

Abstract
The rate limiting step in the steroid synthesis pathway is catalyzed by CYP11A1 through three sequential reactions. The first two steps involve hydroxylations at the 22- and 20-positions, generating 20R,22R-dihydroxycholesterol (20R,22R-DiOHCH), with the third stage leading to a C20-C22 bond cleavage, forming pregnenolone. The present work provides detailed information on active site structure of CYP11A1 in the resting state and substrate-bound ferric forms as well as the CO-ligated adducts. In addition, high quality resonance Raman spectra are reported for the dioxygen complexes, providing new insight into the status of Fe-O-O fragments encountered in the enzymatic cycle. Results show that the three natural substrates of CYP11A1 have quite different effects on the active site structure, including variations of spin state populations, reorientations of heme peripheral groups and, most importantly, substrate-mediated distortions of Fe-CO and Fe-O2 fragments, as revealed by telltale shifts of the observed vibrational modes. Specifically, the vibrational mode patterns observed for the Fe-O-O fragments with the first and third substrates are consistent with H-bonding interactions to the terminal oxygen, a structural feature that tends to promote O-O bond cleavage to form the Compound I intermediate. Furthermore, such spectral data are acquired for complexes with the natural redox partner, adrenodoxin (Adx), revealing protein-protein induced active site structural perturbations. While this work shows that Adx has only a small effect on ferric and ferrous CO states, it has a relatively stronger impact on the Fe-O-O fragments of the functionally relevant oxy complexes.
Graphical Abstract
INTRODUCTION
Steroidogenic cytochromes P450 participate in the synthesis of steroid hormones that are essential for maintenance of mammalian life.1–3 Ultimately, all of these crucial hormones are generated from a common parent hormone, pregnenolone, which is derived from cholesterol in a complex process mediated by the multifunctional steroidogenic P450, CYP11A1. This inner mitochondrial-membrane protein of the adrenal cortex is also known as cytochrome P450scc, a designation which refers to the catalyzed side cleavage reaction occurring between C20 and C22 (Figure 1). This rate limiting step in the steroid synthesis pathway is catalyzed by CYP11A1 through three sequential steps illustrated in Figure 1, where the first two steps convert cholesterol (CH) to 22R-hydrocholesterol (22R-OHCH) and then to 20R,22R-dihydroxycholesterol (20R,22R-DiOHCH), with the third stage leading to a bond cleavage between C20 and C22, thereby forming the key product, pregnenolone, and another small molecule, 4-methylpentanal (also called isocaproaldehyde).4–7
Figure 1.
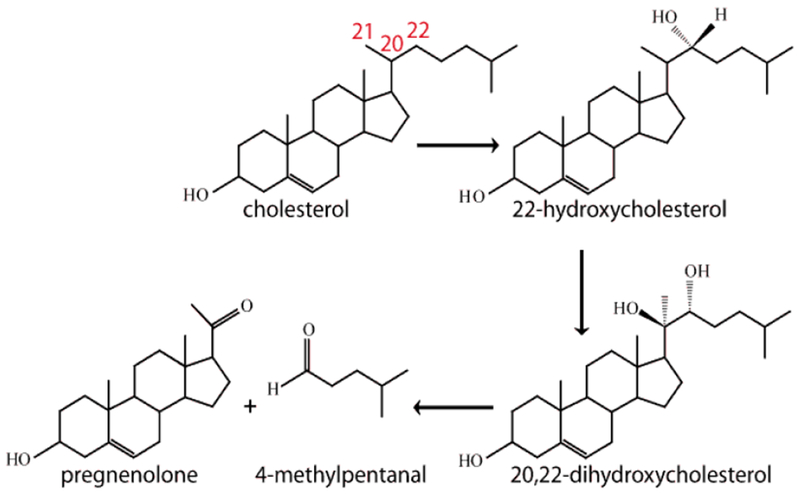
The structures of intermediate products encountered for the conversion of cholesterol to pregnenolone.
The well established enzymatic cycle of cytochromes P450 is initiated by substrate-binding to a low-spin ferric “resting state”, which disrupts a water cluster, displacing a bound axial water ligand, generating a high spin ferric heme, with an elevated reduction potential that triggers acceptance of an electron1,2 In the case of CYP11A1, the electron is provided by NADPH associated adrenodoxin reductase (AdR) and delivered by an Fe-S cluster contained in adrenodoxin (Adx).8,9 Binding of molecular oxygen forms a dioxygen adduct, which is most properly formulated as a ferric superoxide species, Fe(III)-(O-O−).10–13 Delivery of another electron from Adx then produces a ferric peroxo intermediate, Fe(III)-(O-O2−). While there exist special situations where this ferric peroxo species can react directly with electron- deficient fragments of a given substrate,14,15 generally, rapid sequential delivery of two heme pocket protons first forms a transient hydroperoxo- species and then undergoes efficient O-O bond cleavage to generate an extremely potent oxidant, known as Compound I, which is most accurately formulated as a ferryl heme π cation radical; i.e., [(Porphyrin+.) Fe(IV)=O]. 16–22
Interaction with natural or alternative reductases can alter the function and possibly the active site structure of the cytochrome P450.1,2,14,23–25 Adrenodoxin (Adx), which contains a [2Fe-2S] cluster, is generally believed to serve as an electron shuttle between a NADPH containing enzyme, adrenodoxin reductase (AdR), and the active site of CYP11A1.9,23–25 It has been reported that the inherent reduction potential of substrate-bound CYP11A1 is −284 mV, while that for Adx is −273 mV, with protein-protein complex formation causing the reduction potential of Adx to undergo a small negative shift to −291 mV, while that of the CYP11A1 shifts to −312 mV. 26,27 The association of Adx and CYP11A1 is postulated to arise from electrostatic interactions of charged surface residues of Adx and CYP11A1,9 with the recently reported crystal structure of the fusion protein between Adx and CYP11A1 revealing the presence of two salt bridges, consistent with this suggestion: Lys339 (CYP11A1)-Asp72 (Adx) and Lys 343 (CYP11A1)-Asp76(Adx).28
Resonance Raman (rR) spectroscopy has been applied to study various heme proteins and enzymes for decades,29,30 permitting determinations of: spin population change;31 distortions of the heme as reflected in variations of the in-plane and out-of-plane macrocycle modes; 29,30,32–37 and interactions of the vinyl and propionate groups on the heme periphery, as reflected in the behavior of the propionate- and vinyl- bending modes appearing in the low-frequency region and the vinyl group ν(C=C) stretching modes that are observed near 1600-1640 cm−1, both of which are sensitive to out-of-plane displacements of these groups with respect to the mean heme plane.32,38–41 Also, the technique is an especially powerful probe of the disposition of various endogenous and exogenous heme axial ligands. For example, the behavior of the ν(Fe-C) and ν(C-O) for ferrous CO forms reveal detailed information about the polarity of the distal pocket. 42 Of special importance for this work, the ν(Fe-O) and ν(O-O) modes of the oxy form of cytochromes P450 are efficiently rR-enhanced and provide telltale vibrational data that reflect subtle, but functionally important, H-bonding interactions with distal side molecular fragments, including those of bound substrates.42–44 Also important for the present study is the fact that rR is able to probe the status of the key ν(Fe-S) mode. 32,45–48 Moreover, rR measurements can monitor the effects of redox partners on the heme active sites for all of the above forms.48–50
Early rR studies of CYP11A1 were reported mainly by Tsubaki et al., for the ferric and ferrous CO states, providing some valuable insight,50–52 but also leaving some unexplained issues of interpretation, which are further addressed here (vide infra). Most importantly, in the present work we also expand the rR investigation of CYP11A1 to studies of the dioxygen complexes, ordinarily fleeting intermediates that must be prepared and stabilized under low temperature conditions.12,43,44 Studies of such dioxy-intermediates by rR spectroscopy can reveal even subtle active site structural changes that significantly impact the disposition of crucial intermediates and influence alternative reaction pathways that may occur within these enzymatic cycles. 43,44,53 For example, the rR technique can differentiate H-bonding to the proximal oxygen (Fe-Op) in an Fe-Op-Ot fragment, leading to a lower ν(Fe-O)frequency, and H-bonding to the terminal oxygen (Ot), which yields a relatively higher ν(Fe-O) frequency .42−44,54 This is quite an important capability, because computational work for cytochromes P450 and NOS,55,56 as well as experimental work on NOS and truncated hemoglobins,57–59 have indicated that hydrogen bonding to the proximal oxygen will stabilize the peroxo- intermediate, thereby hindering cleavage of the O-O bond, making it more likely that a (susceptible) substrate will be processed through the peroxo intermediate.53,55–59 On the other hand, hydrogen bonding to the terminal oxygen atom favors heterolytic cleavage of the O-O bond, with the reaction inevitably proceeding though the compound I intermediate.16–22
The work presented here provides detailed information about the active site structure of CYP11A1 in the resting state and substrate bound ferric forms, the CO-ligated adducts and the dioxy-intermediates encountered in the enzymatic cycle. In addition, studies are conducted for the above mentioned forms in complex with the natural redox partner, Adx, interrogating protein-protein induced active site structural perturbations. Specifically, the results show that the three natural substrates of CYP11A1 have quite different effects on the active site structure, including variations of spin state populations, reorientations of heme peripheral groups and, most importantly, substrate-mediated distortions of the Fe-XY fragments of bound exogenous ligands, including CO and O2, as revealed by telltale shifts of the ν(X-Y), ν(Fe-X) and δ(Fe-X-Y) vibrational modes. On the other hand, most significantly, our results reveal that the vibrational mode patterns observed for the Fe-O-O fragments of the dioxygan adducts obtained with the first and third substrates are almost identical, being consistent with H-bonding interactions to the terminal oxygen, a structural feature that tends to promote O-O bond cleavage, leading to reactions mediated by a Compound I species, consistent with the conclusions of recent experimental work mentioned above.60–62 Moreover, the effect of adrenodoxin binding to all three forms of CYP11A1 (ferric, ferrous CO and oxygenated) was studied and shows that, although Adx has only a small effect on ferric and ferrous CO states, it is seen for the first time that it has a relatively stronger impact on the Fe-O-O fragments of the functionally relevant oxy complexes.
