

Abstract
We report an experimental and computational analysis of the effects of hydrogen bonding to a metal dinitrogen complex. A series of H-bond donors over a wide pKa range (Δ 20) interact with the nitrogen unit of a ReI-(N2) complex at room temperature. Analysis by 15N NMR, IR spectroscopy, association equilibria, and DFT studies indicates that the H-bonding interaction polarizes and weakens the N−N bond. These results provide insight into the role of the secondary sphere residues in nitrogenase enzymes.
Graphical Abstract
■. INTRODUCTION
The activation and cleavage of N2 is a fundamentally important global process to sustain life and is accomplished in the biosphere by nitrogenases.1 N2 fixation is an energetically demanding process requiring 16 ATP per N2 by nitrogenases2 or high temperatures and pressures (~500 °C, >100 atm) industrially.3–5 Despite extensive effort to mimic the mild operative redox potential, atmospheric pressure, and room temperature (RT) conditions required biologically of nitrogenases, all homogeneous synthetic systems that reduce N2 operate under highly reducing conditions.6–14 One chemical principle that has been proposed to explain how nitrogenase efficiently promotes N2 reduction has been dubbed the “push-pull” mechanism, wherein a “push” of electron density from a reduced metal center is facilitated by a concomitant “pull” from an acidic site. The “push-pull” mechanism, traditionally rationalized through bimetallic activation15 and later refined to include hydrogen bond (H-bond) activation in nitrogenases,2,16 postulates a cooperative activation mode to enable N2 reduction under mild conditions (Figure 1). The recent isolation of a H-bond-stabilized N1H1 nitrogenase intermediate and revised proposals for secondary-sphere interactions along the reduction pathway17 provide evidence for their participation, yet key aspects regarding their role in N2 activation are not known.
Figure 1.

Nitrogenase FeMo-co active site with proposed acidic sites (top left); orbital diagram of push-pull hypothesis (top right); M-(N2) reactivity toward H-bond donors (bottom).
Despite the role that Brønsted acidic sites are proposed to serve in biological N2 reduction, examination of these interactions in model systems has not been possible. When not constrained by a protein matrix, most moderately activated dinitrogen complexes (νNN < 2090 cm−1) undergo protonation at the metal or dinitrogen with weak proton donors, rather than forming an isolable H-bond donor/acceptor adduct.18,19 Due to the inherent incompatibility of reduced metal complexes with Brønsted acidic groups (H-bond donors), previous efforts in our group used Lewis acids (LA) as H-bond donor surrogates to examine acid-mediated activation of an Fe-(N2) complex.20 Unfortunately, the direct translation of this approach to biologically relevant H-bond donors was not possible because of competitive oxidation of the Fe0-(N2) to form FeII-(H) products, even with mild acids.21 Due to these limitations, the role of H-bonding in N2 activation is largely unexplored.
The general incompatibility of proton donors with activated M-N2 complexes can be highlighted with examples where secondary sphere groups (incorporated as either H-bond donors or proton relays) afford oxidized metal products.22–26 The only report to overcome the acid incompatibility uses low temperature (190–220 K) to arrest intermolecular proton transfer and bias association,27 weakly activating W-(N2)2(dppe)2 (ΔνNN ≤ −20 cm−1). To clarify the extent to which the push-pull mechanism of N2 activation can be generally adapted to H-bond donors, we sought to evaluate a complex compatible with a broad range of H-bond donors at RT.
■. RESULTS AND DISCUSSION
Synthesis and Characterization of Lewis Acid and Hydrogen Bond Adducts.
To target a N2 complex compatible with H-bond donors, we initially selected Re(N2)-Cl(PMe2Ph)4 (1),15 because it satisfies the following requirements: (1) high degree of activation of the N2 ligand28 (νNN = 1925 cm−1), (2) stability to alcohol solvents, and (3) mild reduction potential29–32 to limit M-H formation. To assess the extent that acidic groups induce N2 activation, we first evaluated adducts with boranes and alkali cations.33 Addition of 1 equiv of B(C6F5)3 to 1 in fluorobenzene at RT (25–30 °C) resulted in a shift of νNN from 1925 to 1866 cm−1 (ΔνNN = −59 cm−1) (Figure 2B).34 Addition of 1 equiv of alkali cation salts, [Li][BArF4] or [Na][BArF4], afforded distinct results: two ΔνNN shifts (Li: −52 and +19 cm−1; Na: −43 and +18 cm−1). The positive and negative νNN shifts are consistent with two competitive Lewis basic sites: Re-Cl or Re-NNβ, respectively (Figure 2A).35
Figure 2.

(A) Competitive interactions for complexes 1 and 2. Solution IR (PhF) of (B) 1 and (C) 2 with Lewis acids [M][BArF4] or (D) phenol.
