

Abstract
A validated, scalable approach to characterizing (phenotyping) smoking status is needed to facilitate genetic discovery. Using established DNA methylation sites from blood samples as a criterion standard for smoking behavior, we compare three candidate electronic medical record (EMR) smoking metrics based on longitudinal EMR text notes. With data from the Veterans Aging Cohort Study (VACS), we employed a validated algorithm to translate each smoking-related text note into current, past, or never categories. We compared three alternative summary characterizations of smoking: most recent, modal, and trajectories using descriptive statistics and Spearman’s correlation coefficients. Logistic regression and area under the curve analyses were used to compare the associations of these phenotypes with the DNA methylation sites, cg05575921 and cg03636183, which are known to have strong associations with current smoking. DNA methylation data were available from the VACS Biomarker Cohort (VACS-BC), a sub-study of VACS. We also considered whether the associations differed by the certainty of trajectory group assignment (<0.80/≥0.80). Among 140,152 VACS participants, EMR summary smoking phenotypes varied in frequency by the metric chosen: current from 33–53%; past from 16–24%; and never from 24%–33%. The association between the EMR smoking pairs was highest for modal and trajectories (rho=0.89). Among 728 individuals in the VACS-BC, both DNA methylation sites were associated with all three EMR summary metrics (p<0.001), but the strongest association with both methylation sites was observed for trajectories (p<0.001). Longitudinal EMR smoking data support using a summary phenotype, the validity of which is enhanced when data are integrated into statistical trajectories.
keywords/phrases: EMR smoking, smoking methylation, DNA methylation
INTRODUCTION
Twin and family-based studies show that approximately 50% of the risk of tobacco dependence is heritable (Goldman et al. 2005). However, the numerous genetic variants that have been linked to smoking behavior explain only a small proportion of the phenotypic variation. A major challenge for gene discovery is phenotypic ambiguity, especially that stemming from self-reported health behaviors such as smoking in which social desirability bias and/or lack of documentation in the health record may obscure actual behavior (Kinsinger et al. 2017). Additional inaccuracy can result from cross-sectional rather than longitudinal approaches to characterizing behavioral phenotypes. Both factors can contribute to inadequate statistical power to detect small individual genetic effects that characterize complex disorders like smoking.
Several biological markers could be employed to validate self-reported smoking data. Cotinine, a major metabolite of nicotine in tobacco smoke, is the most commonly used biomarker. However, cotinine is elevated from smoking for only days to weeks (Jarvis et al. 1998). Expired carbon monoxide (CO), although sensitive to very recent smoking, lasts for only up to 8 hours (Sandberg, et al. 2011).
DNA methylation is an epigenetic process in which a methyl group is added to a DNA molecule. Environmental factors, such as exposure to cigarette smoke, can result in epigenetic changes (Bollati and Baccarelli 2010), and these changes can serve as biomarkers for long-term environmental exposure (Ladd-Acosta 2015). Epigenome-wide association studies have identified hundreds of DNA methylation sites, referred to as CpG or cg sites, associated with smoking. (Breitling et al. 2011; Harlid et al. 2014; Gao et al. 2015; Ambatipudi et al. 2016, Joehanes et al. 2016). The two sites most frequently associated with smoking are methylation of cg05575921 in the AHRR gene and cg03636183 in the F2RL3 gene (Gao et al. 2015; Ambatipudi, et al. 2016; Lee et al. 2016), and these two CpG sites have been proposed as candidate biomarkers to determine smoking status (Philibert et al. 2013). These sites have been consistently linked to smoking status in multiple population groups (Elliott et al. 2014, Guida et al. 2015, Beach et al. 2017), among both sexes (Dogan et al. 2014), across a wide range of ages (Beach et al. 2015) and different tissues (Stueve et al. 2017), and across different methylation platforms (Breitling et al. 2011, Joehanes et al. 2016).
The performance of cg05575921 as a predictor of smoking had an area under the curve of 0.99 (Philibert et al. 2015) in a small study. A recent study in two independent samples showed that cg05575921 is a robust indicator for smoking, as confirmed by serum cotinine concentrations (Andersen et al. 2017). Methylation at cg05575921 and cg03636183 along with other CpG sites has also been shown to predict lung cancer risk (Baglietto et al. 2017) and lung cancer incidence with an area under of curve (AUC) of approximately 0.8 (Zhang et al. 2016).
Smoking-associated methylation persists after smoking cessation (Joehanes et al. 2016). Methylation at cg05575921 takes approximately 10 years (Fasanelli et al. 2015) and methylation at cg03636183 takes approximately 20 years (Zhang et al. 2014) after quitting smoking to return to a level similar to that of never smokers. Among Framingham Heart Study participants, cg03636183 was one of the top 36 most statistically significant methylation sites that did not return to never-smoker levels, even 30 years after smoking cessation (Joehanes et al. 2016). The recovery of methylation at cg05575921 has been suggested as a quantitative marker for smoking cessation (Philibert et al. 2016) and cigarette consumption (Philibert et al. 2018). These findings suggest that DNA methylation changes at cg05575291 and cg03636183 can be reliably detected and have long-term stability as measures of smoking exposure.
