

SUMMARY
Resistance to asparaginase, an antileukemic enzyme that depletes asparagine, is a common clinical problem. Using a genome-wide CRISPR/Cas9 screen, we found a synthetic lethal interaction between Wnt pathway activation and asparaginase in acute leukemias resistant to this enzyme. Wnt pathway activation induced asparaginase sensitivity in distinct treatment-resistant subtypes of acute leukemia, but not in normal hematopoietic progenitors. Sensitization to asparaginase was mediated by Wnt-dependent stabilization of proteins (Wnt/STOP), which inhibits GSK3-dependent protein ubiquitination and proteasomal degradation, a catabolic source of asparagine. Inhibiting the alpha isoform of GSK3 phenocopied this effect, and pharmacologic GSK3α inhibition profoundly sensitized drug-resistant leukemias to asparaginase. Our findings provide a molecular rationale for activation of Wnt/STOP signaling to improve the therapeutic index of asparaginase.
Keywords: Asparaginase, Wnt signaling, acute leukemia, drug resistance, protein degradation
Graphical Abstract
INTRODUCTION
The genetic concept of synthetic lethality describes an interaction between two mutations that are each well-tolerated individually but are lethal when combined [reviewed in (Ashworth and Lord, 2018)]. Small molecule inhibitors can phenocopy the effect of specific mutations, which led to the search for synthetic lethal drug-mutation interactions that would be selectively toxic to cancer cells harboring specific oncogenic mutations (Hartwell et al., 1997). Proof-of-principle for this idea came from the discovery that PARP inhibitors are profoundly toxic to tumor cells harboring biallelic BRCA1 or BRCA2 mutations but not to normal cells that retain at least one functional allele, thus providing a striking therapeutic index (Bryant et al., 2005; Farmer et al., 2005). Inspired by this concept, we reasoned that drug-drug synthetic lethal interactions had the potential to improve the therapeutic index of cancer therapy, if applied to drugs that were sufficiently selective for cancer cells.
Asparaginase, an exogenous enzyme that deaminates the nonessential amino acid asparagine, has long been recognized to have activity against aggressive hematopoietic neoplasms (Broome, 1961). Asparaginase dose-intensification has improved outcomes for T-cell and B-cell acute lymphoblastic leukemias (T-ALL and B-ALL) (Clavell et al., 1986; DeAngelo et al., 2015; Ertel et al., 1979; Pession et al., 2005). This enzyme also has therapeutic activity in acute myeloid leukemias (AML) and in some non-Hodgkin lymphomas (Alexander et al., 2017; Capizzi et al., 1988; Wells et al., 1993; Yamaguchi et al., 2011). The development of resistance to asparaginase-based treatment regimens has a poor prognosis, and effective therapeutic options are lacking for many of these patients.
The sensitivity of acute leukemia cells to asparaginase is due, at least in part, to low expression of asparagine synthetase (ASNS) in these cells, resulting in their dependence on exogenous asparagine (Haskell and Canellos, 1969; Horowitz et al., 1968). By contrast, physiologic expression of ASNS by most normal cells is thought to explain the favorable therapeutic index of asparaginase (Rizzari et al., 2013). Increased ASNS expression by leukemic blasts can induce asparaginase resistance (Haskell and Canellos, 1969; Horowitz et al., 1968). However, ASNS is not an ideal therapeutic target because its inhibition is likely to worsen asparaginase-induced toxicity to normal tissues. Nevertheless, ASNS expression is not the sole determinant of asparaginase response (Appel et al., 2006; Hermanova et al., 2012; Holleman et al., 2004; Stams et al., 2003), whose biologic basis remains incompletely understood. The goal of this study was to test the hypothesis that asparaginase-resistant leukemia cells harbor gain-of-fitness alterations whose therapeutic targeting would be uniquely toxic to tumor cells upon treatment with the enzyme.
RESULTS
Wnt Pathway Activation Induces Asparaginase Sensitization
To identify molecular pathways that promote leukemic cell fitness upon treatment with asparaginase, we performed a genome-wide CRISPR/Cas9 loss-of-function genetic screen in the T-ALL cell line CCRF-CEM, because this was the most asparaginase-resistant T-ALL cell line in which CRISPR/Cas9 genome editing could be efficiently performed (Figure S1A). We first optimized conditions for a drop-out screen using positive control guide RNAs targeting ASNS (Figure S1B–D) (Van Heeke and Schuster, 1989). We then transduced Cas9-expressing CCRF-CEM cells with the GeCKO genome-wide guide RNA library (Shalem et al., 2014) (Figure 1A, Figure S1E), treated with either vehicle or 10 U/L of asparaginase that lacked detectable toxicity, and guide RNA representation was assessed. ASNS was the gene most significantly depleted in asparaginase-treated cells, followed closely by NKD2 and LGR6 that encode two regulators of Wnt signaling (Figure 1B, Table S1). We prioritized Wnt signaling for further investigation because this was the only pathway known to be regulated by more than one gene among the top “hits” in our screen, making it unlikely this reflected a false-positive result. Analysis of guide RNA-level results revealed that 3 of 6 guide RNAs targeting both NKD2 and LGR6 were significantly depleted in asparaginase-treated cells (Figure S2A and Table S1). We first validated that transduction of CCRF-CEM cells with the top guide RNAs targeting NDK2 or LGR6 yielded significant gene knockdown (Figure S2B–S2C), and sensitized these cells to asparaginase cytotoxicity (Figure S2D). We also performed Sanger sequencing of all of the predicted off-target sites (up to 2 bp mismatches) of the top-scoring gRNAs, which revealed no evidence of off-target mutagenesis (Table S2).
Figure 1. Wnt Pathway Activation Sensitizes Leukemia Cells to Asparaginase.

(A) CCRF-CEM cells were transduced with the GeCKO genome-wide guide RNA library in biologic duplicates, split into treatment with vehicle or asparaginase (10 U/L), and guide RNA representation was assessed after 5 days of treatment.
(B) Significance of gene depletion in asparaginase-treated conditions, as assessed by robust ranking aggregation (RRA) score calculated using MAGeCK analysis. Note that microRNA genes are not shown.
(C) CCRF-CEM cells were transduced with the indicated shRNAs, and knockdown efficiency was assessed by RT-PCR analysis. CT values greater than 36 were defined as not detected (N.D.).
(D) CCRF-CEM cells were transduced with the indicated shRNAs and subjected to Western blot analysis for active (nonphosphorylated) β-catenin or GAPDH.
(E) CCRF-CEM cells transduced with a lentiviral TOPFlash-EGFP (7xTcf-EGFP) reporter of canonical Wnt/β-catenin driven transcription were transduced with the indicated shRNAs, and reporter-driven EGFP fluorescence was assessed.
(F) The indicated cell lines were transduced with the indicated shRNAs and treated with the indicated doses of asparaginase. Relative viability was assessed after 8 days of treatment by counting viable cells. All cell counts were normalized to those in shLuc-transduced, no-asparaginase controls.
(G) CCRF-CEM cells were transduced with the indicated shRNAs, treated with asparaginase (10 U/L) for 48 hr and caspase 3/7 activity assay was assessed.
(H) CCRF-CEM cells were treated as indicated and the number of viable cells after 8 days of treatment was assessed. All cell counts were normalized to those in no-Wnt3a, no-asparaginase controls.
All error bars represent SEM.
See also Figures S1 and S2 and Tables S1 and S2.
NKD2 negatively regulates Wnt signaling by binding and repressing dishevelled proteins (Wharton et al., 2001), whereas LGR proteins function as R-spondin receptors reported to either activate or repress Wnt signaling (de Lau et al., 2011; Nehir et al., 2017; Walker et al., 2011). To test how these molecules regulate Wnt signaling, we transduced CCRF-CEM cells with shRNAs targeting NKD2 or LGR6, or with an shLuciferase control (Figure 1C). Knockdown of NKD2 or LGR6 increased levels of active (nonphosphorylated) β-catenin (Figure 1D), as well as the activity of a TOPFlash (7xTCF-EGFP) reporter of canonical Wnt/β-catenin transcriptional activity (Fuerer and Nusse, 2010) (Figure 1E). Thus, NKD2 and LGR6 are negative regulators of Wnt signaling in T-ALL cells. To validate that loss of NKD2 or LGR6 sensitizes these cells to asparaginase, we again transduced CCRF-CEM cells with these shRNAs and treated them with asparaginase. Knockdown of NDK2 or LGR6 profoundly sensitized several acute leukemia cell lines to asparaginase (Figure 1F), and potentiated asparaginase-induced caspase activation, indicating induction of apoptosis (Figure 1G). This effect was phenocopied by treatment with the physiologic Wnt ligand Wnt3A (Figure 1H). Thus, Wnt pathway activation sensitizes leukemic cells to asparaginase.
GSK3 Inhibition Sensitizes Distinct Acute Leukemia Subtypes to Asparaginase-Induced Cytotoxicity
Inhibition of glycogen synthase kinase 3 (GSK3) activity is a key event in Wnt-induced signal transduction (Nusse and Clevers, 2017; Siegfried et al., 1992; Taelman et al., 2010), prompting us to test whether this effect could be phenocopied by CHIR99021, an ATP-competitive inhibitor of both GSK3 paralogs, GSK3α and GSK3β (Bennett et al., 2002). CHIR99021 induced significant asparaginase sensitization across a panel of cell lines representing distinct subtypes of treatment-resistant acute leukemia, including T-ALL, MLL-rearranged AML and hypodiploid B-ALL (Figure 2A–2B, Table S3). Importantly, CHIR99021 did not increase the toxicity of asparaginase to normal human CD34+ hematopoietic progenitors (Figure 2C), suggesting a selective effect on leukemic cells. Nor did it sensitize leukemic cells to other commonly used antileukemic drugs, including dexamethasone, 6-mercaptopurine, doxorubicin or vincristine (Figure 2D). Thus, GSK3 inhibition can enhance asparaginase toxicity not only in T-ALL, but in other common acute leukemia variants as well.
Figure 2. GSK3 Inhibition Sensitizes Distinct Acute Leukemia Subtypes, but not Normal Hematopoietic Progenitors, to Asparaginase-Induced Cytotoxicity.