Despite the evidence for 1 to interact with weak Lewis acids, the competitive basic site (N2 and Cl) rendered analysis challenging because each donor/acceptor pair has opposing effects on N2 activation. Using 1 as a starting point, we further optimized the other primary sphere ligands to address three limitations: (1) competitive H-bonding to the −Cl ligand, (2) PMe2Ph ligand substitution with nucleophilic H-bond donors (alcohols, carboxylic acids), and (3) sterically limited donor/acceptor interactions. To address (1), we prepared a −Br variant because H-bond acceptor ability decreases down a group.36 To address (2) and (3), we targeted 1,2-bis-(diethylphosphino)ethane (depe) as a donating bis-(phosphine) ligand that features a reduced steric profile (solid cone angle (Θ) for two depe = 434° vs four PMe2Ph = 611.6°).37 trans-Re(N2)Br(depe)2 (2) was prepared in analogy to trans-Re(N2)Cl(depe)230 and structurally characterized (see SI). A voltammetry experiment revealed that the reversible ReI/ReII redox events of 1 and 2 differ by 105 mV (E1/2: 1 −0.555 V; 2 −0.660 V vs Fc/Fc+, 0.1 M [nBu4N][BArF4], PhF), reflecting a small electronic perturbation at the metal center. The solution IR spectrum of 2 (νNN(PhF) = 1940 cm−1; ν15N15N = 1875 cm−1) affords a new νNN of 1850 cm−1 (ΔνNN = −90 cm−1) upon addition of B(C6F5)3.38 Importantly, the addition of 1 equiv of [Li+][BArF4] to 2 resulted in only a single shift of −52 cm−1, indicating that the LA/Br interaction is diminished for hard Lewis acids (Figure 2C).
To assess a H-bond interaction to the coordinated N2 unit, we first examined phenol donors. The addition of 1–5 equiv of phenol to 2 afforded a −29 cm−1 shift and a modest +4 cm−1 shift (Figure 2D). The intensity of the new peaks increased with additional equivalents of phenol (10, 20), consistent with a weak H-bonding interaction.
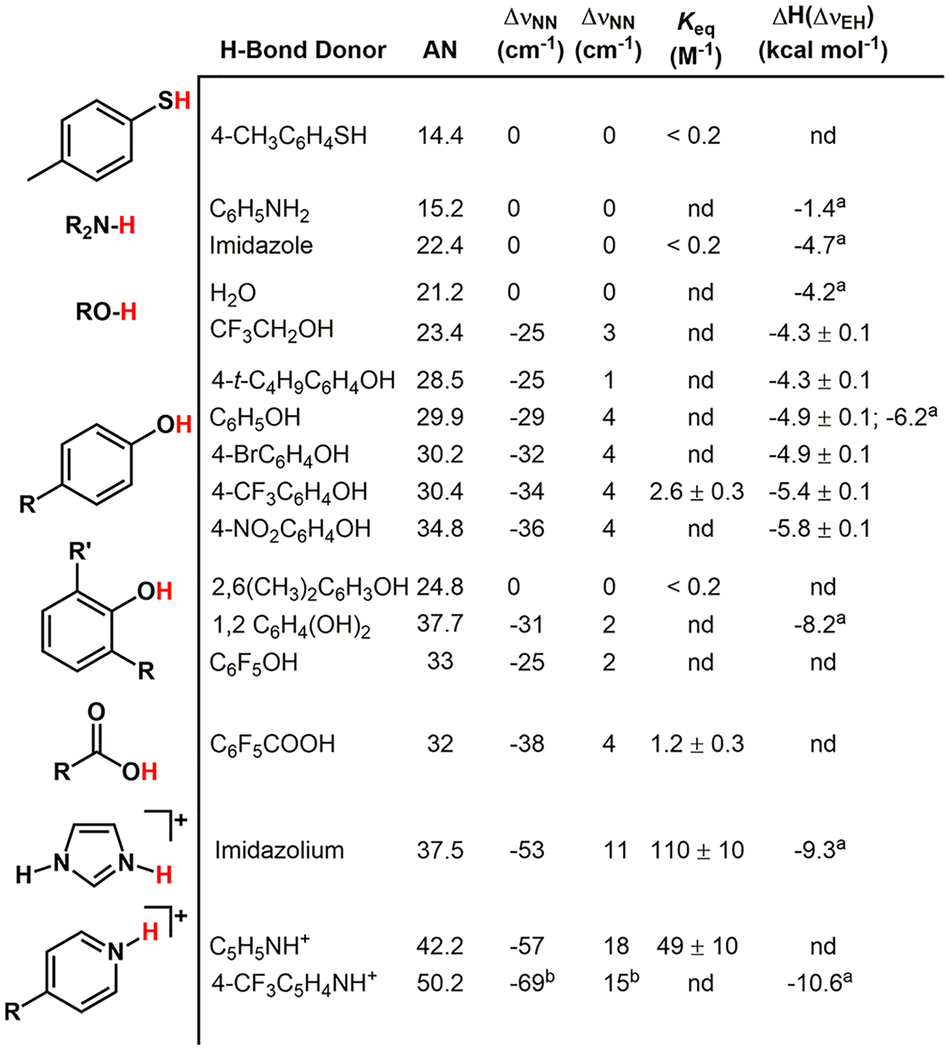
To interrogate the role of H-bond donor strength and steric profile on the activation of the dinitrogen ligand, we evaluated a series of H-bond donors (Figure 3). The H-bond donor strength was quantified as a measure of their Lewis acidity (acceptor number39,40).41 Many of these H-bond donor molecules induce a shift of the νNN that tracks with the measured Lewis acidity (Figure 3). Weak H-bond donors (aniline, imidazole, p-methylthiophenol, and water)42 did not interact with the N2 ligand at RT. In contrast, stronger H-bond donors (alcohols, benzoic acids, and the protonated N-heterocycles; ΔpKa(dmso)43> 20) afforded ΔνNN shifts of increasing magnitude (−25 to −69 cm−1) with their H-bond donor strength (Table 1).44 Phenols with 2,6-substitution exhibited less N2 activation than their H-bond donor strength would predict, which is consistent with a steric limitation for adduct formation with 2, i.e., perfluorophenol ΔνNN = −25cm−1 and no observed interaction for 2,6-dimethylphenol.45 In addition to changes to the H-bond acceptor, the H-bond donor νEH vibration is expected to undergo a shift for H-bond formation.46 The alcohol adducts of 2 feature a new broad νOH peak consistent with H-bond formation.47 Using experimentally derived vibration analysis,46 the ΔνOH corresponds to H-bond strengths ranging from −4.3 ± 0.1 to −5.8 ± 0.1 kcal/mol. To confirm the vibrational assignment of the H-bond adducts, we examined 15N-labeled 2 upon addition of Lewis acids and H-bond donor molecules. The addition of B(C6F5)3, phenol, and [imidazolium][BArF4] to 2-15N exhibited the ΔνNN shifts of the adducts 2-LA offset by 61–65 cm−1, in good agreement with the predicted isotopic shifts for the compounds (Figure S34). Taken together the data support H-bond activation of the −N2 ligand of 2 that can exceed the activation by common alkali metal promoters (Li+ or Na+).3,48
