

Supplemental Digital Content is available in the text
Keywords: lofexidine, opioid use disorder, opioid withdrawal
Abstract
Objectives:
To investigate the safety and efficacy of lofexidine for treating opioid withdrawal syndrome (OWS) and facilitating completion of opioid withdrawal.
Methods:
A multicenter, double-blind, placebo-controlled study was conducted at 18 US centers from June 2013 to December 2014. Participants (n = 603) aged ≥18 years, dependent on short-acting opioids, and seeking withdrawal treatment, randomized 3:3:2 to receive lofexidine 2.88 mg/d (n = 222), lofexidine 2.16 mg/d (n = 230), or placebo (n = 151) for 7 days. Primary outcome was the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop) scores rating withdrawal symptoms over days 1 to 7.
Results:
Participants were of mean age, 35 years; 71% male. Pairwise differences in overall SOWS-Gossop log-transformed least squares means were statistically significant for lofexidine 2.16 mg (difference, −0.21; 95% CI, −0.37 to −0.04; P = 0.02) and 2.88 mg (−0.26; 95% CI, −0.44 to −0.09; P = 0.003) compared with placebo. Fewer than half of participants in both groups completed the study. Completion rates for lofexidine 2.16 mg (41.5%; odds ratio [OR], 1.85; P = 0.007) and 2.88 mg (39.6%; OR, 1.71; P = 0.02) were significantly better compared with placebo (27.8%). Overall adverse event (AE) rates were similar across groups. Common AEs for lofexidine included orthostatic hypotension, hypotension, and bradycardia, but resulted in few study discontinuations.
Conclusions:
Lofexidine 2.16 mg and 2.88 mg significantly reduced symptoms of OWS versus placebo, and increased absolute rates of completing the 7-day study by 14% and 12%, respectively (a relative increase of 85% and 71%). Data suggest that lofexidine is a generally safe and effective nonopioid treatment for opioid withdrawal. Lofexidine could serve as a withdrawal treatment option when a nonopioid agent is preferred or required, when agonist-assisted withdrawal is unavailable, when agonist discontinuation caused OWS, and during induction into maintenance treatment with opioid agonists or antagonists.
Trial Registration:
ClinicalTrials.gov identifier: NCT01863186.
Opioid use disorder (OUD) is associated with well-known morbidity and mortality, and the current US opioid epidemic has been declared a public health emergency. According to recent estimates, there are more than 2.5 million Americans with OUD and more than 33,000 Americans died of an opioid overdose in 2015 (Mattson et al., 2017). Thus, the need for treatment of OUD cannot be overstated.
Opioid withdrawal syndrome (OWS) is a consequence of opioid discontinuation or dose reduction in individuals with prolonged opioid use, whether in the context of OUD or iatrogenic physiologic dependence with analgesic use (Tetrault and O’Connor, 2009). The acute phase of OWS consists of a cluster of characteristic symptoms such as nausea/vomiting, stomach cramps, pain, anxiety, insomnia, hot/cold flashes, and restlessness, which can be extremely uncomfortable, and can vary among individuals in severity, time of onset, duration, and persistence (Tetrault and O’Connor, 2009). OWS is extremely salient to patients and may cause them to continue using opioids. Weiss et al. (2014) reported that among opioid-dependent patients with and without chronic pain, avoidance of withdrawal was the leading reason given for ongoing use.
Given the impact of OWS, its management can be a critical component of patient stabilization, engagement, and facilitation of transition to continuing care. It is important to note that withdrawal management (often referred to as “detoxification”) is not curative, and when used alone in OUD without ongoing relapse-prevention medications, leads to high rates of relapse. But while not sufficient, withdrawal management is often necessary. Although transition to maintenance agonist treatments (such as buprenorphine or methadone) usually prevents or mitigates symptoms of OWS on its own, and may obviate the need for other withdrawal management, in other scenarios withdrawal management is an essential first step towards treatment. Transition to antagonist treatment (such as extended-release naltrexone) is one critical scenario in which full discontinuation of opioids, and withdrawal management, are required. Additionally, it is common (both in residential settings where there is medical monitoring and treatment, such as ASAM Level 3.7-WM, and in ambulatory settings) for patients to enter acute episodes of crisis-driven care in which withdrawal management is initiated pending decisions about appropriate next treatment steps. There are scenarios in which patients choose opioid discontinuation without ongoing relapse-prevention medications, or circumstances limit their availability, such as a lack of clinicians certified to prescribe buprenorphine in some areas. Finally, scenarios of opioid-dose reduction, with or without OUD, may warrant additional treatment of OWS, particularly as recent guidelines and policy changes related to opioid prescribing will result in widespread changes in opioid availability and/or dosage for many patients on chronic opioid pain therapy, putting them at risk of withdrawal.
In addition to their use as maintenance treatments for OUD, opioid agonists such as buprenorphine and methadone are used for OWS management, but have limitations including the potential for misuse and some restrictions on availability because of prejudicial stigma and special regulatory requirements. The α2-adrenergic receptor agonist clonidine, an antihypertensive, is used off-label for OWS management, but is not US Food and Drug Administration (FDA)-approved, has not been adequately studied to inform consistent dosing guidelines, especially for nonspecialists, and may be limited by significant sedation and hypotension at doses effective in alleviating symptoms of OWS (Gowing et al., 2016).