MATERIALS AND METHODS
Materials
The substrates, CH and 22R-OHCH, were purchased from Sigma-Aldrich (USA) and 20R,22R-DiOHCH from Cayman Chemical (USA). The 22R-OHCH and 20R,22R-DiOHCH were dissolved in ethanol solution to make stock solutions with a concentration of 25 mM.
Protein preparation
Adrenodoxin (Adx) and CYP11A1 were purified from adrenal glands purchased from a local harvesting plant (Cargill; Milwaukee, WI). The purification processes were performed, as described in Supporting Information, according to published procedures.23,63,64
Sample preparation for resonance Raman
CYP11A1 purified from the adrenal cortex by the above procedure is recovered in a form that retains the natural substrate, cholesterol. The concentration of CYP11A1 for samples of the ferric and ferrous CO forms was 100 μM in 100 mM phosphate buffer pH 7.4, containing 0.1 mM dithiothreitol (DTT) and 0.1 mM ethylenediaminetetraacetic acid (EDTA). The concentration of enzyme in the samples used for measurement of the ν(Fe-S) stretching frequencies was 200 μM in 10 mM phosphate buffer pH 7.4, 0.1 mM DTT and 0.1 mM EDTA. The ratio of Adx and CYP11A1 was 1.2:1 for those samples containing Adx, noting that the samples containing the redox partner were incubated overnight at 4 °C to ensure full binding of Adx. In generating CYP11A1 samples bound with the other substrates, advantage was taken of the fact that the binding affinity of the other two substrates, 22R-OHCH and 20R,22R-DiOHCH, is much higher than that of cholesterol enabling them to displace the cholesterol from the active site. Thus these substrate-bound samples were made by adding, from an ethanol stock solution, a 3-5 fold excess of the hydroxycholesterol and incubating overnight at 4 °C to ensure full exchange.12,65
Regarding preparation of the various ligated forms, the ferrous CO adducts were made by addition of an excess amount of reducing agent, sodium dithionite (Na2S2O4), from stock solution. A 100 μL aliquot of a 100 μM CYP11A1 was placed in an NMR tube (WG-5 Economy, Wilmad) and sealed with a rubber septum (Sigma-Aldrich, Milwaukee, WI). The tube was connected to a vacuum line with a needle connection. The sample was sparged three times by exchange with Ar gas and then exchanged with CO gas two times to ensure the sample was saturated with CO. A freshly prepared solution of sodium dithionite dissolved in an Ar-degassed buffer was added to the solution of CYP11A1. The ferric sample was reduced by addition of ~2 molar equivalents of sodium dithionite solution (8 μL). The UV spectrum was monitored over the next few minutes, showing a sharp band rising at ~550 nm and a disappearing band at ~645 nm, attributable to the HS ferric form; the solution was allowed to incubate for 15 min to ensure full conversion.
The oxygenated samples of CYP11A1 were prepared from the ferric samples in the following manner. A 100 μL aliquot of a 200 μM ferric sample was placed in an NMR tube and connected to a vacuum line tube adapter. On the vacuum line the sample was alternately degassed and filled with Ar for 3 cycles. Addition of freshly degassed sodium dithionite solution was made by using a gas tight syringe through a rubber septum on the vacuum line system into the ferric sample. The NMR tube was gently tapped to allow the sample to mix well. In practice, the sample was titrated with the sodium dithionite solution until the rising the sharp band at ~550 nm reached a maximum and the band at ~645 nm had disappeared, as monitored by a special device (Model CHEM2000-VIS, Ocean Optics Inc., Dunedin, FL) capable of recording absorption spectra from samples contained in NMR tubes (WG-5 Economy, Wilmad). Then oxygen was added to this ferrous sample by bubbling 16O or 18O gas, using a vortex mixer to gently agitate the sample. During the reduction and oxygenation steps the sample tube was placed in an ice-cold bath at 0°C. The mixing time for oxygenation was different, depending on the substrate being studied; the samples containing the first two substrates, CH w/o Adx and 22R-OHCH w/o Adx, were mixed for 30 seconds, but the sample containing 20R, 22R-DiOHCH w/o Adx required mixing for 90 seconds to complete formation of the oxy-form13. These relatively short mixing times were used, because under these conditions, mixing times longer than a couple minutes gave rise to significant amounts of ferric forms. All samples were frozen by immersing the NMR tube in liquid nitrogen immediately after mixing. Some samples contained ethylene glycol or glycerol, the spectral results obtained being the same as for protein with buffer solution only.
Sample preparation for FTIR
Ferrous CO samples for FTIR measurement were only made for samples bound with 22R-OHCH and 20R, 22R-DiOHCH, the value for the CH bound sample being available from the rR data. The ferrous CO samples were made by adding a minimal amount of Na2S2O4 powder to the CO-saturated ferric sample prepared with 100 mM phosphate buffer containing 0.1 mM EDTA and 0.1 mM DTT in a 2 ml glass vial sealed with a rubber septum, and then incubating for 15 min at 4 °C to ensure full ligation. While addition of solid dithionite can cause pH changes when added in excess, care was taken to add very small amounts; it is noted that the FTIR spectra for these and similarly prepared samples of CO adducts of other P450s yielded spectra identical to those prepared by Tsubaki et al. and others. 30,50–52 Then the ferrous CO sample was transferred as quickly as possible to the FTIR cell, which had already been flushed with CO gas. The concentrations of the protein for the FTIR samples were 200-300 μM.
Resonance Raman measurements
Ferric CYP11A1 samples were measured using the 406.7 nm and 356.4 nm excitation lines, while the Fe(II)-CO adducts were excited by the 441.6 nm line provided by a He-Cd laser (IK Series, Kimmon Koha CO., Ltd.). Multiple excitation lines, including the 356.4, 406.7 and 415.4 nm lines from a Kr+ laser (Coherent Innova Sabre Ion Laser) and 441.6 nm from the He-Cd laser were employed to try to enhance the ν(O-O) modes of the oxy samples, but ν(O-O) and ν(Fe-O) features with adequate signal to noise ratios were obtained only when using the 415.4 nm Krypton lines or the 441.6 nm line from the He-Cd laser; it is noted that in many studies of dioxygen complexes of cytochromes P450, adequate enhancement of the internal modes of Fe-O-Ofragment is obtained with the 413 nm krypton line, but enhancement is slightly better with the 415.4 nm excitation used here. The rR spectra of all samples were collected using a Spex 1269 spectrometer equipped with Spec- 10 LN-cooled detector (Princeton Instruments). The slit width was set at 150 μm, and a 1200 g/mm grating was used; with this grating, the resultant spectral dispersion is 0.46 cm−1/pixel.
The laser power for the ferric sample was adjusted to ~10 mW, whereas for the ferrous CO adducts and oxy samples, it was kept at ~1 mW to minimize photodissociation. Moreover, to avoid laser-induced heating and protein degradation, the samples were contained in spinning NMR tubes. The 180° backscattering geometry was used for all measurements, and the laser beam was focused onto the sample using a cylindrical lens, so as to form a line-image to improve collection efficiency at a given photon flux at the sample surface.66 The ferric and ferrous CO samples were measured at room temperature, while the oxy samples were measured at 77K, employing an in-house designed immersion dewar, fitted with a NMR tube sample spinning device. Spectra were calibrated with fenchone (Sigma-Aldrich, WI), and processed with Grams/32 AI software (Galactic Industries, Salem, NH).
FTIR measurements
The infrared spectra were obtained using a 4020 Galaxy Series FT-IR spectrometer, Matson Instruments. The IR OTTLE33 cell purchased from New Era, when equipped with a Teflon spacer with 0.1 mm width placed between two CaF2 windows, requires only 60 uL of sample for each measurement. The infrared spectrophotometer was run in a single-beam mode. A spectrum was acquired with no sample cell present (i.e., air) to serve as a blank and then 6 separate spectral traces of ferric sample (each with 500 scans) were collected; the same procedure was used for collection of the ferrous-CO samples. The presented data are the difference spectra between the ferrous-CO and ferric samples; as mentioned above, the enzyme concentrations for these samples was 200-300μM.