Figure 3.

Dependence of ΔνNN in 2 on Lewis acids and H-bond donors as quantified by acceptor number (AN) in PhF; H-bond donors ■ (left to right): 4-CH3C6H5SH, C6H5NH2, H2O, imidazole, CF3CH2OH, 1,2-C6H4(OH)2, 2,6-(CH3)2-C6H3OH, C6H5OH, 4-BrC6H4OH, 4-CF3C6H4OH, C6F5COOH, C6F5OH, 4-NO2C6H4OH, [imidazolium][BArF4], 1,2-C6H4(OH)2, [C6H5NH+][BArF4], [4-CF3C6H5NH+][BArF4]; BR3 ● (left to right): B(2,6-C6H3F2)3, B(2,4,6-C6H2F3)3, B(C6F5)3, BF3; M+ ▲ (left to right): [Na][BArF4], [Li][BArF4]
Table 1.
Summary of Lewis Acidities of H-Bond Donors, Activation of 2, Association Constants, and H-Bond Strength
|
Measured in CH2Cl2.
To assess conditions required to form H-bonding interactions to M-(N2) complexes, we evaluated association constants for representative H-bond donors. Variable solubility, vastly different binding constants observed for Lewis acids, and solvent absorption errors for the high loadings of additives precluded the use of titration methods for the entire series of adducts. Thus, transmission IR spectroscopic measurements were used to determine the binding constants of 2 to H-bond donors at a single concentration of 2 and additive in triplicate.49 The neutral H-bond donors (phenols, C6F5COOH) have low association constants of ~1–8 M−1, while the charged N-H donors bind more strongly, >45 M−1.50 The association constant for 2-CF3C6H4OH (2.6 ± 0.2 M−1; CH2Cl2 25 °C) is lower than for pyridine-C6H5OH (17 M−1; CH2Cl2 20 °C),51 indicating a weaker H-bond acceptor strength of the −N2 ligand than pyridine. The low association constants highlight the importance of intramolecular design approaches for H-bond activation with weak H-bond donors.
The large chemical shift range of 15N NMR spectroscopy translates to high sensitivity that provides complementary analysis of Lewis acid and H-bonding interactions to – N2.20,52–55 Upon addition of B(C6F5)3 to 2-15N, the 15N chemical shift of Nβ shifts upfield from −61.3 ppm to −173.3 ppm, while Nα undergoes a modest shift from −91.1 ppm to −93.8 ppm (Figure 4).56 The upfield shift of Nβ is consistent with the increased electron density at the atom: an effect of the push-pull mechanism.20 The addition of 1, 5, 10, and 100 equiv of phenol to 2-15N resulted in a gradual upfield shift of the Nβ resonance to −68.8 ppm with increasing concentration of phenol (Figure 4). The Δδ Nβ for 2 vs 2-C6H5OH is 7.4 ppm and is consistent with interactions at the terminal nitrogen by an electropositive donor/acceptor interaction, i.e., a H-bond. We propose that H-bonding promotes charge transfer via increased π-backbonding, to localize electron density at the terminal nitrogen atom, which affords an upfield shift. The weaker ΔνNN and binding energy of 2-C6H5OH than 2-B(C6F5)3 result in decreased push-pull charge transfer and dynamism on the NMR time scale, respectively. Decreased charge transfer and a time-averaged configuration response between unbound and bound phenol contribute to poor localization of electron density at Nβ and contribute to the modest upfield shift.
