

Abstract
A family of single-component iron precatalysts for the [4+4]-cyclodimerization and intermolecular cross-[4+4]-cycloaddition of monosubstituted 1,3-dienes is described. Cyclooctadiene products were obtained with high regioselectivity, and catalyst-controlled access to either cis- or trans-diastereomers was achieved using 4-substituted diene substrates. Reactions conducted either with single-component precatalysts or with iron dihalide complexes activated in situ proved compatible with common organic functional groups and were applied on multi-gram scale (up to >100 g). Catalytically relevant, S = 1 iron complexes bearing 2-iminopyridine ligands, (RPI)FeL2 (RPI = [2-(2,6-R2-C6H3-N=CMe)-C5H4N] where R = iPR or Me, L2 = bis-olefin) were characterized by single-crystal X-ray diffraction, mößbauer spectroscopy, magnetic measurements, and dft calculations. The structural and spectroscopic parameters are consistent with an electronic structure description comprised of a high spin iron(I) center (Sfe = 3/2) engaged in antiferromagnetically coupling with a ligand radical anion (SPI = −1/2). Mechanistic studies conducted with these single-component precatalysts—including kinetic analyses, 12C/13C isotope effect measurements, and in situ mößbauer spectroscopy— support a mechanism involving oxidative cyclization of two dienes that determines regio- and diastereoselectivity. Topographic steric maps derived from crystallographic data provided insights into the basis for the catalyst-control through stereoselective oxidative cyclization and subsequent, stereospecific allyl-isomerization and C–C bond-forming reductive elimination.
Graphical Abstract
INTRODUCTION
Medium-sized (7–11-membered) rings, by virtue of their ring strain and distinctive conformational preferences,1 are important chemical substructures with applications in the synthesis and properties of polymers,2 fuels,3 fragrances,4,5 and other specialty chemicals (Scheme 1A).6 Among these, eight-membered cyclic olefins are particularly valuable targets as evidenced by ring-opening metathesis polymerization (ROMP) of cyclooctene, 1,5-cyclooctadiene, and their derivatives to access vinyl copolymers and, following hydrogenation, precision polyethylene derivatives.2 While strategies have been developed for the synthesis of eight-membered carbocycles in the context of complex molecule total syntheses,7 most notably through ring-closing olefin metathesis8 and radical cascades,9 alternative methods for the modular and convergent synthesis of these rings with high degrees of regio- and stereocontrol remain desirable for fine-chemical applications.2–6 The [4+4]-cycloaddition of two 1,3-dienes would constitute an atom-economical method to address this need, enabling synthesis of substituted cyclooctadienes from readily accessed, and in many cases, commodity substrates.
Scheme 1.
Demand, conceptual evolution, and design strategy for iron-catalyzed [4+4]-cycloaddition of substituted 1,3-dienes.

Metal-catalyzed [4+4]-cyclodimerization of butadiene to generate 1,5-cyclooctadiene is well established and is practiced industrially using nickel salts in combination with phosphite ligands and trialkylaluminum activators (Scheme 2A).10 Other metal catalysts, most notably an iron(I) α-diimine cyclooctadiene complex, have since been reported to promote this transformation with high turnover numbers and frequencies.11 Enabling metal-catalyzed [4+4]-cycloaddition for substrates beyond butadiene would be attractive due to the availability of substituted 1,3-dienes from sources including biomass and petrochemical feedstocks12 as well as through synthetic methods including enyne metathesis,13 cross-coupling,14 Mizoroki–Heck olefination,15 Wittig olefination,16 and diverted aerobic Heck reactions.17 However, realization of such a method poses several challenges including: (i) the reduced coordination rate of more sterically encumbered substituted dienes relative to butadiene, (ii) the potential for competitive formation of side-products including linear oligomers and [2+2]- and [4+2]-cycloadducts, and (iii) the possibility for formation of multiple regio- and stereoisomeric [4+4] products. Catalyst designs must, therefore, balance competing demands of reactivity and selectivity to achieve synthetic utility.
Scheme 2.
State-of-the-art methods for [4+4]-cycloaddition of 1,3-dienes.
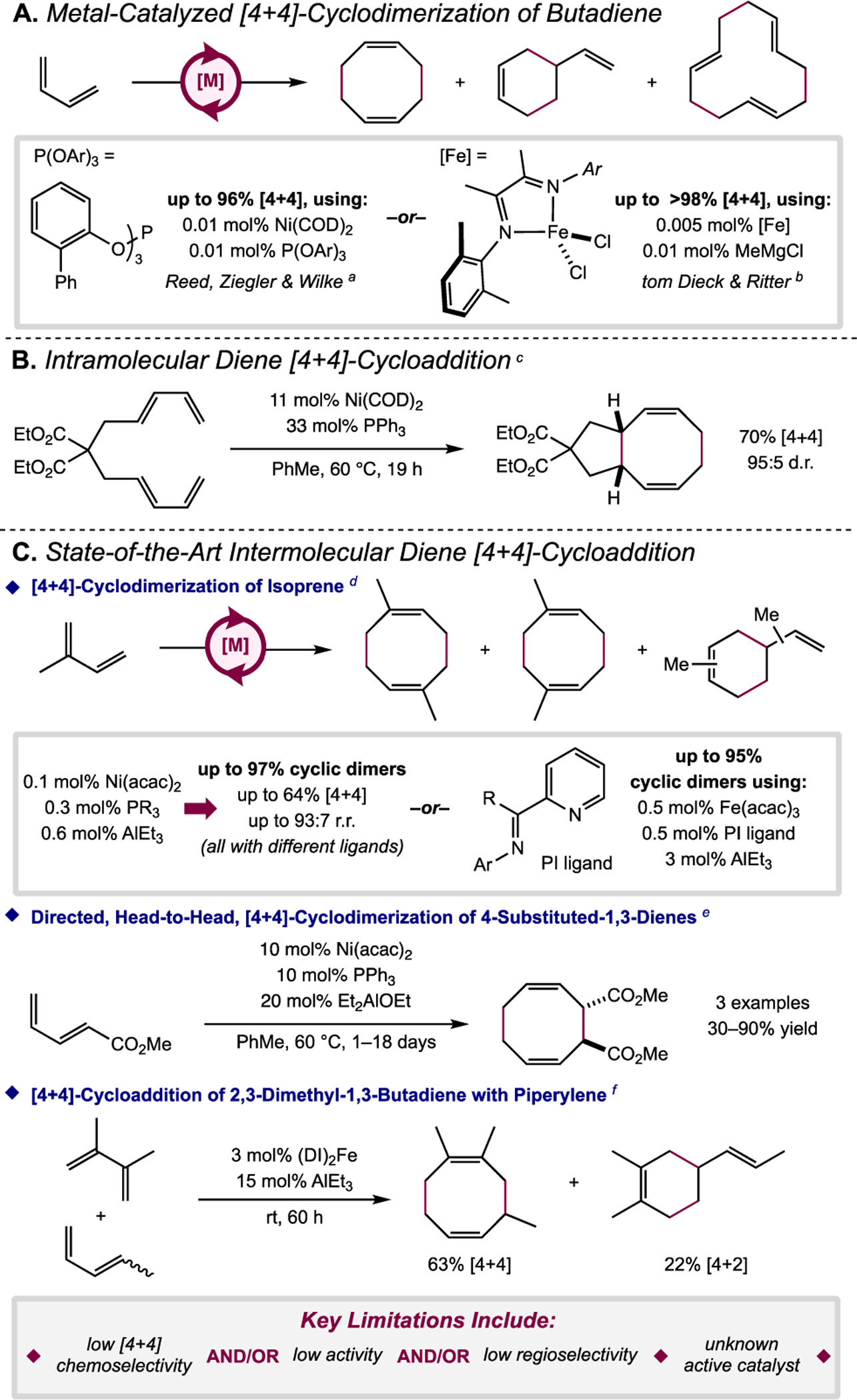
a Ref. 10. b Ref. 11. c Ref. 18. d Refs. 19a & 20b. e Refs. 19c,d. f Ref. 20a.
While high (10–20 mol%) loadings of nickel precatalysts have been reported for intramolecular [4+4]-cycloadditions of tethered dienes (Scheme 2B),18 attempts to apply these conditions for intermolecular coupling of substituted dienes have been met with low activity and/or low chemo- and regioselectivity for the [4+4]-cycloadducts (Scheme 2C).19 Likewise, α-diimine and related ligands used in combination with iron salts and in situ reductants for the cyclodimerization of butadiene have been evaluated for [4+4]- and [4+2]-coupling reactions with other substrates.11a,20 Promising results were obtained with isolated examples (Scheme 2C); however, selectivity for [4+4]-cycloaddition was variable and limited in scope. Moreover, the identity, speciation, and oxidation states of the complexes responsible for C–C bond-formation remained ambiguous.21
Reduced iron complexes bearing tridentate pyridine-2,6-diimine (PDI) ligands catalyze numerous C–C bond-forming reactions22 including the [2+2]-cyclodimerization and hydrovinylation of α-olefins23 as well as the cross-[2+2]-cycloaddition of unactivated alkenes and dienes.23b,24 High degrees of chemo- and regioselectivity were observed, arising from oxidative cyclization of the coupling partners to form metallacyclic intermediates,25,26 where the divergent outcomes reflected the fate of the metallacycle (Scheme 1B). Iron compounds bearing sterically demanding tridentate ligands favored C(sp3)–C(sp3) reductive elimination23b,25 while more open iron complexes underwent preferential β-H elimination followed by C–H reductive elimination.23b,26
These observations suggested that further catalyst evolution could be applied to promote selective [4+4]-cycloaddition of substituted dienes (Scheme 1B). Following oxidative coupling of two dienes, selective [4+4]-cycloaddition would result if allyl isomerization and C–C bond-forming reductive elimination were accelerated relative to competitive diene insertion and β-H elimination pathways. In light of recent mechanistic work providing evidence for the intermediacy of metallacycles in C–C bond-forming reactions catalyzed by α-diimine iron complexes,26 bidentate redox-active ligands seemed well-suited for this purpose. In addition to opening a site for allyl coordination, these ligands could mediate access to high-oxidation-state (iron(III) or iron (IV)) and/or high-spin intermediates to promote C–C reductive elimination.27,28 As such, reexamination of the modular and inexpensive α-diimine (DI) ligands along with the more-open, redox-active 2-iminopyridine (PI) ligands using well-defined, single-component precatalysts was targeted to achieve broadly useful chemo-, regio-, and diastereoselective [4+4]-cycloaddition of substituted dienes.
Here we describe the synthesis and electronic structure determination of iminopyridine iron bis-olefin complexes that, along with select α-diimine iron complexes, are highly active and selective catalysts for the [4+4]-cycloaddition of monosubstituted dienes. Collectively, the single-component precatalysts enabled the regioselective [4+4]-cyclodimerization and intermolecular cross-[4+4]-cycloaddition of 2- and 4-substituted 1,3-dienes (Scheme 1C) with iron loadings as low as 0.025 mol % and substrate ratios as low as 1.5:1. In the case of 4-substituted diene substrates, catalyst-controlled access to either cis- or trans-diastereomers was achieved. Kinetic analyses, 12C/13C isotope effect measurements, and in situ Mößbauer spectroscopic studies provided support for a mechanism in which oxidative cyclization of two dienes determined regio- and diastereoselectivity. The conserved electronic structure of the reduced iminopyridine iron complexes with respect to known α-diimine and pyridine(diimine) iron bis-olefin complexes highlights a potential role for ligand redox activity in selective, iron-catalyzed C–C bond-forming reactions. Thus, not only does this method provide access to modular building blocks likely to find broad application in the synthesis and study of precision polyolefins, lubricants, fuels, fragrances, and other specialty chemicals, the mechanistic insights provide a blueprint for other methods applying metallacyclic intermediates to upgrade unsaturated coupling partners.
RESULTS AND DISCUSSION
Ligand Denticity and Cycloaddition Chemoselectivity.
To evaluate the effect of ligand denticity on the chemoselectivity of iron-catalyzed diene coupling reactions, single-component precatalysts were targeted to minimize the potential for confounding effects arising from ill-defined speciation under in situ activation conditions. Pyridine(diimine) iron dinitrogen complexes have proven to be excellent precatalysts for the [2+2]-cycloaddition of alkenes and dienes,23,24 and α-diimine iron bis-olefin complexes, (RDI)Fe(COD) (RDI = [2,6-R2-C6H3-N=CMe]2 where R = iPr, Me or [2,4,6-Me3-C6H2-N=CMe]2 where R = Mes), were introduced as suitable precatalysts for olefin hydrogenation,29 diene [1,4]-hydrovinylation,26 and butadiene cyclodimerization.11b While the analogous iron(iminopyridine) olefin complexes had been elusive previously,30 (RPI)Fe(COD) and (RPI)Fe(MviMvi) (RPI = [2-(2,6-R2-C6H3-N=CMe)-C5H4N] where R = iPr or Me) were prepared by treating [(RPI)FeCl(μ-Cl)]2 with magnesium butadiene bis(tetrahydrofuran) (Mg(C4H6)•2THF) and 1,5-cyclooctadiene (COD) or 1,3-divinyl-1,1,3,3-tetramethyldisiloxane (MviMvi) in thawing Et2O over 9–20 minutes (Scheme 3; see Supporting Information for additional details). Rapid solvent removal and work-up minimized ligand exchange—which results in formation of the homoleptic bis-ligand complexes, (RPI)2Fe, and iron black—that had frustrated prior efforts. While the (RPI)Fe(COD) complexes were unstable in hydrocarbon or ethereal solvent at ambient temperature (~23 °C), undergoing decomposition to form (RPI)2Fe and iron black, over the span of minutes (where R = Me) or hours (where R = iPr), they were isolated and stored indefinitely at –35 °C as dark brown solids. The (RPI)Fe(MviMvi) complexes proved more robust in solution and were recrystallized as deep red-purple needles from saturated pentane solutions at –35 °C. Additional characterization data are discussed in the “Electronic Structure Determination” section below.