Lofexidine is an α2-adrenergic agonist that has shown evidence in several studies of efficacy in the management of OWS and may have a more favorable side effect profile compared with clonidine (Gowing et al., 2016). It has been approved and used widely for OWS management in the United Kingdom since the early 1990s. At the recommendation of the FDA, in order to meet the agency's criteria for potential approval of lofexidine, this study was undertaken to further define the efficacy and safety of lofexidine for management of opioid withdrawal. The essential design of the study was recommended by the FDA to meet registration requirements. On the basis of results of this study and a previously reported pivotal trial (Gorodetzky et al., 2017), the FDA approved lofexidine in May 2018 with an indication for mitigation of withdrawal symptoms to facilitate abrupt discontinuation of opioids in adults.
METHODS
Trial Overview
This was a phase 3, multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of lofexidine for alleviation of OWS symptoms after abrupt opioid withdrawal. Participants were adults who were dependent on short-acting opioids and seeking treatment for OWS. The protocol was approved by a central institutional review board (IRB; Aspire) or a local IRB at each participating center. All participants provided written informed consent.
Major Enrollment Criteria
Men or women ≥18 years old seeking treatment for opioid use disorder were eligible. Participants were required to have current DSM-IV dependence according to the Mini International Neuropsychiatric Interview (MINI) on any opioid with a half-life similar to heroin or morphine with use for ≥21 of the past 30 days, a baseline score ≥2 on the Objective Opiate Withdrawal Scale (OOWS–Handelsman), and if female, agreement to use an acceptable method of contraception. Pregnant or lactating females were excluded. Participants with use of methadone or buprenorphine in the past 14 days, unstable medical or psychiatric illness (based on the MINI), self-reported human immunodeficiency virus positive status, and use of antihypertensives, antiarrhythmics, psychotropics, or anticonvulsant medications within the past 4 weeks were excluded. All participants were naive to lofexidine exposure.
Study Design
Eligibility was determined during a 7-day screening period. Qualifying participants were randomized (3:3:2 ratio) to receive lofexidine 0.72 mg QID (2.88 mg/d), lofexidine 0.54 mg QID (2.16 mg/d), or matching placebo administered at 8 AM, 1 PM, 6 PM, and 11 PM for 7 days during the double-blind treatment between June 2013 and December 2014. The study was conducted at 18 treatment centers in the United States, all inpatient/residential because of protocol data collection requirements and resultant participant burden. Efficacy assessments were assessed daily 3.5 hours after the 8 AM dose. Vital signs were assessed ≤30 minutes before dosing and at 11:30 AM, 4:30 PM, and 9:30 PM. If vital signs met criteria for hypotension, orthostasis, or bradycardia, the dose was withheld (see eMethods [Supplemental Digital Content 1]). Electrocardiograms (ECGs) were performed during screening (days 1 and 7); before the 8 AM dose on days 1, 2, 4, and 7; and before the 1 PM dose and at 4 PM and 5 PM on days 1 and 7. Participants who completed day 7 were eligible to receive lofexidine for an additional 7 days in an open-label continuation study, to be reported elsewhere. Investigators typically referred participants to ongoing community-based treatment after study completion.
Only the double-blind portion of the study is reported here. Discontinuation criteria necessitated removal from the study for predefined values of QTC prolongation, hypotension, bradycardia, orthostasis, heart rate or blood pressure decrease from baseline, dose holds (>2 per 24 hours, or >6 over days 1–7), syncope, illicit drug use, and prohibited concomitant medication use (see eMethods).
Randomization and Blinding
A stratified randomization procedure was used to assure adequate exposure across sexes. The participants, investigators, staff, sponsor, and contracted clinical research personnel were blinded to treatment-group assignment. Study drug was supplied as blister cards with the appropriate dosing of lofexidine or matched placebo for each day of the double-blind study.
Outcomes
The Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop) was chosen as the primary outcome measure because of its validated ability to assess symptom relief during acute opioid detoxification. It is a self-assessed scale with 10 symptom-related items rated from “none” (0) to “severe” (3) (Vernon et al., 2016). The primary efficacy endpoint was the difference in log-transformed least squares (LS) means for the overall SOWS-Gossop scores performed at 3.5 hours after the 8 AM lofexidine dose on days 1 to 7, to address overall cumulative symptom relief. The principal secondary outcome was proportion of participants completing the study, defined as having received at least 1 dose of study medication on day 7 and completing the 3.5-hour postdose SOWS-Gossop assessment on day 7. SOWS-Gossop and Clinical Opiate Withdrawal Scale (COWS) scores on each of days 1 to 7 were other secondary outcomes. COWS is a clinician-administered assessment of 11 common opioid withdrawal signs and symptoms with scores ranging from 0 (mildest) to 48 (most severe; Wesson and Ling, 2003). Adverse events (AEs) were recorded daily and at a 30-day follow-up phone contact and coded in accordance with Medical Dictionary for Regulatory Activities, version 16.0. Predefined abnormal values for vital signs were recorded as AEs (see eMethods section).
Sample Size and Statistical Analysis
Treatment effect and participant variability with respect to SOWS-Gossop scores were estimated from a prior phase 3 study (Gorodetzky et al., 2017), using the random coefficients model initially planned for this study and estimating the treatment effect of lofexidine 2.88 mg versus placebo with respect to area under the curve (AUC), based on the SOWS-Gossop scores from days 1 to 7 (AUC[1–7]). Accounting for the sequential testing approach, the power to find a statistically significant effect of the lofexidine 2.88-mg dose with respect to AUC(1–7) was 94.6% with a sample size of 600.
Analyses used the modified intent-to-treat (mITT) population, which consisted of all randomized participants who received at least 1 dose of study medication. Due to high dropout rates, a pattern-mixture model approach using a control-based pattern imputation for missing values was implemented for the primary endpoint, also known as a “Jump to Reference” model. As a result, missing observations in the lofexidine treatment groups were constructed from the observed data in the placebo group (Little, 1993; Ratitch et al., 2013). The study completion endpoint was analyzed using a logistic regression model including fixed effects for treatment group and sex. COWS scores were analyzed using mixed model repeated measures modeling. All statistical tests for efficacy were 2-sided (α = 0.05 significance level); all comparisons between treatments were reported with 95% confidence intervals for the difference. Statistical analyses were performed using SAS version 9.3 (SAS Institute).