Figure 4.

15N NMR of 2 (top), 2-B(C6F5)3 (middle), and 2-C6H5OH 100 equiv (bottom) in C6D6.
Importantly the induced upfield shift of Nβ corroborates the proposed H-bond to N2. The 15N NMR analysis of H-bonding to 2 demonstrates association at −Nβ and induced N2 polarization by acidic groups.
Electronic Structure Perturbation and Predicted H-Bond Strengths.
To determine the effect of H-bond donors on M-N2 activation, we used density functional theory (DFT) analyses. Geometry optimization of 2 using the PBE0 functional57 and a combination of 6-311++G(2d,p)58 and def2tzvp59 basis sets with empirical dispersion60 resulted in an N−N bond length of 1.132 Å, in good agreement with our crystallographic data (N−N = 1.137(17) Å). A frequency analysis of the phenol H-bond interactions to either the −Br or −N2 of 2 reproduces the relative magnitude of the ΔνNN shifts (−Br: ΔνNN = +9 cm−1; N2: ΔνNN = −52 cm−1). The computationally modeled donors (aniline, thiophenol, imidazole, H2O, phenol, imidazolium, and 4-trifluoromethylpyridinium) corroborate a ΔνNN shift dependence on H-bond donor strength.61 Noncovalent interaction (NCI) analysis offers a computational methodology to visualize and study weak molecular interactions from calculated electron density.62 The NCI analysis of the H-bond adducts of 2 feature a stabilizing interaction between −Nβ and the H-bond donor (Figure 5A). Together the computational analysis supports the experimental evidence for H-bond interactions between Nβ and H-bond donors.
Figure 5.

(A) Noncovalent interaction between the Nβ of 2 and imidazolium (NCI analysis; supporting ligands omitted for clarity). (B) Re−N and N−N Wiberg bond indices and (C) NBO Nα (red) and Nβ (blue) charges of adducts of 2.
The quantitative analysis of the H-bond interaction provides insight into the energetics and bond metrics associated with the adducts. Biologically relevant H-bond donors (water, amines, thiols) also align with experimentally validated donors. The frequency analysis, vide supra, implies that the N−N bond is activated through the H-bond interaction (Table 2). This activation was reflected in an elongation of the N−N bond across the series of H-bond donors ranging from 1.132 Å (2) to the strongest H-bond donor, 1.151 Å (2-CF3C5H4NH+; ΔN−N = 0.019 Å). The calculated free energy and enthalpy of formation of the H-bond adduct track with donor strength (ΔG = 3.5 to −1.2 kcal/mol; ΔH = −1.4 to −10.6 kcal/mol). Due to multiple contacts between the phenyl-containing donors and 2, the ΔH for H-bond formation was determined using a vibrational analysis of the νE-H bond.27,63 The protonation of imidazole to imidazolium provides a 2.3-fold enhancement in the N−N elongation (ΔN−N = 0.0066 vs 0.0150 Å) and a 2-fold stronger H-bond (ΔH = −4.7 vs −9.3 kcal/mol). The calculated binding enthalpy between the −Br and the −N2 ligand of 2-C6H5OH favors −N2 H-bonding by −0.45 kcal/mol. The small difference in energy accounts for the simultaneous formation of positive and negative ΔνNN shifts in the solution IR measurements.64 For the H-bond adducts of 2, the degree of activation and H-bond strength are dependent on the H-bond donor.
Table 2.
Calculated Parameters for 2 and Adducts
| compound | N-N (Å) |
ΔνNN(cm−1) | HOMO (eV) |
ΔH(ΔνE-H) (kcal/mol) |
NBO Re (e−) |
NBO Nα (e−) |
NBO Nβ (e−) |
Wiberg B.I. Re-N |
Wiberg B.I. N-N |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 1.132 | −4.753 | −1.672 | 0.066 | −0.234 | 1.12 | 2.44 | ||
| 2-C6H5SH | 1.135 | −25 | −4.811 | −2.5 | −1.644 | 0.072 | −0.265 | 1.15 | 2.40 |
| 2-C6H5NH2 | 1.135 | −18 | −4.808 | −1.4 | −1.649 | 0.072 | −0.270 | 1.15 | 2.40 |
| 2-imidazole | 1.139 | −44 | −4.945 | −4.7 | −1.624 | 0.077 | −0.302 | 1.18 | 2.36 |
| 2-H2O | 1.138 | −38 | −4.899 | −4.2 | −1.629 | 0.074 | −0.300 | 1.18 | 2.37 |
| 2-C6H5OH | 1.140 | −52 | −4.964 | −6.2 | −1.612 | 0.083 | −0.320 | 1.20 | 2.34 |
| 2-C6H5OH’ | 1.131 | +9 | −4.888 | −5.8 | −1.658 | 0.691 | −0.223 | 1.12 | 2.45 |
| 2-C6H5OH linear | 1.137 | −25 | −4.957 | −5.1 | −1.622 | 0.087 | −0.317 | 1.19 | 2.36 |
| 2-catechol | 1.141 | −57 | −4.984 | −8.2 | −1.604 | 0.086 | −0.325 | 1.21 | 2.33 |
| 2-imidazoIium | 1.148 | −104 | −5.486 | −9.3 | −1.572 | 0.086 | −0.365 | 1.26 | 2.27 |
| 2-CF3C5H4NH+ | 1.151 | −138 | −5.597 | −10.6 | −1.545 | 0.089 | −0.384 | 1.30 | 2.22 |
| 2-BF3 | 1.174 | −188 | −5.654 | −1.386 | 0.132 | −0.411 | 1.51 | 1.95 |
After validating the computational approach, we evaluated the H-bond-induced changes to the electronic structure of the Re-(N2) unit. We assessed H-bond-induced changes to the Nα−Nβ and Re−Nα bond order via a Wiberg bond index analysis.65 The Nα−Nβ bond index decreases (2.44 to 2.22) while the Re−Nα bond index increases (1.12 to 1.30) with increasing H-bond donor strength (Figure 5B), consistent with H-bond-enhanced π-backbonding. The augmented π-back-donation is enabled by H-bond stabilization of the HOMO π-back-bonding orbital combination (dxz + N2 (π*)) by ≤ −0.84 eV for the representative H-bond donor, 2-CF3C5H4NH+.