Scheme 3.
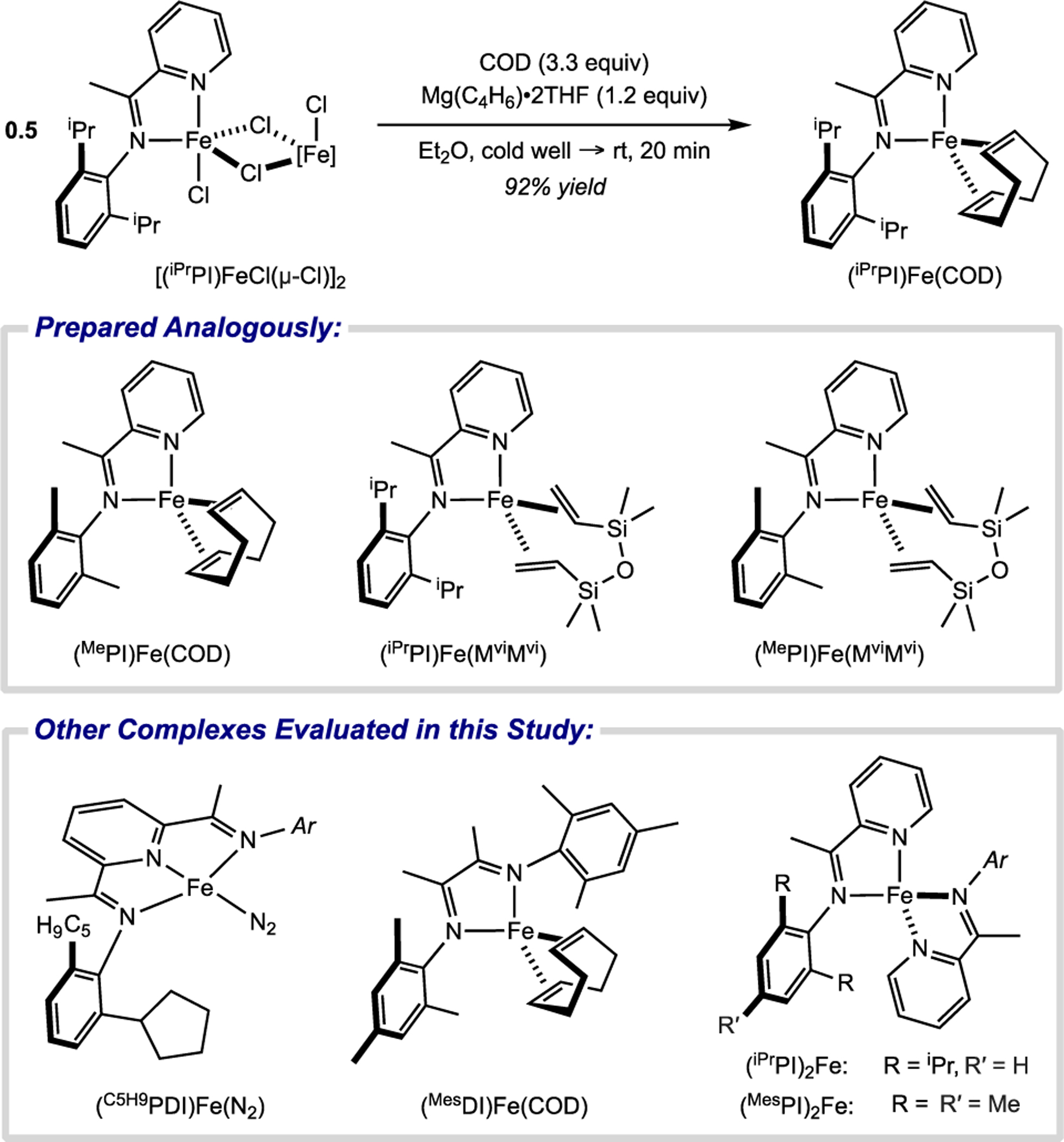
Synthesis of reduced iminopyridine iron bis-olefin complexes.
With access to these well-defined, reduced iron complexes bearing either tridentate (PDI) or bidentate (DI or PI) potentially redox-active ligands, the role of ligand denticity in diene coupling reactions was examined. Piperylene (1a) was selected as a representative 4-substituted-1,3-diene to evaluate iron precatalyst performance. Stirring (E)-1a with 5 mol% of iron complex (C5H9PDI)Fe(N2), which bears a tridentate PDI ligand optimized for alkene [2+2]-cyclodimerization, resulted in selective formation of the analogous diene [2+2]-cyclodimerization product (2a), albeit in low conversion and yield (18%; Scheme 4A). Use of iron complexes with bidentate ligands altered the outcome of the catalytic reaction. With 1 mol% of α-diimine-supported precatalyst (MesDI)Fe(COD), cis-3,7-dimethyl-1,5-cyclooctadiene (3a) was obtained in 93% yield and 95:5 diastereomeric ratio (d.r.; Scheme 4A) as determined by quantitative 13C NMR spectroscopy. Conversely, use of 1 mol% iron(iminopyridine) precatalyst (iPrPI)Fe(COD) generated the opposite diastereomer, trans-3,7-dimethyl-1,5-cyclooctadiene (4a), in 87% yield and >98:2 d.r., demonstrating the influence of the supporting ligand on the chemo-, regio- and diastereoselectivity of the cycloaddition process (Scheme 4A). Furthermore, the high [4+4] activity of these single-component precatalysts stands in contrast to the low reactivity noted previously using in situ activation protocols.11a
Scheme 4.
Ligand denticity controls cycloaddition chemoselectivity.

a Reactions were conducted on 0.5 mmol scale. Yield and d.r. determined from the relative integrals of diagnostic signals for 1a, 2a, 3a, and 4a in the quantitative 13C NMR spectrum of the crude reaction mixture relative to an internal standard added at the end of the reaction. Stereochemical assignment of the diastereomers was corroborated by chiral-phase gas chromatographic analysis (BETA DEX 120, 60 °C isothermal method, 120 min). b Reactions were conducted on 1.0 mmol scale. Yield describes the combined isolated yield of isomeric products; r.r. determined from the relative integrals of diagnostic signals for 6b, head-to-head [4+4]-cycloadduct 7b, and head-to-tail [4+4]-cycloadduct 8b in the quantitative 13C NMR spectrum of the isolated material.
A similarly striking dependence of chemoselectivity on ligand denticity was observed using isoprene (5a) and myrcene (5b) as model 2-substituted-1,3-diene substrates. Stirring 5b with 5 mol% (C5H9PDI)Fe(N2) afforded [2+2]-cycloadduct 6b in 44% isolated yield (94% [2+2] over all other isomers, >98:2 d.r.; Scheme 4B). By contrast, with 1 mol% of iminopyridine iron complex (MePI)Fe(COD), 98% [4+4]-cyclodimerization selectivity was observed, providing 1,6-dimethyl-1,5-cyclooctadiene (7b) in 99% combined isolated yield and 96:4 regioisomeric ratio (r.r.; Scheme 4B). Similarly high selectivity was observed using isoprene as a substrate. Nearly identical results were obtained using (MePI)Fe(COD) and (MePI)Fe(MviMvi) as precatalysts (Scheme 5); however, only very low activity was observed using the isolated, homoleptic bis-ligand complex (MesPI)2Fe. The iron-catalyzed cycloaddition is noteworthy in that it provides selective access to the head-to-head isomer in contrast to nickel catalysis, which has been reported to favor the opposite, head-to-tail, regioisomer (8a) in the cyclodimerization of isoprene (commercially available samples are ~20:80 r.r.).19a,b Taken together these results exemplified remarkable control by ancillary ligands on iron, achieving regio- and diastereoselectivity for the [4+4]-cycloaddition of unactivated dienes without the requirement for directing groups or electronically activating substituents.
Scheme 5.
Evaluation of single-component iminopyridine iron precatalysts for [4+4]-cyclodimerization of isoprene.a

a Reaction conducted on 1 mmol scale at ambient temperature (~23 °C). b Yield and % [4+4] selectivity determined from the calibrated relative integrals of product and cyclooctane signals measured by gas chromatographic analysis of aliquots removed from the reaction mixture at the indicated time. c r.r. determined from the relative integrals of diagnostic signals for 7a and 8a in the quantitative 13C NMR spectrum of the crude reaction mixture.
In Situ Catalyst Activation.
Contrary to the case with piperylene dimerization, several reductants (including Mg(C4H6)•2THF, MeMgCl, and Mg(0) powder) were identified that, upon combination with [(MePI)FeCl(μ-Cl)]2 and isoprene or myrcene, afforded the corresponding [4+4]-cycloadduct (7a or 7b) with comparable reactivity and identical selectivity to that obtained with precatalysts (MePI)Fe(COD) or (MePI)Fe(MviMvi) (see Supplementary Information for details).31 In control experiments, no substrate consumption was observed when FeCl2 was treated with Mg(C4H6)•2THF in the absence of a DI or PI ligand. While the single-component reduced iron precatalysts required handling under rigorously air- and moisture-free conditions, the iron(II) halide precursors ((RDI)FeX2 and [(RPI)FeX(μ-X)]2 where X = Cl or Br) exhibited enhanced robustness, allowing for bench-top manipulation. Using in situ activation of [(MePI)FeCl(μ-Cl)]2, the [4+4]-cyclodimerization of isoprene was conducted in batches as large as 2 mol (168 g) with iron precatalyst loadings as low as 0.025 mol% (175 mg [(MePI)FeCl(μ-Cl)]2). [Caution: The [4+4]-cyclodimerization of 1,3-dienes is highly exothermic. When performing the reaction on a preparative scale, special care should be taken to ensure adequate heat transfer. Maintaining a controlled temperature throughout the course of the reaction is necessary to prevent a pressure build-up.] While rigorously dried diene was required to achieve complete conversion at low catalyst loadings, 7a was alternately obtained in 90% yield using reagents measured and combined outside of a glovebox using modestly increased precatalyst loading (1 mol% [Fe]). While the [4+4]-cyclodimerization was routinely conducted in neat substrate, the in situ activation protocol was also adapted to dilute solutions as a strategy for controlling reaction exotherms. The high chemo- and regioselectivity observed under neat reaction conditions was maintained across reactions conducted in all of the ethereal and hydrocarbon solvents evaluated (Scheme 6). The apparent catalyst fidelity using these in situ activation protocols holds promise for applications to the scalable synthesis of monomers for ROMP.
Scheme 6.
Evaluation of solvents suitable for iron-catalyzed [4+4]-cyclodimerization.a
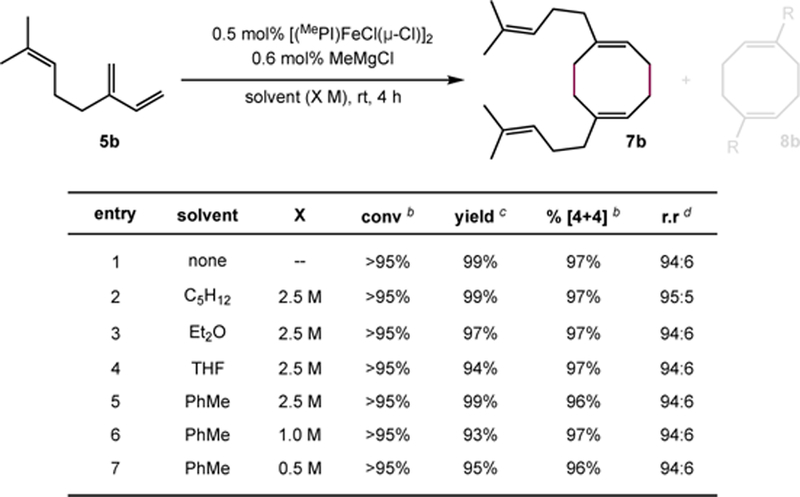
a Reaction conducted on 5 mmol scale at ambient temperature (~23 °C). b Conversion and % [4+4] selectivity determined from relative integrals of starting material and product signals measured by gas chromatographic analysis of aliquots removed from the reaction mixture. c Values describe combined isolated yield of isomeric products d r.r. determined from the relative integrals of diagnostic signals for 7b and 8b in the quantitative 13C NMR spectrum of the isolated material.
Regio- and Diastereoselective [4+4]-Cyclodimerization of Monsubstituted-1,3-Dienes.
With both isolated organometallic precatalysts and in situ activation protocols in hand, the scope of the iron-catalyzed [4+4]-cycloaddition was evaluated with varied 4- (1a-g; Scheme 7) and 2-substituted (5a–l; Scheme 8) 1,3-dienes. While reduced iron complexes are often incompatible with polar functional groups—carbon–heteroatom bond cleavage can lead to irreversible catalyst deactivation32—the product yield and [4+4] selectivity obtained using 1–5 mol% of (MesDI)Fe(COD), (iPrPI)Fe(COD), or (MePI)Fe(COD) proved to be largely insensitive to introduction of heteroatoms. Cyclooctadienes bearing ester (3g, 4g, 7g, 7h), acetal (7e), amide (7i), amine (7j), arene (7k), and heteroarene (7l) functionality were prepared with levels of selectivity comparable to those achieved with the pure hydrocarbon substrates. While residual water and other protic functionality were problematic, resulting in low to no substrate conversion, sensitive functional groups were masked effectively with common protecting groups (e.g. benzyl ethers, silyl ethers, carbamates).
Scheme 7.