The Veterans Health Administration (VHA) benefits from one of the most highly developed health information systems in the world (Corrigan et al. 2002; McQueen et al. 2004). Smoking data from the VHA electronic medical record (EMR) date back to 2000 and have been previously validated against two sources of survey data (McGinnis et al. 2011). An emerging source of large genome-wide association studies (GWAS) data is the Million Veteran Program (MVP) of the Department of Veterans Affairs, which is in the process of obtaining phenotypic and genomic data. One goal is to identify genetic variation contributing to nicotine dependence risk. Thus, the phenotypic refinement effort described here has important implications for achieving this aim. Our goal was to use EMR data to develop and validate a longitudinal phenotype for smoking behavior that could be used to identify novel genetic variants in GWAS. More specifically, we aimed to determine which EMR summary smoking metric (most recent, modal, or trajectory) was most strongly associated with DNA methylation markers (cg05575921 and cg03636183).
MATERIALS AND METHODS
Subjects
The Veterans Aging Cohort Study (VACS) is a large observational cohort study consisting of data from the national VA EMR that includes all HIV-infected (HIV+) patients (over 53,000) in VA care from October 1996 to September 2015 and more than 111,000 uninfected patients matched on region, age, race/ethnicity, and gender. VACS is described in detail elsewhere (Fultz et al. 2006; Justice et al. 2006). The VACS Biomarker Cohort (VACS-BC) is a subsample of VACS that includes 1525 HIV+ and 843 uninfected individuals who provided a blood sample from 2005 to 2007. The VACS-BC is described in detail elsewhere (Armah et al. 2012; Freiberg et al. 2016; Justice et al. 2012). We analyzed genomic DNA data on a subset of 728 of the VACS-BC participants.
Measures
EMR Smoking Data/Phenotype Determination
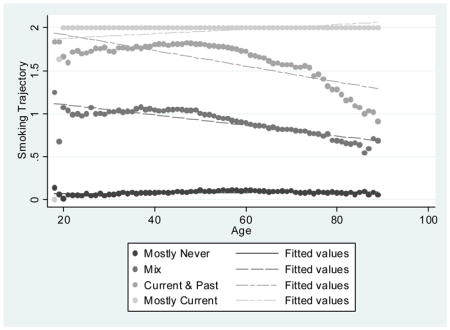
Smoking data were obtained from the VHA Corporate Data Warehouse (CDW). Details on the extraction methods are provided elsewhere (McGinnis et al. 2011). In brief, EMR smoking data are collected from patients nationally and on a yearly basis using the clinical reminder process. EMR smoking data consist of text values that represent responses to specific smoking-related questions asked of patients. These questions can vary by site and over time. Mapping strategies have been created to classify these responses into “never,” “past,” and “current” smoking and can be found on www.vacohort.org (McGinnis et al. 2011). For these analyses we coded never as 0, past as 1, and current as 2. Using all available EMR smoking observations from 2000 to 2015, we created the following smoking phenotypes 1) the most recent time point available (never, past, current); 2) the modal value – the most common value of all data available (never, past, current); and longitudinal smoking trajectory. When there was more than one smoking entry for any year of age, we used the highest smoking value for that year, considering current as highest and never as lowest. Joint trajectory modeling sorts each participant’s smoking values (never, past, current) into “clusters” and estimates distinct trajectories (Marshall et al. 2015). We used age as the time scale to account for possible decreases in smoking with age. The procedure calculates each individual’s probability of belonging to each trajectory and assigns the individual to the trajectory with the highest probability of membership. We used a censored normal model (Jones et al. 2012; Nagin 2005) and evaluated 3-, 4-, and 5-group models and first, second, and third order terms. For maximum precision, trajectories were developed in the full VACS sample. Smoking trajectory fit, as measured by the Bayesian information criterion, improved substantially when increasing from 3 to 4 and 4 to 5 groups. In the 5-group model, one of the groups had a mean probability of group membership of 0.69. In contrast, in the 4-group model with second order terms, the mean probabilities of group membership for the four trajectory groups ranged from 0.81 to 0.99 using second order terms and the smallest trajectory group contained 23.2% of the sample. The 4-group model using third order terms performed similarly in terms of probabilities of group membership and Bayesian information criterion but the groups were less equally distributed; therefore, the four group trajectory model with second order terms was used (Appendix). The smoking trajectory groups were designated as mostly never (0), mix (1), current & past (2), and mostly current (3).
DNA Methylation
Genomic DNA was extracted from whole blood and DNA methylation was profiled using an Infinium Illumina HumanMethylation 450K Beadchip (Illumina, San Diego, CA, USA) at the Yale Center of Genomic Analysis. Array data are deposited in GSE100264. After data quality control and normalization, β values for two CpG sites, cg03636183 and cg05575921, were obtained for each sample and were applied in the subsequent analyses; β values ranged continuously from 0–1, representing the level of methylation at each site. Based on prior research, current smoking status was assigned for methylation β values for two cutoffs of <0.80 and <0.70 for cg05575921 (Philibert et al. 2015) and two cutoff β values of 0<.68 and <0.59 for cg03636183. Two cutoffs were used for each analysis to determine whether the results are dependent on a particular cutoff. These cutoffs were chosen based on the average methylation levels in smoking vs. non-smoking groups in other published studies (Fasanelli F et al. 2015; Gao et al. 2017; Philibert et al. 2015; Philibert et al. 2018; Zhang et al. 2016).
Analyses
Demographic characteristics were summarized for patients in VACS and those in the VACS-BC methylation subset. We compared the following EMR self-reported smoking metric pairs in the VACS data: most recent vs. modal, most recent vs. trajectory, and modal vs. trajectory using crosstab and Spearman’s correlation coefficient.