Examination of the electronic structure of 2 provided insight into the orientation of the acidic interactions to the Re-(N2) unit. Optimization of the H-bond interaction consistently resulted in H-bond formation oriented toward the HOMO N2 (π*) π-backbonding orbital at −Nβ (∠NNH = 125−132°). A constrained end-on linear binding optimization for the phenol H-bond was found to be higher in energy (ΔG = 3.0 kcal/mol), provide a decreased degree of activation (ΔνNN = −25 cm−1), and have a lower binding enthalpy (ΔH = −5.0 kcal/mol) in comparison to the bent geometry. These effects are consistent with decreased facilitated backbonding from the linear acidic interaction relative to the bent configuration and decreased H-bond accepting strength of the N2 σ orbital. The bent binding geometry imparts greater HOMO stabilization and identifies the frontier orbitals as the likely site of reactivity, which is consistent with borane,20 silyl cation,66 and alkyl67 functionalization of other M-N2 complexes. The electronic structure of the complex informs the geometric preference of acidic interactions to the N2 ligands.
To evaluate the influence of increased N2 (π*) population on N2 polarization, we analyzed the natural bond orbital (NBO) charge of Nα and Nβ as a function of H-bond donor strength (Figure 5C). H-bonding to the N2 unit increased the negative charge on the Nβ atom from −0.234 to a maximum at −0.384 (Δe− = −0.150) for 4-trifluoromethylpyridinium with a modest increase in the positive charge of Nα (Δe− < 0.023). The charge localization on Nβ tracks with the difference in charge at Re for each acid (2-CF3C5H4NH+ : Δe− = 0.126), supporting our analysis as a push-pull charge transfer mechanism.68 The induced negative charge on −Nβ with the donor/acceptor interaction implicates increased basicity20 and corroborates the observed 15N NMR shielding of −Nβ. The electronic structure description reflects the “push-pull” mechanism of activation induced by H-bond donors. By this model, acidic H-bond donors serve to pull electron density from the metal d orbitals into a π* antibonding N2 orbital to build up negative charge at −Nβ. Overall, the computational analysis of H-bonding to 2 demonstrates that H-bonding activates the N−N bond by promoting π-backbonding.
■. CONCLUSION
Through a series of experimental and computational studies, we have demonstrated that 2 forms donor/acceptor adducts with a wide variety of acid additives, including H-bond donors at room temperature. The acid tolerance permitted an in-depth study to examine the effects of the H-bonding interaction with the N2 unit. We used a series of H-bond donors to quantify (1) the extent of −N2 activation, (2) enthalpy of H-bond formation, (3) association constants, and (4) imparted N2 polarization. Our study demonstrates that H-bonding to a N2 ligand has important consequences of increasing N−N bond activation by enhancing π-backbonding and also increasing polarization into the N2 unit. These effects may facilitate N2 activation in nitrogenases, and we are working to adapt these principles to synthetic N2 reduction schemes.
Supplementary Material
■. ACKNOWLEDGMENTS
This work was supported by the NIH (Grant No. 1R01GM111486–01A1) and the NSF (Grant No. CHE0840456) for X-ray instrumentation. N.K.S. is a Camille Dreyfus Teacher-Scholar and an Alfred P. Sloan Research Fellow. We thank Dr. Jeff Kampf for crystallographic assistance and Dr. John J. Kiernicki for fruitful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b02288.
X-ray data for compounds (CIF)
X-ray data for compounds (CIF)
Synthetic details, characterization, and computational details (PDF)
The authors declare no competing financial interest.