RESULTS
Among 603 participants who were randomly assigned into the study between June 2013 and December 2014, 1 participant voluntarily withdrew from the trial with mild withdrawal symptoms before first drug administration and did not receive study medication, resulting in an mITT population of 602 participants. A total of 151 participants received placebo, 229 received lofexidine 2.16 mg/d, and 222 received lofexidine 2.88 mg/d (Fig. 1). Baseline characteristics were similar between groups (Table 1). Most participants (83.2%) reported heroin as their primary misused opioid. The mean duration of substance use was 8.7 years. All participants tested positive for opioids at baseline urine screening.
FIGURE 1.
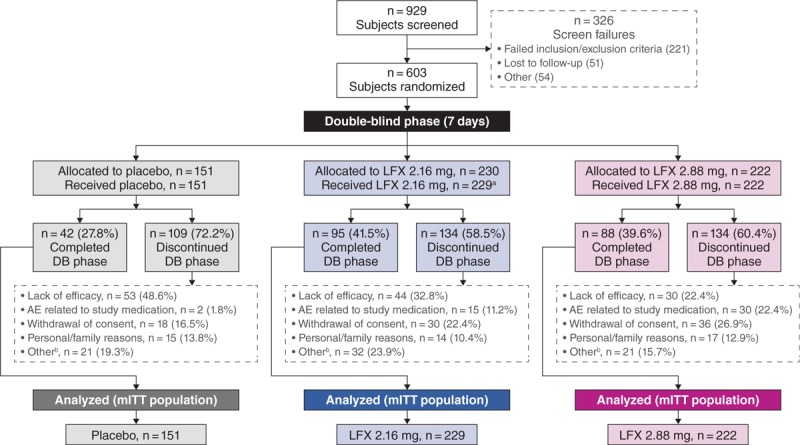
Participant flow diagram. aOne participant was randomly assigned into the study, but not dosed. bIncludes nonrelated adverse events, lack of adherence, evidence of contraband drug use, therapy with exclusionary drug, intensive cravings, did not want to continue inpatient, left against medical advice, completed detoxification, and protocol nonadherence. AE, adverse event; DB, double-blind; LFX, lofexidine; mITT, modified intent-to-treat.
TABLE 1.
Demographics and Drug Use History (Modified Intent-to-Treat Population)
| Characteristics | Placebo (n = 151) | Lofexidine 2.16 mg (n = 229) | Lofexidine 2.88 mg (n = 222) | Overall (n = 602) |
| Age, years | ||||
| Mean (SD) | 36 (11.9) | 35 (10.8) | 35 (10.5) | 35 (11.0) |
| Min, Max | 19, 63 | 19, 74 | 19, 68 | 19, 74 |
| Male, No. (%) | 107 (70.9) | 162 (70.7) | 158 (71.2) | 427 (70.9) |
| Ethnicity, No. (%) | ||||
| Hispanic/Latino | 22 (14.6) | 33 (14.4) | 28 (12.6) | 83 (13.8) |
| Race, No. (%) | ||||
| American Indian or Alaska native | 2 (1.3) | 0 (0.0) | 2 (0.9) | 4 (0.7) |
| Asian | 1 (0.7) | 1 (0.4) | 3 (1.4) | 5 (0.8) |
| Black or African American | 26 (17.2) | 54 (23.6) | 48 (21.6) | 128 (21.3) |
| Native Hawaiian or other Pacific Islander | 3 (2.0) | 0 | 2 (0.9) | 5 (0.8) |
| White | 117 (77.5) | 169 (73.8) | 158 (71.2) | 444 (73.8) |
| Other | 2 (1.3) | 5 (2.2) | 9 (4.1) | 16 (2.7) |
| BMI* | ||||
| Mean (SD) | 25.1 (4.63) | 25.1 (4.71) | 25.1 (5.32) | 25.1 (4.92) |
| Min, Max | 16.0, 51.7 | 15.8, 46.5 | 17.0, 50.1 | 15.8, 51.7 |
| Primary opioid used, No. (%)† | ||||
| Heroin | 122 (80.8) | 197 (86.0) | 182 (82.0) | 501 (83.2) |
| Oxycodone | 9 (6.0) | 10 (4.4) | 18 (8.1) | 37 (6.1) |
| Hydrocodone | 10 (6.6) | 10 (4.4) | 9 (4.1) | 29 (4.8) |
| Other | 9 (6.0) | 12 (5.2) | 11 (5.0) | 32 (5.3) |
| Duration of substance use, mean (SD), years | 8.8 (9.04) | 9.3 (8.97) | 7.9 (7.91) | 8.7 (8.62) |
*Two placebo subjects had missing BMI values.
†One placebo subject and 2 lofexidine 2.88-mg subjects had missing data for primary opioid used.
BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); Max, maximum; Min, minimum.
Efficacy
Both lofexidine groups demonstrated significant differences versus placebo for the primary endpoint. The pairwise difference from placebo in overall log-transformed SOWS-Gossop LS means was −0.21 for lofexidine 2.16 mg (95% CI, −0.37 to −0.04; P = 0.02) and −0.26 for lofexidine 2.88 mg (95% CI, −0.44 to −0.09; P = 0.003), indicating greater OWS relief with lofexidine. Study completion rates were significantly higher in the lofexidine groups: 41.5% in the 2.16-mg group (odds ratio [OR], 1.85; 95% CI, 1.18–2.88; P = 0.007) and 39.6% in the 2.88-mg group (OR, 1.71; 95% CI, 1.09–2.67; P = 0.02) versus 27.8% for placebo.