RESULTS AND DISCUSSION
While the large majority of cytochrome P450-mediated oxidations proceed through the Compound I intermediate arising from proton-assisted O-O bond cleavage of the precursor ferric peroxo- species, any conditions which effectively restrict protonation of the latter can facilitate its attack on susceptible bound substrates.55–57,67–70 Indeed, rR evidence has been recently obtained for the presence of an H-bonding interaction with the proximal oxygen of the (Fe-Op-O) fragment, which supports this mechanism in the case of the C-C bond cleavage reaction that occurs for 17-hydroxy-pregnenolone (17-OH PREG) processing by CYP17A1;43,53 on the other hand, an H-bonding interaction with the terminal oxygen of the Fe-O-Ot fragment disfavors such a reaction, facilitating the multi-step protonation and O-O bond cleavage process that leads to formation of the Compound I species.16–22 Indeed, rR data recently acquired for the dioxy complexes of CYP19 revealed spectral patterns consistent with H-bonding to the distal oxygen atom of the Fe-O-O fragment, suggesting the likely involvement of Compound I mediation of the C-C bond cleavage process that produces estrone product,44 a conclusion which is in essential agreement with recent kinetic studies reported by Sligar and coworkers.71
Given that rR spectroscopy provides an especially effective probe of these functionally important H-bonding interactions with the Fe-O-O fragments of dioxy intermediates, it has herein been used to probe the dioxygen complexes obtained for CYP11A1 bound with each of its three natural substrates, CH, 22R-OHCH and 20R,22R-DiOHCH. However, before proceeding to a discussion of the new results obtained for the dioxygen adducts, in order to help clarify certain nebulous or incomplete results reported in previous studies,50–52 additional rR studies have been undertaken to further define active site structural differences that exist for the ferric and ferrous CO-bound forms with each of the three physiologically relevant substrates. In addition, the present studies provide further definition of the effects of adrenodoxin binding on the heme structure and its interactions with active site structural elements, including the Fe-S bond between the heme and the cysteine residue, a functionally crucial linkage that can be manipulated by slight structural alterations within the proximal heme pocket where adrenodoxin binding occurs.23–27
A. Ferric CYP11A1 and its interaction with adrenodoxin
Early rR studies of this enzyme were reported by Tsubaki et al. for the ferric and ferrous CO states, providing valuable insight into its active site structure.50–52 However, the interpretations of some of these data were hampered by the presence of strong glycerol bands that overlap with some structure-sensitive heme vibrational modes. Compared with previous work, the data presented below was acquired from samples prepared in aqueous conditions without glycerol, ethylene glycol or detergent, all of which were present in the previous study and complicated the interpretation by the presence of the additives’ Raman bands in both the low frequency and high frequency regions; e.g., a high intensity and wide glycerol band appearing at ~1467 cm−1 complicates observation of the ν3 mode of the HS species, which occurs near 1484 cm−1, 50–52 an overlap that can directly lead to the wrong interpretation of the spin population.
Now turning attention to the newly acquired data, the rR spectra of ferric CYP11A1 samples with CH, 22R-OHCH and 20R, 22R-DiOHCH substrates in high and low frequency regions, as well as in the presence of the natural redox partner, adrenodoxin (Adx), are shown in Figure 2. The high frequency spectra were normalized to the ν4 mode at 1370 cm−1 and the low frequency region was normalized to the ν7 mode near 674 cm−1; neither of these strong modes is included in these traces so as to allow a clearer presentation of the lower intensity structure-sensitive heme modes. The assignments of the modes were done according to the previous published data on cytochromes P450s and model compounds of Ni-octaethylporphyrin.29,72
Figure 2.
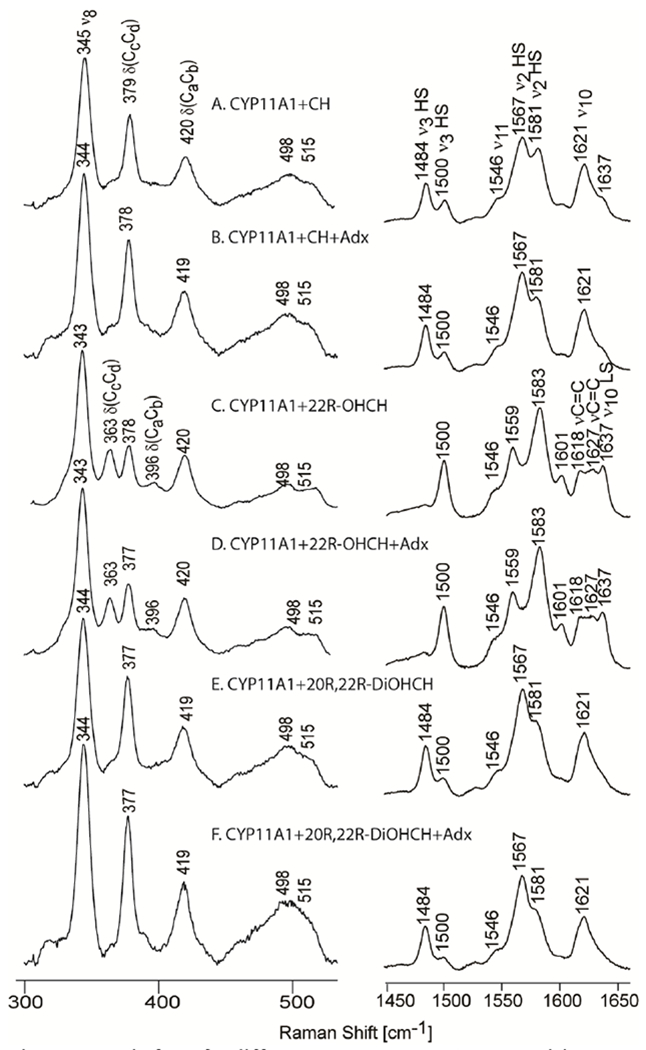
Ferric form for different substrates in buffer comprising 100 mM phosphate pH 7.4, 0.1 mM DTT and 0.1 mM EDTA. Excitation line 406 nm, acquisition time 60 min for low frequency and 30 min for high frequency at room temperature.
High frequency region:
The spectra of cholesterol (CH) bound samples (Figure 2 A, right panel) show components associated with both the high (HS) and low spin (LS) states. The population of high spin state is larger than the LS as seen by higher intensity of the high spin markers ν3 at 1484 cm−1, ν2 at 1567 cm−1 and ν10 at around 1621 cm−1 as compared to the corresponding low spin makers, which are seen at 1500 cm−1, 1581 cm−1 and 1637 cm−1, respectively. It is noted that the HS ν10 mode overlaps with the vinyl stretching modes that are seen more clearly in the spectrum of LS.33 Binding of 22R-OHCH induces an almost complete low spin state conversion, as seen by the presence of the dominant LS state markers ν3 at 1500 cm−1, ν2 ~1582 cm−1, and ν10 at 1637 cm−1. The absence of the HS ν10 mode in this region (now appearing for the LS state at 1637 cm−1) reveals the existence of two vinyl stretching modes at 1618 and 1627 cm−1, the lower frequency one being usually associated with the in-plane conformation of a vinyl group, with the higher frequency one being associated with the out-of-plane conformation.32,38 The fact that the 22R-OHCH substrate does not induce high spin conversion is not surprising, given the fact that the crystal structure of CYP11A1 with this substrate shows that there is a large amount of electron density between the O atom of the hydroxyl group attached to the 22 carbon of the substrate and the heme Fe, with the distance between O and Fe being only 2.56 Å, indicative of a covalent bond between heme iron and substrate.28,73 This observation is entirely consistent with the present rR data and generally consistent with that previously published.50–52 On the other hand, binding of 20R,22R-DiOHCH induces a mixture of spin states (76%HS) similar to, but slightly larger, than that seen for CH-bound sample (63%HS) (Figure 2E); i.e., interestingly, this diol containing substrate does not coordinate to the heme. The stated spin state populations for all three enzyme/substrate complexes were calculated using previously published HS/LS cross section ratios for cytochromes P450.31
Low frequency region:
The low frequency spectrum of the CH-bound sample (Figure 2A, left panel) shows the presence of the propionate bending mode at 379 cm−1 and vinyl bending mode at 420 cm−1. For the 22R-OHCH-bound sample an additional propionate bending mode at 363 cm−1 is activated, a frequency which generally signals the presence of a propionate group experiencing a weaker H-bonding interaction with active site fragments 32,41 Moreover, a small band appearing at 396 cm−1 was also enhanced and can be reasonably associated with weak activation of an in-plane vinyl bending mode. The presence of two vinyl bending modes associated with in-plane (396 cm−1) and out-of-plane (420 cm−1) vinyl group conformations in the low frequency region of the rR spectrum of the 22R-OHCH-bound sample is indeed quite consistent with the two vinyl stretching frequencies observed in the high frequency region, as was discussed above (Figure 2 C). Again, the binding of the third substrate (Figure 2 E, left panel) shows a spectral pattern quite similar to the CH-bound samples.
The effect of adrenodoxin on the ferric state:
As can be seen from the slightly increased intensity of ν3 spin state markers at around 1484 cm−1 from Figure 2B, 2D and 2F in Figure 2, for all three substrates, the addition of Adx to the samples of CYP11A1 causes only very small increases (5-10%) of the high spin population, as determined from the previously referenced procedure,31 with the detailed data being found in Supporting Information, Table S1. It was reported previously, using UV-Vis and resonance Raman spectroscopies, that the redox partner, Adx, promotes formation of more high spin component; e.g., for the CH bound form it was reported that upon binding of Adx the HS population increased from only about 50% to complete transformation to HS.50 While the results presented here did not show such large spin state changes, it has to be noted that the previous rR results were collected on samples that contained glycerol or propylene glycol, whose vibrational modes overlapped the HS state marker band at ~1484 cm−1, making it difficult to properly determine the percentage of HS component.50,51 It is emphasized that with the experimental conditions being used here, the ν3 marker band is quite isolated. In addition, studies we performed using the same experimental condition as previously published with proteins prepared in buffer containing 20% glycerol, still gave no significant changes in the spin state when Adx was added (data not shown). The effect of adrenodoxin binding on the rR spectra in the low frequency region is also insignificant, as seen by the lack of changes in the A/B, C/D and E/F pairs of traces (Figure 2, left panel).
The iron-sulfur linkage:
Obviously, the nature of the linkage between the heme prosthetic group and a given associated protein is quite important for dictating the reactivity patterns of the enzyme. 74–77 Indeed, the Fe-S linkage of cytochromes P450 and some related enzymes plays a key role in enabling these enzymes to mediate such remarkable chemical transformations under normal physiological conditions.1–3 Fortunately, it has been well established that rR spectroscopy, when employing a near UV excitation wavelength (e.g., near 350-360 nm), is one of the most effective probes of the status of this key Fe-S fragment, providing an easily identifiable ν(Fe-S) band in the low frequency rR spectrum.45–47 Specifically, the behavior of this mode can be documented to evaluate the linkage between the heme Fe and the endogenous cysteine thiolate ligand, the strength of which can be modulated by structural alterations within the proximal heme pocket, the most effective perturbations arising by interaction with natural redox partners such as Adx, that typically bind to the proximal side of the active site.