Among individuals in the VACS-BC with methylation marker data, we identified those with low methylation, which is indicative of current smoking (<0.80 or <.70 for cg05575921 and <0.68 or <0.59 for cg03636183, as reported above) for each EMR self-reported smoking metric. Chi-square tests were used to determine whether the methylation markers were statistically significantly associated with each self-reported EMR smoking metric. Analyses for cg03636183 were stratified by ancestry (African or European), because methylation of cg03636183 has been shown to vary by population (Dogan et al. 2015; Zhang et al. 2014).
To determine which EMR smoking metric was most strongly associated with the methylation markers, we generated logistic regression models and calculated corresponding area under the receiver operating characteristics curve (AUROC) and concordance statistics (C-statistics). Linear predictors were generated from each model to use with the roccomp command in Stata to test whether there were statistically significant differences between the AUROCs.
In a sensitivity analysis, we reran the analysis after limiting the sample to patients who had a probability of ≥0.80 for their assigned smoking trajectory category. A high probability reflects a higher percentage of smoking observations falling into their assigned smoking trajectory category, whereas a low probability indicates a lower percentage of smoking observations falling into their designated smoking trajectory category. We also ran the VACS-BC analysis including the smoking value that was closest in time to collection of the blood sample from which the methylation data were obtained. Because methylation of the AHRR gene reflects cannabis smoking, we re-ran the analysis of cg05575921 including only participants who on the confidential VACS survey reported using marijuana or hashish less than once a month or never. Analyses were run in Stata 14.2.
RESULTS
Of the 140,152 patients in VACS with available smoking data, the mean age was 47 years at enrollment, 31% were HIV+, 97% were male, 48% were African American, 40% were white, and 12% were Hispanic or other. Based on modal smoking, 53% were currently smoking, 18% smoked in the past, and 29% never smoked. Of the 728 participants in VACS-BC with methylation data, the mean age was 53 years at the time of the blood draw, 84% were HIV+, 97% were male, 81% were African American, 15% were white, and 4% were Hispanic or other. Based on modal smoking, 68% were currently smoking, 12% smoked in the past, and 20% never smoked.
There were differences in how VACS participants were classified using the three self-reported EMR smoking metrics and the agreement differed between each pair (Table 2). The correlations among the three EMR smoking phenotypes were high: 0.80 for most recent and modal, 0.85 for most recent and trajectory, and 0.89 for modal and trajectory.
Table 2.
Comparison of Self-Reported EMR Smoking Metrics in VACS (n=142,152)
| Most Recent | |||
|---|---|---|---|
| Most Common | Never (0) | Past (1) | Current (2) |
| Never (0) | 37,098 | 2,824 | 1,222 |
| Past (1) | 4,690 | 19,648 | 1,036 |
| Current (2) | 5,407 | 11,067 | 59,160 |
| Correlation: 0.80 | |||
| Most Recent | |||
| Trajectories | Never (0) | Past (1) | Current (2) |
| Mostly Never (0) | 33,133 | 1,428 | 203 |
| Mix (1) | 10,917 | 21,959 | 4,228 |
| Current & Past (2) | 2,579 | 8,955 | 11,335 |
| Mostly Current (3) | 586 | 1,294 | 45,651 |
| Correlation: 0.85 | |||
| Most Common | |||
| Trajectories | Never (0) | Past (1) | Current (2) |
| Mostly Never (0) | 34,678 | 65 | 1 |
| Mix (1) | 6,348 | 23,845 | 6,911 |
| Current & Past (2) | 91 | 1,416 | 21,262 |
| Mostly Current (3) | 27 | 45 | 47,459 |
| Correlation: 0.89 | |||
EMR: Electronic medical record
In VACS-BC, all EMR smoking metrics were associated with both smoking methylation markers. The percentage of patients identified as currently smoking based on both methylation markers increased monotonically from never to past to current smoking for each self-reported EMR smoking metric (Figure 1). The gradient was steepest for the trajectory smoking metric for cg05575921. When we included only patients whose probability of trajectory membership was ≥0.80, we found similar patterns (Figure 1), with only a modest improvement in the association between the EMR smoking measures and methylation markers at the two sites.
Figure 1.