Scope of iron-catalyzed, diastereoselective [4+4]-cyclodimerization of 4-substituted dienes.a
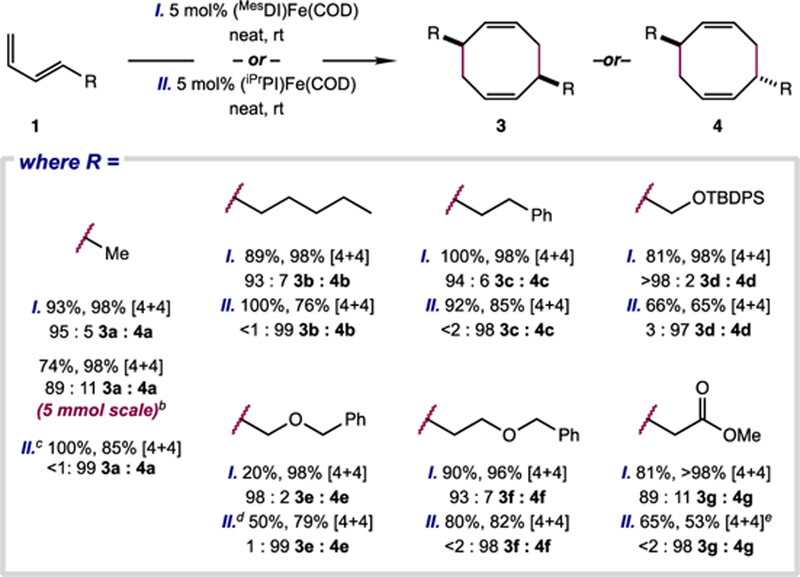
a Unless indicated otherwise, reactions were run on 0.5 mmol scale with 5 mol% [Fe] precatalyst. Values describe combined isolated yield of isomeric products, the portion of this isolated mixture comprised of [4+4]-cycloadducts as determined by gas chromatographic analysis, and the diastereomer ratio of the [4+4]-cycloadducts as determined by relative integration of diagnostic resonances in the quantitative 13C NMR spectrum of the isolated material. TBDPS = tert-butyldiphenylsilyl b with 2.5 mol% [Fe] on 5 mmol scale c with 5 mol% (iPrPI)Fe(MviMvi) d at 40 °C e 28% [4+2]
Scheme 8.
Scope of iron-catalyzed, regioselective [4+4]-cyclodimerization of 2-substituted dienes.a
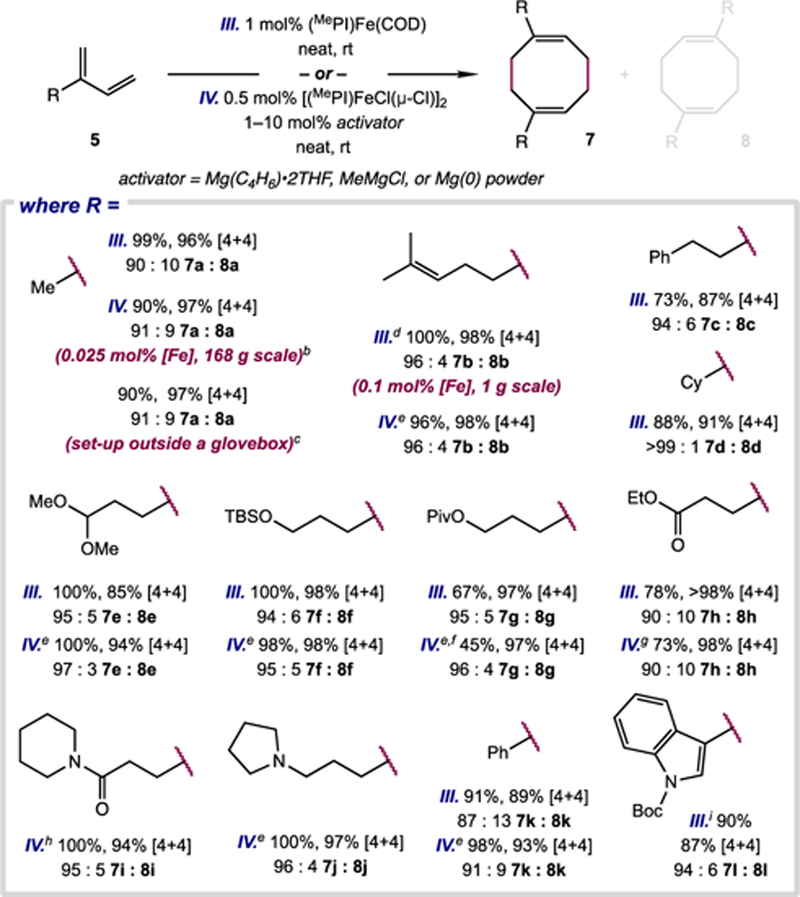
a Unless indicated otherwise, reactions were run on 0.5 mmol (Method III) or 1.0 mmol (Method IV) scale with 1 mol% [Fe] precatalyst. Values describe combined isolated yield of isomeric products, the portion of this isolated mixture comprised of [4+4]-cycloadducts as determined by gas chromatographic analysis, and the regiosomer ratio of the [4+4]-cycloadducts as determined by relative integration of diagnostic resonances in the quantitative 13C NMR spectrum of the isolated material. Mg(C4H6)•2THF = magnesium butadiene bis(tetrahydrofuran), TBS = tert-butyldimethylsilyl b with 0.0125 mol% [(MePI)FeCl(μ-Cl)]2 and 0.06 mol% MeMgCl (3 M in THF) at 10 °C to rt c with 0.5 mol% [(MePI)FeCl(μ-Cl)]2 and 2.4 mol% MeMgCl (3 M in THF) d with 0.1 mol% (MePI)Fe(COD) e with 1 mol% Mg(C4H6)•2THF as activator f additional 0.5 mol% [(MePI)FeCl(μ-Cl)]2 and 1 mol% Mg(C4H6)•2THF added g with 13 mol% Mg(0) powder as activator h with 1.5 mol% [(MePI)FeCl(μ-Cl)]2 and 33 mol% Mg(0) powder i 1 M in PhMe
The conditions for in situ catalyst activation proved similarly robust for [4+4]-cyclodimerization of many 2-substituted dienes. However, the single-component precatalysts afforded improved chemoselectivity over the [(RPI)FeCl(μ-Cl)]2/activator combination for substrates bearing electrophilic functionality prone to competitive reactions with Grignard reagents. Alternatively, magnesium powder was used as a more functional-group-tolerant activator; however the slow activation necessitated extended reaction times and/or increased catalyst loadings (see Supporting Information).
Despite the success of the methods developed for the [4+4]-cyclodimerization of monosubstituted dienes, disubstituted dienes remained challenging substrates by virtue of their increased steric profiles. Subjecting representative disubstituted dienes such as 2,3-dimethyl-1,3-butadiene (9), 3-methyl-1,3-pentadiene (12), and 2-methyl-1,3-pentadiene to Methods I–III outlined above resulted either in little-to-no substrate consumption or in exclusive formation of linear adducts. While in situ activation of iron dichloride and 2,2’-bipyridine (bpy) in the presence of 2,3-dimethyl-1,3-butadiene (9) afforded promising levels of chemoselectivity for [4+4]-cyclodimerization product 10 (Scheme 9A), attempts to prepare and isolate a suitable, well-defined (bpy)FeL2 precatalyst were unsuccessful. Nonetheless, this result highlights the importance of the ligand steric profile in enabling key oxidative cyclization and reductive elimination steps. Further underscoring the fine balance of competing processes determining product formation in these highly encumbered systems, hydroalkenylation products resulting from dimerization of 12 or nopadiene (14) could be obtained in high yield and isomeric purity using iminopyridine precatalyst (iPrPI)Fe(COD) (Scheme 9B). While the formation of these linear products illustrates the challenge of out-competing β-H elimination in sterically encumbered metallacycles, it nonetheless affords access to potentially useful building blocks for further synthetic manipulations.33
Scheme 9.
Iron-catalyzed dimerization of disubstituted-1,3-dienes.a

a Unless noted otherwise, reactions were run on 1.0 mmol scale. Values describe combined isolated yield of isomeric products, the portion of this isolated mixture comprised of [4+4] or hydroalkenylation products with the indicated connectivity as determined by gas chromatographic analysis, and the diastereomer ratio as determined by the relative integration of diagnostic resonances in the quantitative 13C NMR spectrum of the isolated material. b Diene 12 was used as a mixture (70:30 E/Z) of isomers.
Intermolecular Cross-[4+4]-Cycloaddition of 1,3-Dienes.
While high chemo- and regioselectivity for [4+4]-cyclodimerization was observed across varied monosubstitued-1,3-dienes, the relative rates of substrate consumption proved sensitive to subtle differences in substrate substitution pattern and sterics. This feature was leveraged to adapt the conditions to the cross-[4+4]-cycloaddition of two differentially reactive dienes, thereby accessing unsymmetric disubstituted cyclooctadienes.
Using (MePI)Fe(COD) as the precatalyst, the cross-reaction of two different 2-substituted dienes was achieved. This was particularly powerful using an excess of an inexpensive coupling partner such as isoprene or myrcene. Under these conditions, the homocoupled products obtained from the coupling partner used in excess were removed readily under vacuum or by passage of the crude reaction mixture through a silica plug to afford the cross-[4+4]-cycloadduct in high yield, chemo-, and regioselectivity (see Supporting Information for details).
Using precatalyst (MesDI)Fe(COD), the cross-[4+4]-cycloaddition of 4-substituted dienes with 2-substituted dienes was also demonstrated. In this case, the negligible activity of (MesDI)Fe(COD) for the homocoupling of 2-substituted dienes enabled use of substrate ratios as low as 1.5:1. With the portion-wise addition of the 4-substituted diene, these fragment-coupling conditions were employed to unite differentially substituted coupling partners through the cross-[4+4]-cycloaddition, again in high yield, chemo-, and regioselectivity.
Taken together, these methods for the intermolecular, [4+4]-cyclodimerization and cross-[4+4]-cycloaddition of dienes afford access to cyclooctadienes with diverse patterns of functional group incorporation. These strained, cyclic olefins constitute useful building blocks as the sterically and electronically differentiated alkenes are poised to enable diverse transformations including alkene hydrofunctionalization and difunctionalization,5b,34 allylic functionalization,35 and cationic (poly)cyclization.20a,36 Moreover, the potential utility of these products as (co)monomers for ring-opening metathesis was demonstrated through the synthesis of methyl-branched oligomers for investigation as industrial lubricants and plasticizers (see Supporting Information).37 As such, these products hold promise for application in the synthesis and evaluation of novel polymers, fuels, fragrances, and other fine chemicals.
Electronic Structure Determination.
In light of the high activity as well as the remarkable chemo-, regio-, and diastereoselectivity observed for the iron-catalyzed [4+4]-cycloaddition of monosubstituted 1,3-dienes, characterization of catalytically relevant reduced iron species was targeted. Structural and spectroscopic data for (RDI)Fe(COD) (where R = iPr, Me, or Mes) and (iPrDI)Fe(COD) were reported recently; in all cases, the electronic structures of these net S = 1 complexes were best described as high-spin iron(I) anti-ferromagnetically coupled to a ligand-based radical anion.11b,26 As with the α-diimine ligands, the potential for ligand redox activity has been documented for transition metal complexes bearing iminopyridine ligands.38 However, little was known about the electronic structures of the reduced iron complexes relevant to this study.
The solid-state structures of (iPrPI)Fe(COD), (iPrPI)Fe(MviMvi), (iPrPI)2Fe, and (MesPI)2Fe were determined by single-crystal X-ray diffraction, confirming their identities (Figure 1; see also Supporting Information). The latter two complexes, (iPrPI)2Fe and (MesPI)2Fe, are analogous to the (iPrPAI)2Fe complex (iPrPAI = [2,6-iPr2-C6H3-N=CH]2) reported previously by Wieghardt and co-workers,38b and were included for comparison with the heteroleptic complexes. In each case, the coordination geometry about iron is best approximated as tetrahedral.39
Figure 1.
