

Abstract
This review discusses the profound connection between microglia, neuroinflammation, and Alzheimer’s disease (AD)1. Theories have been postulated, tested, and modified over several decades. The findings have further bolstered the belief that microglia-mediated inflammation is both a product and contributor to AD pathology and progression. Distinct microglia phenotypes and their function, microglial recognition and response to protein aggregates in AD, and the overall role of microglia in AD are research areas that have received considerable research attention and yielded significant results. The following article provides a historical perspective of microglia, a detailed discussion of multiple microglia phenotypes including dark microglia, and a review of a number of areas where microglia intersect with AD and other pathological neurological processes. The overall breadth of important discoveries achieved in these areas significantly strengthens the hypothesis that neuroinflammation plays a key role in AD. Future determination of the exact mechanisms by which microglia respond to, and attempt to mitigate, protein aggregation in AD may lead to new therapeutic strategies.
Graphical Abstract
This review will discuss the nexus between microglia, neuroinflammation, and Alzheimer’s disease (AD). Areas of emphasis in the article are distinct microglia phenotypes and their function, microglial recognition and response to amyloid-β (Aβ) aggregates in AD, and the overall contributory role of microglia in AD. The cumulative data point to a dynamic interrelationship between microglial phenotype and function, as well as Aβ accumulation.
Introduction
Brief history of glia
In 1856 Rudolf Virchow coined the term “neuroglia” or “nerve-cement” to describe the brain connective tissue which consists of the non-neuronal or glial cells (Virchow. R 1856, 1858; Kettenmann and Verkhratsky 2008; Kettenmann et al. 2011; Ginhoux et al. 2013). The glial elements were first characterized by Robert Remak in 1838 and Heinrich Muller in 1851 (Muller 1851). Later in the 19th century, they were broadly introduced in various forms by Otto Deiters, Jacob Henle, Gustav Retzius, Camillo Golgi, and Michael von Lenhossek who introduced the term “astrocyte” or “astroglial cell” in 1893 (Deiters 1865; Henle 1869; Lenhossek 1893; Retzius 1894–1916; Golgi 1903). In 1899, Franz Nissl used the term “rod cells” (Stäbchenzellen) to describe the reactive glial elements with migratory, phagocytic, and proliferative functions and mesodermal origin (Nissl 1899). These phagocytic glial elements, which originated from the mesoderm and thus separate from neuronal and neuroglia origins, were termed “mesoglia” by W. Ford Robertson in 1900. He described that mesoglial cells, unlike astrocytes (macroglial cells), were not connected to blood vessels and acquired granular morphology and removed their processes in injured brains (Robertson 1900). Mesoglia, the third element of the central nervous system (CNS), were differentiated from neurons (first element) and astrocytes (second element) by Santiago Ramon y Cajal in 1913. The “microglia” (the plural form of microgliocyte) or “Hortega cells” were thoroughly described by Pio del Rio-Hortega between 1919 and 1927 using a modified silver carbonate impregnation technique. He hypothesized that microglia were phagocytic cells with amoeboid features which migrated from mesoderm, pia mater, to entire brain regions through vessels and white matter tracts during early development. In the more mature brain they acquired a branched, ramified morphology (resting microglia) which transformed to an amoeboid morphological phenotype during pathological conditions (Del Rio-Hortega 1919a, 1919b, 1924, 1925, 1927, 1932). However, another component of mesoglia, neurectodermal oligodendroglia or oligodendrocytes, had no phagocytic activity (Ginhoux et al. 2013). There were controversies over the neuro-ectodermal (Fujita and Kitamura 1975; Kitamura et al. 1984; Hao et al. 1991; Fedoroff et al. 1997) or mesodermal (Murabe and Sano 1982; Hume et al. 1983; Murabe and Sano 1983; Perry et al. 1985) origins of microglia in several investigations. Light microscopy, electron microscopy (EM), and immunohistochemistry techniques supported the mesodermal origin of microglia hypothesis, distinguished the morphological similarities between microglia and macrophages, and supported the monocyte/macrophage-derived microglia hypothesis via phenotypic homologies (Murabe and Sano 1982; Hume et al. 1983; Murabe and Sano 1983; Perry et al. 1985). However, the emergence of immature macrophages within blood islands of the embryonic yolk sac (YS) at fetal day 9 was determined in mouse and rat by Takashi and Naito (Takahashi et al. 1989; Takahashi and Naito 1993). Following circulation formation around E8.5, the primitive macrophages, generated around E8.0, leave the YS blood islands, the only embryonic hematopoietic site at an early stage, and give rise to microglia after neuroepithelium colonization from E9.0/E9.5 (Takahashi and Naito 1993; Alliot et al. 1999; Ginhoux and Prinz 2015).
Spatial and temporal diversity of microglial phenotypes
Although early morphologic studies described microglia as either resting or activated, it has become clear that their behavior is characterized by a remarkable degree of basal plasticity. In vivo imaging of murine microglia has demonstrated that even in a healthy brain microglia are not at rest. Instead these immune cells are actively involved in homeostatic functions. Their highly motile processes continually interact with neuronal and glial cells, blood vessels (Nimmerjahn et al. 2005), as well as synapses (Wake et al. 2009; Tremblay et al. 2010). It is estimated that the entire brain parenchyma is sampled by these homeostatic microglia once every few hours (Nimmerjahn et al. 2005). Indeed, microglia appear to be involved in a spectrum of homeostatic functions including neurogenesis, neuronal migration, axon growth, and modulating synaptic connections (Sato 2015; Frost and Schafer 2016; Reemst et al. 2016; Li and Barres 2018). In addition to this range of normal behavior, studies show that microglia also differ in their phenotype across brain regions and lifespan in an extraordinarily complex fashion (Silvin and Ginhoux 2018). An early description of these differences by Lawson et al. examined microglial morphology and distribution in the adult mouse brain using the F4/80 antiserum. They reported greater microglial abundance in grey matter with highest concentrations in the hippocampus, basal ganglia, substantia nigra, and olfactory telencephalon with reduced immunoreactivity in the brainstem and cerebellum (Lawson et al. 1990). The authors estimated 3.5 × 106 total microglia per mouse brain (Lawson et al. 1990). They also compared microglial morphology noting that this differed depending upon the brain region with round amoeboid cells near areas lacking a blood brain barrier, longitudinally branched cells in fiber tracts, and ramified cells throughout the neuropil (Lawson et al. 1990). A recent study by De Biase et al. increased our understanding of microglial phenotype regulation by characterizing adult mouse microglia via RNA sequencing, electrophysiology, and morphology to demonstrate region specific phenotypes within the basal ganglia (De Biase et al. 2017). Additionally, transient depletion of microglia by treating the animals with a colony stimulating factor receptor 1 (CSF1R) antagonist, PLEX5622, for 2 weeks followed by a 21 day recovery resulted in repopulation of basal ganglia microglia with diverse properties similar to pre-treatment supporting the view that local cues contribute to establishing microglial phenotype (De Biase et al. 2017; O’Neil et al. 2018). Similar findings were observed using 11-12 week old C57BL/6 mice to demonstrate regional diversity of microglial protein expression via flow cytometry (de Haas et al. 2008). Microglia isolated from the striatum, hippocampus, spinal cord, cerebellum, and cerebral cortex demonstrated clear expression of CD11b, CD40, CD45, CD80, CD86, F4/80, TREM-2b, CXCR3, and CCR9 with no evidence of MHCII or CCR7 immunoreactivity (de Haas et al. 2008). However, individual protein levels of these markers varied depending upon the brain region with no clear pattern of overall low or high levels for all proteins relevant to a region (de Haas et al. 2008). It is clear that these regional selective morphologies and phenotypes are not static, as microglia are able to transform from amoeboid to ramified and vice versa in response to varying conditions (Ling and Wong 1993; Ling et al. 2001). It has become clear that microglia also have an altered phenotype associated with normal aging. For instance, increased microglial Iba1/MHCII double immunoreactivity increases throughout the hippocampus in rats with age suggesting a phenotype change occurs (VanGuilder et al. 2011). However, this simple measure of change did not correlate with any behavioral dysfunction suggesting that microglial phenotypes unique to disease or injury states are superimposed upon age-associated changes (VanGuilder et al. 2011). A more recent study corroborates an aging phenotype of microglia using direct RNA sequencing from microglia isolated from 5 and 24-month-old mice (Hickman et al. 2013). The authors demonstrated that microglial are distinct from peripheral macrophages and down regulate gene pathways involved in neurotoxicity while upregulating those involved in neuroprotection with age (Hickman et al. 2013).
Changing role of microglia in Alzheimer’s disease
The age-associated neurodegenerative disease, Alzheimer’s disease (AD), is characterized by a robust increase in morphologically reactive microglia. Moreover, these microglia are often associated with extracellular plaque deposits of amyloid β (Aβ) peptide. In vivo imaging using [11C] (R)-PK11195 positron emission tomography (PET) against the peripheral benzodiazepine receptor, abundantly expressed by mononuclear phagocyte lineage cells, has demonstrated microglial activation in patients with mild to moderate AD-type dementia compared to healthy individuals. Such observations from the living human brain suggests the possibility of microglia phenotypic alteration as an early AD pathological hallmark (Cagnin et al. 2001). A subsequent study of 64 AD patients and 32 controls using an 18-kDa translocator protein PET radiotracer coupled with amyloid imaging using Pittsburgh compound B PET reached a similar conclusion, i.e. that microglial activation appears during prodromal and perhaps preclinical disease (Hamelin et al. 2016). To add to this complexity, the typical late-onset, sporadic form of disease suggests that any disease-associated changes in microglial phenotype are compounded by age-dependent differences. Based upon this plethora of regulatory influences, the study of microglial behavior in AD remains an area of intense study and will be the subject of the remainder of this review. As will be discussed in later sections, the specific nature of Aβ-microglia interactions together with their causes and consequences, remain a significant effort in the field. In addition, there is a remarkable array of microglial phenotypes in AD.
Early work focused on ultrastructural studies of extracellular amyloid-beta (Aβ) plaques and their associated microglia in the cortex of AD patients to reveal Aβ fibrils in altered cisterns of the microglial endoplasmic reticulum. Also, exogenous Aβ fibrils taken up by cultured microglia remained intact in phagosomes up to 20 days. Based on these observations, Wisniewski and Frackowiak hypothesized that Aβ fibrils are produced but not phagocytosed by microglia in AD (Wisniewski et al. 1989; Wisniewski et al. 1990; Frackowiak et al. 1992). In agreement with this, the in vitro production of amyloid-beta precursor protein (APP) by rodent microglia was further reported (Haass et al. 1991; Mönning et al. 1995; LeBlanc et al. 1996). However, lack of APP mRNA in microglia clustering in the vicinity of plaques in postmortem AD brains decreased support for microglial ability to synthesize Aβ as a contribution to plaques (Scott et al. 1993). Subsequently, HLA-DR, a major histocompatibility complex class II (MHC II) cell surface receptor expressed by antigen presenting cells, MHC I to a lesser extent, leucocyte common antigen (LCA), and the Fc gamma RI and Fc gamma RII receptors were all immunohistochemically identified on reactive microglia of postmortem AD brains (McGeer et al. 1987; Itagaki et al. 1988; McGeer et al. 1989; Tooyama et al. 1990). These receptors are indicative of immunoreactivity and phagocytic properties of microglia suggesting that microglia were not producing Aβ but instead responding to it. Subsequently, detection of Aβ in intracytoplasmic membranes, excluding secretory organelles, strengthened a phagocytosis-associated role of microglia in AD (el Hachimi and Foncin 1994). Perhaps most importantly, it has become clear that the type of Aβ and putative cell surface receptors that mediate microglia-Aβ interaction add significant difficulty to understanding how these cells behave in AD. As discussed in detail later in this review, it is now recognized that microglial phenotype is not simply phagocytic in AD. For example, interaction of microglia with Aβ deposits, via scavenger receptors (SRs) and the receptor for advanced glycation end products (RAGE), in an effort to phagocytose Aβ and clear up the extracellular milieu, triggered microglia immobilization, production of cytokines, reactive oxygen species, and nitrogen species, and results in neuronal death supporting a detrimental role for microglia in AD (Banati et al. 1993; El Khoury et al. 1996; Paresce et al. 1996; Lue et al. 2001a).
