating liver injury and carcinogenesis.
BRUCE knockdown U2OS cells are not highly sensitive to replication stressors HU and MMC (Fig. 1). We compared this sensitivity to ATR knockdown U2OS cells. The results indicate that although BRUCE-depleted cells have good sensitivity, it is about 50% of the sensitivity in ATR-depleted cells since ATR is a potent regulator (not shown). The reduced sensitivity better maintains cell survival and could be advantageous to allow time to repair.
Future studies will elucidate how the two ubiqtuitin ligases BRUCE and PRP19 functionally interact to regulate DNA damage response in the liver. In summary, this study illustrates a liver tumor suppressing function of BRUCE and generates a clinically relevant mouse model of Bruce LKO for further studies of HCC pathogenesis under additional oncogene driven genetic backgroud.
Supplementary Material
Acknowledgement
Mass spectrometry data for protein identification were acquired in the University of Cincinnati Proteomics Laboratory under the direction of Ken Greis, PhD on a mass spectrometer funded in part through an NIH S10 shared instrumentation grant (RR027015 Greis-PI). We thank Dr. Li Lan at The University of Pittsburgh for sharing RPA construct.
Financial Support: This work is supported by NIH grants CA158323 and CA158323S (C. Du) and CA129537, GM109768 and funds from HMRI (T. K. Pandita). Additional support was provided by the Center for Environmental Genetics (NIEHS award P30ES006096) to Dr. Liang Niu.
Abbreviations:
- HCC
hepatocellular carcinoma
- MMC
mitomycin C
- HU
hydroxyurea
- DEN
diethylnitrosamine
- DSB
double strand break
Footnotes
Potential conflict of interest: None.
Additional materials and methods can be found in the supplementary information.
References
- 1.Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017;168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauser HP, Bardroff M, Pyrowolakis G, Jentsch S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J Cell Biol 1998;141:1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Z, Naito M, Hori S, Mashima T, Yamori T, Tsuruo T. A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun 1999;264:847–854. [DOI] [PubMed] [Google Scholar]
- 5.Lotz K, Pyrowolakis G, Jentsch S. BRUCE, a Giant E2/E3 Ubiquitin Ligase and Inhibitor of Apoptosis Protein of the trans-Golgi Network, Is Required for Normal Placenta Development and Mouse Survival. Mol Cell Biol 2004;24:9339–9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren J, Shi M, Liu R, Yang QH, Johnson T, Skarnes WC, Du C. The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc Natl Acad Sci U S A 2005;102:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol 2009;11:65–70. [DOI] [PubMed] [Google Scholar]
- 8.Bartke T, Pohl C, Pyrowolakis G, Jentsch S. Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol Cell 2004;14:801–811. [DOI] [PubMed] [Google Scholar]
- 9.Ge C, Che L, Du C. The UBC Domain Is Required for BRUCE to Promote BRIT1/MCPH1 Function in DSB Signaling and Repair Post Formation of BRUCE-USP8-BRIT1 Complex. PLoS One 2015;10:e0144957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge C, Che L, Ren J, Pandita RK, Lu J, Li K, Pandita TK, et al. BRUCE regulates DNA double-strand break response by promoting USP8 deubiquitination of BRIT1. Proc Natl Acad Sci U S A 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer 2015;15:276–289. [DOI] [PubMed] [Google Scholar]
- 12.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003;300:1542–1548. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC. DNA interstrand cross-link repair requires replication-fork convergence. Nat Struct Mol Biol 2015;22:242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011;11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 2004;18:1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 2016;17:337–349. [DOI] [PubMed] [Google Scholar]
- 17.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012;26:1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst) 2014;19:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013;155:1088–1103. [DOI] [PubMed] [Google Scholar]
- 20.Horikoshi N, Pandita RK, Mujoo K, Hambarde S, Sharma D, Mattoo AR, Chakraborty S, et al. beta2-spectrin depletion impairs DNA damage repair. Oncotarget 2016;7:33557–33570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakraborty S, Pandita RK, Hambarde S, Mattoo AR, Charaka V, Ahmed KM, Iyer SP, et al. SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. iScience 2018;2:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez A, D’Andrea A. Fanconi anemia pathway. Curr Biol 2017;27:R986–R988. [DOI] [PubMed] [Google Scholar]
- 23.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet 2005;14:693–701. [DOI] [PubMed] [Google Scholar]
- 24.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012;22:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu J, Su F, Mukherjee S, Mori E, Hu B, Asaithamby A. FANCD2 influences replication fork processes and genome stability in response to clustered DSBs. Cell Cycle 2015;14:1809–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moldovan GL, D’Andrea AD. To the rescue: the Fanconi anemia genome stability pathway salvages replication forks. Cancer Cell 2012;22:5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhury I, Stroik DR, Sobeck A. FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol Cell Biol 2014;34:3939–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001;7:249–262. [DOI] [PubMed] [Google Scholar]
- 29.Chanarat S, Strasser K. Splicing and beyond: the many faces of the Prp19 complex. Biochim Biophys Acta 2013;1833:2126–2134. [DOI] [PubMed] [Google Scholar]
- 30.Mahajan K hPso4/hPrp19: a critical component of DNA repair and DNA damage checkpoint complexes. Oncogene 2016;35:2279–2286. [DOI] [PubMed] [Google Scholar]
- 31.Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, et al. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol Cell 2014;53:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dubois JC, Yates M, Gaudreau-Lapierre A, Clement G, Cappadocia L, Gaudreau L, Zou L, et al. A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Res 2017;45:8859–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao CV, Asch AS, Yamada HY. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis 2017;38:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang SF, Chang CW, Wei RJ, Shiue YL, Wang SN, Yeh YT. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int 2014;2014:153867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006;6:674–687. [DOI] [PubMed] [Google Scholar]
- 36.Tolba R, Kraus T, Liedtke C, Schwarz M, Weiskirchen R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab Anim 2015;49:59–69. [DOI] [PubMed] [Google Scholar]
- 37.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005;5:749–759. [DOI] [PubMed] [Google Scholar]
- 38.Lautt WW: In: Hepatic Circulation: Physiology and Pathophysiology. San Rafael (CA), 2009. [PubMed] [Google Scholar]
- 39.Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, et al. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 2011;43:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silins I, Finnberg N, Stahl A, Hogberg J, Stenius U. Reduced ATM kinase activity and an attenuated p53 response to DNA damage in carcinogen-induced preneoplastic hepatic lesions in the rat. Carcinogenesis 2001;22:2023–2031. [DOI] [PubMed] [Google Scholar]
- 41.Bakiri L, Wagner EF. Mouse models for liver cancer. Mol Oncol 2013;7:206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Affo S, Yu LX, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol 2017;12:153–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet 2004;36:1306–1311. [DOI] [PubMed] [Google Scholar]
- 44.El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology 2014;60:1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.