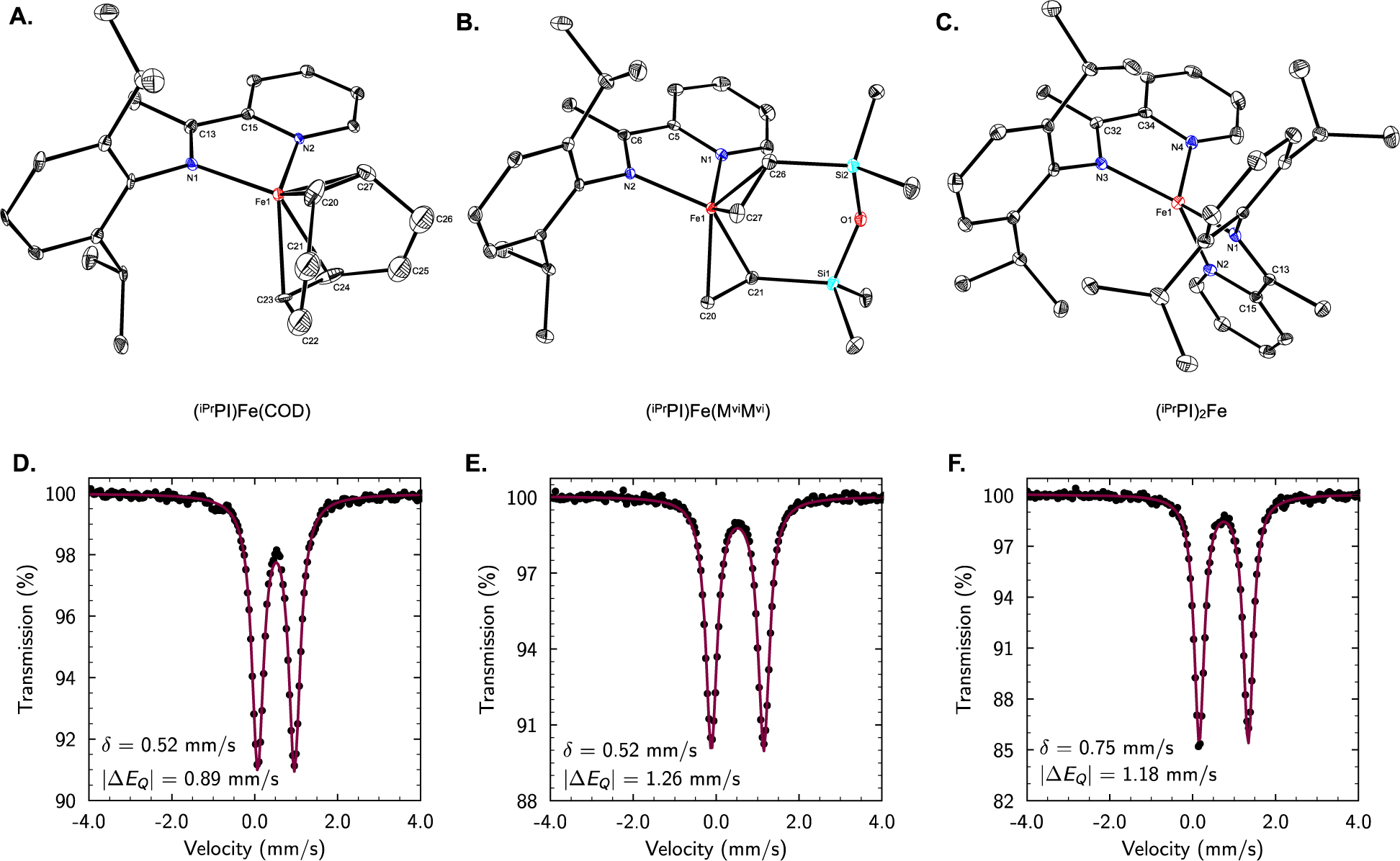
Characterization of Reduced (iPrPI)Fe Complexes. (A–C.) Solid-state structures represented with 30% probability ellipsoids; H atoms omitted for clarity. (D–F.) Zero-field 57Fe Mößbauer spectra collected at 80 K for solid-state samples of (iPrPI)Fe(COD), (iPrPI)Fe(MviMvi), and (iPrPI)2Fe. Red lines represent the fit of the data to the quadrupolar doublets defined by the parameters shown.
Distortion of the metrical parameters of the iminopyridine scaffold is an established reporter of ligand redox activity.38b The pyridine Npyr–Cipso, imine Nim–Cim, and backbone Cim–Cipso bond lengths are particularly diagnostic of the ligand oxidation state. The values obtained for these key bond metrics in each of the aforementioned solid-state structures are summarized in Table 1 along with average values characteristic of a generic iminopyridine in its neutral, radical anion, and closed-shell dianion forms. Altogether, the lengthening of the Npyr–Cipso and Nim–Cim bonds, coupled with the contraction of the Cim–Cipso bonds relative to the lengths expected for the neutral ligand implicate the singly reduced radical anionic form of the ligand across all of these structures. Likewise, the bond lengths and angles of the bis-olefin ligands (COD and MviMvi) exhibit moderate perturbation,40 consistent with partial back-donation from olefin coordination and the Dewar–Chatt–Duncanson model.41
Table 1.
Select Metrical Parameters for Reduced (iPrPI)Fe Complexes and Related Structures. a
| Bond Lengths (Å) | Angles (°) | τ4 b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fe–Nim | Fe–Npyr | Nim–Cim | Npyr–Cipso | Cim–Cipso | C=C | Nim–Fe–Npyr | Nim–Fe–C=C | Npyr–Fe–C=C | ||
| (iPrPI)Fe(COD) c | 1.969(6) | 2.026(6) | 1.341(9) | 1.383(8) | 1.414(8) | 1.351(11) 1.374(12) |
80.23(19) | 134.3 123.1 |
114.1 114.1 |
0.73 |
| BS(3,1) d | 2.013 | 2.097 | 1.349 | 1.397 | 1.427 | 1.393 1.391 |
79.8 | 133.1 126.0 |
117.4 111.9 |
0.72 |
| (iPrPI)Fe(MviMvi) e | 1.984(2) | 2.021(2) | 1.340(3) | 1.376(3) | 1.423(3) | 1.410(3) 1.403(3) |
79.84(8) | 123.2 111.3 |
118.7 109.4 |
0.84 |
| BS(3,1) f | 2.051 | 2.082 | 1.347 | 1.386 | 1.428 | 1.397 1.393 |
79.3 | 121.8 110.8 |
122.6 108.3 |
0.82 |
| (iPrPI)2Fe g | 1.980(3) 1.988(3) |
2.043(3) 2.026(3) |
1.339(4) 1.349(4) |
1.381(5) 1.381(5) |
1.423(5) 1.417(5) |
-- | 79.47(12) 80.09(12) |
-- | -- | 0.70 |
| (iPrPAI)2Fe h,i | 1.988(2) 1.981(2) |
2.043(2) 2.046(2) |
1.342(2) 1.336(3) |
1.387(3) 1.386(3) |
1.411(3) 1.408(3) |
-- | 81.62(7) 82.02(7) |
-- | -- | 0.70 |
| (RPI(ox))0 h,j | -- | -- | 1.28 | 1.35 | 1.47 | -- | -- | -- | -- | -- |
| (RPI•)1– h,j | -- | -- | 1.34 | 1.39 | 1.41 | -- | -- | -- | -- | -- |
| (RPI(red))2– h,j | -- | -- | 1.46 | 1.40 | 1.35 | -- | -- | -- | -- | -- |
Metrical parameters obtained through single crystal X-ray diffraction analysis.
Calculated as described in Ref 39.
See Figure 1A.
Optimized, broken symmetry structure for (iPrPI)Fe(COD) computed using the B3LYP/def2-SVP/def2-TZVP level of density functional theory with application of the ZORA relativistic correction.
See Figure 1B.
Optimized, broken symmetry structure for (iPrPI)Fe(MviMvi) computed using the B3LYP/def2-SVP/def2-TZVP level of density functional theory with application of the ZORA relativistic correction.
See Figure 1C.
Taken from Ref. 38b.
Values listed for one of two structurally similar complexes in the unit cell.
(RPI(ox))0 represents a generic iminopyridine ligand in its neutral form, (RPI•)1– represents a generic iminopyridine ligand in its anionic, one-electron-reduced form, and (RPI(red))2– represents a generic iminopyridine ligand in its dianionic, two-electron-reduced form.
The solid-state magnetic moments measured at 22 °C for the (RPI)Fe(bis-olefin) complexes ((iPrPI)Fe(COD): 2.9 μB; (iPrPI)Fe(MviMvi): 2.7 μB; (MePI)Fe(MviMvi): 2.6 μB;) are all consistent with a net S = 1 ground state. Taken together with the solid-state structural information and analogy to the (DI)Fe(COD) complexes, these results support the assignment of high-spin iron(I) (SFe = 3/2) centers engaged in antiferromagnetic coupling with the iminopyridine ligand radical (SPI = −1/2). These assignments were corroborated by zero-field 57Fe Mößbauer spectroscopic measurements at 80 K of solid-state samples of the (RPI)Fe(bis-olefin) complexes. The isomer shift (δ) and quadrupole splitting (|ΔEQ|) parameters obtained from the fits to these spectra are compiled in Table 2, along with those measured for reference compounds including (RPI)2Fe complexes as well as iron complexes across varied oxidation states bearing α-diimine11b,26,38a,38c or pyridine-2,6-dimine ligands.25,42 Importantly, the isomer shifts observed for all four (RPI)Fe(bis-olefin) complexes (δ = 0.49–0.53 mm/s) were unambiguously within the range typical for high-spin iron(I).43
Table 2.
Select Zero-Field 57Fe Mößbauer Parameters for (RPI)Fe Complexes and Related Structures. a
| [Fe] | δ (mm/s) | |ΔEQ| (mm/s) | Ox. State | [Fe] | δ (mm/s) | |ΔEQ| (mm/s) | Ox. State | |
|---|---|---|---|---|---|---|---|---|
| (iPrPI)Fe(COD) b | 0.52 | 0.89 | Fe(I), SFe = 3/2 | (iPrDI)Fe(η4-C5H8) f | 0.56 | 1.68 | Fe(I), SFe = 3/2 | |
| soln. c | 0.52 | 0.89 | (iPrPI)2Fe h | 0.75 | 1.18 | Fe(II), SFe = 2 | ||
| BS(3,1) d | 0.53 | 1.09 | (MePI)2Fe e | 0.75 | 1.18 | Fe(II), SFe = 2 | ||
| (MePI)Fe(COD) e | 0.49 | 0.92 | Fe(I), SFe = 3/2 | (MesPI)2Fe | 0.74 | 1.33 | Fe(II), SFe = 2 | |
| BS(3,1) d | 0.52 | 1.05 | (iPrDI)2Fe i | 0.77 | 1.81 | Fe(II), SFe = 2 | ||
| (iPrDI)Fe(COD) f | 0.48 | 1.31 | Fe(I), SFe = 3/2 | (iPrPDI)Fe(N2)2 j | 0.39 | 0.53 | Fe(0)–Fe(II) hybrid, SFe = 0 | |
| (iPrPI)Fe(MviMvi) g | 0.52 | 1.26 | Fe(I), SFe = 3/2 | (iPrPDI)Fe(N2) j | 0.38 | 1.72 | Fe(II), SFe = 1 | |
| BS(3,1) d | 0.54 | 1.35 | (iPr(TB)PDI)Fe(N2)2 k | 0.45 | 0.83 | Fe(0)–Fe(II) hybrid, SFe = 0 | ||
| (MePI)Fe(MviMvi) | 0.53 | 1.94 | Fe(I), SFe = 3/2 | (iPr(TB)PDI)Fe | 0.63 | 2.47 | Fe(I), SFe = 3/2 | |
| (CH2CHCH2)2NTs k | ||||||||
| BS(3,1) d | 0.53 | 1.17 | (iPr(TB)PDI)Fe | 0.73 | 1.87 | Fe(I), SFe = 3/2 | ||
| (CH2CHCH2)2 k |
All measurements recorded at 80 K for solid powder samples unless noted otherwise. Complete ligand structures for all listed complexes depicted in the Supporting Information.
See Figure 1D.
Solution state sample frozen in C6H6.
Calculated using the optimized, broken symmetry structure computed at the B3LYP/def2-SVP/def2-TZVP level of density functional theory with the ZORA relativistic correction.
From a sample of 90% (MePI)Fe(COD) and 10% (MePI)2Fe.
Taken from Ref. 26.
See Figure 1E.
See Figure 1F.
Taken from Ref. 38a
Taken from Ref. 42
Taken from Ref. 25.
Full-molecule density functional theory (DFT) calculations were initiated from solid-state structures for (iPrPI)Fe(COD), (iPrPI)Fe(MviMvi), (MePI)Fe(COD),44 and (MePI)Fe(MviMvi) 45 using the B3LYP functional,46,47 and either the unrestricted Kohn–Sham (UKS)48 or broken-symmetry (BS) methods.49 When validated against experimental spectroscopic measurements, these methods have been widely applied for determination of the ground-state electronic structures of first-row transition metals bearing redox-active ligands.38,42,50 Both the UKS and BS methods converged to broken-symmetry BS(3,1) solutions. While the optimized geometries for these structures obtained at the B3LYP/def2-SVP/def2-TZVP level of theory51 were in excellent agreement with the solid-state structures (vide supra), the computed zero-field 57Fe Mößbauer isomer shifts obtained using Neese’s calibration constants50d exhibited a systematic perturbation from the values obtained experimentally (i.e. the computed values were ~0.1 mm/s too large in all cases). Such systematic deviation has been observed previously with other specialized ligand classes.50e,f However, inclusion of an empirical relativistic correction (ZORA)52 for both the geometry optimization and spectroscopic prediction steps afforded improved agreement between experiment and theory.
The qualitative d-orbital splitting diagram and spin-density plot for (iPrPI)Fe(COD) derived from these computational results are depicted in Figure 2, where the spatial overlap (S = 0.47) between primarily iron- and ligand-centered orbitals is indicative of their antiferromagnetic coupling. These depictions are representative of those observed for all four (PI)Fe(bis-olefin) complexes that were examined computationally and highlight the involvement of the iminopyridine ligand in the electronic structure of the catalytically relevant complexes. The computational results thus underscore the similarities in the electronic structures of the iminopyridine, α-diimine, and pyridine-2,6-dimine iron bis-olefin complexes implicated in the mechanisms of C–C bond-formation through oxidative cyclization.25,26
Figure 2.