Heterogenous microglial population in AD
The diversity of the microglial population in AD has been the subject of several studies in recent years and numerous transgenic AD mouse models have contributed to these discoveries. The APP(/)PS1 transgenic strain carries the human transgenes for APP bearing the KM670/671NL Swedish mutation and presenilin-1 (PS1) containing an L166P mutation (Duff et al. 1996; Radde et al. 2006). These mice develop cerebral Aβ deposition within the first few months of life. The 5XFAD strain carries the Swedish and two other APP mutations as well as two PS1 mutations (Oakley et al. 2006). Studies in these mouse models have led to the definition of multiple microglia phenotypes seen under various pathological conditions. One of these phenotypes has been termed disease-associated microglia (DAM), which are prevalent in the 5XFAD mice. DAM were recently characterized using single-cell RNA sequencing (Keren-Shaul et al. 2017). This phenotype is associated with (1) a downregulation of several homeostatic genes such as P2ry12 (purinergic receptor P2RY12), Tmem119 (transmembrane protein 119), and Cx3cr1 (C-X3-C motif chemokine receptor 1); (2) TREM2 (triggering receptor expressed on myeloid cells 2)-dependent upregulation of genes including Axl (receptor tyrosine kinase), Clec7a (C-type lectin domain containing 7A), lpl (lipoprotein lipase), Spp1 (osteopontin) and ITGAX or Cd11c, (integrin subunit alpha X); and (3) TREM2-independent upregulation of ApoE (apolipoprotein E) and Tyrobp (TYRO protein tyrosine kinase binding protein) (Keren-Shaul et al. 2017). The microglia neurodegenerative phenotype (MGnD) is another recently-characterized population that similarly presents with reduced expression of homeostatic genes, including P2ry12 and Tmem119, and increased expression of ApoE, Axl, and Clec7a genes (Krasemann et al. 2017b). These MGnD cells were only observed when neuronal apoptosis also occurred, for instance in the APP/PS1 mouse model, and found to be regulated by TREM2 and ApoE expression (Krasemann et al. 2017a). Another microglial phenotype notably associated with AD and that will be covered in the last part of this review, is dark microglia (Bisht et al. 2016b). Although dark microglia share similar morphological features with typical microglia, pertaining to their size, morphology, association with the extracellular space and relationships with neurons and synapses, they possess distinct characteristics that are observed by EM. Typical microglia strongly express the homeostatic markers CX3CR1, Iba1 and P2RY12. By contrast, dark microglia do not express P2RY12, and downregulate Iba1 and green fluorescent protein (GFP) in CX3CR1-GFP reporter mice (Bisht et al. 2016b). Phenotypic differences between normal and disease-related microglia are summarized in Table 1.
Table 1.
Phenotypic properties of microglia in AD pathology. Current observations and characterizations of normal and distinct phenotypes of microglia. Corresponding references are [1] (Bisht et al. 2016b) [2] (Peters et al. 1991) [3] (Savage et al. 2018) [4] (Tremblay et al. 2010) [5] (Keren-Shaul et al. 2017) [6] (Krasemann et al. 2017b) [7] (Bisht et al. 2016a) [8] (Hui et al. 2018).
| Sample | Typical microglia [1-4] | Dark microglia [1, 7, 8] | DAM and MGnD [5, 6] |
|---|---|---|---|
| AD observation | Clustered and activated around Aβ plaques | APP-PS1 mice | DAM: post mortem AD human brains, 5XFAD mice |
| MGnD: APP-PS1, APP-PS1 Trem2−/− mice | |||
| Morphology | Ramified processes when surveying the parenchyma | Hyper ramified by TEM | Not detemined |
| Short and thick processes when reactive | |||
| Ultrastructural Identification | Distinct chromatin pattern | Loss of chromatin pattern | Not detemined |
| Small oval cell body | Dark appearance possibly due to condensed nucleo- and cytoplasm [7] | ||
| Cytoplasmic phagocytic inclusions, lipofuscin granules and/or lipid bodies | Oxidative stress features (ER and Golgi apparatus dilation) | ||
| Long/narrow ER cisternae | |||
| Immunoreactivity | High immunoreactivity for homeostatic markers (GFP in CX3CR1-GFP mice, P2RY12, IBA1, etc.) | Low immunoreactivity for homeostatic markers (GFP in CX3CR1-GFP mice, IBA1), no expression of P2RY12 | DAM: immunoreactive for IBA1 MGnD: low or no immunoreactivity for P2RY12 |
| Regional Distribution | Throughout the white and grey matter | Median eminence of the hypothalamus | Cerebral cortex |
| Dentate gyrus [8] | Hippocampus (near Aβ plaques) | ||
| Strata radiatum and lacunosum moleculare of the hippocampus CA1 | DAM are not found in the cerebellum | ||
| Basolateral amygdala | |||
Support for immune contribution to AD
The biochemical and histologic characterizations of microglial phenotypes in AD certainly support a role for these cells during disease, but genetic data provides even more compelling evidence of microgliosis contributing to disease. Genome wide association studies (GWAS) have revealed risk loci associated with immune response besides the well-known APOε4 allele (late-onset AD) and pathogenic mutations of APP, PSEN1, and PSEN2 (early-onset autosomal-dominant AD) which are carried by 50% and 2% of individuals with AD, respectively. For example, a large GWAS of 2,032 AD patients and 5,328 controls from France identified loci in CLU or APOJ (clusterin or apolipoprotein J) and CR1 (the complement component (3b/4b) receptor 1), which are involved in Aβ clearance (Lambert et al. 2009). ABCA7 (ATP-binding cassette, sub-family A, member 7), MS4A6A/MS4A4E (membrane-spanning 4-domains, subfamily A), EPHA1 (Ephrin type-A receptor 1), TREM2, HLA-DRB5-DRB1 (MHC II, DR beta 5-1), INPP5D (Inositol polyphosphate-5-phosphatase), MEF2C (myocyte enhancer factor 2C), and CD33, also have polymorphisms associated with risk of late-onset AD (Lambert et al. 2009; Hollingworth et al. 2011a; Naj et al. 2011b; Guerreiro et al. 2013b; Jonsson et al. 2013; Lambert et al. 2013a; Karch et al. 2014). Collectively, there is continuing support for the idea that the immune system, and microglial behavior particularly, contributes to disease progression. Defining the phenotype of microglia during AD, as discussed below, as well as identifying tools for their manipulation, will offer novel intervention strategies.
Immunomodulatory trials for AD
Consistent with the idea of microglial inflammatory contribution to disease, epidemiological studies have revealed a remarkable decline of AD prevalence in individuals with rheumatoid arthritis (RA), most likely as a result of long-term anti-inflammatory therapy for this well-established chronic inflammatory disease (Jenkinson et al. 1989; McGeer et al. 1990; Li et al. 1992; Myllykangas-Luosujärvi and Isomäki 1994). Other epidemiological investigations focused on the contribution of long-term nonsteroidal anti-inflammatory drug (NSAIDs) usage to the risk of AD development and also confirmed an inverse association between NSAID use and AD (Andersen et al. 1995; Breitner et al. 1995; Stewart et al. 1997; Beard et al. 1998; Broe et al. 2000; in ‘t Veld et al. 2001; Lindsay et al. 2002; Zandi et al. 2002). In agreement with the robust biochemical and histologic evidence, an inflammatory hypothesis of AD arose suggesting anti-inflammatory interventions to decrease the incidence and progression of AD. Clinical trials for different NSAIDs with various durations of drug treatment have been performed to validate their potential protective role in mild to moderate AD. Although, 6-month administration of the NSAID indomethacin demonstrated cognitive deficit improvement of AD patients, the statistical data was inconclusive because of the small scale of the clinical trial (Rogers et al. 1993). Furthermore, treatment with a combination of the NSAID diclofenac and the synthetic prostaglandin misoprostol for 25 weeks, the NSAID rofecoxib (a cyclooxygenase-2 (COX-2)-selective inhibitor) or the NSAID naproxen (non-selective COX inhibitor) for 1 year, rofecoxib up to 4 years, the NSAID celecoxib (a COX-2-selective inhibitor) for 52 weeks, and celecoxib or naproxen for 3 years exerted no significant beneficial effect (Scharf et al. 1999; Aisen et al. 2003; Reines et al. 2004; Thal et al. 2005; Group et al. 2007; Soininen et al. 2007; Group et al. 2008). NSAID treatment initiation at the more advanced course of AD, the short durations of treatment, inappropriate dosage to ameliorate the inflammation compared to the epidemiological studies, and side effects, like gastrointestinal dysfunction, fatigue, dizziness, headache, hypertension, and stroke have all likely contributed to clinical trial failure. Similarly, a randomized, placebo-controlled multicenter trial of low-dose prednisone (a synthetic glucocorticoid) treatment reported no significant amelioration effect on cognition of AD patients in primary and secondary analysis (Aisen et al. 2000). Additional study of microglial behavior during AD is required to determine whether immunomodulatory therapies are feasible. The remainder of this review is focused on summarizing current understanding of microglial contributions to AD.
Conformational aspects of Aβ relevant for neuroinflammation
Aβ conformational species
The conformation or aggregation state of Aβ has a substantive impact on the cellular inflammatory response to the peptide. Numerous studies have shown that monomeric Aβ is unstructured and fairly innocuous. Yet, when monomers begin to non-covalently assemble and aggregate, a β-sheet structure develops and predominates. This conversion renders Aβ a more toxic and inflammatory molecule. Aβ aggregation typically does not progress in discrete defined steps and can yield a polydisperse solution of species that differ in size, conformation, and solubility. Several classes of Aβ aggregates have emerged that include oligomers, protofibrils, and fibrils (Figure 1). The most classical and well-characterized Aβ aggregates are fibrils, which make up the core of AD neuritic plaques (Terry 1985). Fibrils are high molecular weight (Mw) rod-like structures greater than 1 μm in length and contain multiple layers of β-sheet linked monomers (Colvin et al. 2016; Walti et al. 2016). Protofibrils also have a defined β-sheet structure, but are smaller in both diameter and length, and are soluble precursors to fibrils (Harper et al. 1999; Walsh et al. 1999; Nichols et al. 2015). Oligomers are also soluble and tend to have less β-sheet structural characteristics than the other more developed Aβ aggregates. Oligomers generally fall into two groups, smaller (dimers through hexamers) (Lambert et al. 1998; Bitan et al. 2003; Jin et al. 2011) and larger Mw species (Hepler et al. 2006).
Figure 1.