Several studies have consistently shown that the precise structure of substrates, all of which bind within the distal pocket of cytochromes P450, have little or no effect on the Fe-S linkage.46–48,78 However, it is anticipated that interactions with redox partners can impact the status of the Fe-S linkage.46,48,79 Results from the present work are shown in Figure 3, where the ν(Fe-S) is observed at 347 cm−1 for CYP11A1 bound with CH, with no detectable change being seen upon binding Adx. This is an unexpected result inasmuch as significant effects on the ν(Fe-S) mode are commonly seen for these types of interactions; e. g., binding of putidaredoxin to P450cam caused a strengthening of Fe-S, as witnessed by a shift of ν(Fe-S) by ~3 cm−1 to higher frequency.46 In addition, a differential effect on the Fe-S bond was seen with CYP2B4 interactions with Cyt b5 vs CPR.48 More recently, studies of nanodisc-associated CYP17 also provided evidence that binding of cytochrome b5 induces a 3 cm−1 shift of the ν(Fe-S) mode to higher frequency. 79
Figure 3.

Low frequency rR spectra of ferric cholesterol-bound CYP11A1 in 10 mM phosphate buffer, pH 7.4, 0.1 mM DTT and 0.1 EDTA. Excitation line was 356.4 nm, acquisition time 60 min at room temperature.
B. Ferrous-CO adduct and its interaction with different substrates and adrenodoxin:
Resonance Raman spectroscopic interrogation of the ferrous CO adducts of heme proteins has now long been established as an effective probe of proximal- and especially distal-pocket active site structure. The internal modes of the Fe-C-O fragment report on the strength of the Fe-L linkage with the trans-axial proximal ligand and simultaneously reflect steric and polar interactions with distal pocket residues, including those presented by enzyme-bound substrate,42,80–82 the latter interaction being crucially important for cytochromes P450, such as the CYP11A1 being studied here. Basically, increases in the degree of dπ(Fe) to CO(π*) back-bonding in the Fe-C-O fragment, arising increases in positive polarity of the distal pocket environment, strengthen the Fe-C bond while simultaneously weakening the C-O bond, resulting in a negative correlation between the ν(Fe-C) and ν(C-O) vibrational modes.42,80–82
While rR spectroscopy has been applied previously to interrogate the active site of the CO adducts of CYP11A1 with various substrates,50–52 one troubling issue occurred with the ferrous CO adducts, where it was reported that the rR spectrum of the 22R-OHCH-bound sample contained only one ν(Fe-C) stretching frequency, while the IR data clearly revealed the presence of two ν(C-O) stretching modes. The most reasonable explanation of this unexpected behavior is unintentional photodissociation of CO of one of the conformers during the Raman experiment. In the present work attempts are made to clarify the interpretation of the vibrational spectra of the ferric and ferrous CO adducts for all three physiologically important substrates and to further evaluate the effect of Adx binding on the Fe-C-O fragment of the CO adducts of CYP11A1.
The rR spectra acquired for the low frequency region are shown in Figure 4.
Figure 4.
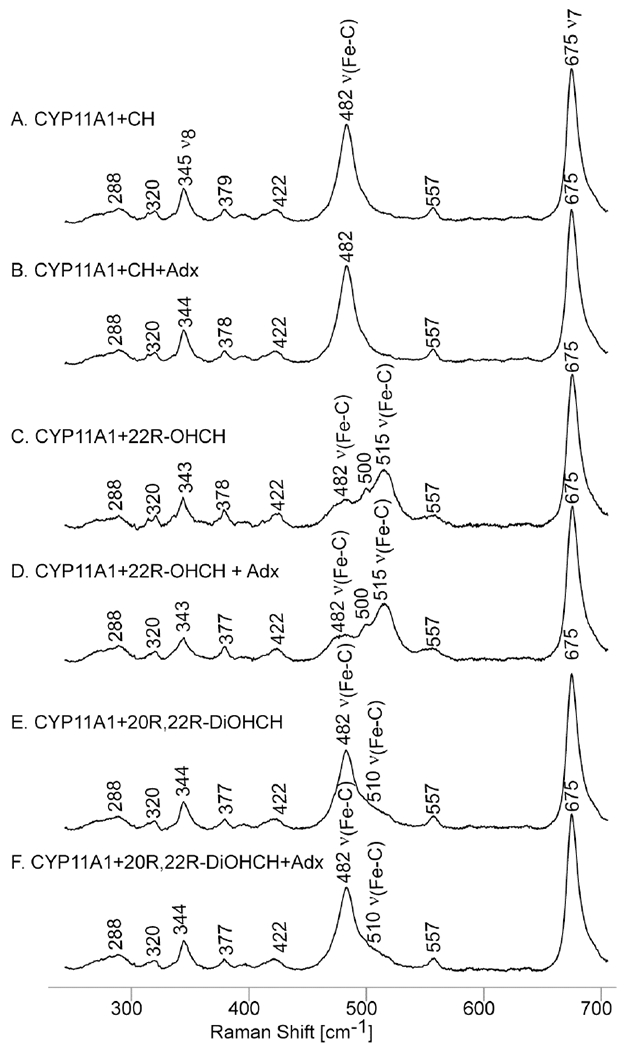
Low frequency rR spectra for CO adducts of CYP11A1 with different substrates in 100 mM phosphate buffer pH 7.4, 0.1 mM DTT and 0.1 EDTA. Excitation line 441.6 nm, acquisition time 60 min at room temperature.
The most obvious finding that is noted upon initial inspection of the data is that there are no observable effects of Adx binding upon the status of the Fe-C-O fragment. In similar studies reported earlier by other workers,52 the changes for Adx binding were reported to be quite small (between 0 −1.5 cm−1). However, the apparent existence of these small effects, along with the presence of some confusing results of those spectral studies, prompted us to reexamine this issue. For example, in those earlier studies it was reported that in some cases, including the sample containing the 22R-OHCH, while two ν(C-O) modes could be observed by FTIR, only a single ν(Fe-C) could be observed (by rR). As will be seen below, these confusing results can be readily explained by the occurrence of unintended photodissociation for the ferrous CO adducts by the rR laser excitation beam.
Now, returning to consider the effects of different substrates on the Fe-C-O fragment, the sample bound with cholesterol, a substrate that presents no hydrophilic R-OH fragments to the bound CO ligand, shows a clear and relatively sharp ν(Fe-C) mode appearing at 482 cm−1, a value in good agreement with the previous work,51 and the corresponding ν(C-O) being observed in the FTIR spectrum at 1952 cm−1, again a value in reasonably good agreement with that reported earlier (i.e., 1954 cm−1).51 In contrast to the relatively simple spectral signature displayed for the CO adduct of the CH-bound enzyme, the CO adduct formed for the enzyme bound with the more polar substrate, 22R-OHCH, whose C22-OH fragment is apparently close enough to the iron binding site to have induced a LS spin state by direct interaction with the heme iron as evidenced in the rR data for the ferric state (Figure 2), gave three observed features appearing in the region where the ν(Fe-C) modes are expected to appear: i.e., at 482 cm−1, 500 cm−1 and 515 cm−1. In order to clarify the interpretation of the spectra acquired for the 22R-OHCH substrate, the Fe(II)-12C16O and Fe(II)-13C16O samples were made and measured with the results being shown in Figure 5. It is clear that both 481 cm−1 and 515 cm−1 bands seen in trace A, shifted down by 5 cm−1 in the spectra of 13C16O isotope, confirming their assignments to ν(Fe-C) modes. On the other hand, the feature observed at 500 cm−1 does not shift upon 12C/13C substitution, securing its assignment to a heme mode. The assignment of the 481cm−1 and 515 cm−1 bands to two different Fe-C-O conformers is supported by observation two high frequency ν(C-O) modes at 1952 cm−1 and 1934 cm−1 in the FTIR, as shown in Figure 6. In previously published work,51 the appearance of only one band at low frequency was reported for this complex, an observation which may be due to accidental photo dissociation perhaps owing to either high laser power (~5 mW) or ineffective spinning during the measurement. Indeed, results of experiments to verify photosensitivity for this complex are shown in the bottom two traces of Figure 5, where it is seen that in the spectra acquired at high photon flux (traces C and D), there is a strong and wide heme mode, with its expected insensitivity to the 13C16O substitution which overlapped with the ν(Fe-C) modes. From previously published data it was reported that only one ν(Fe-C) mode could be detected (at 479 cm−1) for the 22R-OHCH bound CYP11A1 complex. Obviously, this could have arisen from the heme mode seen at 479 cm−1 in the photo-disassociated sample we have generated in the present work (Figure 5 traces C and D).
Figure 5.

Isotope effect for 22R-OHCH CO adducts in buffer comprising 100 mM phosphate pH 7.4, 0.1 mM DTT and 0.1 EDTA. Excitation line 442.4 nm, acquisition time 60 min at room temperature. Ferrous CO adducts with 12CO isotope (A), 13CO (B) and their photodissociated forms, (C) and (D), respectively.
Figure 6.