Figure 2 and eTable 1 (Supplemental Digital Content 2) display SOWS-Gossop geometric mean scores by day. Lofexidine groups showed significant reductions versus placebo on days 1 to 5. Reductions were modestly greater in the lofexidine 2.88-mg group, compared with the 2.16-mg group, but not statistically significant. The placebo treatment curve highlights the natural progression of the withdrawal syndrome, with severity increasing from baseline to a peak on day 2, with gradual reduction through day 7. Mean COWS scores followed a similar pattern, with both lofexidine groups significantly superior to placebo on days 1 to 5 (Fig. 3). Other secondary efficacy outcomes were generally consistent with these results (eTables 2–5; Supplemental Digital Content 3–6).
FIGURE 2.
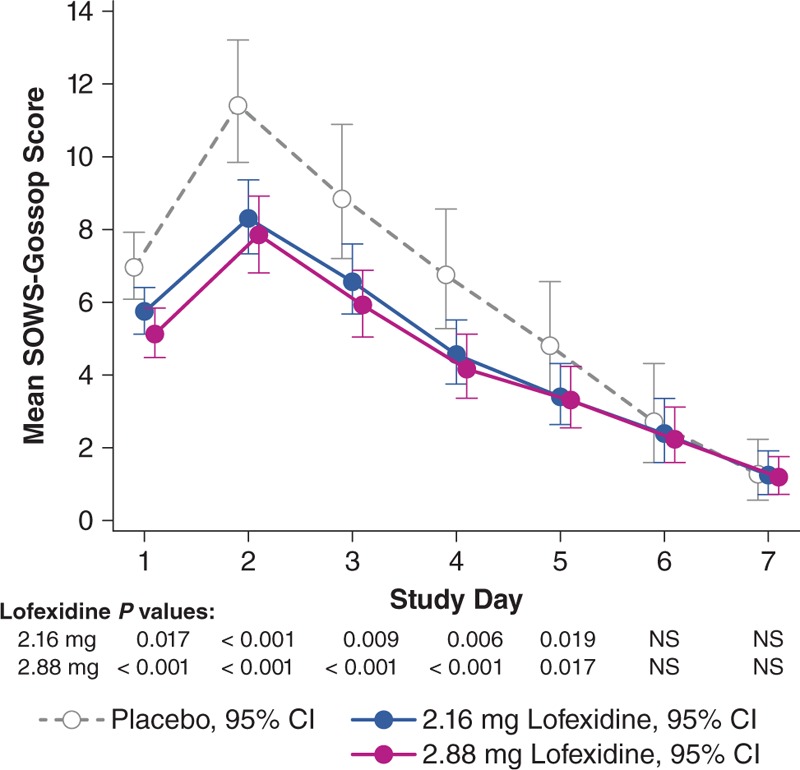
Geometric means (95% confidence interval) for Short Opiate Withdrawal Scale of Gossop score from days 1 to 7 (modified intent-to-treat population). In this pattern-mixture model analysis, the geometric mean SOWS-Gossop score on each study day is the back-transformed least squares mean estimate based on log-transformed data. Lower scores indicate less severe withdrawal symptoms. SOWS-Gossop, Short Opiate Withdrawal Scale of Gossop.
FIGURE 3.
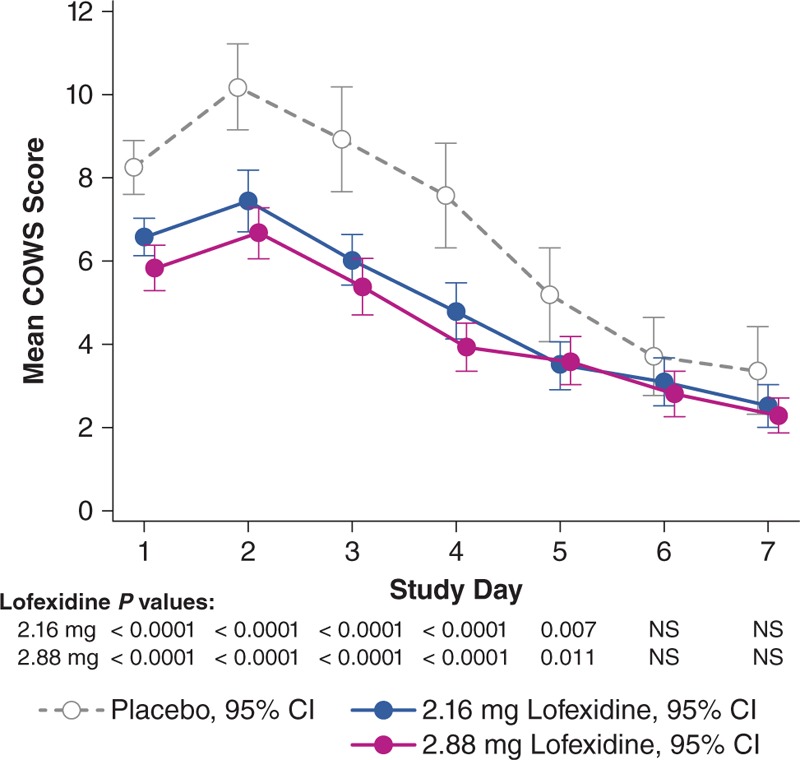
Least square means (95% confidence interval) for COWS score from days 1 to 7 (modified intent-to-treat population). LS means from mixed model repeat measures analysis. Lower scores indicate less severe withdrawal symptoms. COWS, Clinical Opiate Withdrawal Scale; LS, least squares.