Figure 1a. Percent with Current Smoking Based on Cg05575921 for Three Self-Reported Smoking Phenotypes of All Participants (n=728) and Those with Trajectory Probability ≥0.80 (n=583)
Figure 1b. Percent with Current Smoking Based on Cg03636183 for Three Self-Reported Smoking Phenotypes of those with African Ancestry (n=551) and Limited to Those with Trajectory Probability ≥0.80 (n=440)
Figure 1c. Percent with Current Smoking Based on Cg03636183 for Three Self-Reported Smoking Phenotypes of Those with European Ancestry (n=84) and Limited to Those with Trajectory Probability ≥0.80 (n=64)
C-Statistic: Concordance Statistic
AUROC: area under receiver operating curve
EMR: electronic medical record
Agreement (reflected in higher C-statistics) was greater for cg05575921 than for cg03636183 for all EMR smoking metrics (Figure 2). Limiting the sample to patients with ≥0.80 probability of trajectory assignment resulted in higher C-statistics for all comparisons, but did not alter the relative comparisons among metrics. In all comparisons, the smoking trajectory metric had the highest C-statistics (from 0.67–0.89), followed by modal smoking (from 0.67–0.86), most recent smoking (0.64–0.80) and closest smoking (0.63–0.81). For cg05575921, the C-statistics were significantly higher for the trajectories than most recent (p<.001), closest (p<.001), and modal (p=.03) metrics and for modal compared to most recent (p=.002) and closest (p=.01). When we limited the analyses to patients with ≥0.80 probability of trajectory group membership, the differences with cg05575921 were significantly greater for trajectories than for most recent and closest (both p<.001), and for modal than for most recent (p<.001) and closest (p=.02). For cg03636183 among participants of African ancestry limited to those with ≥0.80 probability of trajectory group membership, C-statistics were significantly higher for trajectory than for modal and closest (p=.026 and .049). For cg03636183 among participants of European ancestry, C-statistics were significantly higher for trajectory than for most recent (p=.012) and modal (p=.05). The sample size was too small for comparisons of C-statistics among participants of European ancestry and with ≥0.80 probability of trajectory group membership. Including only the 567 participants who reported using marijuana or hashish less than monthly or never, cg05575921 results were similar to when all 728 were included.
Figure 2.



Figure 2a. Comparison of AUROC C-Statistics from Logistic Regression Models Using Three Phenotypes of EMR Self-Reported Smoking to Predict Current Smoking based on Cg05575921 Among All Participants (n=728) and Those with Trajectory Probability ≥0.80 (n=583)
Figure 2b Comparison of AUROC C-Statistics from Logistic Regression Models Using Three Phenotypes of EMR Self-Reported Smoking to Predict Current Smoking based on Cg03636183 Among All Those of African Ancestry (n=551) and Limited to Those with Trajectory Probability ≥0.80 (n=440)
Figure 2c. Comparison of AUROC C-Statistics from Logistic Regression Models Using Three Phenotypes of EMR Self-Reported Smoking to Predict Current Smoking based on Cg03636183 Among All Those of European Ancestry (n=84) and Limited to Those with Trajectory Probability ≥0.80 (n=64)
*cells too small to calculate c-statistics when limited to those with trajectory probability>0.80.
DISCUSSION
Although methylation-defined smoking status was statistically significantly associated with the single, recent, self-reported smoking status in EMR, there was a greater association of methylation-defined smoking status with longitudinal measures (either modal or trajectory). Using methylation as a criterion standard, regardless of methylation cutoff, we found that longitudinal EMR data provided a valid phenotype for smoking status and the validity was enhanced by the integration of longitudinal data into statistical trajectories compared to modal or most proximal phenotypes. In all cases, associations were strong for the methylation site cg05575921 and for cg03636183 for participants of European ancestry. In addition, associations were stronger for cg03636183 for participants of European ancestry than for those of African ancestry, as expected based on previous studies. These results were robust whether we considered only patients for whom the probability of trajectory group assignment was ≥0.80 or irrespective of the certainty of assignment.
This study both confirms and extends prior work on smoking and DNA methylation levels. This work is in line with prior studies demonstrating that cg05575921 and cg03636183 are associated with smoking status (Gao et al. 2015; Joehanes, et al. 2016) and both have been applied as biomarkers to define smoking status (current, past, and never) (Shenker et al. 2013), smoking intensity (quantity and duration) (Joehanes et al. 2016; Wilson et al. 2017), and smoking cessation (Philibert et al. 2016). To our knowledge, we are the first to use methylation data as a criterion standard for developing a longitudinal phenotype of smoking behavior for genetic discovery. Methylation data are amenable to this application for several reasons. First, they are not subject to the social desirability bias that can confound self-reported smoking status. Second, changes in methylation are only reversed following a long period of smoking abstinence (i.e., decades), so that associations with these biomarkers would be expected to demonstrate an increasing dose-response association between never, past, and current smoking, which we observed in our analyses. Third, the fact that we found similar associations for two separate methylation sites supports the validity of the results.
We demonstrated substantially stronger associations between the criterion standard (cg05575921 and cg0363183) and summary metrics of longitudinal, repeated self-report measures of smoking than with a single report (i.e., most recent assessment of smoking status). As we found in prior analyses of repeated longitudinal self-reported measures of alcohol consumption (Justice et al. 2017; Justice et al resubmitted 2018), summary measures of repeated self-reported smoking also demonstrate stronger associations than single, cross-sectional reports that are often employed as phenotypes in large-scale genetic studies (Sanchez-Roige et al. 2017). Like drinking behaviors, smoking behaviors among middle-aged individuals are typically stable with some decrement with advancing age. In this context, multiple observations, summarized over time and adjusted for age, can reduce the degree of “noise” in the measurement and provide improved phenotypic “signal”. In our prior analyses of self-reported alcohol consumption, an age-adjusted mean AUDIT-C score provided the best association with our criterion standard (the minor allele frequency for an ADH1B polymorphism that has been previously shown to be protective for alcohol use disorder) (Justice et al. in revision).
In the current analysis, statistical trajectories using age as the time scale were superior to the modal response. These findings suggest that it is both important to employ multiple measures of self-reported health behaviors over time and to adjust these for the age of the individual at the time of the report. Otherwise, an older individual’s drinking or smoking behaviors might be misclassified based upon a lower level of use at an advanced age, obscuring higher levels of use at younger ages.