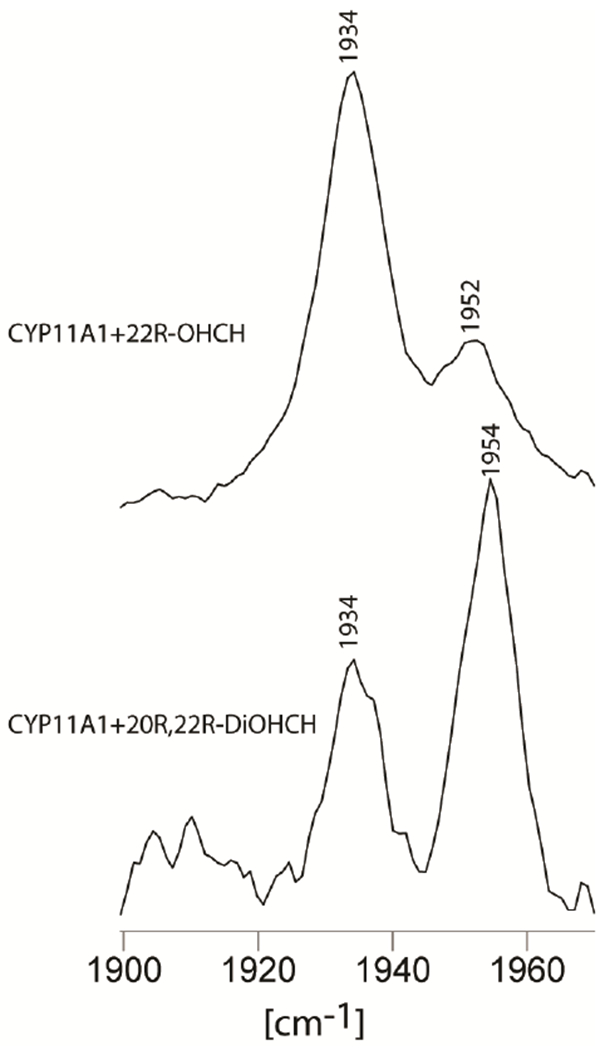
FTIR results for high frequency CYP11A1 with substrate 22R-OHCH and 20R, 22R-DiOHCH ferrous CO forms in 100 mM phosphate buffer pH 7.4, 0.1 mM DTT and 0.1 EDTA.
Given these clarified spectral traces for the 22R-OHCH-bound ferrous CO adduct, the most reasonable conclusion for this species is that this directed C-O-H group interacts with the Fe-C-O fragment to produce two different Fe-C-O conformers. Furthermore, it is also quite reasonable to conclude that the appearance of two ν(Fe-C) and two ν(C-O) modes for the 20R,22R-DiOHCH-bound substrate is also attributable to interactions of one or both C-OH groups with the Fe-C-O fragment; indeed, the two pairs of ν(Fe-C)/ν(C-O) frequencies are quite similar for both complexes. From this collective data set for the three substrates, it seems likely that (for the 2nd and 3rd substrates) the presence of one C-OH group positioned quite near the Fe-C-O fragment leads to two Fe-C-O conformers, one with a disposition similar to that of the “unperturbed” Fe-C-O conformer of the CH-bound enzyme and the other reflecting a reasonably strong H-bonding interaction with the C-OH group present on the substrate.
C. The effects of substrate structure and Adx binding on the Fe-O-O fragment of dioxygen-ligated intermediates of CYP11A1
As was discussed above, recent rR studies of the oxy intermediates of CYP17 and CYP19,43,44 along with analogous studies of NOS,59,67,69,70,83 have shown that this technique is effective in detecting subtle structural differences in H-bond interactions with the Fe-O-O fragment. Specifically, H-bond donation to the proximal oxygen (Op) of the Fe-Op-Ot fragment, detected for the dioxy-adduct of the 17-OH PREG sample,43 which has recently been shown by rR spectroscopy to persist upon reduction to the peroxo intermediate,53 stabilizing it for attack on susceptible substrates, exhibits a relatively low ν(Fe-O) frequency (528 cm−1). On the other hand, H-bonding to the terminal oxygen (Ot) of the Fe-Op-Ot fragment of the 17-OH PROG-bound sample, which exhibits relatively high ν(Fe-O) frequency of 546 cm−1,43 promotes formation of the hydroperoxo intermediate, ultimately leading to O-O bond cleavage and Compound I formation, an intermediate that effectively mediates the more extensively studied hydroxylation reactions. Clearly, the rR technique is an important tool to document these structural variations that, nevertheless, carry profound functional consequences.14,15
Given this demonstrated utility of rR spectroscopy for structural definition of these active site Fe-O-O fragments, efforts were made here, for the first time, to acquire rR spectra of these dioxygen adducts of CYP11A1 with all three natural substrates, seeking to detect any telltale differences in the rR spectra that might reveal important H-bonding differences for the three substrates, as well as allowing detection of rearrangements of such-bonding interactions upon association of CYP11A1 with Adx. In order to study these relatively unstable oxy-intermediates encountered within the catalytic cycles for these three natural substrates of CYP11A1, special procedures were required, as summarized earlier in Methods and Materials. Inasmuch as no differential effects of the three substrates on the Fe-S linkage in the heme proximal pocket were seen in the studies of the ν(Fe-S) modes (vide supra), all effects observed here are presumed to arise from distal side structural perturbations.
Cholesterol bound oxy-CYP11A1:
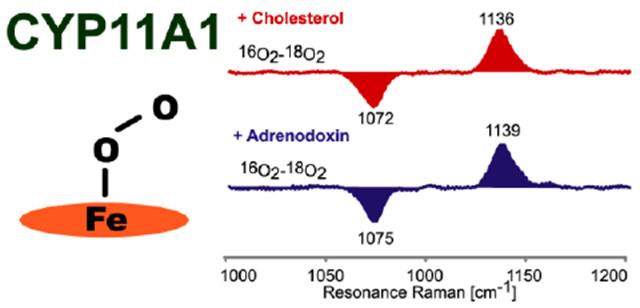
The 415.4 nm line from a Krypton ion laser, which in resonance with the Soret transition of the CH-bound CYP11A1, was used to measure this complex. It is noted that, owing to relatively low S/N attainable for these frozen oxy- CYP samples, definitive identification of the key internal modes of the Fe-O-O fragments is most readily accomplished via generation of the 16O2/18O2 difference traces, which are shown in Figure 7. The ν(O-O) modes are seen in the high frequency region, shown in traces (A) and (B) (large spectrum) for samples with and without Adx, respectively. The ν(Fe-O) modes, seen in the low frequency region are shown in traces (A) and (B) (small spectrum) for oxy-CYP11A1 with and without Adx.
Figure 7.
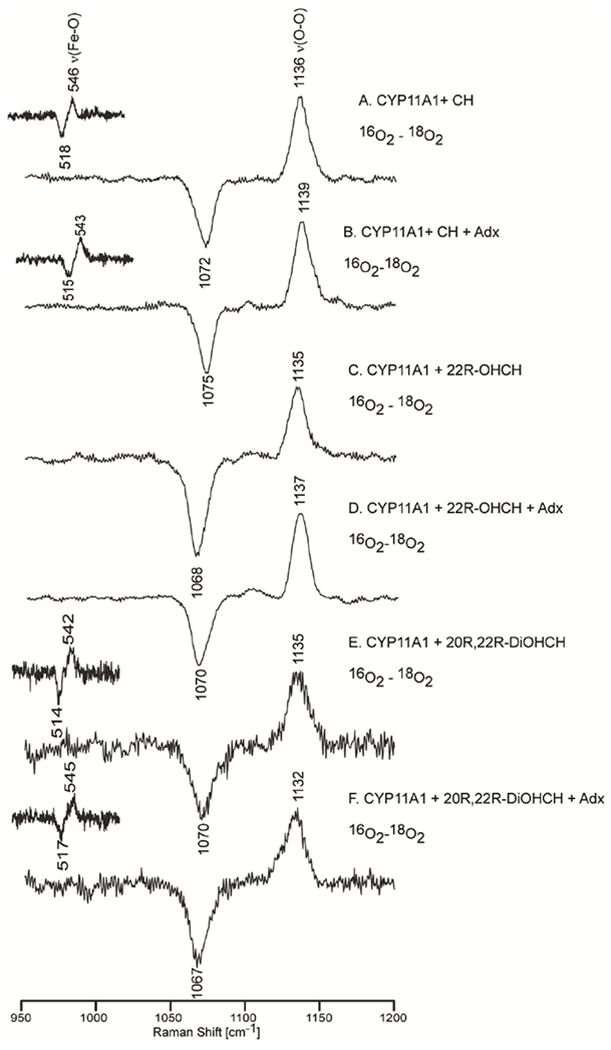
Oxy samples for all the substrates with and without Adx in 100 mM phosphate buffer containing 0.1 mM EDTA and 0.1 mM DTT. The insets above show the ν(Fe-16O) - ν(Fe-18O) difference features.
In the low frequency spectra, the ν(Fe-16O) is assigned at 546 cm−1 and the ν(Fe-18O) at 518 cm−1 for the cholesterol-bound form, a shift of 28 cm−1 (calculated 25 cm−1). As was outlined above in the first paragraphs of this section, this set of values for ν(Fe-O) and ν(O-O) is quite similar to that obtained for camphor-bound oxyCYP101 and for oxyCYP17 and oxyCYP19 with those substrates that are known to be converted via Compound I processing. The conclusion emerging from this is that conversion of CH to 22-OHCH is also likely to proceed through a Compound I intermediate. This finding has long been generally accepted, with recent EPR studies providing results which strongly support the proposed involvement of Compound I in this first step.60 Significantly, the sample prepared including the Adx exhibits a ν(Fe-16O) at 543 cm−1 and a corresponding ν(Fe-18O) at 515 cm−1, this ~3 cm−1 down shift of ν(Fe-O) upon addition of Adx reflects a slight weakening of the Fe-O bond when Adx is present. On the other hand, the ν(16O-16O) (at 1136 cm−1) and ν(18O-18O) (at 1072 cm−1) modes shift up to 1139 cm−1 and 1075 cm−1 upon addition of Adx, the combined results suggesting that the interaction with Adx might slightly weaken the H-bonding interaction with the distal oxygen atom of the O-O bond.