(A-B) T-ALL cells (A), AML cells (OCI-AML2, SKM-1, THP-1) or B-ALL cells (MHH-CALL2) (B) were treated with the GSK3 inhibitor CHIR99021 (CHIR, 1 μM) or vehicle control, together with the indicated doses of asparaginase for 8 days. Relative viability was assessed based on viable cell counts, all of which were normalized to those in vehicle-treated cells. Western blot analysis for the indicated proteins is shown as an inset.
(C) Normal CD34+ human hematopoietic progenitor cells were treated with CHIR99021 (1 μM) or vehicle, together with the indicated doses of asparaginase, and viable cell counts were assessed after treatment for 4 days. Note that these normal hematopoietic progenitors could not be maintained for more than 4 days in culture. Western blot analysis for the indicated proteins is shown as an inset.
(D) CCRF-CEM cells were treated with CHIR99021 (1 μM) or vehicle, and the indicated chemotherapeutic drugs for 8 days. All Western Blots shown indicate levels of active (nonphosphorylated) β-catenin or GAPDH after treatment with CHIR 99021 (1 μM) or vehicle. Viable cells were counted and results normalized to counts in no-CHIR, no-chemotherapy controls. Western blot analysis for the indicated proteins is shown as an inset.
Statistical significance was calculated for the following doses of chemotherapy: 10 U/L asparaginase, 1 μM dexamethasone, 1 μM 6-mercaptopurine, 10 nM doxorubicin and 10 nM vincristine, respectively. Two-way ANOVA with Tukey post-hoc adjustment was performed for each cell line and included an interaction term between asparaginase dose and GSK3 inhibitor.
The p value for the main effect of GSK3 inhibitor vs. vehicle is presented in each plot and all interaction terms were significant (p < 0.0001). Error bars represent SEM.
See also Table S3.
Wnt-Dependent Stabilization of Proteins Mediates Sensitization to Asparaginase
Canonical Wnt signaling is best known as an activator of β-catenin-dependent transcriptional activity (Brunner et al., 1997; van de Wetering et al., 1997), leading us to ask whether β-catenin activation is sufficient to induce asparaginase sensitization. Thus, we transduced CCRF-CEM cells with a constitutively active ΔN90 β-catenin allele (Guo et al., 2012) or shRNAs targeting the β-catenin antagonist APC (Moon and Miller, 1997) and treated them with asparaginase. Surprisingly, these modifications lacked any discernible effect on asparaginase sensitivity despite effective activation of β-catenin-induced transcription, as measured by activity of the TOPFlash reporter, and a transduction efficiency >98% in these cells (Figure 3A–3D). The Wnt pathway also regulates protein synthesis and metabolism by activating both mTOR complexes, mTORC1 (Inoki et al., 2006) and mTORC2 (Esen et al., 2013). However, treatment with the dual mTORC1/2 inhibitor AZD2014 or with the mTORC1 inhibitors rapamycin and RAD001 failed to rescue Wnt-induced sensitization to asparaginase (Figure 3E). On balance, these data indicate that asparaginase sensitization is not mediated by activation of β-catenin or mTOR signaling.
Figure 3. Wnt-induced sensitization to asparaginase is not mediated by β-catenin or mTOR activation.

(A) CCRF-CEM cells transduced with a constitutively active β-catenin were analyzed for expression of the indicated proteins by Western blot analysis (left), activity of a TOPFlash-EGFP reporter of β-catenin dependent transcriptional activity (mid), and transduction efficiency was assessed by immunostaining for β-catenin (right). Statistical significance was calculated using a two-sided Welch t-test. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. n.s., p > 0.05.
(B) CCRF-CEM cells transduced with the indicated constructs were treated with the indicated doses of asparaginase for 8 days, and the number of viable cells was counted. All cell counts were normalized to those in control-transduced, no-asparaginase cells.
(C) CCRF-CEM cells transduced with the indicated shRNAs were analyzed by RT-PCR, and effects on TOPFlash-EGFP reporter activity were assessed. Statistical significance was calculated using a two-sided Welch t-test. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. n.s., p > 0.05.
(D) CCRF-CEM cells transduced with the indicated shRNAs were treated with the indicated doses of asparaginase for 8 days. Viability was assessed as in (B).
(E) CCRF-CEM cells transduced with the indicated shRNAs were treated with vehicle, AZD2014 (100 nM), rapamycin (10 nM) or RAD001 (100 nM), and analyzed by Western blot analysis for the indicated proteins (top). Cells were treated with the indicated doses of asparaginase and viability was assessed as in (B). Note that mTOR inhibition did not rescue Wnt-induced sensitization to asparaginase.
All error bars represent SEM.
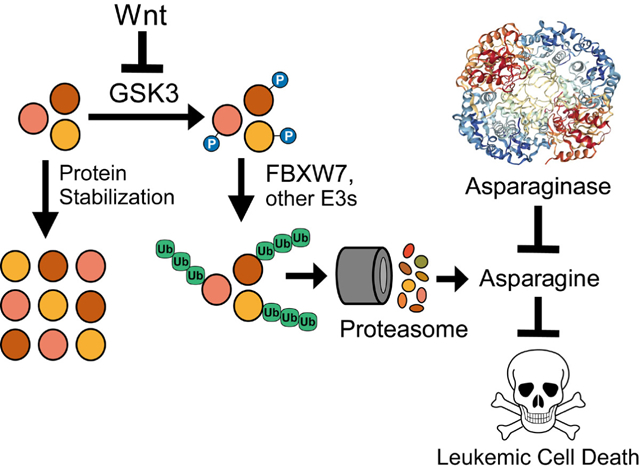
Activation of Wnt signaling increases total cellular protein content and cell size by inhibiting GSK3-dependent protein ubiquitination and proteasomal degradation, an effect termed Wnt-dependent stabilization of proteins (Wnt/STOP) (Acebron et al., 2014; Huang et al., 2015; Taelman et al., 2010). This function of the Wnt pathway appeared relevant to asparaginase sensitization because we observed a measurable decrease in cell size after treatment with the enzyme, even in CCRF-CEM cells that are resistant to asparaginase-induced cytotoxicity (Figure 4A), and this effect was reversed by transduction of these cells with Wnt-activating shRNAs (Figure 4B). Treatment with the Wnt ligand Wnt3A had a similar effect (Figure 4C). We then asked whether Wnt pathway activation inhibits protein degradation in these cells, using a pulse-chase experiment to measure total protein half-life. CCRF-CEM cells were first transduced with Wnt-activating shRNAs targeting NKD2 and LGR6 or with an shLuciferase control. Cells were then incubated with a pulse of the methionine analog azidohomoalanine (AHA) followed by a chase in which the cells were treated with asparaginase and the rate of AHA release due to protein degradation was measured by flow cytometry. Wnt pathway activation did not affect the degree of AHA label incorporation during the pulse period (Figure S3); however, shRNA knockdown of LGR6 or NDK2 increased total protein half-life by approximately 40% in these cells (Figure 4D). These findings indicate that Wnt pathway activation inhibits protein degradation in asparaginase-treated cells.
Figure 4. Wnt-Dependent Stabilization of Proteins Mediates Sensitization to Asparaginase.

(A) CCRF-CEM cells treated with vehicle or asparaginase (10 U/L) for 48 hr, and cell size was assessed by flow cytometry forward scatter height (FSC-H).
(B) CCRF-CEM cells transduced with the indicated constructs were treated with asparaginase (10 U/L) for 48 hr, and cell size was assessed as in (A). Scatter plot (right) depicts results of individual biologic replicates, with horizontal bars indicating mean, and error bars indicating SEM. Differences between groups were analyzed using a one-way ANOVA with Dunnett’s adjustment for multiple comparisons. ** p ≤ 0.01; *** p ≤ 0.001.
(C) CCRF-CEM cells were treated with asparaginase (10 U/L) as well as either vehicle or Wnt3A (100 ng/ml) for 48 hr, and cell size was assessed as in− (A). Scatter plot depicts results of individual biologic replicates, with bars indicating mean +/− SEM. Difference between groups assessed by two-sided Welch t-test. **** p ≤ 0.0001.
(D) CCRF-CEM cells transduced with the indicated shRNAs were incubated with a pulse of the methionine analog AHA, then released from AHA and treated with asparaginase (10 U/L) during the chase period. The degree of AHA label retention was assessed by flow cytometry. Error bars indicate SEM.
(E-F) CCRF-CEM cells were transduced with the indicated constructs, and effects on abundance of Lys-48 ubiquitin chain proteins was assessed by Western blot analysis, in the absence (E) or presence (F) of an FBXW7 expression construct.
(G) CCRF-CEM cells transduced with the indicated constructs were analyzed by Western blot (inset), treated with the indicated doses of asparaginase, and viability was assessed by Trypan blue staining after 8 days. Note that wild-type and mutant FBXW7 were Myc-tagged. Error bars represent SEM.
(H-I) CCRF-CEM cells were transduced with the indicated constructs, the transduction efficiency (H) was assessed by immunostaining for the V5 tag, and their viability (I) upon treatment with the indicated doses of asparaginase was assessed as in (G). Error bars represent SEM.
See also Figures S3, S4 and S5.
The E3 ubiquitin ligase component FBXW7 recognizes its substrates via a canonical phosphodegron that is phosphorylated by GSK3 (Welcker et al., 2004; Welcker et al., 2003) and overexpression of FBXW7 restores the degradation of a subset of proteins stabilized by Wnt/STOP signaling (Acebron et al., 2014; Chen et al., 2016). FBXW7 stimulates lysine 48 (K48)-chain ubiquitination of its target substrates (Davis et al., 2014), a modification that marks target proteins for proteasomal degradation (Kwon and Ciechanover, 2017). If Wnt pathway activation inhibits protein degradation by inhibiting GSK3 mediated phosphorylation of target substrates, then Wnt activation should lead to a measurable decrease in K48-chain ubiquitinated proteins. Indeed, Western blot analysis for total K48-chain ubiquitinated proteins revealed that transduction of CCRF-CEM cells with Wnt-activating shRNAs led to a significant reduction in total K48-chain ubiquitinated proteins (Figure 4E), and this effect was reversed by FBXW7 overexpression (Figure 4F).