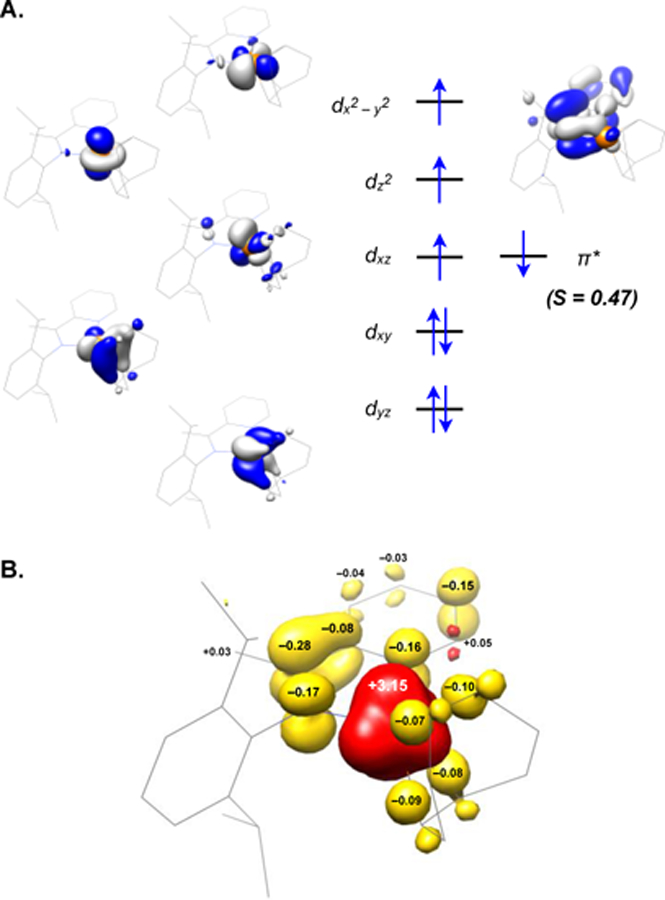
Electronic Structure of (iPrPI)Fe(COD). (A.) Qualitative molecular orbital diagram derived from the BS(3,1) DFT solution for (iPrPI)Fe(COD), representing the corresponding orbitals possessing significant d-orbital character and the singly-occupied, ligand-based orbital. (B.) Spin-density plot for (iPrPI)Fe(COD)obtained from the BS(3,1) Mulliken population analysis (red, positive spin density; yellow, negative spin density). DFT calculations performed at the B3LYP/def2-SVP/def2-TZVP level of theory with the ZORA relativistic correction.
Mechanistic Analysis.
Significant insight was obtained through analysis of the single-component iron bis-olefin precatalysts/model complexes achieving highly selective [4+4]-cycloaddition of 1,3-dienes. However, attempts to isolate and characterize the catalytically active species from the reaction mixture were unsuccessful and invariably yielded the homoleptic bis-ligand complexes, (RPI)2Fe, or the isoprene adduct thereof, (MePI)2Fe(η2-isoprene) (see Supporting Information for single crystal X-ray diffraction data). While (MePI)2Fe did exhibit low activity for the [4+4]-cyclodimerization of 2-substituted dienes, it was not kinetically competent to be the predominant species responsible for catalysis (vida supra; Scheme 5). This is consistent with the observation that (MeDI)2Fe is, likewise, not a competent catalyst for the [4+4]-cyclodimerization of butadiene.11 Given the apparent instability of and difficulty in isolating the active species, kinetic techniques and zero-field 57Fe Mößbauer spectroscopic measurements were employed instead to gain mechanistic insight.
To probe whether catalyst deactivation occurred on the timescale of the catalytic reaction, an experiment analogous to Blackmond’s “same-excess” protocol was performed.53 Substrate consumption was monitored over the entire reaction course by gas chromatographic analysis of aliquots removed from the reaction mixture for two trials in which [Fe]tot ([(MePI)Fe(MviMvi)]tot = 2.5 mM)54 was held constant but the initial myrcene concentration of one was adjusted to mimic initiation at partial conversion. This procedure was conducted for reactions run both in neat substrate ([5b]0 = 5.0 vs. 2.7 M)55 and in dilute toluene solution ([5b]0 = 1.0 vs. 0.5 M). For the reactions conducted in neat substrate, the resulting rate vs. concentration curves (or time-shifted concentration vs. time curves) overlaid within error (Figure 3A–C; blue circles vs. red squares). However, a small but statistically significant deviation between the two was evident for the reactions conducted in toluene (Figure 3D–F). In both cases, good overlay was observed for reactions conducted with (red squares) and without (teal triangles) the addition of exogenous product at the outset of the reaction (i.e. [5b]0 = 2.5 M, [7b]0 = 0 vs. 1.25 M and [5b]0 = 0.5 M, [7b]0 = 0 vs. 0.25 M). Taken together, these results demonstrate that product inhibition does not occur under the standard reaction conditions. While slight catalyst deactivation was observed for reactions conducted in dilute toluene, likely due to ligand exchange and the formation of the homoleptic bis-ligand complex (vide supra), this deactivation process was minimized by performing the reaction in neat substrate. Catalyst turnover numbers were high in both scenarios (TON = 400–2000). Consistent with these observations, dark teal reaction mixtures were observed that persisted throughout the course of product formation but faded to dark orange-brown rapidly following consumption of the diene substrate.
Figure 3.

Kinetic Tests for Catalyst Deactivation and Product Inhibition for Reactions Run in Neat Substrate (A–C) or Toluene Solution (D–F). (A./D.) Concentration of substrate [5b] over time measured by gas chromatographic analysis of aliquots removed from the reaction mixture. (B./E.) Concentration of substrate [5b] over time time-shifted to illustrate overlay with standard conditions. (C./F.) Reaction rate over the course of substrate consumption shown over evenly spaced intervals of Δ[5b] = 0.25 M. Open markers represent data points obtained from individual trials; closed markers represent average values obtained from duplicate trials. [Fe]tot = [(MePI)Fe(MviMvi)]0 = 2.5 mM for all trials. See Supporting Information for additional experimental details.
Additional reaction-progress kinetic analysis provided insight into the nature of the rate dependence on diene [5b]t and iron catalyst [Fe]tot. Consumption of myrcene (5b) and formation of [4+4]-cycloadduct 7b evolved smoothly over the reaction with no change in selectivity over time (Figure 4A). However, a non-integer-order dependence on diene concentration was observed, with apparent zero-order dependence of reaction rate on diene concentration at high [5b]t relative to [Fe]tot (i.e. low conversion or low [Fe] loading) and non-zero-order dependence at lower [5b]t (i.e. high conversion or high [Fe] loading; Figure 4B). Given the absence of competitive catalyst deactivation over the course of the reaction, this observation suggested a change in the catalyst resting state as a function of [5b]. The kinetic order in iron catalyst was thus determined over the linear regime (i.e. where the order in myrcene is zero; 10%–80% conversion) from a series of reactions run at constant [5b]0 wherein the catalyst concentration was varied ([(MePI)Fe(COD)]tot = 2.5–60 mM; 0.02–1.2 mol% [Fe]). A first-order dependence on [Fe]tot was observed (Figure 4C).
Figure 4.

Reaction-Progress Kinetic Analysis. (A.) Concentration of substrate [5b] over time measured by gas chromatographic analysis of aliquots removed from reaction mixtures over varied [Fe]tot. (B.) Reaction rate over the course of substrate consumption shown over evenly spaced intervals of Δ[5b] = 0.25 M obtained from the univariate spline fit of the concentration vs. time plots in A. (C.) The first-order dependence of reaction rate on [Fe]tot illustrated by the linear regression to rate-data obtained at [5b] = 2.5 M, with qualitatively similar dependence observed over the full course of the reaction. Open markers represent data points obtained from individual trials; closed markers represent average values obtained from duplicate trials.
To examine the identity of iron complexes formed under the reaction conditions, zero-field, solution-state 57Fe Mößbauer spectra were recorded at 80 K for frozen solution samples of (iPrPI)Fe(COD) in benzene (see Supporting Information) and myrcene (5b, Figure 5). Because of the high activity of (MePI)Fe(COD) with myrcene, especially at the high concentrations needed for spectroscopic observations, the more sterically encumbered and less active iron precursor, (iPrPI)Fe(COD), was used for the in situ monitoring study. In benzene solution, a single quadrupolar doublet was observed with parameters comparable to those obtained for the solid-state sample, (benzene solution: δ = 0.52 mm/s, |ΔEQ| = 0.89 mm/s; solid state: δ = 0.52 mm/s, |ΔEQ| = 0.89 mm/s, vide supra). By contrast, the Mößbauer spectrum obtained after mixing (iPrPI)Fe(COD) with 25 equivalents of 5b at ambient temperature (~23 °C) for 5 minutes prior to freezing with liquid N2 exhibited features characteristic of at least two similar but distinct iron-containing species. Fitting the data to two species (A and B) yielded parameters (δA = 0.52 mm/s, |ΔEQ|A = 1.28 mm/s (55%); δB = 0.52 mm/s, |ΔEQ|B = 0.65 mm/s (45%)) consistent with two (iPrPI)Fe(L)2 complexes where (L)2 corresponds to one (η4) or two (η2) molecules of the diene, respectively. This assignment is consistent with the observed change in kinetic order as a function of [5b]/[Fe]tot.
Figure 5.

Iron Speciation under Catalytically Relevant Conditions. (A.) Preparation of a solution sample for in situ spectroscopic analysis. (B.) Zero-field 57Fe Mößbauer spectrum collected at 80 K for the frozen solution sample prepared in A. Red and blue lines represent the fit of the data to the quadrupolar doublets defined by the parameters shown. Structures in the gray box represent plausible resting state complexes that are consistent with the kinetic and Mößbauer spectroscopic data; however, their precise identity cannot be assigned definitively.
While the combined kinetic experiments and spectroscopic measurements provided information about the resting state of the iron catalyst, the observed rate law does not distinguish between oxidative cyclization, allyl isomerization, and reductive elimination as rate-determining steps. To gain additional insight into the nature of the selectivity-determining event(s), 12C /13C kinetic isotope effects (KIEs) at all positions were determined. Singleton’s NMR-based technique was applied to measure perturbations of the 12C/13C isotopic ratios obtained for product 7b isolated at low conversion relative to natural abundance material (measured from product 7b obtained after full conversion of substrate 5b).56 Large, primary isotope effects were measured at the site of initial C–C bond-formation (C4, KIE = 1.019(1) averaged over two chemically equivalent sites) and at the vicinal olefinic carbon (C3, KIE = 1.019(4)); a modestly attenuated primary isotope effect was also observed at the opposite terminus (C1, KIE = 1.010(4); Scheme 11). While comparison to high-level computational models is necessary when applying this technique to distinguish between multiple similar mechanisms, natural abundance 12C/13C KIE measurements have been applied successfully in the absence of computational models to provide general information about the identity of the first irreversible product-determining step within an accepted mechanistic pathway.57 With these limitations in mind, the observed KIEs are most consistent with irreversible and therefore regioselectivity-determining oxidative cyclization proceeding through [1,4]-carbometallation from an iron bis-diene complex (see Supporting Information for further discussion). This direct [1,4]-carbometallation also accounts for the high chemoselectivity observed for the iron-catalyzed [4+4]-cycloaddition, as the reaction mechanism bypasses a putative divinyl metallacyclopentane poised to undergo side reactions. A catalytic cycle accounting for the combined kinetic analyses, Mößbauer spectroscopic observations, and KIE measurements is illustrated in Scheme 12. Model reaction time-courses simulated for this mechanistic scenario using COPASI software are consistent with the observed saturation kinetics (see Supporting Information).58
Scheme 11.

13C/12C Isotope Effects.
Scheme 12.
Proposed Catalytic Cycle.a

a (I) Diene coordination. (II) Oxidative cyclization. The observed 13C/12C KIEs do not distinguish between direct oxidative cyclization to afford the σ- or π-allyl metallacycles shown. R.D.S. = rate-determining step. (III) Allyl isomerization. (IV) Ligand-induced reductive elimination or reductive elimination followed by diene coordination.
While the KIE measurements enabled identification of the regioselectivity-determining step, this step is not necessarily also diastereoselectivity-determining. Insight could be drawn from the relationship between the isomeric purity of the diene substrate and the observed product diastereomer ratio. If the product diastereomer ratio were entirely catalyst controlled (i.e. if only (E)-isomer incorporation were possible), the product d.r. would be independent of substrate E/Z ratio (Scheme 13A). This would be possible either through an entirely stereoconvergent mechanism (mechanism i) or if substrate incorporation were entirely isomer-selective (mechanism ii). Alternatively, if incorporation of both (E)- and (Z)-diene isomers were possible (through either stereoconvergent or stereospecific processes, as in mechanisms iii and iv, respectively), the product d.r. would vary as a function of substrate E/Z ratio, resulting from the rate-averaged statistical combination of the substrate isomers (Scheme 13B). Finally, for a mechanism in which only one of the two diene coupling partners were incorporated in an isomer-selective fashion the product d.r. would be related directly to the substrate E/Z ratio (Scheme 13C, mechanisms v and iv).
Scheme 13.
Mechanistic Hypotheses to Account for Catalyst- and Substrate-Controlled Diastereoselectivity.a

a Major products depicted for the cyclodimerization of 4-substituted dienes obtained using iron precatalyst (MesDI)Fe(COD). See Supporting Information for the analogous scheme depicting formation of the major products expected using iron precatalyst (iPrPI)Fe(COD).
To probe these possibilities experimentally, (E)-1b and (Z)-1b were subjected to the [4+4]-cyclodimerization conditions independently. Whereas (E)-1b was converted smoothly to [4+4]-cycloadducts 3b and 4b with high diastereoselectivity, (Z)-1b underwent non-productive isomerization upon exposure to either iron precatalyst (Scheme 14A & B) and cyclodimerization products were not detected. This isomer-dependent reactivity thus ruled out mechanisms iii and iv depicted in Scheme 13 for both the diimine and iminopyridine iron catalysts.
Scheme 14.