It is also important to note that our findings were highly consistent whether the analyses were restricted to patients for whom the statistical trajectory assignments were most certain (trajectory probability ≥0.80 vs. <0.80). Patients for whom the trajectory assignments were most certain had more consistent self-reported smoking behavior (concordance = 52% vs 48% for all reports, p<.001) and/or had more reports (mean = 13.6 vs. 7.0), p<.001). The findings were also similar when we limited the sample to participants who reported using marijuana or hashish less than monthly or never. These findings underscore that our measure is robust to missing data and to multisubstance use.
There are limitations to this study. First, there are relatively few women in the study, potentially reducing generalizability, although sex has not been identified as a moderator of DNA methylation in smokers. In addition, these data lack granular assessments of the dose of cigarette exposure, such as smoking pack years, which have been shown to predict methylation (Joehanes et al. 2016). Similarly, knowing whether a smoker was daily or non-daily would also be important in the assessment of total dose, as non-daily smokers tend to have lower overall exposure (although exposure can still be substantial) (Shiffman et al. 2012). HIV infection results in the depletion of a particular T-cell subset and a shift of overall cell frequencies. To assess whether the reduction of T-cells in HIV+ impacts DNA methylation of cg0755921 and cg03636183 in our sample, we compared DNA methylation of these two loci between HIV+ and uninfected samples and found that they did not differ significantly (cg05575921: t=−1.69, p=0.09 and cg3636183: t=0.14, p=0.88), suggesting that DNA methylation of these loci are associated with smoking but not with HIV status. Finally, the VACS-BC does not contain biomarkers of nicotine metabolism, which is primarily driven by genetic variation in hepatic cytochrome P450 enzymes (Dempsey et al. 2004), and which has been found to influence DNA methylation (Loukola et al. 2015). Although biomarkers may be the most accurate way to identify smoking status, they are impractical on a large scale. We previously developed an algorithm to identify the smoking status of patients in VA care using text fields from EMR smoking data validated against self-reported confidentially collected research survey data (McGinnis et al. 2011). We are currently collecting cotinine concentrations in a separate study of patients and will be able to compare EMR smoking data to cotinine concentrations in future research.
As smoking status is often reported in text fields in non-VA systems as well, these methods can be used outside the VA, so that the findings are generalizable to other health systems. In addition, the VA is rapidly developing a cohort of a million veterans with genetic data in the Million Veterans Program (MVP) (Gaziano et al. 2016), and this study will be an important resource as a potential method to identify smoking status in the MVP.
In conclusion, we have demonstrated that longitudinal trajectories of smoking status based upon repeated assessments of smoking that are recorded in the EMR are strongly associated with two DNA methylation sites, which are sensitive biomarkers of tobacco use. We anticipate that these trajectories will prove to be effective, efficient phenotypes for large-scale genetic and other “omics” discovery efforts.
Table 1.
Characteristics of VACS and VACS-BC with Methylation Data
| VACS (n=142,152) | VACS-BC (n=728) | |
|---|---|---|
|
| ||
| Mean Age (SD) | 47.2 (10.9) | 52.8 (7.9) |
|
| ||
| HIV+ | 31% | 84% |
|
| ||
| Male | 97% | 97% |
|
| ||
| Race/Ethnicity | ||
| African-American | 48% | 81% |
| White | 40% | 15% |
| Hispanic/Other | 12% | 4% |
|
| ||
| Marijuana or Hashish Use | - | |
| Never | 26% | |
| Not in Past Year | 45% | |
| <Once per Month | 9% | |
| ≥Once per Month | 14% | |
| Unknown | 6% | |
|
| ||
| Smoking Variables | ||
|
| ||
| Median # Smoking Observation (IQR) | 6 (3–9) | 15 (8–24) |
|
| ||
| Self-Report from EMR | ||
|
| ||
| Most Recent | ||
| Never (0) | 33% | 23% |
| Past (1) | 24% | 24% |
| Current (2) | 43% | 53% |
|
| ||
| Most Common | ||
| Never (0) | 29% | 20% |
| Past (1) | 18% | 12% |
| Current (2) | 53% | 68% |
|
| ||
| Trajectory | ||
| Mostly Never (0) | 24% | 14% |
| Mix (1) | 26% | 22% |
| Current & Past (2) | 16% | 26% |
| Mostly Current (3) | 33% | 37% |
|
| ||
| Current Smoking Based on Methylation Markers | ||
|
| ||
| Cg05575921<0.80 | 69% | |
|
| ||
| Cg05575921<0.70 | 54% | |
|
| ||
| Cg03636183<0.68 | 82% | |
|
| ||
| Cg03636183<0.59 | - | 52% |
VACS: Veterans Aging Cohort Study
VACS-BC: Veterans Aging Cohort Study - Biomarker Cohort Substudy
EMR: Electronic Medical Record
Acknowledgments
The use of DNA methylation status to determine smoking status is protected by US Patents 8,637,652 and 9,273,358 as well as by pending intellectual property claims owned by Behavioral Diagnostics. This study was funded by a grant from the National Institute on Alcohol Abuse and Alcoholism (U24-AA020794, U01-AA020790, U10 AA013566 (completed), and VHA i01 BX003341. Views presented in the manuscript are those of the authors and do not reflect those of the Department of Veterans Affairs, or the United States Government.