22R-OHCH-bound oxyCYP11A1:
Figure 7 shows the difference spectra for this species, both in the absence and presence of Adx. The high frequency region (right panel, traces C and D) shows the same pattern as was observed for the cholesterol-bound sample, exhibiting the ν(16O-16O) at 1135 cm−1, with the ν(18O-18O) being observed at 1068 cm−1. Also like the CH-bound sample, upon Adx binding, the ν(16O-16O) shifts up to 1137 cm−1, with the corresponding ν(18O-18O) appearing at 1070 cm−1, an isotopic shift in reasonable agreement with that expected for a diatomic O-O oscillator (~65 cm−1). Unfortunately, though numerous attempts to observe the telltale ν(Fe-O) mode were made, employing multiple excitation lines, including the 441.6 nm line from a He:Cd laser, no evidence was obtained for its enhancement. These efforts are summarized in the Supporting Information, along with a discussion of structural and electronic factors that can affect the degree of resonance enhancement of the modes that are essentially isolated to the Fe-O-O fragment. In the absence of observable ν(Fe-O) modes, it is not possible to draw conclusions about the status of the Fe-O-O fragment with respect to its disposition towards the O-O bond cleavage process involved in the hydroxylation pathways. Nevertheless, the well-documented generation of the 20R,22R-DiOHCH product is consistent with the proposal that a Compound I intermediate is involved. Indeed, recent experimental work reported by Hoffman and coworkers, employing cryoradiolysis studies with EPR detection has provided convincing arguments for this assertion.60,61
20R, 22R-DiOHCH bound oxy-CYP11A1:
Figure 7 also shows the difference spectra obtained for the oxy complexes of the 20R, 22R-DiOHCH-bound CYP11A1 complex with and without Adx. The difference patterns for the spectral region containing the ν(O-O) modes were measured with Soret excitation (415.4 nm), as was done for the previous substrates. However, no clear difference pattern for ν(Fe-O) could be observed in the low frequency region. The same efforts were employed as were used to try to enhance the ν(Fe-O) modes of the 22R-OHCH sample (vide supra). In this case, the ν(Fe-O) mode was effectively enhanced using the 441.6 nm excitation line from the He:Cd laser. The data acquired for the enzyme bound with the 3rd substrate, 20R,22R-DiOHCH, reveal the ν(Fe-16O) stretching mode at 542 cm−1 and the ν(16O-16O) mode at 1135 cm−1. Interestingly, the effect of Adx binding on this species is opposite to its effect on the enzyme bound with the first two substrates; i.e., Adx binding to the samples containing the first two substrates increases the strength of the O-O bond, simultaneously decreasing the strength of the Fe-O bond (in the case of the CH-bound form, wherein Raman scattering by ν(Fe-O) is enhanced), while the effect of Adx binding on the sample containing 20R,22R-DiOHCH is to increase the strength of the Fe-O bond and decrease the strength of the O-O bond.
A key finding in the studies of these oxy complexes with the three natural substrates is that the CYP11A1+CH oxy complex yields frequencies for the ν(Fe-O) and ν(O-O) modes that are very similar with those obtained for the oxy complex of CYP11A1 bound to 20R,22R-DiOHCH; i.e., both are quite similar to those seen for the oxy complex of P450cam complexes and those observed for oxy CYP17A1 bearing the 17-OH PROG substrate, all of which encounter hydrogen bonding interactions involving the terminal oxygen of the Fe-O-O fragment, interactions which promote O-O bond cleavage and Compound I formation.18,44,71 This leads to the conclusion that the bond cleavage step likely proceeds through Compound I intermediate, in agreement with previous work.60,61 With the addition of Adx, the ν(Fe-O) increases, indicating the strengthening the Fe-O bond; the observed decrease of the ν(O-O) frequency indicates the corresponding weakening the O-O bond. Both of these shifts are consistent with a slightly stronger hydrogen bond donation to the terminal oxygen, a change that would lower peroxo-reactivity and further promote O-O bond cleavage of the Fe-O-O fragment and formation of Compound I.
D. Relationship of findings by rR spectroscopy to recent EPR/ENDOR work
Finally, it is of interest to point out that results reported for EPR/ENDOR studies of CYP11A1, with all three physiologically important substrates,60,61 provide indirect evidence from analysis of cryoreduced samples, that the enzyme substrate complexes formed with each of these three substrates gave rise to oxy complexes existing in three different conformations, with H-bonded forms generating hydroperoxo species following cryoradiolysis at 77K, while non-H-bonded oxy precursors yielded either a ferric peroxo- species or, in the special case of 220H-CH, a ferrous superoxo species.60,61,84 In support of this argument, detailed analysis of the rR spectra of these oxygenated precursors shown in Supporting Information (Figure S1) is consistent with the presence of three conformers in each of the substrate-bound forms, as had been deduced from analysis of the observed EPR/ENDOR spectra of the cryoreduced samples.60,61 The only somewhat surprising result from the earlier EPR work is that, while a non-H-bonded form in the oxy complex when CH is bound gives rise to a peroxo-species at 77K, the non-H-bonded species in the oxy complex with 22-OH-CH substrate was correlated with a ferrous superoxo- species in the cryoreduced sample;60,61 i.e., the two samples display similar rR spectra of the oxy precursors but give rise to two different formulations of isoelectronic species in the cryoreduced forms. One possible explanation arises in considering subtle but potentially effective H-bonding differences that might exist in these two cases. Thus, while the C22-OH fragment of bound 22-OH-CH is known to coordinate to the heme iron, neither the C20-OH nor the C22-OH groups coordinate to the heme iron when 20,22-DiOH-CH is bound; i.e., there are apparently critical active site H-bonding interactions that position the 22-OH fragment of only the 22-OH-CH substrate in an orientation that directs the unpaired electrons to interact with the heme iron. Such interaction might persist in both the dioxy and cryoreduced sample, such that the lone pair interacts directly with the bound-Fe-O-O superoxo fragment, favoring the generation of the ferrous superoxo rather than the ferric peroxo formulation.
Summary
These newly acquired rR data for the ferric and ferrous CO forms of CYP11A1 are partially consistent with what was published previously, but also clarify certain ambiguities arising from the apparent photo-dissociation of CO during the measurement in the case of the complex between 22R-OHCH and CYP11A1 in the ferrous-CO form.50–52 The present data clearly show that the enzyme bound with the non-H-bonding substrate, CH, yields only one set of vibrational modes for the CO adduct; i.e., ν(Fe-C) at 482 cm−1 and ν(C-O) at 1952 cm−1. On the other hand, for the two substrates possessing hydroxyl substituents that are well positioned to H-bond with the bound Fe-C-O fragment, two sets of Fe-C-O vibrational modes are seen. Thus, for both enzyme/substrate complexes, a ν(Fe-C)/ν(C-O) pair is seen at 482/1954 cm−1, which is quite similar that that observed for the non-H-bonding CH substrate. On the other hand, the presence of one or two H-bonding hydroxyl groups near the Fe-C-O fragment generates a new set of modes having the ν(Fe-C) mode at 515 cm−1 (or 510 cm−1) and the ν(C-O) mode at 1934 cm−1, both sets of data being entirely consistent with H-bonding to the Fe-C-O fragment. The data acquired for the protein-protein complex between CYP11A1 and Adx also show the effect of Adx on the active sites of the ferric and ferrous-CO forms to be minimal.
Finally, this first study of the rR spectra of the dioxygen adducts of CYP11A1 with all three substrates revealed vibrational spectral patterns that are quite similar. Based on conclusions drawn from work on dioxygen adducts of several other cytochromes P450, including CYP101, CYP17 and CYP19, the frequencies of the ν(Fe-O) and ν(O-O) observed here are those expected for Fe-O-O fragment that eventually converts to Compound I intermediates upon reduction. Though it can be argued that the effective H-donor for the various cases might be different (i.e., water for CH and substrate hydroxyl group(s) for the other two substrates), the Fe-O-O fragment in each case is apparently poised to generate the classic Compound I active oxidant. Finally, it is noted that, unlike the CO adducts, binding of adrenodoxin does have a significant impact on the status of the dioxygen complexes, presumably owing to an Adx-induced change in H-bonding to the Fe-O-O fragment, which is known to be more sensitive to this interaction than are Fe-C-O fragments.42
Supplementary Material
Acknowledgments:
This work was supported by National Institutes of Health Grant GM96117 to J.R.K.