Does inhibition of protein degradation in leukemic cells mediate sensitization to asparaginase? We first asked whether FBXW7 overexpression could reverse Wnt-induced sensitization to asparaginase. After transducing CCRF-CEM cells with Wnt-activating shRNAs or an shLuciferase control, we transduced the same cells with expression constructs encoding either wild-type FBXW7 or the FBXW7 R465C mutant that has impaired binding to its canonical phospho-degron (Koepp et al., 2001). Overexpression of wild-type FBXW7 in CCRF-CEM cells blocked the ability of Wnt pathway activation to trigger asparaginase hypersensitivity whereas the R465C mutant had no effect (Figure 4G). We note that CCRF-CEM cells harbor an endogenous FBXW7 mutation (O’Neil et al., 2007; Thompson et al., 2007), but FBXW7 overexpression could also reverse Wnt-induced sensitization to asparaginase in OCI-AML2 and DND41 cells, which do not harbor identifiable FBXW7 mutations (Figure S4A–B) (Barretina et al., 2012; O’Neil et al., 2007; Tate et al., 2018). Thus, FBXW7 overexpression can rescue Wnt-induced sensitization to asparaginase in FBXW7 mutant or wild-type leukemias.
However, FBXW7 is a T-ALL tumor suppressor that can target specific oncoproteins (Davis et al., 2014; Thompson et al., 2007), prompting us to test whether direct stimulation of proteasomal activity is sufficient to reverse Wnt-induced sensitization to asparaginase. Thus, we leveraged a hyperactive open-gate mutant of the proteasomal subunit PSMA4 (DN-PSMA4), whose expression is sufficient to stimulate degradation of a wide range of proteasomal substrates (Choi et al., 2016). CCRF-CEM cells were first transduced with control or Wnt-activating shRNAs, and then transduced with the constitutively active proteasomal subunit ΔN-PSMA4 or a GFP control. Expression of the constitutively active ΔN-PSMA4 proteasomal subunit blocked Wnt-induced sensitization to asparaginase in distinct acute leukemia subtypes (Figure 4H–4I and Figure S4C–D). These findings indicate that Wnt pathway activation sensitizes leukemic cells to asparaginase by inhibiting proteasomal degradation of proteins Protein ubiquitination and proteasomal degradation are important for the degradation of unfolded or damaged proteins (Kwon and Ciechanover, 2017). Thus, we asked whether Wnt/STOP activation might promote death of asparaginase-treated leukemic cells by activating the unfolded protein response (UPR). Activation of the UPR leads to the production of an alternative XPB1 mRNA transcript via non-canonical splicing as well as phosphorylation of PERK (Walter and Ron, 2011). However, treatment of CCRF-CEM T-ALL cells with asparaginase, transduction with Wnt-activating shRNAs, or both in combination did not induce activation of these UPR markers, by contrast with the thapsigargin positive control (Figure S5). These findings thus argue against activation of the unfolded protein response as a mediator of cell death in response to Wnt pathway activation and asparaginase.
GSK3α Inhibition Phenocopies Wnt-Induced Sensitization to Asparaginase and Promotes Leukemic Cell Death by Depleting Asparagine
To explore the therapeutic potential of our studies, we focused on GSK3 as a therapeutic target. Although the pan-GSK3 inhibitor that we used in cell lines lacks suitable pharmacokinetic properties for in vivo studies, isoform-selective inhibitors of each GSK3 paralog with favorable pharmacologic properties have recently been developed (Wagner et al., 2018). We first used shRNAs to selectively deplete GSK3α or GSK3β. Depletion of GSK3α phenocopied Wnt-induced sensitization to asparaginase, whereas GSK3β knockdown had no effect (Figure 5A). Importantly, the effect of GSK3α-targeting shRNAs was rescued by transduction of a GSK3α expression construct that escapes shRNA targeting (Figure 5B). We then tested the aforementioned paralog-selective GSK3 inhibitors and found that the GSK3α-selective inhibitor BRD0705 effectively sensitized CCRF-CEM cells to asparaginase, while the GSK3β-selective inhibitor BRD3731 had only modest effects (Figure 5C).
Figure 5. GSK3α Inhibition Phenocopies Wnt-Induced Sensitization to Asparaginase and Promotes Leukemic Cell Death by Depleting Asparagine.

(A) CCRF-CEM cells were transduced with the indicated shRNAs and analyzed by Western blot (inset) for total GSK3 or GAPDH. Cells were then treated with the indicated doses of asparaginase, and viability was assessed after 8 days of treatment. All cell counts were normalized to those in shLuc-transduced, vehicle-treated controls.
(B) CCRF-CEM cells were transduced with the indicated shRNAs, without or with a GSK3α expression construct that escapes shRNA targeting. Western blot analysis (inset) was performed for total GSK3 or GAPDH. Cells were then treated with the indicated doses of asparaginase and viability was assessed as in (A).
(C) CCRF-CEM cells were treated with vehicle, the GSK3α-selective inhibitor BRD0705, or the GSK3β-selective inhibitor BRD3731, and analyzed by Western blot analysis (inset) for phospho-GSK3 or GAPDH. Cells were then treated with the indicated drugs for 8 days, and viability was assessed as in (A).
(D) CCRF-CEM cells were treated with vehicle, asparaginase (10 U/L), the GSK3α inhibitor BRD0705 (100 nM) or both in combination for 12 hr. Culture media was collected and asparagine levels were quantified by UPLC. Fresh media (RPMI-1640) was used as a control for each UPLC run, and amino acid levels are normalized to those in stock RPMI-1640.
Differences between groups were calculated using a two-sided Welch t-test. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. n.s., p > 0.05.
(E) CCRF-CEM cells were first transduced with shLuc control or Wnt-activating shRNAs targeting NKD2 or LGR6. Cells were then treated with asparaginase (10 U/L) or vehicle and grown in complete growth medium (RPMI-1640 + 10% FBS) or complete growth medium supplemented with 10x L-asparagine or 10x L-glutamine. Fifty percent of the media was removed every 12 hr and replaced with fresh growth medium, supplemented with the appropriate concentration of asparaginase or glutamine. Viability was assessed after 72 hr by counting viable cells.
(F) Viability of cells in (E), normalized to viability in no-asparaginase controls. Difference between groups was analyzed using a two-sided Welch t-test. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. n.s., p > 0.05.
All error bars indicate SEM.
See also Figures S6 and S7 and Table S4.
To gain further insights into mechanisms of cytotoxicity of this combination, we applied unbiased mass spectrometry proteomics to CCRF-CEM cells treated with vehicle, asparaginase alone, the GSK3α inhibitor BRD0705 (which phenocopies Wnt/STOP pathway activation), or the combination of asparaginase and BRD0705. However, mass spectrometry proteomics on an equal amount of proteins from each condition revealed no individual proteins that were significantly up- or down-regulated in any of these experimental conditions (Figure S6, Table S4). While this does not rule out the possibility of changes below the limit of detection of our assay, these findings, together with our data that Wnt-induced sensitization to asparaginase can be reversed by direct stimulation of proteasomal activity, support a model in which Wnt-induced sensitization to asparaginase is mediated by global effects on protein degradation. This model posits that asparaginase-resistant leukemias rely on proteasomal degradation of proteins, a catabolic source of amino acids (Suraweera et al., 2012), to maintain asparagine levels above a critical threshold during treatment with asparaginase, an adaptive pathway blocked by Wnt pathway activation. To test this hypothesis, we quantified free amino acids in CCRF-CEM cells treated with vehicle, the GSK3α inhibitor BRD0705 (which phenocopies Wnt/STOP activation), asparaginase, or asparaginase in combination with BRD0705. While BRD0705 monotherapy had little effect on asparagine concentrations in anabolic (asparagine-replete) conditions, GSK3α inhibition significantly exacerbated asparaginase-induced depletion of asparagine (Figure 5D), with no reduction in glutamine or other amino acids (Figure S7). We then asked whether asparagine depletion is responsible for the cytotoxicity of Wnt pathway activation and asparaginase, by testing whether the toxicity of this combination could be rescued by replenishing free asparagine. CCRF-CEM cells were transduced with control or Wnt-activating shRNAs, and then treated with either vehicle or asparaginase. Cells in each condition were then provided with a 10-fold excess of free asparagine every 12 hr, and viable cell counts were performed after 72 hr of treatment (Figure 5E). Controls included a 10-fold excess of glutamine, whose physiochemical properties resemble those of asparagine but which is not depleted in response to the combination of asparaginase and Wnt pathway activation (Figure S7), or standard growth media. This revealed that Wnt-induced sensitization to asparaginase was rescued by providing an excess of free asparagine, but not glutamine or standard media (Figure 5F). Thus, depletion of free asparagine is a key mediator of the synthetic lethal interaction of Wnt activation and asparaginase.
Synthetic Lethality of GSK3α Inhibition and Asparaginase in Human Leukemia
We then turned our attention to the in vivo potential of our findings. Combined treatment with the GSK3α inhibitor BRD0705 (15 mg/kg via gavage every 12 hr) and asparaginase (1,000 U/kg × 1 dose) was well-tolerated by control immunodeficient NRG mice, with no appreciable weight changes or increases in serum bilirubin levels, an important dose-limiting toxicity of asparaginase in human adults (data not shown). We note that the pharmacologic properties of asparaginase and BRD0705 have been extensively evaluated in mice (Poppenborg et al., 2016; Wagner et al., 2018).
We then injected a cohort of NRG mice with leukemic cells from a primary asparaginase-resistant T-ALL patient-derived xenograft (PDX). Once leukemia engraftment was detected in the peripheral blood, we treated the mice with vehicle, asparaginase, BRD0705, or the combination of asparaginase and BRD0705 (Figure 6A). Although the xenografts proved highly resistant to asparaginase monotherapy, the combination of asparaginase and BRD0705 was both highly efficacious and well-tolerated (Figure 6B and 6C). We also tested the therapeutic activity of this combination in mice engrafted with two additional subtypes of acute lymphoblastic leukemia at high risk of treatment failure, hypodiploid B-ALL and MLL-rearranged B-ALL (Pui et al., 2015). Both of these PDXs also proved completely refractory to asparaginase monotherapy, but the combination of GSK3α inhibition and asparaginase had a highly significant survival advantage (Figure 6D–E).
Figure 6. Synthetic Lethality of GSK3α Inhibition and Asparaginase in Human Leukemia.

(A) Experimental schema. Human patient-derived leukemia xenografts were injected into NRG immunodeficient mice. Once engraftment of leukemia (>5% human leukemic cells in peripheral blood) was confirmed, mice were treated as indicated.