Effect of Diene Geometry on Diastereoselectivity.a
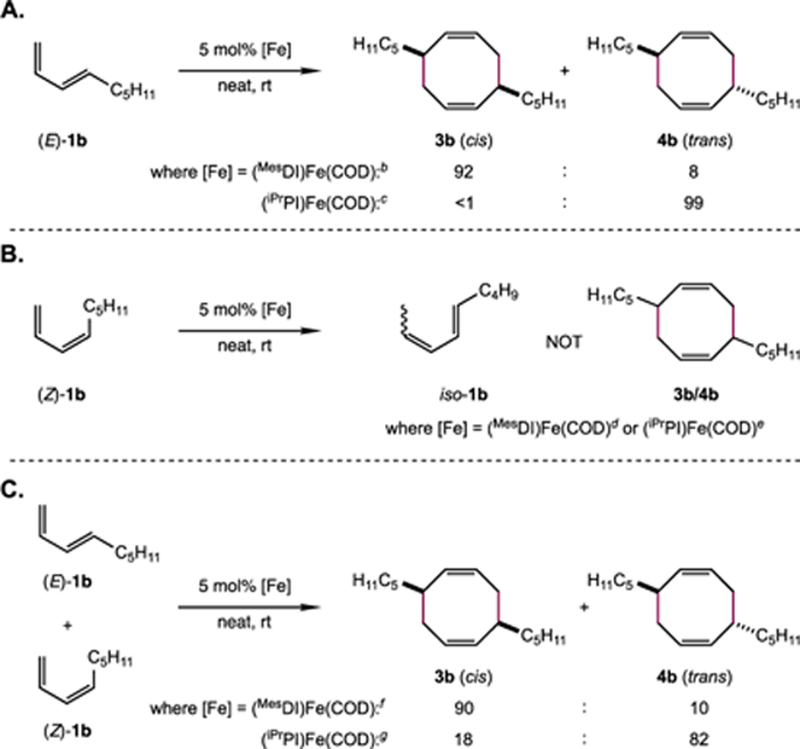
a Compare with Scheme 6. Reactions were run on 0.4 mmol scale with 5 mol% [Fe] precatalyst. Product ratios determined by gas chromatographic analysis and corroborated by relative integration of diagnostic resonances in the quantitative 13C NMR spectrum of the isolated material. b 98% [4+4] selectivity c 77% [4+4] selectivity d no 3b or 4b detected e <10% combined yield of dimeric products with 65% [4+4] selectivity, 74:26 3b : 4b d 30% combined yield dimeric products with 98% [4+4] selectivity; 44% yield iso-1b e 62% combined yield of dimeric products with 60% [4+4] selectivity; 38% yield iso-1b
Subsequently, mixtures of (E)- and (Z)-1b were subjected to the same reaction conditions (Scheme 14C). Where diimine iron complex (MesDI)Fe(COD) was used as a precatalyst, 3b and 4b were obtained in the same ratio as observed with pure (E)-1b substrate. In this case, formation of the [4+4]-cycloadducts was accompanied by the accumulation of iso-1b. These observations suggest that isomer-selective mechanism ii (Scheme 13A) is most likely operative.
In contrast to the results obtained using (MesDI)Fe(COD), both (E)- and (Z)-isomers were consumed upon exposure to (iPrPI)Fe(COD), resulting in the formation of cyclodimerization products with moderately reduced diastereoselectivity and chemoselectivity for the [4+4] products. This isomer-dependent selectivity eliminates mechanisms i and ii depicted in Scheme 13. While these results cannot distinguish between mechanistic possibilities v and vi depicted in Scheme 13, they do provide evidence for a mechanism in which selectivity is determined through the combination of catalyst-controlled diastereoselective/isomer-selective and substrate-controlled stereospecific/isomer-unselective contributions. Moreover, mechanisms ii and v/vi exist on a continuum, and the extent to which isomer-selective reactivity occurs likely varies across substrates. Indeed, exposure of a mixture of piperylene isomers (66:34 (E)-/(Z)-1a) to either precatalyst (MesDI)Fe(COD) or (iPrPI)Fe(COD) results in reduced chemo- and diastereoselectivity (vide supra; indicative of mechanisms v/vi) in both cases
Taken in combination with the mechanism implicated for oxidative cyclization through [1,4]-carbometallation, this finding provided support for a scenario in which the relative stereochemistry of the distal metallacycle substituent and proximal vinyl group is set through diastereoselective and isomer-selective oxidative cyclization. A subsequent, stereospecific allyl-isomerization and C–C bond-forming reductive elimination sequence results in the relative configuration observed in the final product. While alternative mechanisms involving stereoconvergent reductive elimination cannot be ruled out on the basis of the experimental data available, the general mechanistic picture delineated through the studies described herein provided sufficient context to generate a qualitative stereochemical model. To aid in visualization of relevant interactions, the coordinates for the solid-state structures of precatalysts (MesDI)Fe(COD) and (iPrPI)Fe(COD) were analyzed to generate topographic steric maps of the metal-bound, redox-active DI and PI ligands using SambVca 2.0 (Figure 6).59 These plots illustrated a striking difference in the openness of the eastern (as drawn in Figure 6) hemisphere of the iron coordination sphere, with the CS symmetric iPrPI resulting in a lower local percent buried volume (%Vbur) than the effectively C2 MesDI ligand.60,61 To the extent that the transition structures for oxidative cyclization resemble the incipient metallacycles, the ligand-dependent diastereoselectivity observed with 4-substituted diene coupling partners can be justified on the basis of competing repulsive steric interactions between proximal metallacycle substituents and the steric environment of the DI or PI ligand in this hemisphere. Namely the metallacycle en route to trans-diastereomer 4 avoids repulsive gauche-butane interactions but is only accessible when one hemisphere of the iron coordination sphere is open. This arrangement also allows for potential stabilizing π-stacking interactions with the ligand pyridine backbone.62 In contrast, the more symmetrically shielded environment enforced by MesDI favors the metallacycle en route to cis-diastereomer 3.
Figure 6.

Rational for Diastereoselectivity. (A/B.) Topographic steric maps of (MesDI)Fe and (iPrPI)Fe generated from the solid-state structures for the corresponding COD complexes. The Fe atom defines the center of the xyz coordinate system, the N–Fe–N plane defines the xz-plane, and the z-axis bisects the N–Fe–N angle. Colors indicate occupied space toward (−z, blue) or away from (+z, red) the DI or PI ligand. Local percent buried volume (%Vbur) listed for each quadrant of a sphere of radius, r = 3 Å. (C.) Stereochemical rational for ligand-controlled, diastereoselectivity.
CONCLUDING REMARKS
In summary, a family of iron complexes bearing bidentate ligands enabled the [4+4]-cycloaddition of substituted 1,3-dienes with control of chemo- and regioselectivity to afford disubstituted 1,5-cyclooctadienes. Methods to access either of two diastereomeric products selectively were defined. The resultant cyclooctadienes hold promise as modular building blocks in the synthesis of precision polyolefins, lubricants, fuels, and fine chemicals. Determination of the electronic structures of the precatalysts and comparison to in situ spectroscopic measurements implicated catalytically active species in which an iminopyridine ligand radical anion is antiferromagnetically coupled with a high-spin iron(I) center. Mechanistic studies support a mechanism in which stereoselective oxidative cyclization of two dienes from the resting state complex is followed by a stereospecific allyl-isomerization/C–C bond-forming reductive elimination sequence. These insights shed further light on strategies for ligand development to enable selective chemistry that leverages metallacyclic intermediates to upgrade unsaturated coupling partners.
Supplementary Material
Scheme 10.
Scope of iron-catalyzed cross-[4+4]-cycloaddition of substituted dienes.a

a See Methods I–IV in Scheme 6. Reactions were run with 1 or 5 mol% [Fe] precatalyst. For the formation of 16 from two 2-substituted dienes, 0.5 mmol of the limiting, slow-reacting diene and 5 mmol of the fast-reacting diene were employed. For the formation of 17 from the coupling of a 2-substituted and 4-substituted diene, the 4-substituted diene (1; 1.5–2 equiv in total) was added portion-wise to the reaction mixture. b IV: 0.5 mol% [(MePI)FeCl(μ-Cl)]2 and 1 mol% Mg(C4H6)•2THF. c IV: 0.5 mol% [(MePI)FeCl(μ-Cl)]2 and 2.4 mol% MeMgCl (3 M in THF) d IV: 0.5 mol% [(MePI)FeCl(μ-Cl)]2 and 13 mol% Mg(0) powder e III: 5 mol% (MePI)Fe(COD) with 280 μL added PhMe f I: 5 mol% (MesDI)Fe(COD)
ACKNOWLEDGMENT
C.R.K. thanks the NIH for a Ruth L. Kirschstein National Research Service Award (F32 GM126640). Financial support and a gift of piperylene were provided by Firmenich. We thank Dr. István Pelczer (Princeton University) for assistance with NMR experiments, and Dr. Stephan Rummelt for a gift of [(C5H9PDI)Fe(N2)].
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge via the internet at http://pubs.acs.org.
Crystallographic information for (MesDI)Fe(COD), (iPrPI)Fe(COD), (iPrPI)Fe(MviMvi), (iPrPI)2Fe, (MesPI)2Fe, and (MesPI)2Fe(isoprene). (CIF) This metrical parameters and CIFs can also be obtained free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk) under identifiers CCDC 1862087, 1862086, 1877016, 1862088, 1862089, and 1877015.
Experimental details; characterization data including NMR spectra of novel compounds; Mößbauer spectroscopic data; kinetic data; computational methods and results (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Hendrickson JB Molecular Geometry. IV. The Medium Rings. J. Am. Chem. Soc 1964, 86, 4854–4866. [Google Scholar]; (b) Hendrickson JB Molecular Geometry. V. Evaluation of Functions and Conformations of Medium Rings. J. Am. Chem. Soc 1967, 89, 7036–7043. [Google Scholar]
- 2.(a) Martinez H; Ren N; Matta ME; Hillmyer MA Ring-Opening Metathesis Polymerization of 8-Membered Cyclic Olefins. Polym. Chem 2014, 5, 3507–3532. [Google Scholar]; (b) Hill AR; Balogh J; Moncho S; Su H-L; Tuba R; Brothers EN; Al-Hashimi M; Bazzi HS Ring Opening Metathesis Polymerization (ROMP) of Five- to Eight-Membered Cyclic Olefins: Computational, Thermodynamic, and Experimental Approach. J. Polym. Sci. Part A Polym. Chem 2017, 55, 3137–3145. [Google Scholar]
- 3.(a) McAuliffe JC; Paramonov SE; Sanford KJ Fuel Compositions Comprising Isoprene Derivatives WO 2010/148256 Al, 2010.; (b) Meylemans HA; Quintana RL; Harvey BG Efficient Conversion of Pure and Mixed Terpene Feedstocks to High Density Fuels. Fuel 2012, 97, 560–568. [Google Scholar]; (c) Meylemans HA; Baldwin LC; Harvey BG Low-Temperature Properties of Renewable High-Density Fuel Blends. Energy & Fuels, 2013, 27, 883–888. [Google Scholar]; (d) Harvey BG; Merriman WW; Koontz TA High-Density Renewable Diesel and Jet Fuels Prepared from Multicyclic Sesquiterpanes and a 1-Hexene-Derived Synthetic Paraffinic Kerosene. Energy & Fuels 2015, 29, 2431–2436. [Google Scholar]; (e) Leitner W; Klankermayer J; Pischinger S; Pitsch H; Kohse-Höinghaus K Advanced Biofuels and Beyond: Chemistry Solutions for Propulsion and Production. Angew. Chem. Int. Ed 2017, 56, 5412–5452. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kraft P; Bajgrowicz JA; Denis C; Fráter G Odds and Trends: Recent Developments in the Chemistry of Odorants. Angew. Chem. Int. Ed 2000, 39, 2980–3010. [DOI] [PubMed] [Google Scholar]; (b) Birkbeck AA Challenges in the Synthesis of Natural and Non-Natural Volatiles. In The Chemistry and Biology of Volatiles; Herrmann A, Ed.; John Wiley & Sons, Ltd, 2010; pp 173–193. [Google Scholar]
- 5.(a) Vesley JA; Massie SN Acetal Derivatives of Cyclooctyl Carboxaldehydes US 3,985,769, 1976.; (b) Markert T Utilization of Monocyclic Aldehydes as Odoriferous Agents WO 99/54430 A1, 1998.; (c) Fráter G; Bajgrowicz JA; Kraft P Fragrance Chemistry. Tetrahedron, 1998, 54, 7633–7703. [Google Scholar]; (d) Granier T; Bajgrowicz JA; Hanhart A Cyclooct-(En)-Yl Derivatives for Use as Fragrances US 7,888,309 B2, 2011.