References
- (1).Sigel A, Sigel H, and Sigel RKO (2007) “The Ubiquitous Roles of Cytochrome P450 Proteins”, in Metal Ions in Life Sciences. John Wiley & Sons, Ltd. [Google Scholar]
- (2).Ortiz de Montelano PR (2015) Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum Publisher, New York. [Google Scholar]
- (3).Miller WL, and Auchus RJ (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev 32, 81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Stone D, and Hechter O (1954) Studies on ACTH action in perfused bovine adrenals: The site of action of ACTH in corticosteroidogenesis. Arch. Biochem. Biophys 51, 457–469. [DOI] [PubMed] [Google Scholar]
- (5).Simpson ER, and Boyd GS (1966) The cholesterol side-chain cleavage system of the adrenal cortex: A mixed-function oxidase. Biochem. Biophys. Res. Commun 24, 10–17. [DOI] [PubMed] [Google Scholar]
- (6).Burstein S, and Gut M (1976) Intermediates in the conversion of cholesterol to pregnenolone: Kinetics and mechanism. Steroids 28, 115–131. [DOI] [PubMed] [Google Scholar]
- (7).Hume R, Kelly RW, Taylor PL, and Boyd GS (1984) The catalytic cycle of cytochrome P-450scc and intermediates in the conversion of cholesterol to pregnenolone. Eur. J. Biochem 140, 583–591. [DOI] [PubMed] [Google Scholar]
- (8).Lambeth JD, Seybert DW, Lancaster JR Jr., Salerno JC, and Kamin H (1982) Steroidogenic electron transport in adrenal cortex mitochondria. Mol. Cell. Biochem 45, 13–31. [DOI] [PubMed] [Google Scholar]
- (9).Ewen KM, Kleser M, and Bernhardt R (2011) Adrenodoxin: the archetype of vertebrate-type [2Fe-2S] cluster ferredoxins. Biochim Biophys Acta 1814, 111–125. [DOI] [PubMed] [Google Scholar]
- (10).Bangcharoenpaurpong O, Rizos AK, Champion PM, Jollie D, and Sligar SG (1986) Resonance Raman detection of bound dioxygen in cytochrome P-450cam. J Biol Chem 261, 8089–8092. [PubMed] [Google Scholar]
- (11).Hu S, Schneider AJ, and Kincaid JR (1991) Resonance raman studies of oxycytochrome P450cam: effect of substrate structure on v(O-O) and v(Fe-O). J. Am. Chem. Soc 113, 4815–4822. [Google Scholar]
- (12).Tuckey RC, and Kamin H (1982) The Oxyferro Complex of Adrenal Cytochrome P-450ccc. Effect of cholesterol and intermediates on its stability and optical characteristics. J. Biol. Chem 257, 9309–9314. [PubMed] [Google Scholar]
- (13).Tuckey RC, and Kamin H (1983) Kinetics of O2 and CO Binding to Adrenal Cytochrome P-450scc: Effect of Cholesterol, Intermediates and Phosphatidylcholine Vesicles. J. Biol. Chem 258, 4232–4237. [PubMed] [Google Scholar]
- (14).Akhtar M, and Wright JN (2015) Acyl-Carbon Bond Cleaving Cytochrome P450 Enzymes: CYP17A1, CYP19A1 and CYP51A1. Adv. Exp. Med. Biol 851, 107–130. [DOI] [PubMed] [Google Scholar]
- (15).Akhtar M, Wright JN, and Lee-Robichaud P (2011) A Review of Mechanistic Studies on Aromatase (CYP19) and 17α-Hydroxylase-17,20-Lyase (CYP17). J. Steroid Biochem. 125, 2–12. [DOI] [PubMed] [Google Scholar]
- (16).Denisov IG, Makris TM, Sligar SG, and Schlichting I (2005) Structure and chemistry of cytochrome P 450. Chem. Rev. (Washington, DC, United States) 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- (17).Rittle J, and Green MT (2010) Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (18).Groves JT (2006) High-valent iron in chemical and biological oxidations. J. Inorg. Biochem 100, 434–447. [DOI] [PubMed] [Google Scholar]
- (19).Meunier B, de Visser SP, and Shaik S (2004) Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (20).Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, and Green MT (2013) Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McQuarters AB, Wolf MW, Hunt AP, and Lehnert N (2014) 1958-2014: After 56 years of research, cytochrome P 450 reactivity is finally explained. Angew. Chem. Int. Ed 53, 4750–4752. [DOI] [PubMed] [Google Scholar]
- (22).Groves JT (2014) Enzymatic C-H bond activation: Using push to get pull. Nat. Chem 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lambeth JD, Seybert DW, Kamin H, Chem JB, David J, and Seybert W (1979) Ionic effects on adrenal steroidogenic electron transport. The role of adrenodoxin as an electron shuttle. J. Biol. Chem 254, 7255–7264. [PubMed] [Google Scholar]
- (24).Kido T, and Kimura T (1979) The formation of binary and ternary complexes of cytochrome P-450scc with adrenodoxin and adrenodoxin reductase.adrenodoxin complex. J Biol Chem 254, 11806–11815. [PubMed] [Google Scholar]
- (25).Turko IV, Adamovich TB, Kirillova NM, Usanov S,A, and Chashchin V (1989) Cross-linking studies of the cholesterol hydroxylation system from bovine adrenocortical mitochondria. Biochim. Biophys. Acta 996, 37–42. [DOI] [PubMed] [Google Scholar]
- (26).Lambeth JD, and Pemberg OS (1983) Cytochrome P-450scc-Adrenodoxin Complex. J. Biol. Chem 258, 5596–5602. [PubMed] [Google Scholar]
- (27).Lambeth JD, and Kriengsiri S (1985) Cytochrome P-450scc-Adrenodoxin Interactions. J. Biol. Chem 260, 8810–8816. [PubMed] [Google Scholar]
- (28).Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, and Park H-W (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. USA 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kincaid JR (2000) Resonance Raman spectra of heme proteins and model compounds, in The Porphyrin Handbook, (Kadish KM, Smith K, and Guilard R, Eds.), pp 225–291, Academic Press. [Google Scholar]
- (30).Mak PJ (2016) Resonance Raman spectroscopy as a structural probe of cytochrome P450 enzymatic cycle, in Handbook of Porphyrin Science (Kadish KM, Smith K, and Guilard R, Eds.), pp 1–120. World Scientific Publishing Co., Singapore. [Google Scholar]
- (31).Mak PJ, Zhu Q, and Kincaid JR (2013) Using resonance Raman cross-section data to estimate the spin state populations of Cytochromes P450. J. Raman Spectrosc. 44, 1792–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chen Z, Ost TWB, and Schelvis JPM (2004) Phe393 Mutants of Cytochrome P450 BM3 with Modified Heme Redox Potentials Have Altered Heme Vinyl and Propionate Conformations. Biochemistry 43, 1798–1808. [DOI] [PubMed] [Google Scholar]
- (33).Mak PJ, Kaluka D, Manyumwa ME, Zhang H, Deng T, and Kincaid JR (2008) Defining resonance Raman spectral responses to substrate binding by cytochrome P450 from Pseudomonas putida. Biopolymers 89, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Smulevich G, Feis A, Howes BD, Ivancich A, Edited by Kadish Karl M. Smith KM, and Guilard R (2010) Structure-function relationships among heme peroxidases: New insights from electronic absorption, resonance Raman and multifrequency electron paramagnetic resonance spectroscopies. Hand. Porphyr. Sci 6, 367–453. [Google Scholar]
- (35).Friedman JM (1994) Time-resolved resonance Raman spectroscopy as probe of structure, dynamics, and reactivity in hemoglobin. Meth. Enzym 232, 205–231. [DOI] [PubMed] [Google Scholar]
- (36).Kitagawa T, and Mizutani Y (1994) Resonance Raman spectra of highly oxidized metalloporphyrins and heme proteins. Coord. Chem. Rev 135/136, 685–735. [Google Scholar]
- (37).Terner J, Palaniappan V, Gold A, Weiss R, Fitzgerald MM, Sullivan AM, and Hosten CM (2006) Resonance Raman spectroscopy of oxoiron(IV) porphyrin pi-cation radical and oxoiron(IV) hemes in peroxidase intermediates. J. Inorg. Biochem 100, 480–501. [DOI] [PubMed] [Google Scholar]
- (38).Marzocchi MP, and Smulevich G (2003) Relationship between heme vinyl conformation and the protein matrix in peroxidases. J. Raman Spectrosc. 34, 725–736. [Google Scholar]
- (39).Hu S, Smith KM, and Spiro TG (1996) Assignment of Protoheme Resonance Raman Spectrum by Heme Labeling in Myoglobin. J. Am. Chem. Soc 118, 12638–12646. [Google Scholar]
- (40).Rwere F, Mak PJ, and Kincaid JR (2008) The impact of altered protein-heme interactions on the resonance raman spectra of heme proteins. Studies of heme rotational disorder. Biopolymers 89, 179–186. [DOI] [PubMed] [Google Scholar]
- (41).Cerda-Colon JF, Silfa E, and Lopez-Garriga J (1998) Unusual rocking freedom of the heme in the hydrogen sulfide-binding hemoglobin from Lucina pectinata. J. Am. Chem. Soc 120, 9312–9317. [DOI] [PubMed] [Google Scholar]
- (42).Spiro TG, Soldatova AV, and Balakrishnan G (2013) CO, NO and O2 as Vibrational Probes of Heme Protein Interactions. Coord. Chem. Rev 257, 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gregory M, Mak PJ, Sligar SG, and Kincaid JR (2013) Differential hydrogen bonding in human CYP17 dictates hydroxylation versus lyase chemistry. Angew. Chem. Int. Ed. Engl 52, 5342–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mak PJ, Luthra A, Sligar SG, and Kincaid JR (2014) Resonance Raman spectroscopy of the oxygenated intermediates of human CYP19A1 implicates a Compound I intermediate in the final lyase step. J. Am. Chem. Soc 136, 4825–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Champion PM, Stallard BR, Wagner GC, and Gunsalus IC (1982) Resonance Raman detection of an Fe-S bond in cytochrome P450cam. J. Am. Chem. Soc 104, 5469–5473. [Google Scholar]