Safety
The overall incidence of AEs was similar between treatment groups. Incidence rates for AEs related to opioid withdrawal were higher than those for AEs not related to opioid withdrawal, particularly in the placebo group (Table 2). Most events were mild or moderate in severity. The most common AEs were insomnia, orthostatic hypotension, hypotension, bradycardia, and dizziness (Table 2). There were no instances of rebound hypertension as an AE. Common events leading to discontinuation (in ≥2%) in a greater proportion of the total lofexidine group versus the placebo group were dizziness (3% vs 0.7%), hypotension (2% vs 0%), orthostatic hypotension (2% vs 0.7%), and bradycardia (2% vs 0%). Common AEs leading to discontinuation in a greater percentage of placebo participants were diarrhea, pain, nausea, and vomiting (6%–7% vs 1%–2%); insomnia (4% vs 3%); anxiety (3% vs 2%); and abdominal pain, chills, and rhinorrhea (2% vs 0.2%–0.4%). Lack of efficacy was the most frequent reason for study discontinuation (Table 2).
TABLE 2.
Summary of Adverse Events and Study Discontinuation (Modified Intent-to-Treat Population)
| Subject Experience, No. (%) | Placebo (n = 151) | Lofexidine 2.16 mg (n = 229) | Lofexidine 2.88 mg (n = 222) |
| At least 1 AE | 134 (88.7) | 216 (94.3) | 211 (95.0) |
| Opioid withdrawal-related AE* | 128 (84.8) | 181 (79.0) | 177 (79.7) |
| Nonopioid withdrawal-related AE* | 61 (40.4) | 176 (76.9) | 176 (79.3) |
| AE leading to study discontinuation | 44 (29.1) | 43 (18.8) | 55 (24.8) |
| Common AEs (>10%) | |||
| Insomnia | 73 (48.3) | 117 (51.1) | 123 (55.4) |
| Orthostatic hypotension† | 7 (4.6) | 67 (29.3) | 94 (42.3) |
| Bradycardia† | 8 (5.3) | 54 (23.6) | 70 (31.5) |
| Hypotension† | 2 (1.3) | 69 (30.1) | 67 (30.2) |
| Dizziness | 4 (2.6) | 44 (19.2) | 51 (23.0) |
| Diarrhea | 35 (23.2) | 51 (22.3) | 48 (21.6) |
| Pain | 36 (23.8) | 51 (22.3) | 42 (18.9) |
| Headache | 23 (15.2) | 30 (13.1) | 31 (14.0) |
| Somnolence | 8 (5.3) | 25 (10.9) | 29 (13.1) |
| Nausea | 32 (21.2) | 50 (21.8) | 27 (12.2) |
| Sedation | 8 (5.3) | 29 (12.7) | 27 (12.2) |
| Dry mouth | 0 | 22 (9.6) | 24 (10.8) |
| Myalgia | 25 (16.6) | 30 (13.1) | 22 (9.9) |
| Vomiting | 24 (15.9) | 23 (10.0) | 19 (8.6) |
| Study discontinuation | 109 (72.2) | 135 (59.0) | 134 (60.4) |
| Lack of efficacy | 53 (35.1) | 44 (19.1) | 30 (13.5) |
| Study drug-related AE | 2 (1.3) | 15 (6.5) | 30 (13.5) |
| Withdrawal of consent | 18 (11.9) | 30 (13.1) | 36 (16.2) |
| Personal/family reasons | 15 (9.9) | 14 (6.1) | 17 (7.7) |
| Other‡ | 21 (13.9) | 32 (14.0) | 21 (9.5) |
*Study investigators were required to judge whether AEs were related to opioid withdrawal at the time AEs were assessed.
†Orthostatic hypotension, hypotension, and bradycardia were required to be reported as AEs if predefined criteria were met: systolic blood pressure <90 mm Hg, diastolic blood pressure <50 mm Hg, pulse rate <50 beats/min, or >20% decrease from screening; decrease in standing systolic or diastolic blood pressure >25% from recumbent values.
‡Includes nonrelated AEs, lack of adherence, evidence of contraband drug use, therapy with exclusionary drug, intensive cravings, did not want to continue inpatient, left against medical advice, completed detoxification, and protocol nonadherence.
AE, adverse event.
Seven participants (2 [1.3%] receiving placebo and 5 [2.3%] lofexidine 2.88 mg/d) experienced serious AEs (SAEs). Placebo events included acute hepatitis C and QTcF prolongation. Three of the 5 SAEs in lofexidine-treated participants (suicidal ideation 18 days after final lofexidine dose, multiple drug overdose 24 days after final lofexidine dose, and death from multiple drug toxicities 3 days after final lofexidine dose) were considered not related to lofexidine. Two participants receiving lofexidine experienced SAEs of syncope and were discontinued from the study. The syncope events resolved quickly without treatment and were without sequelae.
As participants were in the initial stages of opioid withdrawal at randomization and elevated blood pressures are associated with OWS, vital-sign changes were assessed relative to screening rather than baseline values. Mean blood pressures (seated and standing) were reduced compared with screening values after the 8 AM lofexidine doses, with systolic and diastolic values generally remaining stable throughout the course of the day (eFig. 1 and 2 [Supplemental Digital Content 7 and 8]). Compared with screening, mean seated heart rate showed small reductions whereas standing heart rate was slightly higher at most time points in the lofexidine groups (eFig. 3 [Supplemental Digital Content 9]). Mean blood pressures and heart rate tended to increase slightly over the day in placebo participants.