Appendix 1. Group-based Smoking trajectories among HIV+ and Uninfected Patients in VACS (2000–2015)
Footnotes
Author Contributions
ACJ, HK, KX, and KAM were responsible for the study concept and design. ACJ contributed to the acquisition of the data. ACJ, KAM, JPT, HT, and KX assisted with data analysis and interpretation of findings. XZ performed DNA methylation data processing and quality control. BL performed methylation analysis for smoking. KAM, ACJ, KX, and HK drafted the manuscript. HT, XK, WB, JC, JG, and KC provided critical revisions of the manuscript for important intellectual content. All authors critically reviewed content and approved final version for publication.
Conflict of Interest
Dr. Kranzler has been an advisory board member, consultant, or CME speaker for Alkermes, Indivior and Lundbeck. He is also a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor, and Amygdala Neurosciences. Drs. Kranzler and Gelernter are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. No other authors have conflicts of interest to declare.
References
- Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Calvez-Kelm FL, Kaaks R, Barrdahl M, Boeing H, Aleksandrova K, Trichopoulou A, Lagiou P, Naska A, Palli D, Krogh V, Polidoro S, Tumino R, Panico S, Bueno-de-Mesquita B, Peeters PH, Quiros JR, Navarro C, Ardanaz E, Dorronsoro M, Key T, Vineis P, Murphy N, Riboli E, Romieu I, Herceg Z. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618. doi: 10.2217/epi-2016-0001. [DOI] [PubMed] [Google Scholar]
- Andersen AM, Philibert RA, Gibbons FX, Simons RL, Long J. Accuracy and utility of an epigenetic biomarker for smoking in populations with varying rates of false self-report. Am J Med Genet B Neuropsychiatr Genet. 2017;174(6):641–650. doi: 10.1002/ajmg.b.32555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armah KA, McGinnis K, Baker J, Gibert C, Butt AA, Bryant KJ, Goetz M, Tracy R, Oursler KA, Rimland D, Crothers K, Rodriguez-Barradas M, Crystal S, Gordon A, Kraemer K, Brown S, Gerschenson M, Leaf DA, Deeks SG, Rinaldo C, Kuller LH, Justice A, Freiberg M. HIV status, burden of comorbid disease and biomarker of inflammation, altered coagulation and monocyte activation. Clin Infect Dis. 2012;55:126–136. doi: 10.1093/cid/cis406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglietto L, Ponzi E, Haycock P, Hodge A, Bianca Assumma M, Jung CH, Chung J, Fasanelli F, Guida F, Campanella G, Chadeau-Hyam M, Grankvist K, Johansson M, Ala U, Provero P, Wong EM, Joo J, English DR, Kazmi N, Lund E, Faltus C, Kaaks R, Risch A, Barrdahl M, Sandanger TM, Southey MC, Giles GG, Johansson M, Vineis P, Polidoro S, Relton CL, Severi G. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int J Cancer. 2017;140:50–61. doi: 10.1002/ijc.30431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach SR, Dogan MV, Lei MK, Cutrona CE, Gerrard M, Gibbons FX, Simons RL, Brody GH, Philibert RA. Methylomic Aging as a Window onto the Influence of Lifestyle: Tobacco and Alcohol Use Alter the Rate of Biological Aging. J Am Geriatr Soc. 2015;63:2519–2525. doi: 10.1111/jgs.13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach SRH, Lei MK, Ong ML, Brody GH, Dogan MV, Philibert RA. MTHFR methylation moderates the impact of smoking on DNA methylation at AHRR for African American young adults. Am J Med Genet B Neuropsychiatr Genet. 2017;174(6):608–618. doi: 10.1002/ajmg.b.32544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojesen SE, Timpson N, Relton C, Davey Smith G, Nordestgaard BG. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax. 2017;72:646–653. doi: 10.1136/thoraxjnl-2016-208789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati V, Baccarelli A. Environmental epigenetics. Heredity (Edinb) 2010;105:105–112. doi: 10.1038/hdy.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan JM, Eden J, Smith BM. Leadership by example: Coordinating government roles in improving healthcare quality committee on enhancing federal healthcare quality programs. Washington, DC: National Academy Press; 2002. [Google Scholar]
- Dempsey D, Tutka P, Jacob P, 3rd, Allen F, Schoedel K, Tyndale RF, Benowitz NL. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther. 2004;76:64–72. doi: 10.1016/j.clpt.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Dogan MV, Xiang J, Beach SRH, Cutrona C, Gibbons FX, Simons RL, Brody GH, Stapleton JT, Philibert RA. Ethnicity and smoking associated DNA methylation changes at HIV co-receptor GPR15. Front Psychiatry. 2015;6:132. doi: 10.3389/fpsyt.2015.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, Davey Smith G, Hughes AD, Chaturvedi N, Relton CL. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6:4. doi: 10.1186/1868-7083-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasanelli F, Baglietto L, Ponzi E, Guida F, Campanella G, Johansson M, Grankvist K, Johansson M, Assumma MB, Naccarati A, Chadeau-Hyam M, Ala U, Faltus C, Kaaks R, Risch A, De Stavola B, Hodge A, Giles GG, Southey MC, Relton CL, Haycock PC, Lund E, Polidoro S, Sandanger TM, Severi G, Vineis P. