

Abstract
Study Design:
Narrative review.
Objectives:
To discuss the relationship between degenerative cervical myelopathy (DCM) and vitamin B12 deficiency. Specifically, it is the aim to outline the rational for future research into assessment and therapeutic optimization of vitamin B12 in the treatment of DCM.
Methods:
Literature review.
Results:
DCM is the commonest cause of spinal cord impairment, with an average age of presentation in the sixth decade. Patients at this age have also been reported to have a high prevalence of vitamin B12 deficiency, with estimates of up to 20% in the elderly. Vitamin B12 deficiency can result in subacute combined degeneration of the spinal cord (SACD), and several case reports have pointed to patients with both DCM and SACD. Both SACD and reversible compressive injury due to DCM necessitate remyelination in the spinal cord, a process that requires adequate vitamin B12 levels. Basic science research on nerve crush injuries have shown that vitamin B12 levels are altered after nerve injury and that vitamin B12 along with dexamethasone or nonsteroidal anti-inflammatory drugs can reduce Wallerian degeneration. Furthermore, it has been suggested that a combination of B-vitamins can reduce glutamate-induced neurotoxicity.
Conclusions:
Given the high prevalence of clinical and subclinical vitamin B12 deficiency in the elderly, the role of vitamin B12 in myelination, and vitamin B12 deficiency as a differential diagnosis of DCM, it is important to investigate what role vitamin B12 levels play in patients with DCM in terms of baseline neurological function and whether optimization of vitamin B12 levels can improve surgical outcome. Furthermore, the routine assessment of vitamin B12 levels in patients considered for DCM surgery should be considered.
Keywords: nutrition, anemia, subacute combined degeneration, spinal cord, nitrous oxide, cobalamin
Introduction
Degenerative cervical myelopathy (DCM) encompasses a set of age-related changes of the cervical spine that result in spinal cord impairment through static and dynamic injury mechanisms.1 Patients with DCM typically present with variable degrees of upper and lower limb neurological deficits, including numbness, clumsiness, gait impairment, and motor weakness. Additionally, objective myelopathic signs such as Hoffmann’s sign, Babinski’s reflex, and ankle clonus may be observed.2,3 One of the potential differential diagnoses to consider in these patients is cobalamin or vitamin B12 (B12) deficiency. Neurological deficits encountered with B12 deficiency include peripheral neuropathy, myelopathy, mental status changes, optic neuropathy, or a combination of these.4,5 Patients with both DCM and B12 deficiency are most frequently diagnosed above the age of 50 years, and it has been estimated that the prevalence of B12 deficiency is about 20% in industrialized countries.4 Furthermore, many more patients may have subclinical B12 deficiency.6 Given this high prevalence of B12 deficiency in elderly population, it would seem intuitive that many patients with DCM are also affected. Indeed, there have been some case reports describing patients with DCM and superimposed B12 deficiency.7-9 Investigation of this relationship is important since deficiency of B12 may not only exacerbate myelopathic symptoms in DCM but may also hinder neurological recovery, since B12 is essential for myelination.10 In this review, the mechanism of action, causes of deficiency, and presentation of B12 deficiency will be briefly described and will be followed by the role of routine B12 assessment and its potential role in optimizing surgical outcome in patients with DCM.
Vitamin B12: Mechanism of Action
Vitamin B12 is synthesized exclusively by anaerobic bacteria, and it is obtained in foods of animal origin. Uptake of B12 in the gastrointestinal system requires binding of a glycoprotein called intrinsic factor, which is secreted by gastric parietal cells. The B12-intrinsic factor complex binds to “cubam” receptors expressed on enterocytes in the distal ileum and is absorbed via receptor-mediated endocytosis. Given the critical role of intrinsic factor in B12 uptake, deficiencies in the glycoprotein due to an autoimmune gastritis known as “pernicious anemia” leads to a severe B12 deficiency, with hematological and neurological manifestations.11
Intracellular B12 is stored as 2 active coenzymes: methylcobalamin and deoxyadenosylcobalamin. Methylcobalamin acts as a coenzyme for cytoplasmic methionine synthase, which catalyzes the methylation of homocysteine to methionine. This transmethylation reaction also involves folate (vitamin B9) and is therefore critical for nucleic acid synthesis. Deoxyadenosylcobalamin is a cofactor for methylmalonyl-CoA mutase, which catalyzes the conversion of methylmalonyl-CoA to succinyl-CoA in the mitochondria. Succinyl-CoA subsequently enter the Krebs cycle and is important for the synthesis of lipids and carbohydrates12 (Figure 1).
Figure 1.