QTcF prolongation >60 milliseconds from baseline was reported in 2 participants. A placebo participant had values >500 milliseconds on days 4 and 7 (reported as SAEs) but completed the study. A participant receiving lofexidine 2.88 mg had QTcF between 465 and 489 milliseconds on day 2 and experienced syncope (noted as an SAE above) and was discontinued.
DISCUSSION
In this study of 602 opioid-dependent participants, lofexidine provided relief of OWS at both 2.88 and 2.16-mg/d dosages. Overall reduction in SOWS-Gossop score, and study completion rate, the two principal endpoints, were significantly greater in both lofexidine-dose groups compared with placebo. On days 2 to 4, there was a difference in mean SOWS-Gossop scores between the lofexidine groups and the placebo group of approximately 2 to 4 points, which is considered a clinically meaningful response. As an illustrative example, a 2 to 4-point SOWS-G score decrease corresponds to a decrease of 1 to 2 severity levels on the Likert scale in 2 categories of symptoms, a difference that many patients will find subjectively impactful. This is supported both by the corresponding completion rates, and by psychometric studies (Vernon et al., 2016). Mean SOWS-Gossop and COWS scores were significantly reduced in both lofexidine groups compared with placebo on days 1 to 5, and especially on peak withdrawal days 2 to 3, when participants were most vulnerable to drop-out. Withdrawal symptoms were markedly lower in all 3 treatment groups on days 6 and 7. As reflected in the product labelling, 5 days of treatment may be sufficient for many patients, however, some patients who have persistent symptoms may benefit for longer treatment of up to 14 days. Treatment drop-out rates from withdrawal management in OUD are typically very high in abrupt discontinuation scenarios (Collins et al., 2005). In the present study, the 2.16 and 2.88-mg doses of lofexidine increased the absolute rates of completion by 14% (OR 1.85) and 12% (OR 1.71), respectively, over placebo. Early drop-outs are more likely to have poorer outcomes, and retention through withdrawal management could provide a pathway to next-step continuation treatment.
These findings of efficacy were consistent with earlier controlled studies of lofexidine. Both Yu and Gorodetzky reported significantly lower withdrawal symptom severity associated with lofexidine hydrochloride 3.2 mg (equivalent to lofexidine 2.88 mg) compared with placebo in abruptly withdrawing patients (Yu et al., 2008; Gorodetzky et al., 2017). Gorodetzky also reported a co-primary endpoint, time-to-drop-out, which illustrated that a greater proportion of participants completed treatment and study retention time was longer in the lofexidine group compared with placebo. The Yu study was stopped early because of significant efficacy findings and ethical concerns related to continued administration of placebo but was not considered a pivotal trial by FDA because of its small size and design issues. The current study did not show a statistically significant difference between the 2 doses of lofexidine, and thus the labeling reflects the recommended lower starting dose of 2.16 mg. Patients who do not respond adequately may benefit from increasing to the higher dose. Further comparative results and exploration of dosing will be reported elsewhere.
Our efficacy results were also consistent with a recent meta-analysis of randomized, controlled trials of α2-adrenergic agonists for treatment of OWS, which concluded that lofexidine is safe and effective for OWS management (Gowing et al., 2016).
Lofexidine was well-tolerated with similar overall AE rates across treatment groups and fewer AE-related study discontinuations for lofexidine versus placebo participants. AEs as well as discontinuation because of AEs not related to opioid withdrawal were more common for lofexidine whereas those related to withdrawal were more common for placebo. The common nonwithdrawal AEs in this study included expected drug class effects such as: hypotension, orthostatic hypotension, bradycardia, and dizziness; however, these events were usually managed with dose hold and rarely required discontinuation (2% of lofexidine participants). Two lofexidine participants experienced SAEs of syncope, both considered related to study medication, and required discontinuation per protocol.
The safety profile demonstrated in this study was also congruent with earlier studies (Yu et al., 2008; Gorodetzky et al., 2017). However, rates for hypotension and bradycardia were higher in the current study, presumably because of the protocol requirement of reporting hypotension and bradycardia as AEs if values met predefined criteria, regardless of any symptomatology.
Blood pressure and heart rate reductions are anticipated with use of α2-adrenergic agonists because of their known class mechanism of action. Norepinephrine release is blunted by these agents, thereby counteracting the noradrenergic hyperactivity of opioid withdrawal (Kosten and George, 2002); α2-adrenergic agonism also results directly in lowered blood pressure and heart rate (Kanagy, 2005). Reductions in blood pressure and heart rate can be adequately managed and potentially minimized by withholding a dose of medication, as was the practice in this trial, or lowering the dose, as in another trial (Fishman et al., 2017). In the review of α2-adrenergic agonists for OWS management, hypotension was noted to be more frequent with clonidine than lofexidine at similarly efficacious doses (Gowing et al., 2016). However, this study does not adequately inform the comparative efficacy or safety of lofexidine versus clonidine, which would require a head-to-head trial.
The limitations of this study include some facets of its design. Opioid withdrawal is a proximal treatment goal, often necessary but always insufficient to fully manage physiologic opioid dependence. However, the role of lofexidine investigated in this study did not extend beyond its use in the early stage of withdrawal and it was not evaluated with opioid agonist therapy (OAT) or agonist-assisted withdrawal. The present study also did not include participants with iatrogenic opioid physiologic dependence in pain treatment, an important target population in which scenarios of opioid dose reduction warrant further study. Additionally, our requirement to discontinue participants based on predefined vital sign and ECG values, although necessary as safety precautions for an investigational trial, may be overly conservative for real-world practice. The study results are strengthened by the large number of participants enrolled and the use of the SOWS-Gossop, a sensitive, precise assessment that captures the participant's experience of OWS (Vernon et al., 2016). The use of a placebo control group, as required by the FDA, raises ethical issues regarding depriving patients of known efficacious withdrawal treatments. This was partially addressed by use of ancillary medications (such as bismuth sulfate, acetaminophen, and zolpidem) and by offering rescue procedures (such as buprenorphine) to those that terminated early.