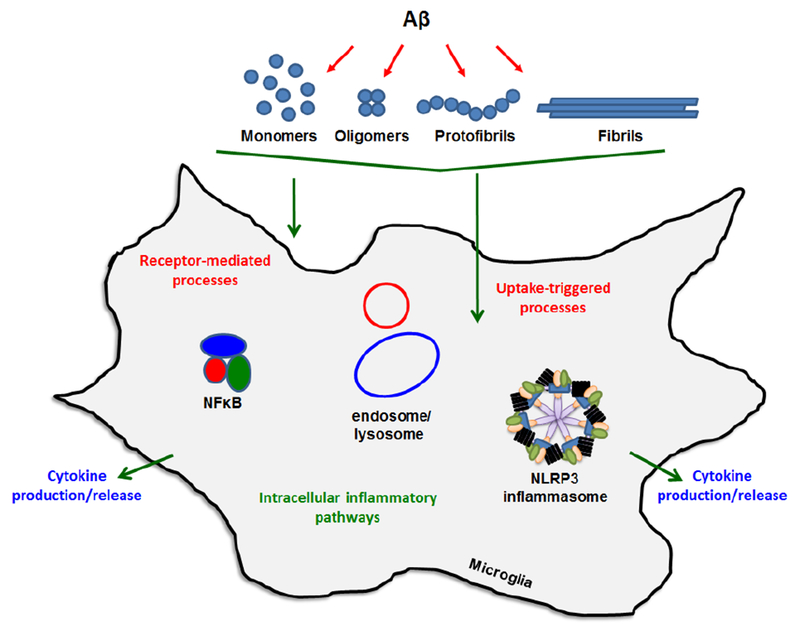
Mechanisms of Aβ-mediated neuroinflammation. Interactions between microglia and various conformational forms of Aβ involve stimulation of cell-surface receptors and internalization by non-receptor mechanisms. Either interaction can provoke pro-inflammatory cytokine release through multiple intracellular pathways. Two key processes involve NFκB-mediated transcription/translation of cytokines such as TNFα and pro-IL-1β followed by NLRP3 inflammasome-mediated cleavage of pro-IL-1β to mature IL-1β. Internalized Aβ can trigger inflammasome activity and this may occur via endosomal or lysosomal factors.
Technical considerations for the preparation of Aβ for cellular studies
It is challenging to directly compare studies of Aβ biological activity when there is frequently a lack of rigorous definition of the Aβ solution species. In many cases, these solutions contain multiple Aβ species, which complicates interpretation. Numerous studies over the years have identified solution conditions that promote formation of one Aβ species over another. The combination of these conditions and additional separation steps can be successfully employed to isolate a particular species. Reconstitution of dry Aβ peptide will most assuredly yield a mixed solution. Thus, additional steps are usually required to obtain monomeric Aβ. This is especially the case for Aβ42 due to its significantly higher aggregation propensity than Aβ40 (Jarrett et al. 1993). Pretreatment of dry Aβ peptide with hexafluoroisopropanol and/or in alkaline conditions decreases the amount of preformed aggregates (O’Nuallain et al. 2006; Teplow 2006), but the best method for Aβ monomer purification is size exclusion chromatography (SEC) (Walsh et al. 1997). A well-established method for preparing oligomers entails resuspension of Aβ in dimethyl sulfoxide, dilution into cold Ham’s F-12 cell culture medium without phenol red, and incubation at 4 °C (Stine et al. 2003). Conditions for preparing, isolating, and characterizing soluble protofibrils were initially reported 20 years ago (Walsh et al. 1997; Harper et al. 1999; Walsh et al. 1999) and thoroughly chronicled in subsequent years (Lashuel and Grillo-Bosch 2005). Insoluble Aβ fibrils have commonly been prepared by 37 °C incubation at neutral (Harper et al. 1997) or acidic pH (Stine et al. 2003). Higher ionic strength encourages Aβ fibril formation (Harper et al. 1999; Nichols et al. 2002; Nichols et al. 2015) and the fibril insolubility allows isolation in the pellet fraction after centrifugation of the sample at 17,000g (Walsh et al. 1997). Thoughtful preparation and rigorous characterization for a given Aβ preparation is important to fully interpret the interactions between Aβ and biological targets.
Aβ conformation and inflammatory receptor interaction
Due to the different conformation states and variable pathogenicity of Aβ, several studies have focused on how different species interact with neuroinflammation-associated receptors (Figure 1). These receptors have been found in pre-synaptic and post-synaptic neurons as well as intracellularly and on the cell surface of astrocytes and microglia (Jarosz-Griffiths et al. 2016). Microglial receptors continue to be of particular interest due to the cells’ key role in the spread of inflammation (Doens and Fernandez 2014). The inclusion of Aβ as a pathogen-associated molecular pattern (PAMP) and danger-associated molecular pattern (DAMP) further expands the number of cell-surface Aβ receptors and adds intracellular receptors such as nod-like receptors (NLRs) to the growing list (Heneka 2017). Although there are numerous Aβ receptors, this section will highlight a few that interact with different forms of Aβ and mediate inflammatory processes. Early studies provided evidence for a receptor complex comprised of CD36, α6β1 integrins, and CD47 that recognizes fibrillar Aβ and initiates a pro-inflammatory cascade involving tyrosine kinases. Individual components were determined by systematic testing of selective antagonists for each component (Bamberger et al. 2003). In addition to the integrin receptor complex, an impressive number of reports have implicated Toll-like receptors (TLRs) as major mediators of inflammation induced by fibrillar Aβ (Walter et al. 2007; Jana et al. 2008; Udan et al. 2008; Reed-Geaghan et al. 2009; Stewart et al. 2010). TLRs 2, 4, and 6 along with CD36 and the TLR accessory protein CD14 have all been shown to contribute substantially to the overall proinflammatory response. As interest grew in soluble aggregated forms of Aβ, receptors were examined for their ability to recognize these species. Terrill-Usery et al demonstrated that TLRs (via the adaptor protein MyD88) were the major mediators of tumor necrosis α (TNFα) and interleukin-1β (IL-1β) production induced by protofibrillar Aβ42 in primary wild-type and MyD88−/− mouse microglia (Terrill-Usery et al. 2014). RAGE is another microglia receptor involved in neuroinflammation. RAGE was implicated as a mediator of Aβ-induced synaptic depression in entorhinal cortex slices after bath application of the slices with a solution of Aβ42 monomers and small oligomers (dimers and trimers) (Origlia et al. 2010). NLRs are potential intracellular receptors for Aβ, and although their direct interaction has not been demonstrated, it has been known for some time that Aβ can enter microglia, trigger NLRP3 inflammasome activation, and produce mature IL-1β (Halle et al. 2008). Furthermore, CD36/TLR4/6-mediated uptake of soluble Aβ triggered inflammasome activation and conversion of the Aβ into a fibrillar form (Sheedy et al. 2013). This finding provided evidence that the NLRP3 inflammasome contributes to Aβ nucleation and aggregation inside the cell. An intriguing receptor for Aβ is APP itself. A recent report showed that oligomeric Aβ42 oligomers and fibrils stimulated tyrosine kinase pathways and TNFα production at similar levels in wild-type microglia, yet only fibrils induced an inflammatory response in APP−/− mouse microglia (Manocha et al. 2016). The finding suggested that APP may serve as a receptor for oligomeric Aβ. Additional Aβ receptors that may be sensitive to Aβ conformation include formyl-peptide-receptor-like-1 (FPRL1), additional SRs, complement receptors, and TREM2 (Doens and Fernandez 2014; Zhong et al. 2018).
Aβ conformation and cytokine production
The relationship between Aβ aggregation structure and cellular stimulation of pro-inflammatory cytokine release (Figure 1) has been explored by numerous groups. An early study demonstrated that fibrillar forms of Aβ40, and the fragment Aβ25-35, stimulated IL-1β release from lipopolysaccharide (LPS)-treated THP-1 human monocytes (Lorton et al. 1996) While non-aggregated Aβ was not stimulatory. This paradigm of LPS-primed immune cells continues to be used as a model for infection in neurodegeneration or to study two-signal activation of the inflammasome. Despite the utility of LPS priming, it was subsequently shown that fibrillar Aβ40 alone stimulated tyrosine kinase-mediated pathways leading to increased TNFα and IL-1β gene expression in THP-1 cells (Combs et al. 2001). Stimulation of cytokines (TNFα and IL-1β) by fibrillar Aβ40 was also corroborated in primary murine microglia (Yates et al. 2000). More recent work has focused on Aβ42 and other types of aggregated Aβ species. Golenbock and colleagues demonstrated production of IL-1β in LPS-treated primary murine microglia by fibrillar, but not nonfibrillar reverse Aβ peptide (Halle et al. 2008). To further probe the impact of Aβ conformation, White el al. directly compared the stimulatory effect of oligomeric and fibrillar Aβ42 in rat astrocytes and found that the two species stimulate different types of inflammatory responses. They observed that Aβ42 oligomers were more potent than fibrils and induced a profound, early inflammatory response. Fibrils caused a delayed, more chronic response (White et al. 2005). Similar findings were obtained in a later study where both low and high Mw Aβ42 oligomers (L-Aβol and H-Aβol respectively) were compared to Aβ42 fibrils (Heurtaux et al. 2010). Both oligomeric species induced similar levels of TNFα in MMGT12 murine microglial cells, and these levels were significantly higher than those induced by fibrils. This robust proinflammatory activity by soluble Aβ42 aggregates has also been observed in another report that directly compared TNFα secretion from primary murine microglia in response to carefully isolated Aβ42 monomers, protofibrils, and fibrils (Paranjape et al. 2012). Monomers and fibrils triggered very low levels of TNFα, while protofibrils provoked ~20-fold higher TNFα levels than the other species. This low level of TNFα production by fibrils was despite the finding that fibrils displayed significantly higher thioflavin T fluorescence, a marker of β-sheet structure. Interestingly, Aβ40 protofibrils were much less effective than Aβ42 protofibrils. The normal aging process may play a role in the responsiveness of microglia to different conformations of Aβ. In a study comparing TNFα secretion in postnatal and adult murine microglia, both oligomeric and fibrillar Aβ42 stimulated TNFα in young microglia. However, only Aβ42 oligomers induced TNFα in adult microglia (Floden and Combs 2006). Alternate Aβ conformations may impact levels of particular inflammatory molecules differently. Combs and colleagues demonstrated that oligomers were better than fibrils at stimulating IL-6 in murine microglia, yet the reverse was true for stimulating keratinocyte chemoattractant chemokine (Sondag et al. 2009). The cumulative data emphasizes the importance of Aβ conformation in multiple mechanisms of microglia interaction and phenotypic transformation. This is particularly true for Aβ42, with its increased aggregation propensity, ability to form multiple aggregation species, and high relevancy in neuroinflammatory processes linked to AD pathology.
Aβ conformation and microglial internalization
Microglia are the primary phagocytic cell in the brain. The interaction between microglia and extracellular brain Aβ is a significant area of AD research. Much of this focus is on the ability of microglia to internalize Aβ as this would provide an efficient way to clear Aβ and reduce or prevent accumulation. However, even this interaction is sensitive to Aβ conformation or aggregation state. There are many reports of microglial internalization of different forms of Aβ (monomer, oligomer, protofibril, fibril), but fewer have directly compared these species within a single published report. Maxfield and colleagues utilized radio- and fluorescent-labeling to study primary murine microglial internalization of soluble (presumably monomeric) and fibrillar Aβ at low concentrations (0.2-1.0 μM) (Chung et al. 1999). For Aβ fibrils, they observed saturable uptake, minimal degradation, and slow release. Soluble Aβ was internalized by a non-saturable process and was released rapidly from microglia also with minimal degradation. Two major mechanistic classes of cellular internalization include phagocytosis for particles greater than 0.5 μm, and macropinocytosis for extracellular fluid containing smaller, soluble particles (Gordon 2003). The two processes have commonalities in receptors and signaling pathways, particularly the formation of cups in the plasma membrane (Swanson 2008). The mechanistic aspects of Aβ internalization by microglia were further probed by Landreth and colleagues who again delineated between fibrillar and soluble Aβ. In their study using both BV-2 and primary murine microglia, it was determined that fibrillar Aβ was internalized by phagocytosis and soluble Aβ through fluid-phase macropinocytosis (Mandrekar et al. 2009). The concept of “soluble” Aβ was investigated more closely by inclusion of soluble aggregated species such as oligomers and protofibrils. Grutzendler and colleagues demonstrated in an in vivo study that microglia were highly effective at protofibrillar Aβ uptake but were incapable of phagocytosis of fibrillar congophilic Aβ (Liu et al. 2010). Further differences between Aβ species were subsequently reported in another study using primary murine microglia (Taneo et al. 2015). Aβ42 oligomers and fibrils were internalized more readily than monomers, but only the oligomers induced endo/phagolysosome rupture releasing cathepsin B into the cytoplasm. In another study conducted by Nichols and colleagues, a rigorous separation of “soluble” Aβ was implemented. Size-exclusion chromatography was used to isolate fluorescently-labeled monomeric and protofibrillar Aβ42. This methodology produced a more nuanced picture of Aβ internalization and showed that primary mouse microglia internalize protofibrils rapidly and robustly compared to monomers (Gouwens et al. 2016). Additional experimentation by the group concluded that very little degradation of the protofibrils occurred. The overall data indicate that microglia-mediated internalization is greatly dependent on Aβ conformation or aggregation state. The molecular details of the interactions driving and directing the internalization processes remain to be elucidated (Figure 1). One fate of internalized Aβ may be packaging into microvesicles and release from the cells back into the extracellular space. Microvesicles are spherical structures ranging in size from 100-1000 nm that bleb from the cell surface carrying a variety of biomolecules (Budnik et al. 2016). Several studies have reported the release of previously internalized Aβ in association with microvesicles (Joshi et al. 2014; Sollvander et al. 2016; Gouwens et al. 2018). This process appears to be sensitive to Aβ conformation and has implications for spreading of Aβ and proliferation of neuroinflammation.