(B) Body weights of mice injected with the chemotherapy-resistant T-ALL PDX in the experiment shown in (C). Error bars represent SEM.
(C-E) Survival curves of mice injected with xenografts from patients with a primary asparaginase-resistant T-ALL (C), hypodiploid B-ALL (D), or chemotherapy-refractory MLL-rearranged B-ALL (E), and treated as indicated in (A). Treatment start and end points denoted by arrowheads on each graph.
DISCUSSION
Using a genome-wide CRISPR/Cas9 screen, we identified a synthetic lethal interaction between activation of Wnt signaling and asparaginase in acute leukemias resistant to this enzyme. Wnt activation results in inhibition of GSK3 activity (Siegfried et al., 1992; Taelman et al., 2010), and pharmacologic GSK3 inhibition sensitized a panel of treatment resistant acute leukemias to asparaginase. Wnt-induced inhibition of GSK3 is best known for its ability to activate β-catenin driven transcription, which is oncogenic (Nusse and Clevers, 2017). While classic Wnt/β-catenin activating mutations (such as those affecting APC or CTNNB1, which encodes β-catenin) are rare in human leukemias, physiologic Wnt/β-catenin signaling regulates normal and malignant hematopoietic development [reviewed in (Lento et al., 2013; Richter et al., 2017; Staal et al., 2016)]. However, we found that Wnt-induced sensitization to asparaginase was independent of β-catenin, and was instead mediated by Wnt-dependent stabilization of proteins (Wnt/STOP), which inhibits protein ubiquitination and proteasomal degradation. Indeed, expression of a hyperactive PSMA4 proteasomal subunit that broadly increases proteasomal degradation (Choi et al., 2016) completely blocked Wnt-induced sensitization to asparaginase. Proteasomal degradation of proteins is a catabolic source of amino acids (Suraweera et al., 2012), and we found that Wnt/STOP activation exacerbated asparaginase-induced depletion of free asparagine. Moreover, the toxicity of this combination was rescued by providing an excess of free asparagine, but not glutamine. Thus, asparagine depletion mediates the synthetic lethal interaction of Wnt/STOP activation and asparaginase (Figure 7).
Figure 7. Proposed model.

Asparaginase structure is from (Yun et al., 2007).
Transduction of FBXW7 was sufficient to rescue leukemic cells from Wnt-induced sensitization to asparaginase, which is perhaps surprising because E3 ubiquitin ligases are generally considered to have a narrow set of protein targets. However, these findings are in line with previous data demonstrating that overexpression of this E3 ligase blocks the ability of Wnt pathway activation to trigger protein stabilization and regulate cell size (Acebron et al., 2014). These findings raise the possibility that FBXW7 is the primary E3 ligase that functions in Wnt-regulated protein degradation under physiologic conditions. However, an alternative possibility is that FBXW7 functions as one-of-many E3 ligases in Wnt/STOP signaling, but its overexpression promotes degradation of some particularly abundant cellular proteins, thus providing a sufficient catabolic source of asparagine to reverse the toxicity of asparaginase. Distinguishing among these and other possibilities will require additional investigation.
We found that inhibition of GSK3α was sufficient to phenocopy asparaginase sensitization, and the combination of GSK3α inhibitors and asparaginase has profound activity in drug-resistant human leukemias. While widespread inhibition of GSK3 activity could cause toxicity due to activation of oncogenic β-catenin signaling (Nusse and Clevers, 2017), this effect requires inhibition of both GSK3 paralogs, at least in some contexts (Banerji et al., 2012; Doble et al., 2007; Wagner et al., 2018). Thus, the application of selective GSK3α inhibitors may reduce toxicity from widespread stabilization of β-catenin. We note that GSK3A, as well as some genes reported to induce asparaginase sensitization such as EIF2AK4 (also known as GCN2) and ATF4 (Gwinn et al., 2018; Nakamura et al., 2018), failed to score in our genome-wide screen. We believe this reflects the limitations of available genome-wide guide RNA libraries, which are currently biased towards guide RNAs targeting early (5’-terminal) protein-coding exons, rather than those encoding functional protein domains. We anticipate that ongoing efforts to refine these libraries will allow future screens to approach saturation.
Our findings demonstrate that synthetic lethal drug-drug interactions can be leveraged to improve the therapeutic index of cancer therapy. This approach requires drug combinations with selective toxicity to cancer cells. We anticipate several classes of therapeutics that may be particularly well-suited for such an approach: drugs devised from the principles of drug-mutation synthetic lethal interactions, such as PARP inhibitors for BRCA1 or BRCA2 mutant tumors (Bryant et al., 2005; Farmer et al., 2005); drugs that target mutant oncoproteins that are uniquely present in cancer cells, such as selective inhibitors for neomorphic IDH1 or IDH2 mutations (Wang et al., 2013); or drugs that target genetic vulnerabilities induced by “passenger” gene mutations, such as those targeting MTAP deletions acquired due to its proximity to the CDKN2A tumor suppressor (Kryukov et al., 2016). It is perhaps less predictable that such an approach would be successful for conventional chemotherapeutics, which generally target cellular factors that are required for both normal and malignant cells. Nevertheless, the fact that these drugs are the primary therapeutic option with curative potential for a broad range of human cancers demonstrates the existence of a substantial therapeutic window, which can be further improved as demonstrated here.
It is of considerable interest that the combination of Wnt/STOP activation and asparaginase is potently toxic to leukemic cells, whereas normal cells appear to have effective compensatory mechanisms. One possibility is that normal cells have an improved capacity for asparagine biosynthesis and are thus not dependent on regulated protein degradation to tolerate asparaginase. However, several of the asparaginase-resistant cells utilized in our studies express asparagine synthetase, suggesting a role for additional factors. These may include checkpoints that normally couple proliferation to asparagine availability, a link that could be readily disrupted by oncogenic mutations. If so, the combination of Wnt/STOP activation and asparaginase could have efficacy against a broad range of tumors harboring relevant gene mutations.
STAR★ Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Alejandro Gutierrez (alejandro.gutierrez@childrens.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient-Derived Xenografts
Acute leukemia clinical specimens were collected from patients with newly diagnosed or relapsed acute leukemias enrolled on Dana-Farber Cancer Institute clinical trials, with written informed consent in accordance with the Declaration of Helsinki. Human subject studies were approved by the Dana-Farber Cancer Institute Institutional Review Board. Patient-derived xenografts were generated by engrafting viably frozen leukemic cells into immunodeficient mice, as described (Townsend et al., 2016). Mouse studies were approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee and performed in accordance with all relevant regulatory standards.
Cell Lines and Cell Culture
293T cells, T-ALL cell lines, AML cell lines and B-ALL cell lines were obtained from ATCC (Manassas, VA, USA), DSMZ (Braunschweig, Germany), the A. Thomas Look laboratory (Boston, MA, USA) or Alex Kentsis laboratory (New York, NY, USA) and cultured in DMEM, RPMI-1640 or MEM alpha (Thermo Fisher Scientific) with 10% or 20% fetal bovine serum (FBS, Sigma-Aldrich, Saint Louis, MO) or TET system approved FBS (Clontech, Mountain View, CA) and 1% penicillin/streptomycin (Thermo Fisher Scientific) at 37°C, 5% CO2. Human CD34 + progenitor cells from mobilized peripheral blood of healthy donors were obtained from Fred Hutchinson Cancer Research Center (Seattle, WA, USA). CD34+ progenitors were cultured in IMDM (Thermo Fisher Scientific) supplemented with 20% FBS and recombinant human interleukin-3 (R&D systems, Minneapolis, MN), recombinant human interleukin-6 (R&D systems, Minneapolis, MN) and recombinant human stem cell factor (R&D systems, Minneapolis, MN) to a final concentration of 50 ng/ml each.
Cell line identities were validated using STR profiling at the Dana-Farber Cancer Institute Molecular Diagnostics Laboratory (most recently in June 2018), and mycoplasma contamination was excluded using the MycoAlert Mycoplasma Detection Kit according to the manufacturer’s instructions (Lonza, Portsmouth, NH; most recently in March 2018).
Mice
NOD rag gamma (NRG) mice were purchased from the Jackson Laboratories (Bar Harbor, ME; Stock # 007799). 6–8 weeks-old male NRG mice were used for experiments and littermates were kept in individual cages. Mice were randomly assigned to experimental groups. Mice were handled in strict accordance with Good Animal Practice as defined by the Office of Laboratory Animal Welfare. All animal work was done with Boston Children’s Hospital (BCH) Institutional Animal Care and Use Committee approval (protocol # 18-09-3784R).
Lentiviral and Retroviral Transduction
Lentiviruses were generated by co-transfecting pLKO.1 plasmids of interest together with packaging vectors psPAX2 (a gift from Didier Trono; Addgene plasmid # 12260) and VSV.G (a gift from Tannishtha Reya; Addgene plasmid # 14888) using OptiMEM (Invitrogen, Carlsbad, CA) and Fugene (Promega, Madison, WI), as previously described (Burns et al., 2018). Retrovirus was produced by co-transfecting plasmids with packaging vectors gag/pol (a gift from Tannishtha Reya; Addgene plasmid # 14887) and VSV.G.
Lentiviral and retroviral infections were performed by spinoculating T-ALL cell lines with virus-containing media (1,500 g × 90 minutes) in the presence of 8 μg/ml polybrene (Merck Millipore, Darmstadt, Germany). Selection with antibiotics was started 24 hr after infection with neomycin (700 μg/ml for a minimum of 5 days; Thermo Fisher Scientific), puromycin (1 μg/ml for a minimum of 48 hr; Thermo Fisher Scientific), or blasticidin (15 μg/ml for a minimum of 5 days; Invivogen).
Transient transfection was performed using Lipofectamine 2000 reagent (Invitrogen). Briefly, 800,000 cells were seeded in 2 ml of growth medium in 24 well plates. Five mg plasmid of interest and 10 ml lipofectamine were mixed with 300 μl OptiMEM, incubated for 10 minutes and added to the wells. Antibiotic selection was begun after 48 hr of incubation.