In the United States, FDA-approved medications for treatment of OUD currently consist of an opioid agonist (methadone), a partial opioid agonist (buprenorphine), and an opioid antagonist (naltrexone). Although methadone and buprenorphine are effective OATs for management of OUD, they are controlled substances with limitations that include restricted access and availability (Sharma et al., 2017), potential for misuse and diversion (Soyka, 2014), and barriers to acceptability in certain arenas, such as criminal justice settings (Nunn et al., 2009). These treatments have also been discouraged and stigmatized among some patient subpopulations, including those involved in 12-step mutual help programs (Frank, 2011; Rieckmann et al., 2014). Furthermore, patients may have residual symptoms during initiation of OAT or during agonist-assisted withdrawal, and may benefit from augmentation (Diaper et al., 2014), and augmentation with an α2-adrenergic agonist has been used to prolong abstinence in buprenorphine-maintained patients with a putative mechanism of amelioration of stress-induced relapse, consistent with its mechanism as a sympatholytic (Kowalczyk et al., 2015). Nonopioid treatment of OWS is helpful before transition to naltrexone in order to limit risk of precipitating OWS and may shorten the time to initiation of naltrexone (Sullivan et al., 2017).
Lofexidine may have advantages over opioid-based medications for withdrawal management when these are not suitable for certain patients and may provide synergistic benefits when they are inadequate alone. Clonidine will likely have cost advantages over lofexidine (as does any older, generic medication over a newly introduced medication), but although it is routinely prescribed for OWS, it is not FDA-approved for OWS, and therefore does not have OWS labeling, and has not been well-studied to determine appropriate dosing guidelines (Gowing et al., 2016). Although clonidine likely has similar efficacy, its key clinical limitations include the need for individualized “trial-and-error” dosing to balance efficacy and side effects, which include significant sedation and hypotension and potential for rebound hypertension (Clonidine hydrochloride package insert, 2015; Gowing et al., 2016). By contrast, the current study suggests that lofexidine doses up to 2.88 mg/d are well-tolerated without the need for dose titration, sedation rates are low, and rebound hypertension was not a significant clinical concern requiring treatment in the lofexidine clinical trials.
CONCLUSIONS
Data from this study suggest that lofexidine is a safe and effective treatment for patients undergoing acute withdrawal from opioids following abrupt cessation. Lofexidine could add to the armamentarium for withdrawal management and expand treatment access, especially when a nonopioid agent is desired, or when agonist-assisted withdrawal is not easily obtainable or adequate as a stand-alone option for a patient's individual treatment goal. Forthcoming results of other trials should help to further elaborate clinical utility and applicability in a wider range of clinical settings. Given its efficacy in reducing the severity of OWS, which has a consistent underlying mechanism in dependent patients regardless of the opioid used (Kosten and George, 2002), lofexidine may also be useful for managing OWS associated with discontinuation or dose reduction of OAT or opioids prescribed for analgesia (although these were not studied in the current trial). The demonstration of improved withdrawal symptoms and increased withdrawal completion rates supports that lofexidine can play a role in helping to overcome the OWS barrier. Further research is warranted to improve overall low retention rates in patients with OUD during opioid withdrawal treatment. The formal FDA withdrawal management indication for lofexidine, with well-known α2-adrenergic agonist class effects, will provide patients and practitioners an important new tool to help facilitate continuing care.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
Conflicts of interest: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Dr Fishman is a consultant for US WorldMeds, LLC, and for Alkermes, and reported receiving study funding from NIDA, Arnold Foundation, and Media Rez. Dr Tirado reported serving on speaker panels for Indivior and Alkermes plc. Dr Alam is a consultant for US WorldMeds, LLC, and for Alkermes, and reported receiving research support from Allergan, Otsuka, Janssen, and Alkermes. Ms Gullo is an employee and shareholder of US WorldMeds, LLC. Mr Clinch is an employee and shareholder of US WorldMeds, LLC. Dr Gorodetzky is a consultant to US WorldMeds, LLC. This work was supported by US WorldMeds, LLC, and the National Institute on Drug Abuse (grant U01DA033276).
Role of funder/sponsor: US WorldMeds was involved in the design and general oversight of the study but not in the collection and management of the data. Analysis and interpretation of the data included contributions from statisticians employed by US WorldMeds. Authors employed by US WorldMeds were involved in the preparation, review, approval, and decision to submit the manuscript for publication. The authors made the final decision to submit the manuscript for publication.
The CLEEN-SLATE Team includes the following study group investigators: Danesh Alam, MD (Northwestern Medicine Central DuPage Hospital, Winfield, Illinois); Jason Andersen, DO (Mountain View Hospital, Payson, Utah); Mohammed Bari, MD (Pacific Health Systems, National City, California); Bernadette D'Souza, MD (Midwest Clinical Research Center, Dayton, Ohio); Marc Fishman, MD, FASAM (Johns Hopkins School of Medicine, Baltimore, Maryland); Richard Guzzetta, MD (Touchstone Medical Group, Clovis, California); Robert Molpus, MD (Orlando Clinical Research, Orlando, Florida); Ricky Mofsen, DO (St Louis Clinical Trials, St Louis, Missouri); Robert Riesenberg, MD (Atlanta Center for Medical Research, Atlanta, Georgia); Ihsan M. Salloum, MD, MPH, DFAPA (University of Miami Miller School of Medicine, Miami, Florida); Rajinder Shiwach, MD (InSite Clinical Research, DeSoto, Texas); Carlos Tirado, MD (CARMA Health, PLLC, Austin, Texas); Kashinath Yadalam, MD (Lake Charles Memorial Health System, Lake Charles, Louisiana).