Another major microglia-mediated immune pathway that is also sensitive to Aβ conformation or aggregation state is the classical complement pathway. A long-established finding showed that the assembly of Aβ aggregates was critical for binding of C1q and activation of complement (Jiang et al. 1994). Furthermore, Aβ, complement-dependent pathways, and microglia cause elevated synaptic pruning and synaptic loss in AD (Hong et al. 2016). There are numerous aspects to the relationship between microglial activation and neurodegeneration. While many of these mechanisms have been elucidated in vitro, studies of AD patients have shown that the degree of microglial activation was positively correlated with clinical dementia rating, Braak staging, and neuritic plaque development (reviewed in (Mrak 2012). Further discussion of this relationship is provided in the next section.
The relationship of microglia and Aβ plaque deposition
Aβ-induced alterations of the microglial phenotype
Microglia readily react to disturbances of their environment; accordingly, the deposition of Aβ as insoluble plaques in the brain parenchyma of AD patients changes microglial function. Early studies already described pronounced microglial accumulation around amyloid plaques (microgliosis) in AD brain tissue (Dickson et al. 1988; Haga et al. 1989; Itagaki et al. 1989), combined with dramatic morphological changes of plaque-associated microglia (Akiyama et al. 2000). These features are recapitulated in the aforementioned mouse models of AD pathology, which reproduce amyloid plaque deposition. These models are useful for studying the relationship between amyloid plaque deposition and microglial phenotypes, allowing also for experimental manipulation (Ashe and Zahs 2010; Jucker 2010).
In line with the clear increase in microglial numbers in the direct vicinity of amyloid plaques, recent studies confirmed increased microglial proliferation rates in living mice in response to cerebral β-amyloidosis (Fuger et al. 2017). Additional in vivo mouse studies demonstrate that CSF1R signaling appears to play a critical role in regulating this proliferation (Kamphuis et al. 2012; Gomez-Nicola et al. 2013; Olmos-Alonso et al. 2016). In addition, long-term two-photon imaging of microglia in APP/PS1 mice revealed that plaque-distant microglia proliferate and subsequently migrate to amyloid plaques (Fuger et al. 2017), providing a mechanism for the commonly observed microgliosis in AD models and indicating that soluble factors (rather than aggregated amyloid itself) may be the trigger for microglial proliferation.
Alterations of microglia in the vicinity of Aβ plaques are not restricted to changes in morphology and cell number. For example, various studies demonstrated impairments in microglial phagocytosis in AD models (Floden and Combs 2011; Krabbe et al. 2013; Wildsmith et al. 2013). Such changes in microglial clearance of Aβ are of immense interest to the field of AD research, as microglial phagocytic activity would in principle be able to degrade Aβ aggregates and may therefore alleviate amyloid-induced pathology (D’Andrea et al. 2004; Heneka and O’Banion 2007; Heneka et al. 2015). As such, improving microglial phagocytosis using Aβ-directed antibodies is still being intensely studied as a therapeutic strategy in clinical trials for AD patients (Fu et al. 2010; Graham et al. 2017). For example, the anti-Aβ antibody aducanumab was shown to strongly enhance recruitment of microglia to amyloid plaques and simultaneously reduce the amount of plaques in AD patients, suggesting that improving microglial clearance of Aβ might indeed represent a promising therapeutic avenue (Sevigny et al. 2016).
In addition to reduced phagocytosis, extensive studies suggest upregulated pro-inflammatory microglial activity (Rogers et al. 1992; Lue et al. 2001b; Butovsky et al. 2005; Lindberg et al. 2005; Floden and Combs 2006). Aβ induces complex changes in the microglial phenotype since it acts on a diverse array of microglial signaling receptors, including TLRs and SRs (El Khoury et al. 1996; Paresce et al. 1996; Landreth and Reed-Geaghan 2009), and activates complex signaling cascades such as the NLRP3 inflammasome (Halle et al. 2008). The considerable heterogeneity in gene expression between individual microglia and particularly between microglia associated with plaques and in plaque-free areas was highlighted by recent single-cell transcriptional profiling studies (Keren-Shaul et al. 2017; Korin et al. 2017; Krasemann et al. 2017b; Mathys et al. 2017). In these studies, microglial subtypes associated with neurodegenerative disease, including DAM and MGnD (as described in Table 1), implicated TREM2 and ApoE as key mediators of these phenotypes (Keren-Shaul et al. 2017; Krasemann et al. 2017b). Furthermore, it was shown that a switch in the microglial activation state is necessary for their response to amyloid deposits. For example, microglia in a homeostatic state (e.g. shown by their expression of P2RY12) do not cluster around amyloid plaques. Moreover, altering the balance between microglial disease-associated and homeostatic phenotypes significantly affects the microglial reaction towards amyloid pathology by altering microglial clustering, phagocytosis, and cytokine secretion (Jay et al. 2015a). For example, a signaling role for DAM markers ApoE and TREM2 in the induction of microglial activation in response to Aβ deposition has now been demonstrated through analysis of microglial gene expression profiles in knockout mouse models (Keren-Shaul et al. 2017; Krasemann et al. 2017b; Ulrich et al. 2018). Interestingly, genetic variants of both proteins have been linked to AD susceptibility, with variants in APoE and TREM2 being the two strongest known risk factors for late-onset AD (Corder et al. 1993; Guerreiro et al. 2013a; Jonsson et al. 2013). In the future, single-cell studies will aid in identifying heterogeneous subpopulations of microglia and define disease-specific subgroups. Correlations with functional phenotypes could shed new light on therapeutic targets to specifically manipulate microglia in areas of pathology.
While these studies have provided considerable evidence that Aβ plaques alter microglial activity and function, changes in microglial function are not a mere bystander effect of Aβ deposition. Instead, many studies demonstrate that the microglial functional state itself modulates amyloid pathology, demonstrating a bi-directional relationship between microglial function and amyloid plaque deposition (Figure 2).
Figure 2.
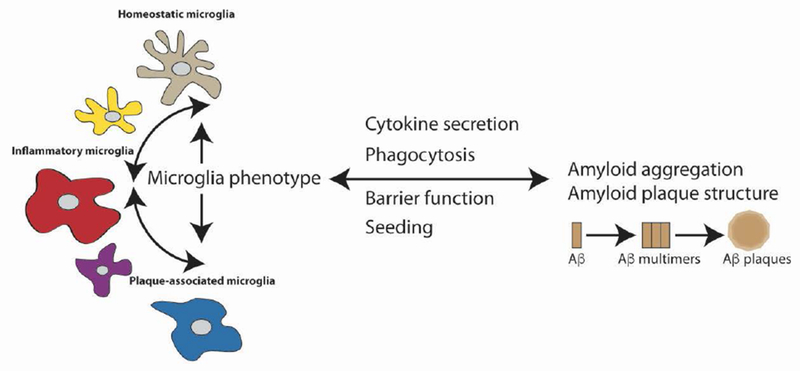
Bi-directional relationship between microglia and plaque deposition. Significant evidence indicates that Aβ aggregation and accumulation in plaque deposits leads to changes in microglia phenotype and microglia-mediated neuroinflammation. However, many studies also demonstrate that the microglial functional state can, in turn, modulate Aβ aggregation, plaque deposition, and pathology. Mechanisms by which microglia may negatively modulate Aβ aggregation or spread may include phagocytosis or the formation of a tight barrier around plaques, while positive modulation may involve secretion of cytokines or seeding of Aβ by microglial cellular factors.
Modulation of amyloid pathology by microglia
Strong evidence for the idea that microglia may directly impact Aβ aggregation was provided by genome-wide association studies that linked variants in microglial genes to an increased risk for the development of late-onset AD (Hollingworth et al. 2011b; Naj et al. 2011a; Lambert et al. 2013b; Zhang et al. 2013; Gagliano et al. 2016; Efthymiou and Goate 2017; Huang et al. 2017). These studies have intensified the interest in studying the microglial contribution to AD pathogenesis.
Due to the identification of TREM2 as a strong genetic risk factor for AD, its role in AD is being intensely studied. TREM2 binds a variety of membrane lipids and is involved in microglial phagocytosis of damaged neurons and Aβ (Wang et al. 2015b; Wang et al. 2015a; Zhao et al. 2018). In addition, TREM2 has recently been shown to directly bind oligomeric Aβ (Zhao et al. 2018). As mentioned above, recent studies also demonstrated that TREM2 is controlling the balance between homeostatic and pathology-associated microglial states (Keren-Shaul et al. 2017; Krasemann et al. 2017b; Mazaheri et al. 2017). However, while knockout of TREM2 robustly impairs microglial clustering around amyloid plaques in mice, it has produced inconsistent results on amyloid plaque load (Jay et al. 2015b; Wang et al. 2015b; Wang et al. 2015a), which may be due to different roles of TREM2 in different stages of disease (Jay et al. 2017). However, a recent study found that overexpressing human TREM2 in mouse microglia leads to a robust reduction in cerebral amyloidosis, clearly indicating that activation of this receptor may be therapeutically useful (Lee et al. 2018).
Recently, a previously unappreciated role for TREM2 in the formation of a protective microglial barrier around amyloid plaques has been suggested. A number of studies showed that microglia, which cluster around Aβ plaques, use their processes to form a tight barrier to shield the surrounding environment from toxic Aβ species. Interestingly, TREM2 appeared to be specifically expressed by microglial processes contacting amyloid plaques and TREM2 knockout abolished microglial clustering. As a result, TREM2 knockout mice and also human TREM2 R47H cases of late-onset AD had increased amounts of diffuse forms of Aβ surrounding amyloid plaques (Wang et al. 2016; Yuan et al. 2016). Importantly, this change in Aβ plaque structure was associated with increased neuronal damage, indicating that, in addition to the total amount of amyloid, the particular structure of amyloid deposits could also impact disease progression and can be influenced by microglial function. Similarly, other factors, such as deletion of CX3CR1 or anti-Aβ immunization, also modulate amyloid plaque structure and neuronal damage (Condello et al. 2015), implicating microglial barrier function as a novel mechanism in amyloid-plaque compaction and the restriction of amyloid-associated toxicity. Deciphering cause or consequence remains difficult in this context. In the brains of AD patients, a variety of plaque structures occur, ranging from dense core to diffuse plaques (D’Andrea et al. 2004; Yuan et al. 2016; Rasmussen et al. 2017). The amount of microglial accumulation differs markedly between dense core and diffuse plaques; however, it is unclear whether the change in plaque structure is caused by differences in microglial barrier function or whether different plaque conformations trigger differential migration/turnover of microglia. Adding to the complexity, it was previously demonstrated that even the dense core fibrillar plaques may possess Aβ conformational diversity by serving as reservoirs of oligomeric Aβ (Koffie et al. 2009).