METHOD DETAILS
Pooled ASNS/AAVS1 Library
A pooled guide RNA library with 3 unique guide RNAs targeting genomic loci encoding the catalytic domain of ASNS (http://www.uniprot.org/uniprot/P08243), and 3 unique guide RNAs targeting the safe-harbor AAVS1 locus located in intron 1 of the PPP1R12C gene (Sadelain et al., 2011), were designed as described (Sanjana et al., 2014). The oligos were cloned to a modified version of lentiGuide-Puro (a gift from Feng Zhang, Addgene plasmid # 52963) in which the guide RNA scaffold was replaced by a structurally optimized form (A-U flip and stem extension, called combined modification) previously reported to increase the efficiency of Cas9 targeting (Chen et al., 2013). Briefly, guide RNAs targeting the relevant genomic loci were designed using the Zhang lab CRISPR design tool (http://crispr.mit.edu/). One μl of 100 μm forward and reverse oligo was mixed with 1 μl 10X T4 DNA Ligation Buffer (NEB, Ipswich, MA), 6.5 μl ddH2O, 0.5 μl T4 PNK (NEB) and annealed for 30 minutes at 37°C and 5 min 95°C. 1 μl phosphoannealed oligo (diluted 1:500) was then ligated into 1 μl of BsmBI-digested lentiviral pHK09-puro plasmid using 1 μl Quick ligase (NEB), 5 μl 2x Quick Ligase buffer (NEB) and 2 μl ddH2O for 5 minutes at room temperature. Guide RNA target sequences are provided in Table S5.
Pooled lentivirus was produced by co-transfecting equal amounts of each of these guide RNAs together with the psPAX2 and VSV.G vectors as described above. Virus was concentrated using a Beckmann XL-90 ultracentrifuge (Beckman Coulter) at 100,000 g (24,000 rpm) for 2 hr at 4°C. Viral titers were determined using alamarBlue staining, as described (https://portals.broadinstitute.org/gpp/public/resources/protocols). CCRF-CEM cells (40,000 per well in 100 ml RPMI medium) infected with lentiCas9-blast (a gift from Feng Zhang; Addgene plasmid # 52962) were plated in 96-well format and transduced with lentivirus at multiplicity of infection (MOI) of 0.3. Infected cells were selected 48 hr post-infection with puromycin at 1 μg/ml for 7 days. Infected cells were treated with vehicle (PBS) or asparaginase in 24-well format (400,000 cell per well in 1 ml RPMI medium). Cells were harvested 5 days after start of asparaginase treatment, and genomic DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen). Guide RNA sequences were PCR-amplified using pHKO9 sequencing primers (Table S2), PCR-purified using the QIAquick PCR purification Kit (Qiagen), and next-generation “CRISPR sequencing” was performed at the MGH CCIB DNA Core facility (https://dnacore.mgh.harvard.edu/new-cgi-bin/site/pages/crispr_sequencing_main.jsp). Cutting efficiency was assessed using CrispRVariantsLite v1.1 (Lindsay et al., 2016).
Genome-Wide Loss of Function Screen
The genome-wide loss-of function screen was performed using the GeCKO v2 human library (a gift from Feng Zhang; Addgene #1000000048 and 1000000049), as described (Sanjana et al., 2014; Shalem et al., 2014). CCRF-CEM cells were first transduced with lentiCas9-blast, selected with blasticidin, and Cas9 activity was confirmed using a self-excising GFP construct, pXPR_011 (a gift from John Doench and David Root; Addgene plasmid # 59702). GeCKO v2 consists of two half-libraries (A and B), each of which was transduced in biologic duplicates into 1.8 × 108 Cas9-expressing CCRF-CEM cells at MOI = 0.3. Cells were selected with puromycin (1 μg/ml) beginning 24 hr post transduction, which was continued for 8 days. Based on the number of cells that survived selection and estimated growth rate, coverage was estimated at 663x for GeCKO half-library A, and 891x for GeCKO half-library B. Cells were split every other day, and the minimum number of cells kept at each split was 84 million for each half-library, in order to minimize loss of guide RNA coverage. Cells were treated with 10 U/L asparaginase beginning on day 10, and cells were harvested after 5 days of treatment. Genomic DNA was extracted using the Blood & Cell Culture DNA Maxi Kit (Qiagen). Samples were sequenced using Next-Generation Sequencing (NextSeq 500) at the Molecular Biology Core Facilities at Dana Farber Cancer Institute (http://mbcf.dfci.harvard.edu/genomics/next-generation-sequencing/). Significance of gene depletion based on guide RNA drop-out was calculated using MAGeCK software (https://sourceforge.net/projects/mageck/) (Li et al., 2014).
Analysis of gRNA Off-Target Sites
Oligos were cloned into a modified version of lentiGuide-Puro as described above. Cas9-expressing CCRF-CEM cells were infected with gRNAs targeting NKD2 or LGR6. DNA was extracted after puromycin selection and genomic DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen). Off-target sites were identified using the CRISPOR algorithm (Haeussler et al., 2016), http://crispor.tefor.net/). Predicted off-target loci with ≤ 2 base pair mismatches were PCR-amplified and PCR-purified using the QUIAquick PCR purification Kit (Qiagen). Sanger sequencing was performed at Genewiz (https://www.genewiz.com/).
shRNA and Expression Plasmids
The following lentiviral shRNA vectors in pLKO.1 with puromycin resistance were generated by the RNAi Consortium library of the Broad Institute, and obtained from Sigma-Aldrich: shLuciferase (TRCN0000072243), shNKD2#1 (TRCN0000187580), shNKD2#3 (TRCN0000428381), shLGR6#2 (TRCN0000063619) shLGR6#4 (TRCN0000063621), shAPC#1 (TRCN0000010296), shAPC#2 (TRCN0000010297), shGSK3α#1 (TRCN0000010340), shGSK3α#4 (TRCN0000038682), shGSK3β#2 (TRCN0000039564), shGSK3β#6 (TRCN0000010551).
The construct encoding a constitutively active β-catenin (ΔN90) allele was a gift from Bob Weinberg (https://www.Addgene.org/36985/). Expression constructs expressing wild-type FBXW7 (also known as CDC4) or its R465C mutant were a gift from Bert Vogelstein (https://www.Addgene.org/16652/ and https://www.Addgene.org/16653/). The 7TGC (7xTcf-eGFP//SV40-mCherry) TOPFlash reporter was a gift from Roel Nusse (https://www.Addgene.org/24304/). A hyperactive open-gate mutant of the human proteasomal subunit PSMA4, termed ΔN-PSMA4, was designed by deleting the cDNA sequences encoding amino acids 2 to 10 (SRRYDSRTT) of PSMA4 isoform NP_002780.1, encoded by the transcript NM_002789.6, based on the data of Choi and colleagues (Choi et al., 2016). This ΔN-PSMA4 coding sequence was synthesized by gene synthesis, and cloned into the pLX304 lentiviral expression vector in-frame with the C-terminal V5 tag provided by this vector, by GeneCopoeia (Rockville, MD). The GSK3α pWZL expression vector was previously described (Banerji et al., 2012).
Assessment of Chemotherapy Response and Apoptosis
Cells (25,000 per well) were seeded in 100 ml of complete growth medium in 96-well plates and incubated with chemotherapeutic agents or vehicle. T-ALL cells were split every 48 hr and AML cells were split every 72 hr. Cell viability was assessed by counting viable cells based on trypan blue vital dye staining (Invitrogen), according to the manufacturer’s instructions. Chemotherapeutic drugs included: asparaginase (pegaspargase, Shire, Lexington, MA), dexamethasone (Sigma-Aldrich), vincristine (Selleckchem, Houston, TX), doxorubicin (Sigma-Aldrich), 6-mercaptopurine (Abcam, Cambridge, UK), CHIR99021 (Selleckchem), rapamycin (Selleckchem), RAD001 (Selleckchem), AZD2014 (Selleckchem), thapsigargin (Sigma-Aldrich) and Wnt3A (R&D systems, Minneapolis, MN). BRD0705 and BRD3731 were synthesized as described (Wagner et al., 2018). Caspase 3/7 activity was assessed using the Caspase Glo 3/7 Assay (Promega, Madison, WI) according to the manufacturer’s instructions.
TOPFlash Reporter
Briefly, 400,000 CCRF-CEM cells were transduced with the TOPFlash reporter 7TGC (Fuerer and Nusse, 2010), as described in the lentiviral infection section above. Cells expression the constitutive mCherry selection marker were selected by fluorescence activated cell sorting (FACS), treated with the Wnt ligand Wnt3A for 5 days, and cells with Wnt-inducible GFP expression were selected by sorting for mCherry and GFP double-positive cells. Selected cells were released from the Wnt ligand for a minimum of 7 days and then further manipulated with expression plasmids. GFP positivity of the mCherry positive cell fraction was assessed on a Beckton-Dickinson LSR-II instrument.
Western Blot Analysis
Cells were lysed in RIPA buffer (Merck Millipore) supplemented with cOmplete protease inhibitor (Roche, Basel, Switzerland) and PhosSTOP phosphatase inhibitor (Roche). Laemmli sample buffer (Bio-Rad, Hercules, CA) and β-mercaptoethanol (Sigma-Aldrich) were mixed with 20 μg of protein lysate before being run on a 4–12% bis-tris polyacrylamide gel (Thermo Fisher Scientific). Blots were transferred to PVDF membrane (Thermo Fisher Scientific) and blocked with 5% BSA (New England Biolabs) in phosphate-buffered saline with 0.1% Tween (Boston Bioproducts, Ashland, MA) and probed with the following antibodies: Non-phospho β-catenin (Ser33/37/Thr41) antibody (1:1000, Cell Signaling Technologies #8814), total β-catenin antibody (1:1000, Cell Signaling Technologies #8480), total GSK3α/β antibody (1:1000, Cell Signaling Technologies #5676), phospho-GSK3α/β (Tyr279/216) antibody (Thermo Fisher Scientific #OPA1–03083), Myc-tag antibody (1:1000, Cell Signaling Technologies #2272), phospho-p70 S6 kinase (Thr389) antibody (1:1000 Cell Signaling Technologies #9234), anti-ubiquitin antibody (1:1000, Millipore, Lys-48 specific, clone Apu2 #05–1307), phospho-PERK (Thr980) antibody (1:1000, Cell Signaling Technologies #3179) or GAPDH (1:1000, Cell Signaling Technologies #2118). Detection of horseradish peroxidase-linked secondary antibodies (1:2000, Cell Signaling Technologies #7074S) with horseradish peroxidase substrate (Thermo Fisher Scientific) was visualized using Amersham Imager 600 (GE Healthcare Life Sciences, Marlborough, MA).