■ REFERENCES
- (1).Hoffman BM; Dean DR; Seefeldt LC Climbing Nitrogenase: Toward a Mechanism of Enzymatic Nitrogen Fixation. Acc. Chem. Res 2009, 42 (5), 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hoffman BM; Lukoyanov D; Yang Z-Y; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114 (8), 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Connor GP; Holland PL Coordination chemistry insights into the role of alkali metal promoters in dinitrogen reduction. Catal. Today 2017, 286, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Crossland JL; Tyler DR Iron-dinitrogen coordination chemistry: Dinitrogen activation and reactivity. Coord. Chem. Rev 2010, 254 (17–18), 1883–1894 [Google Scholar]
- (5).Hazari N Homogeneous iron complexes for the conversion of dinitrogen into ammonia and hydrazine. Chem. Soc. Rev 2010, 39 (11), 4044–4056. [DOI] [PubMed] [Google Scholar]
- (6).Yandulov DV; Schrock RR Reduction of dinitrogen to ammonia at a well-protected reaction site in a molybdenum triamidoamine complex. J. Am. Chem. Soc 2002, 124 (22), 6252–6253. [DOI] [PubMed] [Google Scholar]
- (7).Yandulov DV; Schrock RR Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301 (5629), 76–78. [DOI] [PubMed] [Google Scholar]
- (8).Arashiba K; Miyake Y; Nishibayashi Y A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Chem 2011, 3 (2), 120–125. [DOI] [PubMed] [Google Scholar]
- (9).Anderson JS; Rittle J; Peters JC Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501 (7465), 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Creutz SE; Peters JC Catalytic Reduction of N2 to NH3 by an FeN2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc 2014, 136 (3), 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ung G; Peters JC Low-Temperature N2 Binding to Two-Coordinate L2Fe0 Enables Reductive Trapping of L2FeN2 and NH3 Generation. Angew. Chem.-Int. Ed 2015, 54 (2), 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Challdey MJ; Del Castillo TJ; Matson BD; Roddy JP; Peters JC Catalyt6ic N2 to NH3 Conversion by Fe at Lower Driving Force: A Proposed Role for Metallocene-Mediated PCET. ACS Cent. Sci 2017, 3 (3), 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hill PJ; Doyle LR; Crawford AD; Myers WK; Ashley AE Selective Catalytic Reduction of N-2 to N2H4 by a Simple Fe Complex. J. Am. Chem. Soc 2016, 138 (41), 13521–13524. [DOI] [PubMed] [Google Scholar]
- (14).Kuriyama S; Arashiba K; Nakajima K; Tanaka H; Kamaru N; Yoshizawa K; Nishibayashi Y Catalytic Formation of Ammonia from Molecular Dinitrogen by Use of Dinitrogen-Bridged Dimolybdenum-Dinitrogen Complexes Bearing PNP-Pincer Ligands: Remarkable Effect of Substituent at PNP-Pincer Ligand. J. Am. Chem. Soc 2014, 136 (27), 9719–9731 [DOI] [PubMed] [Google Scholar]
- (15).Chatt J; Dilworth JR; Richards RL; Sanders JR Chemical evidence concerning function of molybdenum in nitrogenase. Nature 1969, 224 (5225), 1201–1202. [DOI] [PubMed] [Google Scholar]
- (16).Hoffman BM; Lukoyanov D; Dean DR; Seefeldt LC Nitrogenase: A Draft Mechanism. Acc. Chem. Res 2013, 46 (2), 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359 (6383), 1484–1489. [DOI] [PubMed] [Google Scholar]
- (18).Studt F; Tuczek F Theoretical, spectroscopic, and mechanistic studies on transition-metal dinitrogen complexes: Implications to reactivity and relevance to the nitrogenase problem. J. Comput. Chem 2006, 27 (12), 1278–1291. [DOI] [PubMed] [Google Scholar]
- (19).Connor GP; Lease N; Casuras A; Goldman AS; Holland PL; Mayer JM Protonation and electrochemical reduction of rhodium- and iridium-dinitrogen complexes in organic solution. Dalton Trans. 2017, 46 (41), 14325–14330. [DOI] [PubMed] [Google Scholar]
- (20).Geri JB; Shanahan JP; Szymczak NK Testing the Push-Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc 2017, 139 (16), 5952–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).H-bond donors tested include 1,2-diaminobenzene, squar-amide, and CF3CH2OH. See SI for details. The resulting oxidized FeII-(N2)(H) complex has a weakly activated N2 ligand (νNN 2097 cm−1) and is insufficiently basic at the terminal nitrogen atom to interact with either [Na+][BArF4] or C6F5OH in PhF (see SI).