- 6.(a) Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]; (b) Aldeghi M; Malhotra S; Selwood DL; Chan AWE Two- and Three-Dimensional Rings in Drugs. Chem. Biol. Drug Des 2014, 83, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bauer RA; Wenderski TA; Tan DS Biomimetic Diversity-Oriented Synthesis of Benzannulated Medium Rings via Ring Expansion. Nat. Chem. Biol 2012, 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Yet L Metal-Mediated Synthesis of Medium-Sized Rings. Chem. Rev 2000, 100, 2963–3008. [DOI] [PubMed] [Google Scholar]; (b) Yu Z-X; Wang Y; Wang Y Transition-Metal-Catalyzed Cycloadditions for the Synthesis of Eight-Membered Carbocycles. Chem. Asian J 2010, 5, 1072–1088. [DOI] [PubMed] [Google Scholar]
- 8.(a) Blanchard N; Eustache J Synthesis of Natural Products Containing Medium-Size Carbocycles by Ring-Closing Alkene Metathesis. In Metathesis in Natural Product Synthesis; Cossy J, Arseniyadis S, Meyer C, Eds.; John Wiley & Sons, Ltd, 2010; pp 1–43. [Google Scholar]; (b) Yet L Olefin Ring-Closing Metathesis. In Organic Reactions; Wiley, 2016; pp 1–1304. [Google Scholar]; (c) Fürstner A Catalysis for Total Synthesis: A Personal Account. Angew. Chem. Int. Ed 2014, 53, 8587–8598. [DOI] [PubMed] [Google Scholar]
- 9.(a) Brill ZG; Grover HK; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science, 2016, 352, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Farney EP; Feng SS; Schäfers F; Reisman SE Total Synthesis of (+)-Pleuromutilin. J. Am. Chem. Soc 2018, 140, 1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Oenbrink G; Schiffer T Cyclododecatriene, Cyclooctadiene, and 4-Vinylcyclohexene. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2009; pp 37–40. [Google Scholar]; (b) Reed HWB The catalytic cyclic polymerization of butadiene. J. Chem. Soc 1954, 1931–1941.; (c) Brenner W; Heimbach P; Hey H; Müller EW; Wilke G Liebigs Ann. Chem 1969, 727, 161–182. [Google Scholar]
- 11.(a) tom Dieck H; Dietrich J Selectivity and Mechanism of Diene Cyclodimerization on Iron(0) Complexes. Angew. Chem. Int. Ed 1985, 24, 781–783. [Google Scholar]; (b) Lee H; Campbell MG; Hernández Sánchez R; Börgel J; Raynaud J; Parker SE; Ritter T Mechanistic Insight Into High-Spin Iron(I)-Catalyzed Butadiene Dimerization. Organometallics, 2016, 35, 2923–2929. [Google Scholar]
- 12.(a) Schmidt R; Griesbaum K; Behr A; Biedenkapp D; Voges H-W; Garbe D; Paetz C; Collin G; Mayer D; Höke H Hydrocarbons. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH, 2014; pp 1–74. [Google Scholar]; (b) Behr A; Johnen L Myrcene as a Natural Base Chemical in Sustainable Chemistry: A Critical Review. Chem. Sus. Chem 2009, 2, 1072–1095. [DOI] [PubMed] [Google Scholar]; (c) Morais ARC; Dworakowska S; Reis A; Gouveia L; Matos CT; Bogdał D; Bogel-Łukasik R Chemical and Biological-Based Isoprene Production: Green Metrics. Catal. Today, 2015, 239, 38–43. [Google Scholar]; (d) Sun R; Zheng M; Li X; Pang J; Wang A; Wang X; Zhang T Production of Renewable 1,3-Pentadiene from Xylitol via Formic Acid-Mediated Deoxydehydration and Palladium-Catalyzed Deoxygenation Reactions. Green Chem 2017, 19, 638–642. [Google Scholar]
- 13.Diver ST; Giessert AJ Enyne Metathesis (Enyne Bond Reorganization). Chem. Rev 2004, 104, 1317–1382. [DOI] [PubMed] [Google Scholar]
- 14.(a) Nunomoto S; Kawakami Y; Yamashita Y Synthesis of 2-Substituted 1,3-Butadienes by Cross-Coupling Reaction of 2-(1,3-Butadienyl)Magnesium Chloride with Alkyl or Aryl Iodides. Bull. Chem. Soc. Jpn 1981, 54, 2831–2832. [Google Scholar]; (b) Sahlberg C; Quader A; Claesson A Synthesis of Conjugated Dienes by Nickel-Catalyzed Reactions of 1,3-Alkadien-2-yl Phosphates with Grignard Reagents. Tetrahedron Lett 1983, 24, 5137–5138. [Google Scholar]; (c) Karlström ASE; Rönn M; Thorarensen A; Bäckvall J-E A Versatile Route to 2-Substituted Cyclic 1,3-Dienes via a Copper(I)-Catalyzed Cross-Coupling Reaction of Dienyl Triflates with Grignard Reagents. J. Org. Chem 1998, 63 , 2517–2522. [DOI] [PubMed] [Google Scholar]; (d) Karlström ASE; Itami K; Bäckvall J-E Nickel-Catalyzed Cross-Coupling of Dienyl Phosphates with Grignard Reagents in the Synthesis of 2-Substituted 1,3-Dienes. J. Org. Chem 1999, 64, 1745–1749. [DOI] [PubMed] [Google Scholar]; (e) Cahiez G; Habiak V; Gager O Efficient Preparation of Terminal Conjugated Dienes by Coupling of Dienol Phosphates with Grignard Reagents under Iron Catalysis. Org. Lett 2008, 10, 2389–2392. [DOI] [PubMed] [Google Scholar]; (f) Wang G; Mohan S; Negishi E Highly Selective Synthesis of Conjugated Dienoic and Trienoic Esters via Alkyne Elementometalation–Pd-Catalyzed Cross-Coupling. Proc. Natl. Acad. Sci. USA, 2011, 108, 11344–11349. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Fiorito D; Folliet S; Liu Y; Mazet C A General Nickel-Catalyzed Kumada Vinylation for the Preparation of 2-Substituted 1,3-Dienes. ACS Catal 2018, 8, 1392–1398. [Google Scholar]; (h) Olivares AM; Weix DJ Multimetallic Ni- and Pd-Catalyzed Cross-Electrophile Coupling To Form Highly Substituted 1,3-Dienes. J. Am. Chem. Soc 2018, 140, 2446–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Nguyen VT; Dang HT; Pham HH; Nguyen VD; Flores-Hansen C; Arman HD; Larionov OV Highly Regio- and Stereoselective Catalytic Synthesis of Conjugated Dienes and Polyenes. J. Am. Chem. Soc 2018, 140, 8434–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Hansen AL; Skrydstrup T Regioselective Heck Couplings of α,β-Unsaturated Tosylates and Mesylates with Electron-Rich Olefins. Org. Lett 2005, 7, 5585–5587. [DOI] [PubMed] [Google Scholar]; (b) Hansen AL; Ebran J-P; Ahlquist M; Norrby P-O; Skrydstrup T Heck Coupling with Nonactivated Alkenyl Tosylates and Phosphates: Examples of Effective 1,2-Migrations of the Alkenyl Palladium(II) Intermediates. Angew. Chem. Int. Ed 2006, 45, 3349–3353. [DOI] [PubMed] [Google Scholar]; (c) Zhou P; Jiang H; Huang L; Li X Acetoxypalladation of Unactivated Alkynes and Capture with Alkenes to Give 1-Acetoxy-1,3-Dienes Taking Dioxygen as Terminal Oxidant. Chem. Commun 2011, 47, 1003–1005. [DOI] [PubMed] [Google Scholar]; (d) Zheng C; Wang D; Stahl SS Catalyst-Controlled Regioselectivity in the Synthesis of Branched Conjugated Dienes via Aerobic Oxidative Heck Reactions. J. Am. Chem. Soc 2012, 134, 16496–16499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Delcamp JH; Gormisky PE; White M, Oxidative C Heck Vinylation for the Synthesis of Complex Dienes and Polyenes. J. Am. Chem. Soc 2013, 135, 8460–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Molloy JJ; Seath CP; West MJ; McLaughlin C; Fazakerley NJ; Kennedy AR; Nelson DJ; Watson AJB Interrogating Pd(II) Anion Metathesis Using a Bifunctional Chemical Probe: A Transmetalation Switch. J. Am. Chem. Soc 2018, 140, 126–130. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ideses R; Shani A The Wittig Reaction: Comments on the Mechanism and Application as a Tool in the Synthesis of Conjugated Dienes. Tetrahedron 1989, 45, 3523–3534. [Google Scholar]; (b) Dong D-J; Li H-H; Tian S-K A Highly Tunable Stereoselective Olefination of Semistabilized Triphenylphosphonium Ylides with N-Sulfonyl Imines. J. Am. Chem. Soc 2010, 132, 5018–5020. [DOI] [PubMed] [Google Scholar]
- 17.McAlpine NJ; Wang L; Carrow BP A Diverted Aerobic Heck Reaction Enables Selective 1,3-Diene and 1,3,5-Triene Synthesis through C–C Bond Scission. J. Am. Chem. Soc 2018, 140, 13634–13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Wender PA; Ihle NC Nickel-Catalyzed Intramolecular [4+4]-Cycloadditions: A New Method for the Synthesis of Polycycles Containing Eight-Membered Rings. J. Am. Chem. Soc 1986, 108, 4678–4679. [Google Scholar]; (b) Wender PA; Snapper ML Intramolecular Nickel Catalyzed Cycloadditions of Bis-Dienes: 3 Approaches to the Taxane Skeleton. Tetrahedron Lett 1987, 28, 2221–2224. [Google Scholar]; (c) Wender PA; Ihle NC Nickel-Catalyzed Intramolecular [4+4] Cycloadditions: 2. Allylic Stereoinduction and Modelling Studies in the Preparation of Bicyclo[6.4.0]Dodecadienes. Tetrahedron Lett 1987, 28, 2451–2454. [Google Scholar]; (d) Wender PA; Ihle NC; Correia CRD Nickel-Catalyzed Intramolecular [4+4] Cycloadditions. 4. Enantioselective Total Synthesis of (+)-Asteriscanolide. J. Am. Chem. Soc 1988, 110, 5904–5906. [Google Scholar]; (e) Wender PA; Tebbe MJ Nickel(0)-Catalyzed Intramolecular [4+4] Cycloadditions: 5. The Type II Reaction in the Synthesis of Bicyclo[5.3.1]Undecadienes. Synthesis 1991, 1089–1094.; (f) Wender PA; Nuss JM; Smith DB; Suárez-Sobrino A; Vågberg J; Decosta D; Bordner J Transition Metal Catalyzed Cycloadditions: An Intramolecular [4 + 4] Cycloaddition Strategy for the Efficient Synthesis of Dicyclopenta[a,d]Cyclooctene 5−8−5 Ring Systems. J. Org. Chem 1997, 62, 4908–4909. [Google Scholar]
- 19.(a) van Leeuwen PWNM; Roobeek CF On the Mechanism of the Nickel-Catalysed Regioselective Cyclodimerization of Isoprene. Tetrahedron, 1981, 37, 1973–1983. [Google Scholar]; (b) Jolly PW Nickel Catalyzed Oligomerization of 1,3-Dienes and Related Reactions. In Comprehensive Organometallic Chemistry; Wilkinson G, Stone FGA, Abel EW, Eds.; Pergamon: Oxford, 1982; pp 671–711. [Google Scholar]; (c) Brun P; Tenaglia A; Waegell B Regio and Stereo Selective Synthesis of Disubstituted Cyclooctadienes. Tetrahedron Lett 1983, 24, 385–388. [Google Scholar]; (d) Tenaglia A; Brun P; Waegell B Nickel-Catalyzed Oligomerization of Functionalized Conjugated Dienes. J. Organomet. Chem 1985, 285, 343–357. [Google Scholar]
- 20.(a) Mallien M; Haupt ETK; tom Dieck H Catalytic Isomerization of Methylated 1,5-Cyclooctadienes. Angew. Chem. Int. Ed 1988, 27, 1062–1064. [Google Scholar]; (b) Wu C-Y; Swift HE Diolefin Reactions Catalyzed by Transition Metal-Schiff Base Complexes. J. Catal 1972, 24, 510–520. [Google Scholar]
- 21. Mechanistic study of the iron-catalyzed [4+4]-cyclodimerization of butadiene was described recently, implicating (MeDI)Fe(COD) as the on-cycle resting state complex (see ref 11b). However, given the discrepancy between selectivities and rates reported for the [4+4]-cycloaddition of substituted 1,3-dienes relative to butadiene, it was not clear at the outset of this study whether these insights would be transferrable.
- 22.(a) Small BL Discovery and Development of Pyridine-Bis(Imine) and Related Catalysts for Olefin Polymerization and Oligomerization. Acc. Chem. Res 2015, 48, 2599–2611. [DOI] [PubMed] [Google Scholar]; (b) Chirik PJ Carbon–Carbon Bond Formation in a Weak Ligand Field: Leveraging Open-Shell First-Row Transition-Metal Catalysts. Angew. Chem. Int. Ed 2017, 56 , 5170–5181. [DOI] [PubMed] [Google Scholar]
- 23.(a) Bouwkamp MW; Bowman AC; Lobkovsky E; Chirik PJ Iron-Catalyzed [2π + 2π] Cycloaddition of α,ω-Dienes: The Importance of Redox-Active Supporting Ligands. J. Am. Chem. Soc 2006, 128, 13340–13341. [DOI] [PubMed] [Google Scholar]; (b) Hoyt JM; Schmidt VA; Tondreau AM; Chirik PJ Iron-Catalyzed Intermolecular [2+2] Cycloadditions of Unactivated Alkenes. Science, 2015, 349, 960–963. [DOI] [PubMed] [Google Scholar]
- 24.Russell SK; Lobkovsky E; Chirik PJ Iron-Catalyzed Intermolecular [2π + 2π] Cycloaddition. J. Am. Chem. Soc 2011, 133, 8858–8861. [DOI] [PubMed] [Google Scholar]
- 25.Hoyt JM; Sylvester KT; Semproni SP; Chirik PJ Synthesis and Electronic Structure of Bis(Imino)Pyridine Iron Metallacyclic Intermediates in Iron-Catalyzed Cyclization Reactions. J. Am. Chem. Soc 2013, 135, 4862–4877. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt VA; Kennedy CR; Bezdek MJ; Chirik PJ Selective [1,4]-Hydrovinylation of 1,3-Dienes with Unactivated Olefins Enabled by Iron Diimine Catalysts. J. Am. Chem. Soc 2018, 140, 3443–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Chirik PJ Preface: Forum on Redox-Active Ligands. Inorg. Chem 2011, 50, 9737–9740. [DOI] [PubMed] [Google Scholar]; (b) Lyaskovskyy V; de Bruin B Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal 2012, 2, 270–279. [Google Scholar]; (c) Luca OR; Crabtree RH Redox-Active Ligands in Catalysis. Chem. Soc. Rev 2013, 42, 1440–1459. [DOI] [PubMed] [Google Scholar]
- 28.(a) Lau W; Huffman J; Kochi J Electrochemical oxidation-reduction of organometallic complexes. Effect of the oxidation state on the pathways for reductive elimination of dialkyliron complexes. Organometallics, 1982, 1, 155–169. [Google Scholar]; (b) Joannou M; Darmon J; Bezdek M; Chirik P Exploring C(sp3)–C(sp3) Reductive Elimination from an Isolable Iron Metallacycle. Polyhedron, 2018, 159, 308–317. [Google Scholar]
- 29.Bart SC; Hawrelak EJ; Lobkovsky E; Chirik PJ Low-Valent α-Diimine Iron Complexes for Catalytic Olefin Hydrogenation. Organometallics, 2005, 24, 5518–5527. [Google Scholar]
- 30.McNeill E; Ritter T 1,4-Functionalization of 1,3-Dienes With Low-Valent Iron Catalysts. Acc. Chem. Res 2015, 48, 2330–2343. [DOI] [PubMed] [Google Scholar]
- 31. Grignard reagents, including Mg(C4H6)•2THF, were reported previously as competent reductants/activators for the iron precatalysts employed for the [4+4]-cyclodimerization of butadiene. See Ref. 11.