Assessment of Cell Size
Cells (400,000 per well) were plated in 1 ml of complete growth medium, containing a final concentration of 10 U/L asparaginase or 100 ng/ml Wnt3A ligand in a 24-well format. After 48 hr of treatment, forward scatter height (FSC-H) was assessed by flow cytometry.
Assessment of Transduction Efficiency by Immunostaining
Cells (1 million) were collected by centrifugation and fixed with 4% formaldehyde in PBS for 15 minutes at room temperature. Subsequently, cells were permeabilized by adding ice-cold methanol to a final concentration of 90% and incubated for 30 minutes on ice. Permeabilized cells were resuspended in 100 μL of incubation buffer (0.5% BSA in PBS) containing the primary antibody and incubated for 1 hr at room temperature. Cells were washed twice and resuspended in incubation buffer containing the fluorochrome-conjugated secondary antibody (1:250, Cell Signaling, Anti-Rabbit IgG (H+L), F(ab’)2 Fragment Alexa Fluor 488 Conjugate #4412). Fluorescence positivity was assessed by flow cytometry.
Quantitative Reverse Transcriptase PCR (qRT-PCR)
RNA was isolated using RNeasy kit (Qiagen) and cDNA was made using SuperScript III first-strand cDNA synthesis kit (Thermo Fisher Scientific). qRT-PCR was performed using Power SYBR® Green PCR Master Mix (Thermo Fisher Scientific) and 7500 real-time PCR system (Applied Biosystems). Primers used are described in Table S2.
Assessment of Protein Stability
Protein degradation was assessed using a non-radioactive quantification of the methionine analog L-azidohomoalanine (AHA) AlexaFluor488 (Thermo Fisher), as previously described (Wang et al., 2017). Briefly, 400,000 CCRF-CEM cells were seeded in 1 ml of methionine-free RPMI containing 10% dialyzed FBS. After 30 minutes, the pulse step was performed by replacing this media with 1 ml RPMI supplemented with 10% dialyzed FBS and AHA at a final concentration of 50 μM for 18 hr. In the chase step, cells were released from AHA by replacing media with RPMI containing 10% dialyzed FBS and 10x L-methionine (2 mM) for 2 hr. Subsequently, media was replaced with regular growth medium and cells were treated with a final concentration of 10 U/L asparaginase, followed by fixation of cells. AHA labeled proteins were tagged using TAMRA alkyne click chemistry, and fluorescence intensity was measured by flow cytometry. A sample without AHA labeling but TAMRA alkyne tag was included as a negative control to account for background fluorescence.
Proteomics
CCRF-CEM cells were seeded in 1 ml of complete growth medium, containing a final concentration of 10 U/L asparaginase and/or 100 nM BRD0705 in a 24-well format. To each well, the caspase inhibitor Z-VAD-FMK (Promega) was added at a final concentration of 20 μM. After 48 hr of treatment, protein lysates were made using RIPA buffer (Merck Millipore) supplemented with cOmplete protease inhibitor (Roche, Basel, Switzerland) and PhosSTOP phosphatase inhibitor (Roche). Laemmli-sample buffer and β-mercaptoethanol were mixed with 20 μg of protein lysate and run on a 4–12% bis-tris polyacrylamide gel. Electrophoresis was run at 140V and stopped after 30 minutes. The gel was stained with GelCode Blue Stain Reagent (ThermoFisher) under constant agitation at room temperature for 1 hr. Subsequently, each lane of the polyacrylamide gel was cut into three slices. Excised gel bands were cut into approximately 1 mm3 pieces, subjected to in-gel trypsin digestion (Shevchenko et al., 1996), washed and dehydrated in acetonitrile, dried in a speed-vac, and re-hydrated in 50 mM ammonium bicarbonate solution with 12.5 ng/μl of sequencing-grade trypsin (Promega, Madison, WI). After 45 min, the excess trypsin was removed and replaced with 50 mM ammonium bicarbonate solution, followed by overnight incubation at 37°C. Peptides were extracted into a solution of 50% acetonitrile and 1% formic acid and dried in a speedvac. Samples were then reconstituted, subjected to reverse-phase chromatography on a C18 HPLC column, and analyzed by electrospray ionization mass spectrometry using an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific, Waltham, MA) (Eng et al., 1994). All databases include a reversed version of all the sequences and the data was filtered to <2% peptide false discovery rate.
Rescue of Wnt Activation and Asparaginase with Amino Acid Supplementation
CCRF-CEM cells transduced with the indicated shRNAs (25,000 per well) were seeded in 100 mL of complete growth medium, containing a final concentration of 10 U/L asparaginase (or PBS vehicle control) in a 96-well format. Growth medium (RPMI-1640 with 10% fetal bovine serum) was supplemented with L-asparagine (Sigma-Aldrich) at a final concentration of 3.78 mM (10X) or L-glutamine at a final concentration of 20.5 mM (10X). Every 12 hr, 50 μl of complete growth medium was removed and replaced with 50 μl fresh growth media, supplemented with the appropriate concentration of asparaginase. After 72 hr of treatment, viability was assessed by trypan blue viable cell staining.
Amino Acid Quantification
CCRF-CEM cells (400,000 per well) were seeded in 1 ml of complete growth medium in a 24-well format. The growth medium was supplemented with final concentrations of 10 U/L asparaginase and/or 100 nM BRD0705. After 12 hr of treatment, media was collected and stored at −80C until amino acid quantification. Amino acids were subjected to derivatization using 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate and quantified by Ultra Performance Liquid Chromatography using the Waters MassTrack Amino Acid Analysis Solution according to the manufacturer’s instructions (Waters Corporation, Milford, MA).
In Vivo Drug Treatment of Patient-Derived Xenografts
NRG mice were exposed to a sub-lethal dose of radiation (4.5 Gy) and subsequently injected with human leukemia cells. All injections were performed by tail vein injection. Blood collections were performed to monitor leukemia onset in mice injected with leukemic cells, which was assessed by staining for human CD45 expression using anti-human CD45-PE-Cy7 (1:400, BD# 560915) antibody staining. As soon as leukemia engraftment was confirmed based on ≥5% human leukemia cells in the peripheral blood, treatment of mice was begun. Asparaginase (1,000 U/kg) or PBS were injected by tail vein injection as a single dose on day 1 of drug treatment, and BRD0705 (15 mg/kg) or vehicle were given every 12 hr for 12 days by oral gavage. Vehicle was formulated as previously described (Wagner et al., 2018). Mice were anesthetized with isoflurane (Patterson Veterinary, Greeley, CO) prior to gavage. After start of treatment, body weight was monitored every third day. To assess bilirubin levels, retro-orbital blood collection was performed, and bilirubin levels were measured in the Boston Children’s Hospital clinical laboratory. The primary endpoint of this experiment was survival. Mice were euthanized as soon as they developed weight loss greater than 15% or signs/symptoms of progressing disease. Post-mortem analysis of leukemic burden was performed by extracting bone marrow cells, which were filtered through a 40 μM mesh filter and red blood cells were lysed using the BD Biosciences Red Blood Cell Lysis reagent (BD #555899). Isolated bone marrow cells were stained for assessment of leukemic burden using following antibodies from BD Biosciences: Anti-human CD4-APC-Cy7 (1:100, BD# 561839) and anti-human CD8-PerCP-Cy5.5 (1:100, BD# 560662).
Data and Software Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD013061.
Quantification and Statistical Analysis
For two-group comparisons of continuous measures, a two-tailed Welch unequal variances t-test was used. For 3-group comparisons, a one-way analysis of variance model (ANOVA) was performed and a Dunnett adjustment for multiple comparisons was used. For analysis of two effects, a two-way ANOVA model was constructed and included an interaction term between the two effects. Post-hoc adjustment for multiple comparisons for two-way ANOVA included Tukey-Kramer adjustment. The log rank test was used to test for differences in survival between groups, and the method of Kaplan and Meier was used to construct survival curves. Data shown as bar graphs represent the mean +/− standard error of the mean (SEM) of a minimum of 3 biologic replicates, unless otherwise indicated. Differential expression for proteomic data was determined using the voom-limma method with an empirical Bayes statistic (eBayes (trend=TRUE, robust=TRUE)) in R. Counts were normalized and filtered prior to analysis using the functions calcNormFactors (method=“upperquartile”) and filterByExpr. Quality of filtering was assessed using plots including voom (plot=TRUE) and plotSA (fit) where the fit was output from lmFit and the eBayes functions, and p values were adjusted using the method of Benjamini-Hochberg (or false discovery rate (FDR)). All p values reported are two-sided and considered as significant if <0.05.
Supplementary Material
Table S1, related fo Figure 1. Significance of gene and gRNA depletion or enrichment in asparaginase-treated cells.
Table S3, related to Figure 2. Characteristics of T-ALL, B-ALL and AML cell lines utilized.