- (22).Mock MT; Chen S; O’Hagan M; Rousseau R; Dougherty WG; Kassel WS; Bullock RM Dinitrogen Reduction by a Chromium(0) Complex Supported by a 16-Membered Phosphorus Macrocycle. J. Am. Chem. Soc 2013, 135 (31), 11493–11496. [DOI] [PubMed] [Google Scholar]
- (23).Egbert JD; O’Hagan M; Wiedner ES; Bullock RM; Piro NA; Kassel WS; Mock MT Putting chromium on the map for N2 reduction: production of hydrazine and ammonia. A study of cis-M(N2)2 (M = Cr, Mo, W) bis(diphosphine) complexes. Chem. Commun 2016, 52 (60), 9343–9346. [DOI] [PubMed] [Google Scholar]
- (24).Weiss CJ; Egbert JD; Chen S; Helm ML; Bullock RM; Mock MT Protonation Studies of a Tungsten Dinitrogen Complex Supported by a Diphosphine Ligand Containing a Pendant Amine. Organometallics 2014, 33 (9), 2189–2200. [Google Scholar]
- (25).Creutz SE; Peters JC Exploring secondary-sphere interactions in Fe-NxHy complexes relevant to N2 fixation. Chem. Sci 2017, 8 (3), 2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Labios LA; Heiden ZM; Mock MT Electronic and Steric Influences of Pendant Amine Groups on the Protonation of Molybdenum Bis(dinitrogen) Complexes . Inorg. Chem 2015, 54(9), 4409–4422. [DOI] [PubMed] [Google Scholar]
- (27).Kireev NV; Filippov OA; Pavlov AA; Epstein LM; Makhaey VD; Dyadchenko VP; Shubina ES; Belkova NV Steric and Acidity Control in Hydrogen Bonding and Proton Transfer to trans-W(N2)2(dppe)2. Inorg. Chem 2018, 57 (3), 1656–1664. [DOI] [PubMed] [Google Scholar]
- (28).Chatt J; Dilworth JR; Leigh GJ A series of nitrogen complexes of rhenium(i). J. Chem. Soc. D 1969, 13, 687–688. [Google Scholar]
- (29).Chatt J; Elson CM; Hooper NE; Leigh GJ Charge-distribution in complexes. J. Chem. Soc., Dalton Trans 1975, 22, 2392–2401. [Google Scholar]
- (30).Chatt J; Hussain W; Leigh GJ; Ali HM; Pickett CJ; Rankin DA The preparation and properties of some diphosphines R2PCH2CH2PR2 (R = alkyl or aryl) and of their rhenium(i) dinitrogen derivatives. J. Chem. Soc., Dalton Trans 1985, 6, 1131–1136. [Google Scholar]
- (31).Chatt J; Dilworth JR; Leigh GJ Dinitrogen complexes of rhenium(i) and rhenium(ii). J. Chem. Soc., Dalton Trans 1973, 6, 612–618. [Google Scholar]
- (32).Chatt J; Dilworth JR; Leigh GJ; Richards RL Polynuclear Dinitrogen Complexes. J. Chem. Soc. D 1970, 15, 955–956. [Google Scholar]
- (33).The boranes and alkali cations have significantly lower Lewis acidity than the electron-deficient metal complexes previously investigated for bimolecular activation of 1 (ref 32).
- (34).The degree of activation is lower, compared to Fe(N2)(depe)2 (ΔνNN = −129 cm−1) and likely due to the increased steric environment of the bulky PMe2Ph ancillary ligands in a rigid octahedral geometry for 1.
- (35).The IR shifts can be interpreted as two distinct Lewis acid binding modes that shift Re electron density away from and to the NN antibonding π* orbitals, consistent with a Lewis acid interaction.
- (36).Brammer L; Bruton EA; Sherwood P Understanding the Behavior of Halogens as Hydrogen Bond Acceptors. Cryst. Growth Des 2001, 1 (4), 277–290. [Google Scholar]
- (37).Bilbrey JA; Kazez AH; Locklin J; Allen WD Exact ligand cone angles. J. Comput. Chem 2013, 34 (14), 1189–1197. [DOI] [PubMed] [Google Scholar]
- (38).The addition of BF3(Et2O) induced the largest ΔνNN, 124 cm−1, while weaker polyfluorinated boranes B(2,6-C6H3F2)3 and B(2,4,6-C6H2F3)3 produced modest activation (ΔνNN 31-50 cm−1).
- (39).Mayer U; Gutmann V; Gerger W Acceptor number - Quantitative Empirical Parameter for Electrophilic Properties of Solvents. Monatsh. Chem 1975, 106 (6), 1235–1257. [Google Scholar]
- (40).Beckett MA; Strickland GC; Holland JR; Varma KS A convenient NMR method for the measurement of Lewis acidity at boron centres: Correlation of reaction rates of Lewis acid initiated epoxide polymerizations with Lewis acidity. Polymer 1996, 37 (20), 4629–4631. [Google Scholar]
- (41).Alternative common metrics were inappropriate for the system investigated; pKa is a metric for proton transfer rather than hydrogen-bond donor strength and does not appropriately describe many H-bond donors studied.
- (42).With 100 equivalents of imidazole or 4-methylthiophenol in CH2Cl2, low-intensity shoulders are observed and fit to corresponding peaks at 1906 and 1898 cm−1, respecitvely (ΔνNN= −29 and −38 cm−1). Due to the low intensity of the peaks, the assignment as the νNN for a H-bond adduct is not definitive. The absence of H-bonding to 2 from water can alternatively be attributed to the poor miscibility of water with PhF or competitive H-bonding to bulk solvent in THF.
- (43).Bordwell FG Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res 1988, 21 (12), 456–463. [Google Scholar]
- (44).2 is stable to air in solution for days at 25 °C; the addition of H-bond donors or Lewis acids accelerates the oxidation to [ReII(N2) Br(depe)2]+ (t1/2 < 3 h at 25 °C for B(C6F5)3). Strong H-bond donors (imidazolium, pyridinium) decompose 2 over 1 h in PhF or 15 min in CH2Cl2 to yield [ReII(N2)Br(depe)2]+ or complex mixtures (C6F5COOH) (see SI).