- 32.Trovitch RJ; Lobkovsky E; Bouwkamp MW; Chirik PJ Carbon−Oxygen Bond Cleavage by Bis(imino)pyridine Iron Compounds: Catalyst Deactivation Pathways and Observation of Acyl C−O Bond Cleavage in Esters. Organometallics, 2008, 27, 6264–6278. [Google Scholar]
- 33.Hirano M Recent Advances in the Catalytic Linear Cross-Dimerizations. ACS Catal 2019, 9, 1408–1430. [Google Scholar]
- 34.(a) Franke R; Selent D; Börner A Applied Hydroformylation. Chem. Rev 2012, 112, 5675–5732. [DOI] [PubMed] [Google Scholar]; (b) Shen X; Gong H; Zhou Y; Zhao Y; Lin J; Chen M Unsymmetrical Difunctionalization of Cyclooctadiene under Continuous Flow Conditions: Expanding the Scope of Ring Opening Metathesis Polymerization. Chem. Sci 2018, 9, 1846–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Bayeh L; Tambar UK Catalytic Asymmetric Intermolecular Allylic Functionalization of Unactivated Internal Alkenes. ACS Catal 2017, 7, 8533–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Andrus MB; Zhou Z Highly Enantioselective Copper−Bisoxazoline-Catalyzed Allylic Oxidation of Cyclic Olefins with tert-Butyl p-nitroperbenzoate. J. Am. Chem. Soc 2002, 124, 8806–8807. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi S; Pitet LM; Hillmyer MA Regio- and Stereoselective Ring-Opening Metathesis Polymerization of 3-Substituted Cyclooctenes. J. Am. Chem. Soc 2011, 133, 5794–5797. [DOI] [PubMed] [Google Scholar]; (d) Zang Z-L; Zhao S; Karnakanti S; Liu C-L; Shao P-L; He Y Catalytic Multisite-Selective Acetoxylation Reactions at sp2 vs sp3 C–H Bonds in Cyclic Olefins. Org. Lett 2016, 18, 5014–5017. [DOI] [PubMed] [Google Scholar]; (e) Bayeh L; Le PQ; Tambar UK Catalytic allylic oxidation of internal alkenes to a multifunctional chiral building block. Nature, 2017, 547, 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.(a) Baldenius K-U; tom Dieck H; König WA; Icheln D; Runge T Enantioselective Syntheses of Cyclopentanoid Compounds from Isoprene and Trans-1,3-Pentadiene. Angew. Chem. Int. Ed 1992, 31, 305–307. [Google Scholar]; (b) Tabushi I; Fujita K; Oda R Stereochemistry of Cationic Addition -π,π Transannular Cyclization of 1,5-Cyclooctadiene. Tetrahedron Lett 1967, 8, 3815–3819. [Google Scholar]
- 37.(a) Mang T; Noll S; Bartels T Lubricants, 1. Fundamentals of Lubricants and Lubrication. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2011; pp 383–411. [Google Scholar]; (b) Ray S; Rao PVC; Choudary NV Poly-α-Olefin-Based Synthetic Lubricants: A Short Review on Various Synthetic Routes. Lubr. Sci 2011, 24, 23–44. [Google Scholar]
- 38.(a) Muresan N; Lu C; Ghosh M; Peters J; Abe M; Henling L; Weyhermöller T; Bill E; Wieghardt K Bis(α-diimine)iron Complexes: Electronic Structure Determination by Spectroscopy and Broken Symmetry Density Functional Theoretical Calculations. Inorg. Chem 2008, 47, 4579–4590. [DOI] [PubMed] [Google Scholar]; (b) Lu CC; Bill E; Weyhermüller T; Bothe E; Wieghardt K Neutral Bis(α-Iminopyridine)Metal Complexes of the First-Row Transition Ions (Cr, Mn, Fe, Co, Ni, Zn) and Their Monocationic Analogues: Mixed Valency Involving a Redox Noninnocent Ligand System. J. Am. Chem. Soc 2008, 130, 3181–3197. [DOI] [PubMed] [Google Scholar]; (c) Khusniyarov M; Weyhermüller T; Bill E; Wieghardt K Tuning the Oxidation Level, the Spin State, and the Degree of Electron Delocalization in Homo- and Heteroleptic Bis(α-diimine)iron Complexes J. Am. Chem. Soc 2009, 131, 1208–1221. [DOI] [PubMed] [Google Scholar]; (d) Lu CC; Weyhermüller T; Bill E; Wieghardt K Accessing the Different Redox States of α-Iminopyridines within Cobalt Complexes. Inorg. Chem 2009, 48, 6055–6064. [DOI] [PubMed] [Google Scholar]
- 39.The τ4 parameter describes variations in the geometry of 4-coordinate complexes on a spectrum from square planar to tetrahedral: Yang L; Powell DR; Houser RP Structural Variation in Copper(I) Complexes with Pyridylmethylamide Ligands: Structural Analysis with a New Four-Coordinate Geometry Index, τ4. Dalt. Trans 2007, 955–964. [DOI] [PubMed]
- 40.Allen FH; Kennard O; Watson DG; Brammer L; Orpen AG; Taylor R Tables of Bond Lengths Determined by X-Ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans 2, 1987, S1–S19. [Google Scholar]
- 41.(a) Dewar MJS A Review of π Complex Theory. Bull. Soc. Chim. Fr 1951, 18, 71–79. [Google Scholar]; (b) Chatt J; Duncanson LA 586. Olefin Co-Ordination Compounds. Part III. Infra-Red Spectra and Structure: Attempted Preparation of Acetylene Complexes. J. Chem. Soc 1953, 2939–2947.
- 42.(a) Bart SC; Chłopek K; Bill E; Bouwkamp MW; Lobkovsky E; Neese F; Wieghardt K; Chirik PJ Electronic Structure of Bis(Imino)Pyridine Iron Dichloride, Monochloride, and Neutral Ligand Complexes: A Combined Structural, Spectroscopic, and Computational Study. J. Am. Chem. Soc 2006, 128, 13901–13912. [DOI] [PubMed] [Google Scholar]; (b) Stieber SCE; Milsmann C; Hoyt JM; Turner ZR; Finkelstein KD; Wieghardt K; DeBeer S; Chirik PJ Bis(Imino)Pyridine Iron Dinitrogen Compounds Revisited: Differences in Electronic Structure Between Four- and Five-Coordinate Derivatives. Inorg. Chem 2012, 51, 3770–3785. [DOI] [PubMed] [Google Scholar]
- 43.Gütlich P; Bill E; Trautwein AX Hyperfine Interactions. In Mössbauer Spectroscopy and Transition Metal Chemistry; Springer-Verlag: Berlin Heidelberg, 2011; pp 73–135. [Google Scholar]
- 44. Calculations for (MePI)Fe(COD) initiated following truncation of the isopropyl groups from the solid-state structure of (iPrPI)Fe(COD).
- 45. Calculations for (MePI)Fe(MviMvi) initiated from a solid-state structure that revealed connectivity but was of insufficient quality for publication.
- 46.(a) Neese F The ORCA Program System. WIREs Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]; (b) Neese F Software Update: The ORCA Program System, Version 4.0. WIREs Comput Mol Sci 2018, 8, e1327. [Google Scholar]
- 47.(a) Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A, 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]; (b) Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B, 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 48.Pople JA; Gill PMW; Handy NC Spin-Unrestricted Character of Kohn-Sham Orbitals for Open-Shell Systems. Int. J. Quantum Chem 1995, 56, 303–305. [Google Scholar]
- 49.(a) Ginsberg AP Magnetic Exchange in Transition Metal Complexes. 12. Calculation of Cluster Exchange Coupling Constants with the Χα-Scattered Wave Method. J. Am. Chem. Soc 1980, 102, 111–117. [Google Scholar]; (b) Noodleman L Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimers. J. Chem. Phys 1981, 74, 5737–5743. [Google Scholar]; (c) Noodleman L; Peng CY; Case DA; Mouesca J-M Orbital Interactions, Electron Delocalization and Spin Coupling in Iron-Sulfur Clusters. Coord. Chem. Rev 1995, 144, 199–244. [Google Scholar]; (d) Neese F Definition of Corresponding Orbitals and the Diradical Character in Broken Symmetry DFT Calculations on Spin Coupled Systems. J. Phys. Chem. Solids, 2004, 65, 781–785. [Google Scholar]
- 50.(a) Neese F Prediction and Interpretation of the 57Fe Isomer Shift in Mössbauer Spectra by Density Functional Theory. Inorg. Chim. Acta 2002, 337, 181–192. [Google Scholar]; (b) Sinnecker S; Slep LD; Bill E; Neese F Performance of Nonrelativistic and Quasi-Relativistic Hybrid DFT for the Prediction of Electric and Magnetic Hyperfine Parameters in 57Fe Mössbauer Spectra. Inorg. Chem 2005, 44, 2245–2254. [DOI] [PubMed] [Google Scholar]; (c) Neese F A Critical Evaluation of DFT, Including Time-Dependent DFT, Applied to Bioinorganic Chemistry. J. Biol. Inorg. Chem 2006, 11, 702–711. [DOI] [PubMed] [Google Scholar]; (d) Römelt M; Ye S; Neese F Calibration of Modern Density Functional Theory Methods for the Prediction of 57Fe Mössbauer Isomer Shifts: Meta-GGA and Double-Hybrid Functionals. Inorg. Chem 2009, 48, 784–785. [DOI] [PubMed] [Google Scholar]; (e) Bjornsson R; Neese F; DeBeer S Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem 2017, 56, 1470–1477 [DOI] [PubMed] [Google Scholar]; (f) McWilliams SF; Brennan-Wydra E; MacLeod KC; Holland PL Density Functional Calculations for Prediction of 57Fe Mössbauer Isomer Shifts and Quadrupole Splittings in β-Diketiminate Complexes. ACS Omega, 2017, 2, 2594–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.(a) Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]; (b) Weigend F Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 52.(a) van Wüllen C Molecular Density Functional Calculations in the Regular Relativistic Approximation: Method, Application to Coinage Metal Diatomics, Hydrides, Fluorides and Chlorides, and Comparison with First-Order Relativistic Calculations. J. Chem. Phys 1998, 109, 392–399. [Google Scholar]; (b) Pantazis DA; Chen X-Y; Landis CR; Neese F All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput 2008, 4, 908–919. [DOI] [PubMed] [Google Scholar]
- 53.(a) Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem. Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- 54. Qualitatively similar results were obtained using [(MePI)Fe(COD)]. Results obtained using precatalyst [(MePI)Fe(MviMvi)] shown due to the improved precision obtained through the use of a uniform precatalyst stock solution.
- 55. For reactions conducted in neat substrate, the total reaction volume changes as a function of conversion by virtue of the differing densities of substrate and product. This was accounted for by assuming that the substrate/product mixture behaved as an ideal solution (see Supporting Information for details). The qualitative agreement between experiments conducted in neat substrate and dilute solution validates this assumption.
- 56.Singleton DA; Thomas AA High-Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc 1995, 117, 9357–9358. [Google Scholar]
- 57.(a) Coombs JR; Haeffner F; Kliman LT; Morken JP Scope and Mechanism of the Pt-Catalyzed Enantioselective Diboration of Monosubstituted Alkenes. J. Am. Chem. Soc 2013, 135, 11222–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang JY; Strom AE; Hartwig JF Mechanistic Studies of Palladium-Catalyzed Aminocarbonylation of Aryl Chlorides with Carbon Monoxide and Ammonia. J. Am. Chem. Soc 2018, 140, 7979–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wendlandt AE; Vangal P; Jacobsen EN Quaternary Stereocentres via an Enantioconvergent Catalytic SN1 Reaction. Nature, 2018, 556, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Grélaud S; Cooper P; Feron LJ; Bower JF Branch-Selective and Enantioselective Iridium-Catalyzed Alkene Hydroarylation via Anilide-Directed C–H Oxidative Addition. J. Am. Chem. Soc 2018, 140, 9351–9356. [DOI] [PubMed] [Google Scholar]
- 58.Hoops S; Sahle S; Gauges R; Lee C; Pahle J; Simus N; Singhal M; Xu L; Mendes P; Kummer U COPASI: a COmplex PAthway SImulator. Bioinformatics, 2006, 22, 3067–3074. [DOI] [PubMed] [Google Scholar]
- 59.Falivene L; Credendino R; Poater A; Petta A; Serra L; Oliva R; Scarano V; Cavallo L Organometallics, 2016, 35, 2286–2293. [Google Scholar]
- 60.Clavier H; Nolan SP Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun 2010, 46, 841–861. [DOI] [PubMed] [Google Scholar]
- 61. While the NMR spectroscopic data obtained for (MesDI)Fe(COD) at 25 °C in C6D6 were consistent with a C2v ligand, the solid-state structure exhibited slight canting of the flanking aryl rings, rendering the C2 description more accurate.
- 62.(a) Krenske EH; Houk. KN Aromatic Interactions as Control Elements in Stereoselective Organic Reactions. Acc. Chem. Res 2013, 46, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neel AJ; Hilton MJ; Sigman MS; Toste FD Exploiting non-covalent π interactions for catalyst design. Nature, 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.