In addition to the recent interest in TREM2, a multitude of studies have analyzed the effect of overexpression or knockout of certain cytokines on amyloid pathology. However, these studies have again shown conflicting results (Chakrabarty et al. 2010a; Chakrabarty et al. 2010b; Vom Berg et al. 2012). For example, overexpression of IL-1β was found to exert beneficial effects on Aβ deposition (Shaftel et al. 2007), while other studies demonstrated ameliorated disease progression upon blockage of IL-1 signaling or knockout of the IL-1β-producing NLRP3 inflammasome (Kitazawa et al. 2011; Heneka et al. 2013). Recently, the inflammasome adaptor protein ASC, which forms aggregates (so called specks) upon inflammasome activation and can subsequently be released from microglia, was implicated in cross-seeding Aβ pathology (Venegas et al. 2017), suggesting another microglial mechanism in the modulation of cerebral β-amyloidosis. Similarly, a two-photon in vivo study indicated that microglial uptake of aggregated Aβ, followed by microglial death and release of residual Aβ, could lead to the spreading of amyloid deposition (Baik et al. 2016).
Another factor that needs to be considered when studying microglial function in AD is the extraordinarily long lifetime of individual microglia. While an early study already indicated that microglia have very low turnover rates (Lawson et al. 1992), recent studies have readdressed this question (Askew et al. 2017; Fuger et al. 2017; Tay et al. 2017). Two studies employing genetic labeling of individual microglia reported very long lifetimes of more than 15 months for cortical microglia (Fuger et al. 2017; Tay et al. 2017). Of note, the turnover of microglia varies between brain regions, proving once more the heterogeneity among individual microglia (Askew et al. 2017; Tay et al. 2017). This extremely long lifetime of microglia has important implications for microglial function. In aged mice, microglial are skewed towards adopting a pro-inflammatory phenotype with impaired homeostatic functions (Sierra et al. 2007; Norden and Godbout 2013; Hefendehl et al. 2014; Grabert et al. 2016; Galatro et al. 2017). Furthermore, some studies suggest that microglial senescence occurs with age and might contribute to the development of neurodegenerative diseases (Streit et al. 2004; Streit et al. 2009).
Due to their extraordinarily long lifespan, alterations of the microglial phenotype in response to disturbances in the environment may change the microglial reaction to much later occurring stimuli, a concept known as innate immune memory (Wendeln et al. 2018). Accordingly, it was recently shown that microglia are capable of immune memory, proving that external stimuli can induce long-lasting alterations of the microglial epigenome and thereby alter the microglial reaction to secondary stimuli, including Aβ deposition (Wendeln et al. 2018). Importantly, the concept of innate immune memory might also explain the many confounding studies on the role of microglia in AD, since a combination of stimuli over a long period of time (possibly across the whole microglial lifetime) might impact the microglial phenotype in disease, but this is a field that is only beginning to be explored.
Effects of microglia depletion
The studies described above convincingly demonstrate that alterations of microglial activity impact Aβ deposition. An alternative approach to study the contribution of microglia to amyloid pathology is the use of microglia-depletion models. For example, using expression of a ganciclovir-activated suicide gene in microglia, a 90% reduction in the number of microglia was achieved, but this reduction in microglial numbers did not alter amyloid plaque load in APP/PS1 mice over a four week incubation period (Grathwohl et al. 2009). Moreover, upon discontinuation of ganciclovir treatment, peripheral myeloid cells rapidly repopulate the brain in this model (Varvel et al. 2012; Varvel et al. 2015). However, the resulting replacement of microglia with peripherally derived myeloid cells did not alter amyloid plaque load (Prokop et al. 2015; Varvel et al. 2015), suggesting that the impairment of microglial clearance function in AD cannot be rescued by the infiltration of peripheral cells, as these are exposed to the same altered brain environment. Interestingly, a recent study using diphtheria toxin based ablation of microglia in APP/PS1 mice reported no changes to plaque number over 2 weeks, but a 13% increase in plaque size, indicating once more that the microglial barrier contributes to amyloid plaque compaction (Zhao et al. 2017).
A different approach to achieve microglial depletion is based on the inhibition of microglial CSF1R signaling. With certain doses of CSF1R inhibitors, a nearly complete ablation of microglia can be achieved (Elmore et al. 2014). Depletion of microglia using CSF1R inhibitor in a mouse models of AD did not alter Aβ load, but improved cognition (Dagher et al. 2015). In line with this finding, using a CSF1R inhibitor at a dose that blocks microglial proliferation without ablating microglia also improved cognitive measures without altering amyloid load in an AD mouse model (Olmos-Alonso et al. 2016), indicating that targeting of microglial turnover with CSF1R modulators might exert beneficial effects independent of amyloid load. In the latter study, CSF1R inhibition shifted microglia to adopt an anti-inflammatory phenotype, indicating once more the importance of the microglial activity state in modulating disease progression. Strikingly, a recent study indicated that early long-term depletion of microglia in the 5XFAD mouse model of Alzheimer’s pathology led to a dramatic decrease in plaque load (Sosna et al. 2018), implying that in addition to amyloid plaque compaction, microglia may also play a significant role in inducing plaque formation.
Dark microglia: a new phenotype predominantly associated with pathological states
Similar to the DAM and MgND, dark microglia are a disease-associated phenotype that is abundant in AD pathology. These cells were described in EM and their ultrastructural features will be discussed below. They are rare in healthy young adult mice, but drastically increase in number in the APP/PS1 mouse model of AD, and under conditions considered risk factors for AD, i.e. aging and chronic stress (Bisht et al. 2016b). Previous studies have indicated that chronic stress is both a cause and consequence of sporadic AD, which represents approximately 95% of all cases (for more on this topic, see the extended review by (Machado et al. 2014). Using paradigms of chronic unpredictable stress, an increased density of dark microglia was observed after two weeks of stress in wild-type mice compared to controls housed without any disturbance (Bisht et al. 2016b). Upon stress, dark microglia accounted for about one-fourth of the typical microglial population (Bisht et al. 2016b). Additionally, at 14 months of age, which corresponds to middle age in mice, dark microglia became highly prevalent, and even more in APP/PS1 mice of the same age. In this context of AD pathology, dark microglia accounted for two-third of the typical microglial population (Bisht et al. 2016b). In these APP/PS1 mice, where Aβ plaques are seen as early as 4 months of age (Malm et al. 2011), TREM2-positive dark microglia were detected nearby the amyloid plaques and in regions of neuronal dystrophy, showing engulfed amyloid deposits, as well as juxtaposing synaptic elements, which contained an accumulation of autophagosomes (Bisht et al. 2016b). As discussed above, TREM2 has been associated with the modulation of amyloid pathology (Wang et al. 2015a), while variants of this gene were also identified as an important risk factor for sporadic AD (Guerreiro et al. 2013a; Jonsson et al. 2013). The importance of TREM2 in dark microglia in AD is still undetermined. However, during early postnatal development, where dark microglia are also observed, these cells required the expression of TREM2, being strongly reduced in numbers in the hippocampus of TREM2 knockout mice (versus wild-type controls) (Bisht et al. 2017).
Distinct properties of dark microglia
Dark microglia are recognized by their electron-dense cytoplasm and nucleoplasm, making them appear as “dark” as mitochondria by EM (Bisht et al. 2016b). Bisht et al. hypothesized that this distinctive feature of the dark microglia could reflect cellular shrinkage, associated with oxidative stress in other cell types (Bisht et al. 2016a). Indeed, dark microglia display various features of oxidative stress including the dilation of their endoplasmic reticulum and Golgi apparatus, modifications related to their nucleus (condensation of the nucleoplasm, partial to complete lack of heterochromatin pattern), and alterations to mitochondria’s integrity, all of which might be attributed to oxidative stress (Bisht et al. 2016b). Under homeostatic conditions, enzymes and other antioxidant molecules are produced to neutralize reactive species (Rahal et al. 2014). However, this equilibrium is lost in non-homeostatic conditions, due to elevated reactive species levels or an altered production of antioxidants. The toxic reactive species can compromise cellular functions by affecting lipids, proteins, and nucleic acids, thus resulting in organelle impairments as well as morphological alterations (Rahal et al. 2014).
Another feature of dark microglia is their highly ramified and extremely thin processes that encircle various types of synaptic elements, i.e. synaptic clefts, pre-synaptic axon terminals, as well as post-synaptic dendritic branches and spines (Bisht et al. 2016b). By comparison, typical microglia make focal contacts with synaptic elements (Tremblay et al. 2010). The classical complement pathway might underlie dark microglia’s interactions with synapses considering that they strongly express CD11b, a component of the complement receptor 3 (CR3), in their distal processes encircling synaptic elements (Bisht et al. 2016b). As discussed above, previous studies have demonstrated a crucial involvement of this complement pathway, involving binding of CR3 to different complement components, in synaptic loss under various conditions (Stephan et al. 2013; Hong et al. 2016; Vasek et al. 2016). In AD pathology, 4 month-old APP/PS1 mice lacking the component 3 (C3) showed reduced synaptic loss in the hippocampus when compared to age-matched APP/PS1 mice (Hong et al. 2016). Likewise, 16 month-old APP/PS1 mice deficient in C3 had less extensive synaptic loss in the CA3 region of the hippocampus compared to age-matched APP/PS1 mice (Shi et al. 2015). Additionally, dark microglia associate with enlarged pockets of extracellular space (Bisht et al. 2016b), even more than typical microglia (Tremblay et al. 2010). This finding suggests an increased remodeling of the extracellular matrix by dark microglia, which could modulate the concentration of various glio- and neuroactive substances within the perisynaptic environment (Tremblay et al. 2011).
Beyond these ultrastructural differences, dark microglia’s distribution seems to be quite distinct from typical microglia as well. Typical microglia are localized both in the gray and white matter, being most present in the former, in young adult female BALB/c mice (Lawson et al. 1990), and they are found throughout the brain (Lawson et al. 1990). Dark microglia, however, appear so far to be restricted to specific brain regions, e.g. stratum lacunosum moleculare of the hippocampus CA1, the subgranular layers of prefrontal cortex, the basolateral nucleus of the amygdala, and the median eminence of the hypothalamus (Bisht et al. 2016b). Interestingly, the stratum lacunosum moleculare of the hippocampus CA1 display a pronounced loss of dendritic spines in the APP/PS1 mouse model (Siskova et al. 2014). While the brain regions where dark microglia are prevalent exert distinct functions, structurally, they have one characteristic in common: the presence of large blood vessels. Interestingly enough, dark microglia were found to tightly juxtapose the vasculature (Bisht et al. 2016b), with more than half of these cells directly touching one blood vessel in ultrathin section (Bisht et al. 2016b). Whether these cells could help to maintain blood brain barrier integrity, which is compromised in AD (Zenaro et al. 2017) still remains undetermined. Furthermore, dark microglia were mostly found within clusters, a microglial feature usually seen increasingly with aging (Hefendehl et al. 2014). An abnormal distribution of microglial parenchymal surveillance could lead to areas that are more prone to damage and pathology. In addition, clusters may also be the result of fast repopulating microglial cells (Bruttger et al. 2015). Bruttger et al. found after microglial ablation, through the administration of diphtheria toxin into 8-week old mice in which these cells selectively express the diphtheria toxin receptor, that microglia formed clusters during repopulation at the peak of proliferation (i.e. day 7 post-injection) then spread out to their specific territory (at day 14 post-injection). Dividing astrocytes were also shown using EM to associate with the vasculature and proliferate in response to acute injury (Bardehle et al. 2013). These overall observations raise the intriguing possibility that dark microglia may be newly formed microglial cells that have yet to spread out to their designated area.