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun. 2015;6:10192. doi: 10.1038/ncomms10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiberg MS, Bebu I, Tracy R, So-Armah K, Okulicz J, Ganesan A, Armstrong A, O’Bryan T, Rimland D, Justice AC, Agan BK Infectious Disease Clinical Research Program Working Group HIV. D-dimer levels before HIV seroconversion remain elevated even after viral suppression and are associated with an increased risk of non-AIDS events. PLoS One. 2016;11:e0152588. doi: 10.1371/journal.pone.0152588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fultz SL, Skanderson M, Mole LA, Gandhi N, Bryant K, Crystal S, Justice AC. Development and verification of a “virtual” cohort using the National VA Health Information System. Medical Care. 2006;44:S25–S30. doi: 10.1097/01.mlr.0000223670.00890.74. [DOI] [PubMed] [Google Scholar]
- Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7:113. doi: 10.1186/s13148-015-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaziano M, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, Guarino P, Aslan M, Anderson D, LaFleur R, Hammond T, Schaa K, Moser J, Huang G, Muralidhar S, Przygodzki R, O’Leary TJ. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70:214–233. doi: 10.1016/j.jclinepi.2015.09.016. [DOI] [PubMed] [Google Scholar]
- Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nat Rev Genet. 2005;6:521–532. doi: 10.1038/nrg1635. [DOI] [PubMed] [Google Scholar]
- Guida F, Sandanger TM, Castagne R, Campanella G, Polidoro S, Palli D, Krogh V, Tumino R, Sacerdote C, Panico S, Severi G, Kyrtopoulos SA, Georgiadis P, Vermeulen RC, Lund E, Vineis P, Chadeau-Hyam M. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24:2349–2359. doi: 10.1093/hmg/ddu751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlid S, Xu Z, Panduri V, Sandler DP, Taylor JA. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the Sister Study. Environ Health Perspect. 2014;122:673–678. doi: 10.1289/ehp.1307480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MJ, Russel MA, Benowitz NL, Feyerabend CF. Elimination of cotinine from body fluids: implications for noninvasive measurement of tovacco smoke exposure. Am J Public Health. 1998;78:696–698. doi: 10.2105/ajph.78.6.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CU, Aslibekyan S, Moreno-Macias H, Smith JA, Brody JA, Dhingra R, Yousefi P, Pankow JS, Kunze S, Shah SH, McRae AF, Lohman K, Sha J, Absher DM, Ferrucci L, Zhao W, Demerath EW, Bressler J, Grove ML, Huan T, Liu CC, Mendelson MM, Yao C, Kiel DP, Peters A, Wang-Sattler R, Visscher PM, Wray NR, Starr JM, Ding J, Rodriguez CJ, Wareham NJ, Irvin MR, Zhi D, Barrdahl M, Vineis P, Ambatipudi S, Uitterlinden AG, Hofman A, Schwartz J, Colicino E, Jou L, Vokonas PS, Hernandez DG, Singleton AB, Bandinelli S, Turner ST, Ware EB, Smith AK, Klengel T, Binder EB, Psaty BM, Taylor KD, Gharib SA, Swenson BR, Liang L, Dmeo DL, O’Connor GT, Herceg Z, Ressler KJ, Conneely KN, Sotoodehnia N, Kardia SLR, Melzer D, Baccarelli AA, vanMeurs JBJ, Romieu I, Arnett DK, Ong KK, Liu Y, Waldenberger M, Deary IJ, Fornage M, Levy D, London SJ. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BL, Nagin DS. A note on a Stata plug in for estimating group-based trajectory models. Sociol Method Res. 2013;42:608–613. [Google Scholar]
- Justice AC, Dombrowski E, Conigliaro J, Fultz SL, Gibson D, Madenwald T, Goulet J, Simberkoff M, Butt AA, Rimland D, Rodriguez-Barradas MC, Gibert CL, Oursler KA, Brown S, Leaf DA, Goetz MB, Bryant K. Veterans Aging Cohort Study (VACS): Overview and description. Medical Care. 2006;44:S13–S24. doi: 10.1097/01.mlr.0000223741.02074.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice AC, Freiberg MS, Tracy R, Kuller L, Tate JP, Goetz MB, Fiellin DA, Vanasse GK, Butt AA, Rodriguez-Barradas MC, Gibert C, Oursler KA, Deeks SG, Bryant K the VACS Project Team. Does an index composed of clinical data reflect effects of inflammation, coagulation, and monocyte activation on mortality among those aging with HIV? Clin Infect Dis. 2012;54:984–994. doi: 10.1093/cid/cir989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice AC, McGinnis KA, Tate JP, Xu K, Becker WC, Zhao H, Gelernter J, Kranzler HR. Validating Harmful Alcohol Use as a Phenotype for Genetic Discovery Using Phosphatidylethanol and a Polymorphism in ADH1B. Alcohol Clin Exp Res. 2017;41:998–1003. doi: 10.1111/acer.13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice AC, Smith RV, Tate JP, McGinnis KA, Xu K, Becker WC, Lee KY, Lynch K, Sun N, Concato J, Fiellin DA, Zhao H, Gelernter J, Kranzler HR. AUDIT-C and ICD codes as phenotypes for harmful alcohol use: association with ADH1B polymorphisms in two U.S. populations. Addiction. 2018 doi: 10.1111/add.14374. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsinger LS, Anderson C, Kim J, Larson M, Chan SH, King HA, Rice KL, Slatore CG, Tanner NT, Pittman K, Monte RJ, McNeil RB, Grubber JM, Kelley MJ, Provenzale D, Datta SK, Sperber NS, Barnes LK, Abbott DH, Sims KJ, Whitley RL, Wu RR, Jackson GL. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Internal Medicine. 177:399–406. doi: 10.1001/jamainternmed.2016.9022. [DOI] [PubMed] [Google Scholar]