Table S4, related to Figure 5. Peptide counts from proteomics analysis of CCRF-CEM cells, and comparisons across conditions.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Active (non-Ser33/37/Thr41-phosphorylated) β-catenin | Cell Signaling | Cat#8814 |
| Total β-catenin | Cell Signaling | Cat#8480 |
| GAPDH | Cell Signaling | Cat#2118 |
| Phospho-p70S6K (Thr389) | Cell Signaling | Cat#9234 |
| Anti-Myc tag | Cell Signaling | Cat#2272 |
| Phospho-GSK3αβ(Tyr279/216) | Thermo Fisher | Cat#OPA1–03083 |
| Total GSK3αβ | Cell Signaling | Cat#5676 |
| anti-ubiquitin antibody (Lys-48 specific, clone Apu2) | Millipore | Cat#05–1307 |
| Anti-Rabbit IgG (H+L), F(ab’)2 Fragment Alexa Fluor 488 Conjugate | Cell Signaling | Cat#4412 |
| Phospho-PERK (Thr980) | Cell Signaling | Cat#3179 |
| Secondary HRP-linked antibody | Cell Signaling | Cat#7074 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| T-ALL Patient-Derived Xenograft T-ALL 9 | This paper | N/A |
| B-ALL Patient-Derived Xenograft CBAB-21952 | This paper | N/A |
| Hypodiploid B-ALL Patient-Derived Xenograft 05–577 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Asparaginase (pegaspargase) | Shire Pharmaceuticals | Oncaspar |
| Human Wnt3A | R&D systems | Cat#5036-WN-010 |
| Trypan blue | Invitrogen | Cat#T10282 |
| CHIR99021 | Selleckchem | Cat#S1263 |
| Human interleukin-6 | R&D systems | Cat#206-IL-010 |
| Human stem cell factor | R&D systems | Cat#255-SC |
| Puromycin | Thermo Fisher | Cat#A1113803 |
| Blasticidin | Thermo Fisher | Cat#R21001 |
| Neomycin | Thermo Fisher | Cat#21810031 |
| Polybrene | Merck Millipore | Cat#TR-1003-G |
| Lipofectamine 2000 | Invitrogen | Cat#11668030 |
| Dexamethasone | Sigma-Aldrich | Cat#D4902 |
| Vincristine | Selleckchem | Cat#S1241 |
| Doxorubicin | Sigma-Aldrich | Cat#D1515 |
| 6-Mercaptopurine | Abcam | Cat#ab142389 |
| Rapamycin | Selleckchem | Cat#S1039 |
| RAD001 | Selleckchem | Cat#S1120 |
| AZD2014 | Selleckchem | Cat#S2783 |
| Thapsigargin | Sigma-Aldrich | Cat#T9033 |
| BRD0703 | (Wagner et al., 2018) | N/A |
| BRD3731 | (Wagner et al., 2018) | N/A |
| RIPA buffer | Merck Millipore | Cat#20–188 |
| PhosSTOP phosphatase inhibitor | Roche | Cat#4906845001 |
| Laemmli sample buffer | Bio-Rad | Cat#161–0737 |
| b-mercaptoethanol | Sigma-Aldrich | Cat#M6250 |
| 4–12% bis-tris polyacrylamide gel | Thermo Fisher | Cat#NP0336 |
| PVDF membrane | Thermo Fisher | Cat# LC2005 |
| Azidohomoalanine (AHA) AlexaFluor488 | Thermo Fisher | Cat#C10289 |
| TAMRA alkyne | Thermo Fisher | Cat#T10183 |
| L-Asparagine | Sigma-Aldrich | Cat#A4159–25G |
| L-Glutamine | Sigma-Aldrich | Cat#G8540 |
| GelCode Blue Stain Reagent | Thermo Fisher | Cat#24590 |
| RPMI-1640 | Thermo Fisher | Cat#11875119 |
| Critical Commercial Assays | ||
| Caspase Glo 3/7 Assay | Promega | Cat#G8090 |
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69504 |
| QIAquick PCR purification kit | Qiagen | Cat#28104 |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | Cat#51194 |
| Z-VAD-FMK Caspase Inhibitor | Promega | Cat #G7231 |
| Deposited Data | ||
| Proteomics of CCRF-CEM cells treated with asparaginase and/or BRD0705 | ProteomeXchange | Dataset ID: PXD013061 |
| Experimental Models: Cell Lines | ||
| CCRF-CEM cells | ATCC | Cat#CCL-119 |
| Jurkat cells | ATCC | Cat#TIB-152 |
| PF382 cells | DSMZ | Cat#ACC-38 |
| DND41 cells | DSMZ | Cat# ACC-525 |
| MOLT4 cells | ATCC | Cat#CRL-1582 |
| OCI-AML2 cells | Alex Kentsis lab | N/A |
| SKM-1 cells | Alex Kentsis lab | N/A |
| THP-1 cells | Alex Kentsis lab | N/A |
| MHH CALL2 cells | DSMZ | Cat#ACC-341 |
| Normal human CD34+ hematopoietic progenitors | Fred Hutchinson Cancer Research Center | https://sharedresources.fredhutch.org/products/cd34-cells |
| Experimental Models: Organisms/Strains | ||
| NRG mice (NOD.Cg-Rag1<tm1Mom> Il2rg<tm1Wjl>/SzJ) | Jackson labs | 007799 |
| Oligonucleotides | ||
| See Table S5. | ||
| Recombinant DNA | ||
| LentiCas9-Blast | Addgene.org | Cat#52962 |
| LentiGuide-Puro | Addgene.org | Cat#52963 |
| Gecko v2 guide RNA library | Addgene.org | Cat#1000000048 and 1000000049 |
| shLuciferase | Broad RNAi Consortium | Cat#TRCN0000072243 |
| shNKD2 #1 | Broad RNAi Consortium | Cat#TRCN0000187580 |
| shNKD2 #3 | Broad RNAi Consortium | Cat#TRCN0000428381 |
| shLGR6 #2 | Broad RNAi Consortium | Cat#TRCN0000063619 |
| shLGR6 #4 | Broad RNAi Consortium | Cat#TRCN0000063621 |
| shAPC #1 | Broad RNAi Consortium | Cat#TRCN0000010296 |
| shAPC #2 | Broad RNAi Consortium | Cat#TRCN0000010297 |
| shGSK3α #1 | Broad RNAi Consortium | Cat#TRCN0000010340 |
| shGSK3α #4 | Broad RNAi Consortium | Cat#TRCN0000038682 |
| shGSK3β #2 | Broad RNAi Consortium | Cat#TRCN0000039564 |
| shGSK3β #6 | Broad RNAi Consortium | Cat#TRCN0000010551 |
| psPAX2 | Addgene.org | Cat#12260 |
| VSV.G | Addgene.org | Cat#14888 |
| Gag/pol | Addgene.org | Cat#14887 |
| pXPR_011 | Addgene.org | Cat#59702 |
| Constitutively active β-catenin (ΔN90) | Addgene.org | Cat#36985 |
| FBXW7 wild-type | Addgene.org | Cat#16652 |
| FBXW7 R465C mutant | Addgene.org | Cat#16653 |
| FBXW7 wild-type (CS-T3614-LX304–01-B) | Genecopoeia.com | |
| FBXW7 R465C mutant (EX-OL03574-LX304-B) | Genecopoeia.com | |
| 7TGC TOPFlash reporter | Addgene.org | Cat#24304 |
| Hyperactive ΔN-PSMA4 | This paper | N/A |
| GSK3α in pWZL expression vector | (Banerji et al., 2012) | N/A |
| Software and Algorithms | ||
| CrispRVariantsLite v1.1 | (Lindsay et al., 2016) | Version 1.1 |
| MAGeCK v.0.5.7 | (Li et al., 2014) | https://sourceforge.net/projects/mageck |
| CRISPOR | (Haeussler et al., 2016) | http://crispor.tefor.net/ |
| RStudio | v. 1.1.463 | http://www.rstudio.org |
| Other | ||
SIGNIFICANCE.
The intensification of asparaginase-based therapy has improved outcomes for several subtypes of acute leukemia, but the development of treatment resistance has a poor prognosis. We hypothesized, from the concept of synthetic lethality, that gain-of-fitness alterations in drug-resistant cells had conferred a survival advantage that could be exploited therapeutically. We found a synthetic lethal interaction between activation of Wnt-dependent stabilization of proteins (Wnt/STOP) and asparaginase in acute leukemias resistant to this enzyme. Inhibition of the alpha isoform of GSK3 was sufficient to phenocopy this effect, and the combination of GSK3α- selective inhibitors and asparaginase had marked therapeutic activity against leukemias resistant to monotherapy with either agent. These data indicate that drug-drug synthetic lethal interactions can improve the therapeutic index of cancer therapy.
Highlights for Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias.
Hinze et al.
A genetic screen revealed synthetic lethality of Wnt activation and asparaginase
Wnt signaling induces asparaginase sensitivity by inhibiting protein degradation
GSK3α inhibition phenocopies Wnt-induced sensitization to asparaginase
This combination has potent activity against asparaginase-resistant leukemias
Hinze et al. find that asparaginase treatment is synthetically lethal in acute leukemias with the activation of Wnt-dependent stabilization of proteins, which reduces GSK3-dependent protein degradation to limit Asn availability. Inhibiting GSK3α sensitizes asparaginase-resistant leukemias to the treatment.
ACKNOWLEDGEMENTS
We thank Alex Kentsis, Marc Mansour, Ralph DeBerardinis, and Min Jae Lee for advice and discussion, and Meaghan McGuinness, Roxane Labrosse, Ronald Mathieu, Mahnaz Paktinat, Zachary Herbert, Ross Tomaino, Mark Kellogg and Peter Blanding for experimental assistance. We thank Jennifer Perry, Andrei Krivtsov and Scott Armstrong for B-ALL PDX models. We thank John Gilbert for editorial assistance. This work was supported by NIH/NCI grant 1R01CA193651, the Boston Children’s Hospital Translational Investigator Service, and the Victoria Ann Schuerch Memorial Foundation and Victoria’s Walk. L.H. was supported by the German National Academic Foundation and the Biomedical Education Program. A.G. was supported by a CHPA Investigatorship at Boston Children’s Hospital. K.S. was supported by the NIH/NCI grant R35 CA210030 and a Leukemia and Lymphoma Society Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Boston Children’s Hospital has filed US provisional patent application 62/697,053, entitled “Method for treating cancer”, filed July 12, 2018, on the subject matter of this publication. F.F.W. has consulted for a biotechnology company on a GSK3-related project, and the Broad Institute of MIT and Harvard has filed US patents US20160375006, WO2014059383 and WO2018187630 on BRD0705 and related GSK3 inhibitors. K.S. has consulted for Novartis and Rigel Pharmaceuticals and receives grant support from Novartis for research unrelated to this study. The authors declare no other competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven supplemental figures and five supplemental tables and can be found with this article online.