- (45).Bifurcated hydrogen-bond donors were examined, but due to low acceptor numbers and low solubility of squaramides and ureas, no activation was observed. Catechol showed a minor 2 cm−1 improvement, compared to phenol, while diphenylthiourea afforded a modest IR sideband (see SI).
- (46).Iogansen AV Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta, Part A 1999, 55 (7), 1585–1612. [Google Scholar]
- (47).The new νOH peak is not observed for 2,6-dimethylphenol. Shifts for stronger O−H and N−H donors are not possible to measure in PhF due to overlap with the C−H subtraction region of the solvent.
- (48).McWilliams SF; Rodgers KR; Lukat-Rodgers G; Mercado BQ; Grubel K; Holland PL Alkali Metal Variation and Twisting of the FeNNFe Core in Bridging Diiron Dinitrogen Complexes. Inorg. Chem 2016, 55 (6), 2960–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Using 1H NMR as an alternative approach does not deconvolute the two binding modes (N2−H bond vs Br−H bond).
- (50).The Lewis acids have association constants of 8.4–1300 M−1, which are much lower than previously observed for Fe(N2)(depe)2; see SI for details.
- (51).Rubin J; Panson GS Hydrogen Bonding. II. Phenol Interactions with Substituted Pyridines1a. J. Phys. Chem 1965, 69 (9), 3089–3091. [Google Scholar]
- (52).Schuster II; Dyllick-Brenzinger C; Roberts JD Nitrogen-15 nuclear magnetic resonance spectroscopy. Effects of hydrogen bonding and protonation on nitrogen chemical shifts of pyrazoles. J. Org. Chem 1979, 44 (11), 1765–1768. [Google Scholar]
- (53).Casewit C; Roberts JD; Bartsch RA Nitrogen-15 nuclear magnetic resonance studies of benzenediazonium ions. Effects of solvent, substituent, anion, and 18-crown-6. J. Org. Chem 1982, 47(15), 2875–2878. [Google Scholar]
- (54).Morishima I; Inubushi T 15N nuclear magnetic resonance studies of iron-bound C15N− in ferric low-spin cyanide complexes of various porphyrin derivatives and various hemoproteins. J. Am. Chem. Soc 1978, 100 (11), 3568–3574. [Google Scholar]
- (55).Donovanmtunzi S; Richards RL; Mason J N-15 Nuclear Magnetic Resonance Spectroscopy of Dinitrogen-Bridged Complexes. J. Chem. Soc., Dalton Trans 1984, 11, 2429–2433. [Google Scholar]
- (56).The observed shielding of Nβ is similar to the reported shifts for the 15N NMR of 1 with trialkylalanes (ref 55).
- (57).Adamo C; Barone V Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys 1999, 110 (13), 6158–6170. [Google Scholar]
- (58).Rassolov VA; Ratner MA; Pople JA; Redfern PC; Curtiss LA 6–31G* basis set for third-row atoms. J. Comput. Chem 2001, 22 (9), 976–984. [Google Scholar]
- (59).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7 (18), 3297–3305. [DOI] [PubMed] [Google Scholar]
- (60).Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132 (15), 154104. [DOI] [PubMed] [Google Scholar]
- (61).The computationally and experimentally determined ΔνNN activations are linearly dependent, reflecting a scaling relation that overestimates the ΔνNN of the 2-H bond donor interactions.
- (62).Johnson ER; Keinan S; Mori-Sánchez P; Contreras-García J; Cohen AJ; Yang W Revealing Noncovalent Interactions. J. Am. Chem. Soc 2010, 132 (18), 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Due to multiple contacts between the phenyl-containing donors and 2, the ΔH for H-bond formation was determined using a vibrational analysis of the νE−H (see ref 27 and SI for full details and complementary methods).
- (64).Computational analysis of catechol does not support a bifurcated H-bond donor configuration resulting in ΔνNN −57 or −115 cm−1, respectively, supported by the modest shift observed by solution IR relative to phenol.
- (65).Wiberg KB Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24 (3), 1083–1096. [Google Scholar]
- (66).Piascik AD; Hill PJ; Crawford AD; Doyle LR; Green JC; Ashley AE Cationic silyldiazenido complexes of the Fe(diphosphine)2(N2) platform: structural and electronic models for an elusive first intermediate in N2 fixation. Chem. Commun 2017, 53 (54), 7657–7660. [DOI] [PubMed] [Google Scholar]
- (67).Cherry J-PF; Stephens FH; Johnson MJA; Diaconescu PL; Cummins CC Terminal Phosphide and Dinitrogen Molybdenum Compounds Obtained from Pnictide-Bridged Precursors. Inorg. Chem 2001, 40 (27), 6860–6862. [DOI] [PubMed] [Google Scholar]
- (68).DFT analysis of imidazole and imidazolium H-bonds to Fe(N2) (depe)2 features increased charge transfer and H-bond enthalpy (ΔH ≤ −10.6 kcal/mol) from the larger “push” of the more reducing metal center. See SI for full details and analysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.