Additional contributions: Editorial support was provided by The Curry Rockefeller Group, LLC, and was funded by US WorldMeds.
Author contributions: Dr Gorodetzky had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Concept and design: Gorodetzky, Gullo, Clinch. Acquisition, analysis, or interpretation of data: Fishman, Tirado, Alam, Gullo, Clinch, Gorodetzky. Drafting of the manuscript: Fishman, Alam. Critical revision of the manuscript for important intellectual content: Fishman, Tirado, Alam, Gullo, Clinch, Gorodetzky. Statistical analysis: Clinch. Administrative, technical, or material support: Gullo, Clinch. Supervision: Gorodetzky, Gullo.
REFERENCES
- Clonidine hydrochloride [package insert]. Actavis Elizabeth LLC: Elizabeth, NJ; 2015. [Google Scholar]
- Collins ED, Kleber HD, Whittington RA, et al. Anesthesia-assisted vs buprenorphine- or clonidine-assisted heroin detoxification and naltrexone induction: a randomized trial. JAMA 2005; 294:903–913. [DOI] [PubMed] [Google Scholar]
- Diaper AM, Law FD, Melichar JK. Pharmacological strategies for detoxification. Br J Clin Pharmacol 2014; 77:302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman M, Gullo K, Clinch T, et al. Open label tolerability study of lofexidine HCl. Poster presented at: 28th American Academy of Addiction Psychiatry Annual Meeting and Scientific Symposium; December 7–10, 2017; San Diego, CA. [Google Scholar]
- Frank D. The trouble with morality: the effects of 12-step discourse on addicts’ decision-making. J Psychoactive Drugs 2011; 43:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodetzky CW, Walsh SL, Martin PR, et al. A phase III, randomized, multi-center, double blind, placebo controlled study of safety and efficacy of lofexidine for relief of symptoms in individuals undergoing inpatient opioid withdrawal. Drug Alcohol Depend 2017; 176:79–88. [DOI] [PubMed] [Google Scholar]
- Gowing L, Farrell M, Ali R, et al. Alpha2-adrenergic agonists for the management of opioid withdrawal. Cochrane Database Syst Rev 2016; 5:CD002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagy NL. α2-Adrenergic receptor signalling in hypertension. Clin Sci (Lond) 2005; 109:431–437. [DOI] [PubMed] [Google Scholar]
- Kosten TR, George TP. The neurobiology of opioid dependence: implications for treatment. Sci Pract Perspect 2002; 1:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk WJ, Phillips KA, Jobes ML, et al. Clonidine maintenance prolongs opioid abstinence and decouples stress from craving in daily life: a randomized controlled trial with ecological momentary assessment. Am J Psychiatry 2015; 172:760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little RJ. Pattern-mixture models for multivariate incomplete data. J Am Stat Assoc 1993; 88:125–134. [Google Scholar]
- Mattson CL, Schieber L, Scholl L, et al. Annual surveillance report of drug-related risks and outcomes—United States, 2017 [Centers for Disease Control and Prevention web site]. August 31, 2017. Available at: https://www.cdc.gov/drugoverdose/pdf/pubs/2017-cdc-drug-surveillance-report.pdf Accessed November 6, 2017. [Google Scholar]
- Nunn A, Zaller N, Dickman S, et al. Methadone and buprenorphine prescribing and referral practices in US prison systems: results from a nationwide survey. Drug Alcohol Depend 2009; 105:83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratitch B, O’Kelly M, Tosiello R. Missing data in clinical trials: from clinical assumptions to statistical analysis using pattern mixture models. Pharm Stat 2013; 12:337–347. [DOI] [PubMed] [Google Scholar]
- Rieckmann TR, Abraham AJ, Kovas AE, et al. Impact of research network participation on the adoption of buprenorphine for substance abuse treatment. Addict Behav 2014; 39:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Kelly SM, Mitchell SG, et al. Update on barriers to pharmacotherapy for opioid use disorders. Curr Psychiatry Rep 2017; 19:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyka M. Buprenorphine use and risk of abuse and diversion. Adv Pharmacoepidemiol Drug Saf [serial online] 2014; 3:145.Available from: OMICS International, Henderson, NV. Accessed March 28, 2018. [Google Scholar]
- Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry 2017; 174:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetrault JM, O’Connor PG. Ries RK, Fiellin DA, Miller SC, Saitz R. Management of opioid intoxification and withdrawal. Principles of Addiction Medicine 4th ed.Philadelphia: Lippincott Williams & Wilkins; 2009. 589–606. [Google Scholar]
- Vernon MK, Reinders S, Mannix S, et al. Psychometric evaluation of the 10-item Short Opiate Withdrawal Scale-Gossop (SOWS-Gossop) in patients undergoing opioid detoxification. Addict Behav 2016; 60:109–116. [DOI] [PubMed] [Google Scholar]
- Weiss RD, Potter JS, Griffin ML, et al. Reasons for opioid use among patients with dependence on prescription opioids: the role of chronic pain. J Subst Abuse Treat 2014; 47:140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesson DR, Ling W. The Clinical Opiate Withdrawal Scale (COWS). J Psychoactive Drugs 2003; 35:253–259. [DOI] [PubMed] [Google Scholar]
- Yu E, Miotto K, Akerele E, et al. A phase 3 placebo-controlled, double-blind, multi-site trial of the alpha-2-adrenergic agonist, lofexidine, for opioid withdrawal. Drug Alcohol Depend 2008; 97:158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.