REFERENCES
- Acebron SP, Karaulanov E, Berger BS, Huang YL, and Niehrs C (2014). Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell 54, 663–674. [DOI] [PubMed] [Google Scholar]
- Alexander TB, Wang L, Inaba H, Triplett BM, Pounds S, Ribeiro RC, Pui CH, and Rubnitz JE (2017). Decreased relapsed rate and treatment-related mortality contribute to improved outcomes for pediatric acute myeloid leukemia in successive clinical trials. Cancer 123, 3791–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel IM, den Boer ML, Meijerink JP, Veerman AJ, Reniers NC, and Pieters R (2006). Up-regulation of asparagine synthetase expression is not linked to the clinical response L-asparaginase in pediatric acute lymphoblastic leukemia. Blood 107, 4244–4249. [DOI] [PubMed] [Google Scholar]
- Ashworth A, and Lord CJ (2018). Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nature reviews Clinical oncology. [DOI] [PubMed] [Google Scholar]
- Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, Kakoza RM, Chow KT, Ross L, Alexe G, et al. (2012). The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest 122, 935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CN, Ross SE, Longo KA, Bajnok L, Hemati N, Johnson KW, Harrison SD, and MacDougald OA (2002). Regulation of Wnt signaling during adipogenesis. J Biol Chem 277, 30998–31004. [DOI] [PubMed] [Google Scholar]
- Broome JD (1961). Evidence that the L-Asparaginase Activity of Guinea Pig Serum is responsible for its Antilymphoma Effects. Nature 191, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner E, Peter O, Schweizer L, and Basler K (1997). pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila. Nature 385, 829–833. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Burns MA, Liao ZW, Yamagata N, Pouliot GP, Stevenson KE, Neuberg DS, Thorner AR, Ducar M, Silverman EA, Hunger SP, et al. (2018). Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capizzi RL, Davis R, Powell B, Cuttner J, Ellison RR, Cooper MR, Dillman R, Major WB, Dupre E, and McIntyre OR (1988). Synergy between high-dose cytarabine and asparaginase in the treatment of adults with refractory and relapsed acute myelogenous leukemia--a Cancer and Leukemia Group B Study. J Clin Oncol 6, 499–508. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, and Huang B (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li Y, Xue J, Gong A, Yu G, Zhou A, Lin K, Zhang S, Zhang N, Gottardi CJ, and Huang S (2016). Wnt-induced deubiquitination FoxM1 ensures nucleus beta-catenin transactivation. EMBO J 35, 668–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WH, de Poot SA, Lee JH, Kim JH, Han DH, Kim YK, Finley D, and Lee MJ (2016). Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nature communications 7, 10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavell LA, Gelber RD, Cohen HJ, Hitchcock-Bryan S, Cassady JR, Tarbell NJ, Blattner SR, Tantravahi R, Leavitt P, and Sallan SE (1986). Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N Engl J Med 315, 657–663. [DOI] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, and Clurman BE (2014). Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, et al. (2011). Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297. [DOI] [PubMed] [Google Scholar]
- DeAngelo DJ, Stevenson KE, Dahlberg SE, Silverman LB, Couban S, Supko JG, Amrein PC, Ballen KK, Seftel MD, Turner AR, et al. (2015). Long-term outcome of a pediatric-inspired regimen used for adults aged 18–50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia 29, 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, and Woodgett JR (2007). Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 12, 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Ertel IJ, Nesbit ME, Hammond D, Weiner J, and Sather H (1979). Effective dose of L-asparaginase for induction of remission in previously treated children with acute lymphocytic leukemia: a report from Childrens Cancer Study Group. Cancer Res 39, 3893–3896. [PubMed] [Google Scholar]
- Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, and Long F (2013). WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab 17, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fuerer C, and Nusse R (2010). Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PloS one 5, e9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, and Sweet-Cordero EA (2018). Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 33, 91–107 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Haskell CM, and Canellos GP (1969). l-asparaginase resistance in human leukemia--asparagine synthetase. Biochemical pharmacology 18, 2578–2580. [DOI] [PubMed] [Google Scholar]
- Hermanova I, Zaliova M, Trka J, and Starkova J (2012). Low expression of asparagine synthetase in lymphoid blasts precludes its role in sensitivity to L-asparaginase. Exp Hematol 40, 657–665. [DOI] [PubMed] [Google Scholar]
- Holleman A, Cheok MH, den Boer ML, Yang W, Veerman AJ, Kazemier KM, Pei D, Cheng C, Pui CH, Relling MV, et al. (2004). Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med 351, 533–542. [DOI] [PubMed] [Google Scholar]
- Horowitz B, Madras BK, Meister A, Old LJ, Boyes EA, and Stockert E (1968). Asparagine synthetase activity of mouse leukemias. Science 160, 533–535. [DOI] [PubMed] [Google Scholar]
- Huang YL, Anvarian Z, Doderlein G, Acebron SP, and Niehrs C (2015). Maternal Wnt/STOP signaling promotes cell division during early Xenopus embryogenesis. Proceedings of the National Academy of Sciences of the United States of America 112, 5732–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. (2006). TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–968. [DOI] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, and Elledge SJ (2001). Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294, 173–177. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, and Ciechanover A (2017). The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends in biochemical sciences 42, 873–886. [DOI] [PubMed] [Google Scholar]
- Lento W, Congdon K, Voermans C, Kritzik M, and Reya T (2013). Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H, Burger A, Biyong B, Felker A, Hess C, Zaugg J, Chiavacci E, Anders C, Jinek M, Mosimann C, and Robinson MD (2016). CrispRVariants charts the mutation spectrum of genome engineering experiments. Nat Biotechnol 34, 701–702. [DOI] [PubMed] [Google Scholar]
- Moon RT, and Miller JR (1997). The APC tumor suppressor protein in development and cancer. Trends Genet 13, 256–258. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, Tomita D, Fujimoto J, Kurasawa O, Takahara C, et al. (2018). Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proceedings of the National Academy of Sciences of the United States of America 115, E7776–E7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehir K, Cosgun N, Hecht A, Yang X, Mangolini M, Aghajanirefah A, Chen Z, Xiao G, Klemm L, Hong C, et al. (2017). Lgr5 Functions As Negative Regulator of Wnt Signaling in B Cells and Is Critical for Self-Renewal of Normal and Transformed B Cells. Blood (ASH annual meeting abstracts) 130, 3989. [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. (2007). FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med 204, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pession A, Valsecchi MG, Masera G, Kamps WA, Magyarosy E, Rizzari C, van Wering ER, Lo Nigro L, van der Does A, Locatelli F, et al. (2005). Long-term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukemia. J Clin Oncol 23, 7161–7167. [DOI] [PubMed] [Google Scholar]
- Poppenborg SM, Wittmann J, Walther W, Brandenburg G, Krahmer R, Baumgart J, and Leenders F (2016). Impact of anti-PEG IgM antibodies on the pharmacokinetics of pegylated asparaginase preparations in mice. Eur J Pharm Sci 91, 122–130. [DOI] [PubMed] [Google Scholar]
- Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, Vora A, Baruchel A, Silverman LB, Schmiegelow K, et al. (2015). Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J Clin Oncol 33, 2938–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Traver D, and Willert K (2017). The role of Wnt signaling in hematopoietic stem cell development. Crit Rev Biochem Mol Biol 52, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzari C, Conter V, Stary J, Colombini A, Moericke A, and Schrappe M (2013). Optimizing asparaginase therapy for acute lymphoblastic leukemia. Curr Opin Oncol 25 Suppl 1, S1–9. [DOI] [PubMed] [Google Scholar]
- Sadelain M, Papapetrou EP, and Bushman FD (2011). Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer 12, 51–58. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Siegfried E, Chou TB, and Perrimon N (1992). wingless signaling acts through zeste-white 3, the Drosophila homolog of glycogen synthase kinase-3, to regulate engrailed and establish cell fate. Cell 71, 1167–1179. [DOI] [PubMed] [Google Scholar]
- Staal FJ, Chhatta A, and Mikkers H (2016). Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp Hematol 44, 451–457. [DOI] [PubMed] [Google Scholar]
- Stams WA, den Boer ML, Beverloo HB, Meijerink JP, Stigter RL, van Wering ER, Janka-Schaub GE, Slater R, and Pieters R (2003). Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood 101, 2743–2747. [DOI] [PubMed] [Google Scholar]
- Suraweera A, Munch C, Hanssum A, and Bertolotti A (2012). Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 48, 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, and De Robertis EM (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2018). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, and Aifantis I (2007). The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 204, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. (2016). The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell 29, 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A, et al. (1997). Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88, 789–799. [DOI] [PubMed] [Google Scholar]
- Van Heeke G, and Schuster SM (1989). The N-terminal cysteine of human asparagine synthetase is essential for glutamine-dependent activity. J Biol Chem 264, 19475–19477. [PubMed] [Google Scholar]
- Wagner FF, Benajiba L, Campbell AJ, Weiwer M, Sacher JR, Gale JP, Ross L, Puissant A, Alexe G, Conway A, et al. (2018). Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker F, Zhang HH, Odorizzi A, and Burgess AW (2011). LGR5 is a negative regulator of tumourigenicity, antagonizes Wnt signalling and regulates cell adhesion in colorectal cancer cell lines. PloS one 6, e22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang J, Lee YM, Ng S, Shi Y, Hua ZC, Lin Q, and Shen HM (2017). Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat Protoc 12, 279–288. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, and Clurman BE (2004). The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America 101, 9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, and Roberts JM (2003). Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell 12, 381–392. [DOI] [PubMed] [Google Scholar]
- Wells RJ, Woods WG, Lampkin BC, Nesbit ME, Lee JW, Buckley JD, Versteeg C, and Hammond GD (1993). Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Childrens Cancer Group. J Clin Oncol 11, 538–545. [DOI] [PubMed] [Google Scholar]
- Wharton KA Jr., Zimmermann G, Rousset R, and Scott MP (2001). Vertebrate proteins related to Drosophila Naked Cuticle bind Dishevelled and antagonize Wnt signaling. Dev Biol 234, 93–106. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Kwong YL, Kim WS, Maeda Y, Hashimoto C, Suh C, Izutsu K, Ishida F, Isobe Y, Sueoka E, et al. (2011). Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-Cell Tumor Study Group study. J Clin Oncol 29, 4410–4416. [DOI] [PubMed] [Google Scholar]
- Yun MK, Nourse A, White SW, Rock CO, and Heath RJ (2007). Crystal structure and allosteric regulation of the cytoplasmic Escherichia coli L-asparaginase I. J Mol Biol 369, 794–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related fo Figure 1. Significance of gene and gRNA depletion or enrichment in asparaginase-treated cells.
Table S3, related to Figure 2. Characteristics of T-ALL, B-ALL and AML cell lines utilized.
Table S4, related to Figure 5. Peptide counts from proteomics analysis of CCRF-CEM cells, and comparisons across conditions.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD013061.