Even though dark microglia’s distinct ultrastructural features and interactions with synapses as well as the vasculature were described and their regional distribution within the brain is being characterized, some of their essential properties including their nature are still shrouded in mystery. However, some pieces of puzzle have been put together to unravel the riddle behind their origin. In particular, the first clue that dark microglia originate from the embryonic yolk sac comes from the fact that these cells are found in the hippocampus of adult CCR2 knockout mice (Bisht et al. 2016b). In this model, most of the monocytes remain in the bone marrow, due to the lack of CCR2, a chemokine receptor that is required for their egress and recruitment to inflammatory sites (Tsou et al. 2007). CCR2 is also considered necessary for the infiltration of monocytes into the brain (Mildner et al. 2009). Nevertheless, as mentioned by Bisht et al. (2016b), the presence of dark microglia in the CCR2 knockout mice does not preclude the possibility that these cells could be monocytes that infiltrate the brain via CCR2-independent pathways. Dark microglia do, however, express the microglia-specific marker 4D4 (Krasemann et al. 2017b), which provides support for a microglial origin (Bisht et al. 2016b). In addition, dark microglia do not express 4C12, which is considered a marker of inflammatory monocytes (Bisht et al. 2016b). Although the physiological significance of dark microglia has yet to be elucidated, the findings presented in this section together suggest that dark microglia could represent a subset of microglial cells that become stressed as a result of their hyperactive involvement with the maintenance of brain physiology.
Summary/Conclusion
Microglia survey the central nervous system and are responsible for a multitude of immune, inflammatory, and clearance functions. Their role in neurodegenerative disease has been clearly established and this role may be a potential avenue of therapeutic intervention. The presence of different phenotypes of microglia and how they change or respond to pathologic changes during disease processes is of significant importance in the understanding of complex etiologies. Much attention and research has focused on Aβ, as the primary component of neuritic plaques in AD, and a potent stimulator of microglial activation. The extent, manner, and mechanism by which microglia respond to the presence and accumulation of protein aggregates such as Aβ ultimately may determine their impact and the overall outcome. A multitude of research demonstrates a bidirectional relationship between amyloid plaque deposition and microglial phenotypes, proving that changes in immune-associated molecules alone are sufficient to modulate Aβ pathology. These effects on Aβ pathology are mediated by alterations of microglial phenotypes and functions, e.g. by impacting microglial phagocytosis, barrier function, or cytokine secretion. Recent studies also suggest that some of the molecules implicated in microglial modulation of Aβ pathology might act together in previously unappreciated ways and converge on common microglial signaling pathways, e.g. pathways controlling metabolism (Chan et al. 2015; Ip et al. 2017; Krasemann et al. 2017b; Ulland et al. 2017). Future studies will reveal whether a common theme of clearly definable master regulators underlies the complex interplay between different microglial functions and thereby impacts neurological disease progression.
--Human subjects --.
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
⇒ if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
No
⇒ if it is a Review or Editorial, skip complete sentence ⇒ if No, include a statement: “ARRIVE guidelines were not followed for the following reason:” (edit phrasing to form a complete sentence as necessary).
⇒ if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Acknowledgements / Conflict of Interest Disclosure
This review was, in part, aided by work conducted under the support of National Institutes of Health Grants R15AG033913 (MRN). Marie-Ève Tremblay is recipient of a Canada Research Chair (Tier 2) of Neuroimmune Plasticity in Health and Therapy. Her work for this project was supported by a Foundation Grant from the Canadian Institutes of Health Research (CIHR). Marie-Kim St-Pierre is a recipient of CIHR and Fonds de recherche du Québec-Santé (FRQS) master’s scholarships. We respectfully include a conflict of interest statement disclosing that Colin K. Combs is an author on this publication and also serves as an editor for the Journal of Neurochemistry.
Footnotes
Abbreviations used – ABCA7, ATP-binding cassette sub-family member 7; AD, Alzheimer’s disease; Aβ, amyloid-β protein; ApoE, apolipoprotein E; ApoJ, apolipoprotein J; APP, amyloid precursor protein; ASC, apoptosis-associated speck-like protein containing a CARD; CARD, caspase recruitment domain; CLU, clusterin; CNS, central nervous system; CSF1R, colony stimulating factor receptor 1; COX-2, cyclooxygenase-2; CR1, complement component (3b/4b) receptor 1; CR3, complement receptor 3; DAM, disease-associated microglia; DAMP, danger-associated molecular pattern; EPHA1, ephrin type-A receptor 1; FPRL1, formyl-peptide-receptor-like-1; GFP, green fluorescent protein; GWAS, genome-wide association study; Iba1, ionized calcium binding adaptor molecule 1; IL-1β, interleukin-1β; INPP5D, inositol polyphosphate-5-phosphatase; LCA, leucocyte common antigen; LPS, lipopolysaccharide; MEF2C, myocyte enhancer factor 2C; MGnD, microglia neurodegenerative phenotype; MHCII, major histocompatibility complex class II; MS4A6A/MS4A4AE, membrane-spanning 4-domains subfamily 4; Mw, molecular weight; NFκB, nuclear factor kappa B; NLR, nod-like receptor; NSAID, nonsteroidal anti-inflammatory drug; RAGE, receptor for advanced glycation end products; PAMP, pathogen-associated molecular pattern; PET, positron emission tomography; PS1, presenilin-1; RA, rheumatoid arthritis; SR, scavenger receptor; SEC, size exclusion chromatography; EM, electron microscopy; TLR, Toll-like receptor; TNFα, tumor necrosis factor α; TREM2, triggering receptor expressed on myeloid cells 2; YS, embryonic yolk sac.
REFERENCES
- Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Thal LJ (2000) A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 54, 588–593. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Alzheimer’s Disease Cooperative S (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot F, Godin I, and Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Research. Developmental Brain Research 117, 145–152. [DOI] [PubMed] [Google Scholar]
- Andersen K, Launer LJ, Ott A, Hoes AW, Breteler MM, and Hofman A (1995) Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer’s disease? The Rotterdam Study. Neurology 45, 1441–1445. [DOI] [PubMed] [Google Scholar]
- Ashe KH and Zahs KR (2010) Probing the Biology of Alzheimer’s Disease in Mice. Neuron 66, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Gomez-Nicola D (2017) Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Reports 18, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik SH, Kang S, Son SM, and Mook-Jung I (2016) Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 64, 2274. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, and Landreth GE (2003) A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci 23, 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, and Kreutzberg GW (1993) Cytotoxicity of microglia. Glia 7, 111–118. [DOI] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Gotz M (2013) Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16, 580–586. [DOI] [PubMed] [Google Scholar]
- Beard CM, Waring SC, O’Brien PC, Kurland LT, and Kokmen E (1998) Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease: a case-control study in Rochester, Minnesota, 1980 through 1984. Mayo Clinic Proceedings 73, 951–955. [DOI] [PubMed] [Google Scholar]
- Bisht K, Sharma K, Lacoste B, and Tremblay MECP (2016a) Dark microglia: Why are they dark? Commun Integr Biol 9, e1230575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht K, Picard K, Vernoux N, Sharma K, Grinberg RP, Faustino JV, Tremblay M-E (2017) Dark microglia: a follow-up story across the lifespan Society for Neuroscience, Washington DC. [Google Scholar]
- Bisht K, Sharma KP, Lecours C, Gabriela Sanchez M, El Hajj H, Milior G, Tremblay ME (2016b) Dark microglia: A new phenotype predominantly associated with pathological states. Glia 64, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, and Teplow DB (2003) Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 100, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, Saunders AM (1995) Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiology of aging 16, 523–530. [DOI] [PubMed] [Google Scholar]
- Broe GA, Grayson DA, Creasey HM, Waite LM, Casey BJ, Bennett HP, Halliday GM (2000) Anti-inflammatory drugs protect against Alzheimer disease at low doses. Archives of Neurology 57, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, Waisman A (2015) Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 43, 92–106. [DOI] [PubMed] [Google Scholar]
- Budnik V, Ruiz-Canada C, and Wendler F (2016) Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben-Yaakov K, and Schwartz M (2005) Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-γ and IL-4 render them protective. Mol Cell Neurosci 29, 381. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Banati RB (2001) In-vivo measurement of activated microglia in dementia. Lancet (London, England) 358, 461–467. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, Dickson D, Golde TE, and Das P (2010a) IFN-γ promotes complement expression and attenuates amyloid plaque deposition in amyloid-β precursor protein transgenic mice. J Immunol 184, 5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Das P (2010b) Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. The FASEB Journal 24, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, White CC, Winn PA, Cimpean M, Replogle JM, Glick LR, De Jager PL (2015) CD33 modulates TREM2: convergence of Alzheimer loci. Nat Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, and Maxfield FR (1999) Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer’s amyloid β-peptide by microglial cells. Journal 274, 32301–32308. [DOI] [PubMed] [Google Scholar]
- Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Griffin RG (2016) Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J Am Chem Soc 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, and Landreth GE (2001) β-amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci 21, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, and Grutzendler J (2015) Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun 6, 6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (New York, N.Y.) 261, 921. [DOI] [PubMed] [Google Scholar]
- D’Andrea MR, Cole GM, and Ard MD (2004) The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Journal 25, 675. [DOI] [PubMed] [Google Scholar]
- Dagher NN, Najafi AR, Kayala KMN, Elmore MRP, White TE, Medeiros R, Green KN (2015) Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation 12, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Bonci A (2017) Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 95, 341–356 e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas AH, Boddeke HW, and Biber K (2008) Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 56, 888–894. [DOI] [PubMed] [Google Scholar]
- Deiters O (1865) Untersuchungen uber Gehirn und Ruckenmark des Menschen und der Saugethiere, Vieweg. [Google Scholar]
- Del Rio-Hortega P (1919a) El tercer elemento de los centros nerviosos I La microglia en estado normal II Intervencíon de la microglia en los procesos patológicos III Naturaleza probable de la microglia. Bol de la Soc esp de biol 9, 69–120. [Google Scholar]
- Del Rio-Hortega P (1919b) Poder fagocitario y movilidad de la microglia. Bol de la Soc esp de biol 9, 154. [Google Scholar]
- Del Rio-Hortega P (1924) Lo que debe entenderse por tercer elemento de los centros nerviosos. Bol de la Soc esp de biol 11, 33. [Google Scholar]
- Del Rio-Hortega P (1925) Participacíon de la microglia en la formacíon de los cuerpos amiláceos del tejido nervioso. Bol de la Soc esp de hist nat 27, 127. [Google Scholar]
- Del Rio-Hortega P (1927) Lesiones elementales de los centros nerviosos. Rev méd de Barcelona 8, 36. [Google Scholar]
- Del Rio-Hortega P (1932) Microglia. In: Penfield W, editor. Cytology and Cellular Pathology of the Nervous System. New York: Hoeber, 482–1924–1534. [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, and Yen SH (1988) Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 132, 86. [PMC free article] [PubMed] [Google Scholar]
- Doens D and Fernandez PL (2014) Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J Neuroinflammation 11, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Younkin S (1996) Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. [DOI] [PubMed] [Google Scholar]
- Efthymiou AG and Goate AM (2017) Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Hachimi KH and Foncin JF (1994) Do microglial cells phagocyte the beta/A4-amyloid senile plaque core of Alzheimer disease? Comptes rendus de l’Academie des sciences. Serie III, Sciences de la vie 317, 445–451. [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, and Loike JD (1996) Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 382, 716–719. [DOI] [PubMed] [Google Scholar]
- Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Green KN (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff S, Zhai R, and Novak JP (1997) Microglia and astroglia have a common progenitor cell. Journal of Neuroscience Research 50, 477–486. [DOI] [PubMed] [Google Scholar]
- Floden AM and Combs CK (2006) β-amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci 26, 4644–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floden AM and Combs CK (2011) Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J Alz Dis 25, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, and Wang KC (1992) Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta neuropathologica 84, 225–233. [DOI] [PubMed] [Google Scholar]
- Frost JL and Schafer DP (2016) Microglia: Architects of the Developing Nervous System. Trends Cell Biol 26, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu HJ, Liu B, Frost JL, and Lemere CA (2010) Amyloid-β immunotherapy for Alzheimer’s disease. CNS Neurol Disord Drug Targets 9, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuger P, Hefendehl JK, Veeraraghavalu K, Wendeln A-C, Schlosser C, Obermuller U, Jucker M (2017) Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci 20, 1371. [DOI] [PubMed] [Google Scholar]
- Fujita S and Kitamura T (1975) Origin of brain macrophages and the nature of the so-called microglia. Acta Neuropathologica. Supplementum Suppl 6, 291–296. [DOI] [PubMed] [Google Scholar]
- Gagliano SA, Pouget JG, Hardy J, Knight J, Barnes MR, Ryten M, and Weale ME (2016) Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol 3, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, Eggen BJL (2017) Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. [DOI] [PubMed] [Google Scholar]
- Ginhoux F and Prinz M (2015) Origin of microglia: current concepts and past controversies. Cold Spring Harbor Perspectives in Biology 7, a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D, and Huber T (2013) Origin and differentiation of microglia. Frontiers in Cellular Neuroscience 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golgi C (1903) Opera Omnia, Hoepli.