- Ladd-Acosta C. Epigenetic Signatures as Biomarkers of Exposure. Curr Environ Health Rep. 2015;2:117–125. doi: 10.1007/s40572-015-0051-2. [DOI] [PubMed] [Google Scholar]
- Lee MK, Hong Y, Kim SY, London SJ, Kim WJ. DNA methylation and smoking in Korean adults: epigenome-wide association study. Clin Epigenetics. 2016;8:103. doi: 10.1186/s13148-016-0266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukola A, Buchwald J, Gupta R, Palviainen T, Hallfors J, Tikkanen E, Korhonen T, Ollikainen M, Sarin AP, Ripatti S, Lehtimaki T, Raitakari O, Salomaa V, Rose RJ, Tyndale RF, Kaprio J. A Genome-Wide Association Study of a Biomarker of Nicotine Metabolism. PLoS Genet. 2015;11:e1005498. doi: 10.1371/journal.pgen.1005498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis KA, Brandt CA, Skanderson M, Justice AC, Shahrir S, Butt AA, Brown ST, Freiberg MS, Gibert CL, Goetz MB, Kim JW, Pisani MA, Rimland DT, Rodriguez-Barradas MC, Sico JJ, Tindle HA, Crothers K. Validating smoking data from the verteran’s affairs health factors dataset, an electronic data source. Nicotine Tob Res. 2011;13:1233–1239. doi: 10.1093/ntr/ntr206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen L, Mittman BS, Demakis JG. Overview of the Veterans Health Administration (VHA) quality enhancement research initiative (QUERI) Journal of the American Medical Informatics Association. 2004;11:339–343. doi: 10.1197/jamia.M1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagin DS. Group-based modeling of development. Cambridge, MA: Harvard University Press; 2005. [Google Scholar]
- Philibert RA, Beach SR, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5(1):19. doi: 10.1186/1868-7083-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert R, Dogan M, Noel A, Miller S, Krukow B, Papworth E, Cowley J, Long JD, Beach SRH, Black DW. Dose response and prediction characteristics of a methylation sensitive digital PCR assay for cigarette consumption in adults. Front Genetics. 2018;9:137. doi: 10.3389/fgene.2018.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert R, Hollenbeck N, Andersen E, McElroy S, Wilson S, Vercande K, Beach SRT, Osborn T, Gerrard M, Gibbons FX, Wang K. Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front Psychiatry. 2016;7:55. doi: 10.3389/fpsyt.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert R, Hollenbeck N, Andersen E, Osborn T, Gerrard M, Gibbons FX, Wang K. A Quantitative epigenetic approach for the assessment of cigarette consumption. Front Psychol. 2015;6:656. doi: 10.3389/fpsyg.2015.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Roige S, Fontanillas P, Elson SL, Gray JC, de Wit H, Davis LK, MacKillop J, Palmer AA. Genome-wide association study of alcohol use disorder identification test (AUDIT) scores in 20 328 research participants of European ancestry. Addict Biol. 2017 Oct 23; doi: 10.1111/adb.12574. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg A, Magnus S, Grunewald J, Eklund A, Wheelock AM. Assessing recent smoking status by measuring exhaled carbon monoxide levels. PLoS One. 2011;6:e28864. doi: 10.1371/journal.pone.0028864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker NS, Ueland PM, Polidoro S, van Veldhoven K, Ricceri F, Brown R, Flanagan JM, Vineis P. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24:712–716. doi: 10.1097/EDE.0b013e31829d5cb3. [DOI] [PubMed] [Google Scholar]
- Shiffman S, Tindle H, Li X, Scholl S, Dunbar M, Mitchell-Miland C. Characteristics of smoking patterns of intermittent smokers. Exp Clin Psychopharmocol. 2012;20:264–77. doi: 10.1037/a0027546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stueve TR, Li WZ, Shi J, Marconett CN, Zhang T, Yang C, Mullen C, Yan C, Wheeler W, Hua X, Zhou B, Borok Z, Caporaso NE, Pesatori AC, Duan J, Laird-Offringa IA, Landi MT. Epigenome-wide analysis of DNA methylation in lung tissue shows concordance with blood studies and identifies tobacco smoke-inducible enhancers. Hum Mol Genet. 2017;26:3014–3027. doi: 10.1093/hmg/ddx188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Wahl S, Pfeiffer L, Ward-Caviness CK, Kunze S, Kretschmer A, Reischl E, Peters A, Gieger C, Waldenberger M. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics. 2017;18:805. doi: 10.1186/s12864-017-4198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Elgizouli M, Schottker B, Holleczek B, Nieters A, Brenner H. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics. 2016;8:127. doi: 10.1186/s13148-016-0292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Schottker B, Ordonez-Mena J, Holleczek B, Yang R, Burwinkel B, Butterbach K, Brenner H. F2RL3 methylation, lung cancer incidence and mortality. Int J Cancer. 2015;137:1739–1748. doi: 10.1002/ijc.29537. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang R, Burwinkel B, Breitling LP, Brenner H. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ Health Perspect. 2014;122:131–137. doi: 10.1289/ehp.1306937. [DOI] [PMC free article] [PubMed] [Google Scholar]