- (46).Unno M, Christian JF, Benson DE, Gerber NC, Sligar SG, and Champion PM (1997) Resonance Raman investigations of cytochrome P450cam complexed with putidaredoxin. J. Am. Chem. Soc 119, 6614–6620. [Google Scholar]
- (47).Mak PJ, Yang Y, Im S, Waskell LA, and Kincaid JR (2012) Experimental Documentation of the Structural Consequences of Hydrogen-Bonding Interactions to the Proximal Cysteine of a Cytochrome P450. Angew. Chem. Int. Ed 51, 10403–10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mak PJ, Im S-C, Zhang H, Waskell LA, and Kincaid JR (2008) Resonance Raman studies of cytochrome P450 2B4 in its interactions with substrates and redox partners. Biochemistry 47, 3950–3963. [DOI] [PubMed] [Google Scholar]
- (49).Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SG, Hoffman BM, and Champion PM (2001) Resonance Raman and EPR Investigations of the D251N Oxycytochrome P450 cam/Putidaredoxin Complex. Biochemistry 4, 6852–6859. [DOI] [PubMed] [Google Scholar]
- (50).Tsubaki M, Hiwatashi A, and Ichikawa Y (1986) Effects of cholesterol and adrenodoxin binding on the heme moiety of cytochrome P-450scc: a resonance Raman study. Biochemistry 25, 3563–3569. [DOI] [PubMed] [Google Scholar]
- (51).Tsubaki M, Hiwatashi A, and Ichikawa Y (1987) Effects of cholesterol analogues and inhibitors on the heme moiety of cytochrome P-450scc: a resonance Raman study. Biochemistry 26, 4535–4540. [DOI] [PubMed] [Google Scholar]
- (52).Tsubaki M, Yoshikawa JS, Ichikawa Y, and Yuli N (1992) Effects of Cholesterol Side-Chain Groups and Adrenodoxin Binding on the Vibrational Modes of Carbon Monoxide Bound to Cytochrome P-450scc: Implications of the Productive and Nonproductive Substrate Bindings. Biochemistry 31, 8991–8999. [DOI] [PubMed] [Google Scholar]
- (53).Mak PJ, Gregory MC, Denisov IG, Sligar SG, and Kincaid JR (2015) Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. USA 112, 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Spiro TG, and Soldatova AV (2012) Ambidentate H-bonding of NO and O2 in heme proteins. J. Inorg. Biochem 115, 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sen K, and Hackett JC (2010) Peroxo–Iron Mediated Deformylation in Sterol 14α-Demethylase Catalysis. J. Am. Chem. Soc 132, 10293–10305. [DOI] [PubMed] [Google Scholar]
- (56).Cho K-B, and Gauld JW (2005) Second half-reaction of nitric oxide synthase: computational insights into the initial step and key proposed intermediate. J. Phys. Chem. B 109, 23706–23714. [DOI] [PubMed] [Google Scholar]
- (57).Pant K, and Crane BR (2006) Nitrosyl - Heme Structures of Bacillus subtilis Nitric Oxide Synthase Have Implications for Understanding Substrate Oxidation. Biochemistry 45, 2537–2544. [DOI] [PubMed] [Google Scholar]
- (58).Lu C, Egawa T, Wainwright LM, Poole RK, and Yeh S-R (2007) Structural and functional properties of a truncated hemoglobin from a food-borne pathogen Campylobacter jejuni. J. Biol. Chem 282, 13627–13636. [DOI] [PubMed] [Google Scholar]
- (59).Chartier FJM, and Couture M (2007) Substrate-specific interactions with the heme-bound oxygen molecule of nitric-oxide synthase. J. Biol. Chem 282, 20877–20886. [DOI] [PubMed] [Google Scholar]
- (60).Davydov R, Gilep AA, Strushkevich NV, Usanov SA, and Hoffman BM (2012) Compound I Is the Reactive Intermediate in the First Monooxygenation Step during Conversion of Cholesterol to Pregnenolone by Cytochrome P450scc: EPR/ENDOR/Cryoreduction/Annealing Studies. J. Am. Chem. Soc 134, 17149–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Davydov R, Strushkevich N, Smil D, Yantsevich A, Gilep A, Usanov S, and Hoffman BM (2015) Evidence That Compound I Is the Active Species in Both the Hydroxylase and Lyase Steps by Which P450scc Converts Cholesterol to Pregnenolone: EPR/ENDOR/Cryoreduction/Annealing Studies. Biochemistry 54, 7089–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yoshimoto FK, Jung IJ, Goyal S, Gonzalez E, and Guengerich FP (2016) Isotope-Labeling Studies Support the Electrophilic Compound I Iron Active Species, FeO3+, for the Carbon-Carbon Bond Cleavage Reaction of the Cholesterol Side-Chain Cleavage Enzyme, Cytochrome P450 11A1. J. Am. Chem. Soc 138, 12124–12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tuckey RC, and Stevenson PM (1984) Properties of Bovine Luteal Cytochrome P450scc Incorporated into Artificial Phospholipid Vesicles. Int. J. Biochem 16, 497–503. [DOI] [PubMed] [Google Scholar]
- (64).Tuckey RC, and Stevenson PM (1984) Properties of Ferredoxin Reductase and Ferredoxin from the Bovine Corpus Luteum. Int. J. Biochem 16, 489–495. [DOI] [PubMed] [Google Scholar]
- (65).Lambeth JD, Kitchen E, Farooqui AA, Tuckey R, and Kamin H (1982) Cytochrome P-450scc-Substrate Interactions. J. Biol. Chem 257, 1876–1884. [PubMed] [Google Scholar]
- (66).Shriver DF, and Dunn JBR (1974) Backscattering geometry for Raman spectroscopy of colored materials. Appl. Spectrosc 28, 319–323. [Google Scholar]
- (67).Li D, Kabir M, Stuehr DJ, Rousseau DL, and Yeh S (2007) Substrate- and Isoform-Specific Dioxygen Complexes of Nitric Oxide Synthase. J. Am. Chem. Soc 129, 6943–6951. [DOI] [PubMed] [Google Scholar]
- (68).Khatri Y, Gregory MC, Grinkova YV, Denisov IG, and Sligar SG (2014) Active site proton delivery and the lyase activity of human CYP17A1. Biochem. Biophys. Res. Commun 443, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Couture M, Stuehr DJ, and Rousseau DL (2000) The Ferrous Dioxygen Complex of the Oxygenase Domain of Neuronal Nitric-oxide Synthase. J. Biol. Chem 275, 3201–3205. [DOI] [PubMed] [Google Scholar]
- (70).Rousseau DL, Li D, Couture M, and Yeh S-R (2005) Ligand-protein interactions in nitric oxide synthase. J. Inorg. Biochem 99, 306–323. [DOI] [PubMed] [Google Scholar]
- (71).Khatri Y, Luthra A, Duggal R, and Sligar SG (2014) Kinetic solvent isotope effect in steady-state turnover by CYP19A1 suggests involvement of Compound 1 for both hydroxylation and aromatization steps. FEBS Lett. 588, 3117–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Li XY, Czernuszewicz RS, Kincaid JR, Stein P, Spiro TG, Su YO, Li X-Y, Spiro TG, Kitagawa T, Abe M, Ogoshi H, and Kyogoku Y (1978) Resonance Raman spectra of octaethylporphyrinatonickel(II) and meso-deuterated and nitrogen-15 substituted derivatives. II. A normal coordinate analysis. J. Chem. Phys (Spiro TG, Ed.) 69, 47–61. [Google Scholar]
- (73).Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD, and Pikuleva I. a. (2011) Structural basis for three-step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J. Biol. Chem 286, 5607–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Antonini E, and Brunori M (1971) Hemoglobin and Myoglobin in Their Reactions with Ligands. North Holland Publishing Company: Amsterdam. [Google Scholar]
- (75).Pruitt K, and Tenovuo J (1985) The Lactoperoxidase System: Chemistry and Biological Significance. Marcel Dekker: New York. [Google Scholar]
- (76).Reiter B, and Perraudin JP (1991) Peroxidases in Chemistry and Biology (Everse J, Everse KE, and Grisham MB, Eds.), pp 143–180. CRC Press: Boca Raton, FL. [Google Scholar]
- (77).Sivaramakrishnan S, Ouellet H, Matsumura H, Guan S, Moenne-Loccoz P, Burlingame AL, and Montellano P. R. O. de. (2012) Proximal Ligand Electron Donation and Reactivity of the Cytochrome P450 Ferric–Peroxo Anion. J. Am. Chem. Soc 134, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Mak PJ, Gregory MC, Sligar SG, and Kincaid JR (2014) Resonance Raman spectroscopy reveals that substrate structure selectively impacts the heme-bound diatomic ligands of CYP17. Biochemistry 53, 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Duggal R, Liu Y, Gregory MC, Denisov IG, Kincaid JR, and Sligar SG (2016) Evidence that cytochrome b5 acts as a redox donor in CYP17A1 mediated androgen synthesis. Biochem. Biophys. Res. Commun 477, 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ibrahim M, Xu C, and Spiro TG (2006) Differential sensing of protein influences by NO and CO vibrations in heme adducts. J. Am. Chem. Soc 128, 16834–16845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Ray GB, Li XY, Ibers JA, Sessler JL, and Spiro TG (1994) How far can proteins bend the FeCO unit? Distal polar and steric effects in heme proteins and models. J. Am. Chem. Soc 116, 162–176. [Google Scholar]
- (82).Li XY, and Spiro TG (1988) Is bound carbonyl linear or bent in heme proteins? Evidence from resonance Raman and infrared spectroscopic data. J. Am. Chem. Soc 110, 6024–6033. [DOI] [PubMed] [Google Scholar]
- (83).Chartier FJM, Blais SP, and Couture M (2006) A weak Fe-O bond in the oxygenated complex of the nitric-oxide synthase of Staphylococcus aureus. J. Biol. Chem 281, 9953–9962. [DOI] [PubMed] [Google Scholar]
- (84).Davydov R, Satterlee JD, Fujii H, Sauer-Masarwa A, Busch DH, and Hoffman BM (2003) A Superoxo-Ferrous State in a Reduced Oxy-Ferrous Hemoprotein and Model Compounds. J. Am. Chem. Soc 125, 16340–16346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.