- Gomez-Nicola D, Fransen NL, Suzzi S, and Perry VH (2013) Regulation of Microglial Proliferation during Chronic Neurodegeneration. J Neurosci 33, 2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3, 23–35. [DOI] [PubMed] [Google Scholar]
- Gouwens LK, Makoni NJ, Rogers VA, and Nichols MR (2016) Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res 1648, 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens LK, Ismail MS, Rogers VA, Zeller NT, Garrad EC, Amtashar FS, Nichols MR (2018) Aβ42 protofibrils interact with and are trafficked through microglial-derived microvesicles. ACS Chem Neurosci 9, 1416–1425. [DOI] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, McColl BW (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nature Neurosci 19, 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham WV, Bonito-Oliva A, and Sakmar TP (2017) Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu Rev Med 68, 413. [DOI] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Jucker M (2009) Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat Neurosci 12, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group AR, Lyketsos CG, Breitner JCS, Green RC, Martin BK, Meinert C, Sabbagh M (2007) Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808. [DOI] [PubMed] [Google Scholar]
- Group AR, Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JCS, Mullan M (2008) Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Archives of Neurology 65, 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Hardy J (2013a) TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Alzheimer Genetic Analysis G (2013b) TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Hung AY, and Selkoe DJ (1991) Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci 11, 3783–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga S, Akai K, and Ishii T (1989) Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol 77, 569. [DOI] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, Clinical I. t. (2016) Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264. [DOI] [PubMed] [Google Scholar]
- Hao C, Richardson A, and Fedoroff S (1991) Macrophage-like cells originate from neuroepithelium in culture: characterization and properties of the macrophage-like cells. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience 9, 1–14. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lieber CM, and Lansbury PT Jr. (1997) Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chem. Biol 4, 951–959. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, and Lansbury PT Jr. (1999) Assembly of Aβ amyloid peptides: an in vitro model for a possible early event in Alzheimer’s disease. Biochemistry 38, 8972–8980. [DOI] [PubMed] [Google Scholar]
- Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, and Jucker M (2014) Homeostatic and injury-induced microglia behavior in the aging brain. Aging cell 13, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT (2017) Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol 27, 220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT and O’Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184, 69. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Golenbock DT (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henle J. a. M. F. (1869) Uber die sogenannte Bindesubstanz der Centralorgane des Nervensystems. Z. Med 34, 49–82. [Google Scholar]
- Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Joyce JG (2006) Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 45, 15157–15167. [DOI] [PubMed] [Google Scholar]
- Heurtaux T, Michelucci A, Losciuto S, Gallotti C, Felten P, Dorban G, Heuschling P (2010) Microglial activation depends on beta-amyloid conformation: role of the formylpeptide receptor 2. J Neurochem 114, 576–586. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, and El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P Harold D Sims R Gerrish A Lambert J-C Carrasquillo MM, Williams J (2011a) Common variants in ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P Harold D Sims R Gerrish A Lambert J-C Carrasquillo MM, Williams J (2011b) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Stevens BCP (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K-L, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Goate AM (2017) A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui CW, St-Pierre A. l., El Hajj H, Remy Y, Hebert S. b. S., Luheshi GN, Tremblay M-E (2018) Prenatal Immune Challenge in Mice Leads to Partly Sex-Dependent Behavioral, Microglial, and Molecular Abnormalities Associated with Schizophrenia. Front Mol Neurosci 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume DA, Perry VH, and Gordon S (1983) Immunohistochemical localization of a macrophage-specific antigen in developing mouse retina: phagocytosis of dying neurons and differentiation of microglial cells to form a regular array in the plexiform layers. The Journal of Cell Biology 97, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in ‘t Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Stricker BH (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med 345, 1515–1521. [DOI] [PubMed] [Google Scholar]
- Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R (2017) Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, and Akiyama H (1988) Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neuroscience Letters 91, 259–264. [DOI] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, and Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24, 173. [DOI] [PubMed] [Google Scholar]
- Jana M, Palencia CA, and Pahan K (2008) Fibrillar amyloid-β peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol 181, 7254–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz-Griffiths HH, Noble E, Rushworth JV, and Hooper NM (2016) Amyloid-β receptors: the good, the bad, and the prion protein. J Biol Chem 291, 3174–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, and Lansbury PT Jr. (1993) The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32, 4693–4697. [DOI] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Landreth GE (2017) Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci 37, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Lamb BT (2015a) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med 212, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Lamb BT (2015b) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson ML, Bliss MR, Brain AT, and Scott DL (1989) Rheumatoid arthritis and senile dementia of the Alzheimer’s type. British Journal of Rheumatology 28, 86–88. [DOI] [PubMed] [Google Scholar]
- Jiang H, Burdick D, Glabe CG, Cotman CW, and Tenner AJ (1994) β-amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol 152, 5050–5059. [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, and Selkoe DJ (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Stefansson K (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Turola E, Ruiz A, Bergami A, Libera DD, Benussi L, Verderio C (2014) Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ 21, 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M (2010) The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med 16, 1210. [DOI] [PubMed] [Google Scholar]
- Kamphuis W, Orre M, Kooijman L, Dahmen M, and Hol EM (2012) Differential cell proliferation in the cortex of the appsweps1de9 alzheimer’s disease mouse model. Glia 60, 615. [DOI] [PubMed] [Google Scholar]
- Karch CM, Cruchaga C, and Goate A (2014) Alzheimer’s Disease Genetics: From the bench to the clinic. Neuron 83, 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, Amit I (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276. [DOI] [PubMed] [Google Scholar]
- Kettenmann H and Verkhratsky A (2008) Neuroglia: the 150 years after. Trends in Neurosciences 31, 653–659. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch U-K, Noda M, and Verkhratsky A (2011) Physiology of microglia. Physiological Reviews 91, 461–553. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Miyake T, and Fujita S (1984) Genesis of resting microglia in the gray matter of mouse hippocampus. The Journal of Comparative Neurology 226, 421–433. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, LaFerla FM (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol 187, 6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, … Spires-Jones T. L. (2009) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA 106, 4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, Rolls A (2017) High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci 20, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Heppner FL (2013) Functional Impairment of Microglia Coincides with β-Amyloid Deposition in Mice with Alzheimer-Like Pathology. PLoS ONE 8, e60921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Butovsky O (2017a) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Butovsky O (2017b) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Amouyel P (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41, 1094–1099. [DOI] [PubMed] [Google Scholar]
- Lambert J-C Ibrahim-Verbaas CA Harold D Naj AC Sims R Bellenguez C, Amouyel P (2013a) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC Verbaas CA Harold D Naj AC Sims R Bellenguez C, Amouyel P (2013b) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreth GE and Reed-Geaghan EG (2009) Toll-Like Receptors in Alzheimer’s Disease, in Curr Top Microbiol Immunol, Vol. 336, p 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA and Grillo-Bosch D (2005) In vitro preparation of prefibrillar intermediates of amyloid-β and α-synuclein. Methods Mol Biol 299, 19–33. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, and Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48, 405. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, and Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Xue R, and Gambetti P (1996) Amyloid precursor protein metabolism in primary cell cultures of neurons, astrocytes, and microglia. Journal of Neurochemistry 66, 2300–2310. [DOI] [PubMed] [Google Scholar]
- Lee CYD, Daggett A, Gu X, Jiang L-L, Langfelder P, Li X, Yang XW (2018) Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 97, 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhossek MV (1893) Der feinere Bau des Nervensystems im Lichte neuester Forschung. Fischer’s Medicinische Buchhandlung H. Kornfield. [Google Scholar]
- Li G, Shen YC, Li YT, Chen CH, Zhau YW, and Silverman JM (1992) A case-control study of Alzheimer’s disease in China. Neurology 42, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Li Q and Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18, 225–242. [DOI] [PubMed] [Google Scholar]
- Lindberg C, Selenica M-LB, Westlind-Danielsson A, and Schultzberg M (2005) β-amyloid protein structure determines the nature of cytokine release from rat microglia. J Mol Neurosci 27, 1. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, and McDowell I (2002) Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology 156, 445–453. [DOI] [PubMed] [Google Scholar]
- Ling EA and Wong WC (1993) The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 7, 9–18. [DOI] [PubMed] [Google Scholar]
- Ling EA, Ng YK, Wu CH, and Kaur C (2001) Microglia: its development and role as a neuropathology sensor. Prog Brain Res 132, 61–79. [DOI] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, and Grutzendler J (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci 30, 17091–17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorton D, Kocsis JM, King L, Madden K, and Brunden KR (1996) β-amyloid induces increased release of interleukin-1β from lipopolysaccharide-activated human monocytes. J Neuroimmunol 67, 21–29. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Yan SD (2001a) Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Experimental Neurology 171, 29–45. [DOI] [PubMed] [Google Scholar]
- Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Rogers J (2001b) Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 35, 72. [DOI] [PubMed] [Google Scholar]
- Machado A, Herrera AJ, de Pablos RM, Espinosa-Oliva AM, Sarmiento M, Ayala A, Cano J (2014) Chronic stress as a risk factor for Alzheimer’s disease. Rev Neurosci 25, 785–804. [DOI] [PubMed] [Google Scholar]
- Malm T, Koistinaho J, and Kanninen K (2011) Utilization of APPswe/PS1dE9 transgenic mice in research of Alzheimer’s disease: Focus on gene therapy and cell-based therapy applications. Int J Alzheimers Dis 2011, 517160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, and Landreth GE (2009) Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci 29, 4252–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manocha GD, Floden AM, Rausch K, Kulas JA, McGregor BA, Rojanathammanee L, Combs CK (2016) APP regulates microglial phenotype in a mouse model of Alzheimer’s disease. J Neurosci 36, 8471–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M,T Sai L-H (2017) Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 21, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Haass C (2017) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, and McGeer EG (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neuroscience Letters 79, 195–200. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Akiyama H, Itagaki S, and McGeer EG (1989) Immune system response in Alzheimer’s disease. The Canadian Journal of Neurological Sciences. Le Journal Canadien Des Sciences Neurologiques 16, 516–527. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer E, Rogers J, and Sibley J (1990) Anti-inflammatory drugs and Alzheimer disease. Lancet (London, England) 335, 1037. [DOI] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, Prinz M (2009) CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132, 2487–2500. [DOI] [PubMed] [Google Scholar]
- Mönning U, Sandbrink R, Weidemann A, Banati RB, Masters CL, and Beyreuther K (1995) Extracellular matrix influences the biogenesis of amyloid precursor protein in microglial cells. J Biol Chem 270, 7104–7110. [DOI] [PubMed] [Google Scholar]
- Mrak RE (2012) Microglia in Alzheimer brain: a neuropathological perspective. Int J Alzheimers Dis 2012, 165021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H (1851) Zur Histologie der Netzhaut. Z. Wiss. Zool 3, 234–237. [Google Scholar]
- Murabe Y and Sano Y (1982) Morphological studies on neuroglia. VI. Postnatal development of microglial cells. Cell and Tissue Research 225, 469–485. [DOI] [PubMed] [Google Scholar]
- Murabe Y and Sano Y (1983) Morphological studies on neuroglia. VII. Distribution of “brain macrophages” in brains of neonatal and adult rats, as determined by means of immunohistochemistry. Cell and Tissue Research 229, 85–95. [DOI] [PubMed] [Google Scholar]
- Myllykangas-Luosujärvi R and Isomäki H (1994) Alzheimer’s disease and rheumatoid arthritis. British Journal of Rheumatology 33, 501–502. [DOI] [PubMed] [Google Scholar]
- Naj A Jun G Beecham G Wang L-S Vardarajan BN Buros J, Schellenberg GD (2011a) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC Jun G Beecham GW Wang L-S Vardarajan BN Buros J, Schellenberg GD (2011b) Common variants in MS4A4/MS4A6E, CD2uAP, CD33, and EPHA1 are associated with late-onset Alzheimer’s disease. Nature genetics 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols MR, Colvin BA, Hood EA, Paranjape GS, Osborn DC, and Terrill-Usery SE (2015) Biophysical comparison of soluble amyloid-β(1-42) protofibrils, oligomers, and protofilaments. Biochemistry 54, 2193–2204. [DOI] [PubMed] [Google Scholar]
- Nichols MR, Moss MA, Reed DK, Lin WL, Mukhopadhyay R, Hoh JH, and Rosenberry TL (2002) Growth of β-amyloid(1-40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 41, 6115–6127. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Nissl F (1899) Ueber einige Beziehungen zwischen Nervenzellerkrankungen und gliiSsen Erscheinungen bei verschiedenen Psychosen. Arch Psychiat 32, 1–21. [Google Scholar]
- Norden DM and Godbout JP (2013) Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropath Appl Neuro 39, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil SM, Witcher KG, McKim DB, and Godbout JP (2018) Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun 6, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, Thiagarajan G, and Wetzel R (2006) Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol 413, 34–74. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Vassar R (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos-Alonso A, Schetters STT, Sri S, Askew K, Mancuso R, Vargas-Caballero M, Gomez-Nicola D (2016) Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 139, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, and Domenici L (2010) Microglial receptor for advanced glycation end product-dependent signal pathway drives β-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci 30, 11414–11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjape GS, Gouwens LK, Osborn DC, and Nichols MR (2012) Isolated amyloid-β(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem Neurosci 3, 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, and Maxfield FR (1996) Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron 17, 553. [DOI] [PubMed] [Google Scholar]
- Perry VH, Hume DA, and Gordon S (1985) Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15, 313–326. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, and Webster HD (1991) The fine structure of the nervous system : neurons and their supporting cells. Oxford University Press, New York. [Google Scholar]
- Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, Heppner FL (2015) Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J Exp Med, jem.20150479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Jucker M (2006) Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7, 940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S, and Dhama K (2014) Oxidative Stress, Prooxidants, and Antioxidants: The Interplay. Biomed Res Int 2014, 761264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen J, Mahler J, Beschorner N, Kaeser SA, Häsler LM, Baumann F, Jucker M (2017) Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc Natl Acad Sci USA 114, 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, and Landreth GE (2009) CD14 and Toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci 29, 11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reemst K, Noctor SC, Lucassen PJ, and Hol EM (2016) The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front Hum Neurosci 10, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Rofecoxib Protocol 091 Study G. (2004) Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62, 66–71. [DOI] [PubMed] [Google Scholar]
- Retzius G (1894–1916) Die Neuroglia des Gehirns beim Menschen und bei Saeugethieren, Verlag von Gustav Fischer. . Biol Untersuchungen. [Google Scholar]
- Robertson F (1900) A Microscopic Demonstration of the Normal and Pathological Histology of Mesoglia Cells. Journal of Mental Science 46, 724–724. [Google Scholar]
- Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Ward P (1992) Complement activation by β-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 89, 10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, … Willson P (1993) Clinical trial of indomethacin in Alzheimer’s disease. Neurology 43, 1609–1611. [DOI] [PubMed] [Google Scholar]
- Sato K (2015) Effects of Microglia on Neurogenesis. Glia 63, 1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage JC, Picard K, Gonzalez-Ibanez F, and Tremblay ME (2018) A brief history of microglial ultrastructure: Distinctive features, phenotypes, and functions discovered over the past 60 years by electron microscopy. Front Immunol 9, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf S, Mander A, Ugoni A, Vajda F, and Christophidis N (1999) A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 53, 197–201. [DOI] [PubMed] [Google Scholar]
- Scott SA, Johnson SA, Zarow C, and Perlmutter LS (1993) Inability to detect beta-amyloid protein precursor mRNA in Alzheimer plaque-associated microglia. Experimental Neurology 121, 113–118. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Sandrock A (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller J-n H, Johnson RE, and O’Banion MK (2007) Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117, 1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Moore KJ (2013) CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 8, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Lemere CA (2015) Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci 35, 13029–13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, McEwen BS, and Bulloch K (2007) Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412. [DOI] [PubMed] [Google Scholar]
- Silvin A and Ginhoux F (2018) Microglia heterogeneity along a spatio-temporal axis: More questions than answers. Glia 66, 2045–2057. [DOI] [PubMed] [Google Scholar]
- Siskova Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Remy S (2014) Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84, 1023–1033. [DOI] [PubMed] [Google Scholar]
- Soininen H, West C, Robbins J, and Niculescu L (2007) Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders 23, 8–21. [DOI] [PubMed] [Google Scholar]
- Sollvander S, Nikitidou E, Brolin R, Soderberg L, Sehlin D, Lannfelt L, and Erlandsson A (2016) Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol Neurodegener 11, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondag CM, Dhawan G, and Combs CK (2009) Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosna J, Philipp S, Albay R, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, … Glabe C. G. (2018) Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Barres BACP (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33, 13460–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Moore KJ (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, and Metter EJ (1997) Risk of Alzheimer’s disease and duration of NSAID use. Neurology 48, 626–632. [DOI] [PubMed] [Google Scholar]
- Stine WBJ, Dahlgren KN, Krafft GA, and LaDu MJ (2003) In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J Biol Chem 278, 11612–11622. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, and Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45, 208. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue Q-S, and Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA (2008) Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol 9, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K and Naito M (1993) Development, differentiation, and proliferation of macrophages in the rat yolk sac. Tissue and Cell 25, 351–362. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamamura F, and Naito M (1989) Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: a light-microscopic, enzyme-cytochemical, immunohistochemical, and ultrastructural study. Journal of Leukocyte Biology 45, 87–96. [DOI] [PubMed] [Google Scholar]
- Taneo J, Adachi T, Yoshida A, Takayasu K, Takahara K, and Inaba K (2015) Amyloid β oligomers induce interleukin-1β production in primary microglia in a cathepsin B- and reactive oxygen species-dependent manner. Biochem Biophys Res Commun 458, 561–567. [DOI] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar, Prinz M (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci. [DOI] [PubMed] [Google Scholar]
- Teplow DB (2006) Preparation of amyloid β-protein for structural and functional studies. Methods Enzymol 413, 20–33. [DOI] [PubMed] [Google Scholar]
- Terrill-Usery SE, Mohan MJ, and Nichols MR (2014) Amyloid-β(1-42) protofibrils stimulate a quantum of secreted IL-1β despite significant intracellular IL-1β accumulation in microglia. Biochim Biophys Acta 1842, 2276–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD (1985) Textbook of Neuropathology, pp 824–841. Williams & Wilkins, Baltimore. [Google Scholar]
- Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Rofecoxib Protocol 078 study g. (2005) A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology 30, 1204–1215. [DOI] [PubMed] [Google Scholar]
- Tooyama I, Kimura H, Akiyama H, and McGeer PL (1990) Reactive microglia express class I and class II major histocompatibility complex antigens in Alzheimer’s disease. Brain Research 523, 273–280. [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, and Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8, e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, and Nimmerjahn A (2011) The role of microglia in the healthy brain. J Neurosci 31, 16064–16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Charo IFCP (2007) Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117, 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan ML, Ajit D, Crouse NR, and Nichols MR (2008) Toll-like receptors 2 and 4 mediate Aβ(1-42) activation of the innate immune response in a human monocytic cell line. J Neurochem 104, 524–533. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, … Colonna M. (2017) TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nystrom S, Nilsson KP, Song WM, Holtzman DM (2018) ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Bixler GV, Brucklacher RM, Farley JA, Yan H, Warrington JP, Freeman WM (2011) Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J Neuroinflammation 8, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, Degenhardt K, Resch C, Bosch A, Jucker M, and Neher JJ (2015) Replacement of brain-resident myeloid cells does not alter cerebral amyloid-deposition in mouse models of Alzheimer’s disease. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Jucker M (2012) Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc. Natl. Acad. Sci. USA 109, 18150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Klein RSCPCN (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Heneka MT (2017) Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355. [DOI] [PubMed] [Google Scholar]
- Virchow R (1856) Gesammelte Abhandlungen zyr wissenschaftlischen Medizin. Frankfurt: Verlag von Meidinger Sohn & Comp. [Google Scholar]
- Virchow R(1858) Die Cellularpathologie in ihrer Begründung auf physiologische and pathologische Gewebelehre. Zwanzig Vorlesungen gehalten während der Monate Februar, März und April 1858 im pathologischen Institut zu Berlin. Berlin: August Hirschwald, 440. [Google Scholar]
- Vom Berg J, Prokop S, Miller KR, Obst J, Kälin R. E., Lopategui-Cabezas I., Heppner F. L. (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med 18, 1812. [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, and Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29, 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, and Teplow DB (1997) Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem 272, 22364–22372. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Teplow DB (1999) Amyloid β-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem 274, 25945–25952. [DOI] [PubMed] [Google Scholar]
- Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Fassbender K (2007) Role of the Toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20, 947–956. [DOI] [PubMed] [Google Scholar]
- Walti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Bockmann A, Riek R (2016) Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc Natl Acad Sci USA 113, E4976–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Colonna MC (2015a) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich Jason D, Young Katherine L, Robinette Michelle L, Colonna M (2015b) TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 160, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Colonna M (2016) TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G,. Neher J J (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JA, Manelli AM, Holmberg KH, Van Eldik LJ, and Ladu MJ (2005) Differential effects of oligomeric and fibrillar amyloid-β1–42 on astrocyte-mediated inflammation. Neurobiol Dis 18, 459–465. [DOI] [PubMed] [Google Scholar]
- Wildsmith KR, Holley M, Savage JC, Skerrett R, and Landreth GE (2013) Evidence for impaired amyloid-β clearance in Alzheimer’s disease. Alzheimers Res Ther 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, Wang KC, Kujawa M, and Lach B (1989) Ultrastructural studies of the cells forming amyloid fibers in classical plaques. The Canadian Journal of Neurological Sciences. Le Journal Canadien Des Sciences Neurologiques 16, 535–542. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Vorbrodt AW, Wegiel J, Morys J, and Lossinsky AS (1990) Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. American Journal of Medical Genetics. Supplement 7, 287–297. [DOI] [PubMed] [Google Scholar]
- Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, … Brunden KR (2000) Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J. Neurochem 74, 1017–1025. [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Grutzendler J (2016) TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 90, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JCS, and Cache County Study I (2002) Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology 59, 880–886. [DOI] [PubMed] [Google Scholar]
- Zenaro E, Piacentino G, and Constantin G (2017) The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis 107, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Emilsson V (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Hu W, Tsai J, Li W, and Gan W-B (2017) Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer’s disease. Mol Neurodegener 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Li X, Jiang L-L, Gui X, Liu Y, Xu H (2018) TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 97, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Wang Z, Wang D, Wang Z, Martens YA, Wu L, Chen XF (2018) Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol Neurodegener 13, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]