Vitamin B12 coenzyme function. B12 acts as a coenzyme in the conversion of homocysteine to methionine in the cytosol, and the conversion of methylmalonyl-CoA to succinyl-CoA in the mitochondrion. The cytoplasmic reaction requires folate, as the methyl group that is added to homocysteine is removed from 5-methyl tetrahydrofolate. Tetrahydrofolate is a precursor in the synthetic pathway for purines and pyrimidines, while succinyl-CoA enters the Krebs cycle and is important for lipid and carbohydrate synthesis. Reprinted with permission from Springer: Nature Reviews Gastroenterology and Hepatology (Nielsen et al11).
Methylcobalamin is also important for the synthesis and maintenance of the myelin sheath. A number of studies report the development of white-matter lesions or retarded myelination in patients with B12 deficiency.13-15 Although the precise molecular mechanisms underlying methylcobalamin-mediated myelination are unknown, a number of models have been suggested, including increased synthesis of lecithin (the primary component of myelin sheath lipids)16,17; downregulation of Erk1/2 and upregulation of myelin basic protein18; increased synthesis of myelinotrophic cytokines and growth factors, such as IL-6 and EGF19; upregulation of neurotrophic gene factors20; and regulation of normal prion protein concentration in the central nervous system.21
Vitamin B12 Deficiency: Anemia, Neuropathy, and Myelopathy
Vitamin B12 deficiency is a significant health concern in the United States; it is estimated that 5% to 40% of the elderly population have low serum B12 levels.4,22-24 Due to enterohepatic circulation and kidney reabsorption, humans have extensive stores of B12 and require several years of inadequate intake to present with a clinical deficiency. As a result, with the exception of unsupplemented populations of vegans, B12 deficiency occurs primarily through gastrointestinal malabsorption.11,25 The most direct measurement of B12 status is the measurement of total serum B12. Laboratory ranges for normal (>221 pmol/L), low (148-221 pmol/L), and acute deficiency (<148 pmol/L) have been established and are used in most clinical settings.22 However, a major limitation of this assay is that it assesses total circulating B12, about 80% of which is bound to haptocorrin, a transcobalamin protein, and not bioavailable.25 Furthermore, a number of studies have shown that serum B12 does not reliably represent levels of cellular B12. As a result, assessing serum B12 alone does not allow for an accurate diagnosis of deficiency.25 A more effective method of diagnosis is to use serum B12 measurements in conjunction with other biomarkers, namely, homocysteine (Hcy), methylmalonic acid (MMA), and holo-transcobalamin (holo-TC). Hcy and MMA accumulation occurs as a result of inactivation of the 2 B12-dependent enzymes, methionine synthase and methylmalonyl-CoA mutase, respectively. Most studies set the upper limit of normal plasma Hcy to 15 μmol/L; higher levels are indicative of a nutritional deficiency.26 However, since the conversion of Hcy to methionine via methionine synthase also depends on the availability of folate, nutritional deficiencies in either folate or B12 could result in increased levels of Hcy. MMA, on the other hand, is not affected by other vitamins and is therefore considered a more specific biomarker of B12 deficiency. Serum levels of MMA that are greater than 260 nmol/L indicate an elevated reading26 (Table 1). Notably, certain pathologies such as renal dysfunction may also present with increased levels of MMA; as a result, the use of this marker in elderly patients with renal disease should be done cautiously.27 Last, Holo-TC, in contrast to haptocorrin, is the readily bioavailable form of B12 transport, and is therefore a more accurate biomarker of B12 status. The normal range of holo-TC is 20 to 125 pmol/L.28 An algorithm for the diagnosis of B12 deficiency using these 3 biomarkers in addition to serum B12 was presented by Hannibal et al.25
Table 1.
Diagnostic Parameters, References Ranges, and Potential Confounding Factors for Assessment of Vitamin B12 Deficiencya.
| Parameter | Reference Range | Confounding Factors |
|---|---|---|
| B12 | >148 pmol/L | Renal insufficiency (↑) |
| Hcy | <15 μmol/L | Renal insufficiency(↑) |
| Folate deficiency (↑) | ||
| Vitamin B6 deficiency (↑) | ||
| MMA | <260 nmol/L | Renal insufficiency (↑) |
| Age (↑) | ||
| Intestinal bacterial overgrowth (↑) |
Abbreviations: B12, vitamin B12; Hcy, homocysteine; MMA, methylmalonic acid.
aAdapted from Hermann W, Obeid R. Cobalamin deficiency. In: Stranger O, ed. Water Soluble Vitamins: Clinical Research and Future Application. Berlin, Germany: Springer; 2012:301-322.
The Schilling test, an assay for pernicious anemia in which radiolabeled vitamin B12 is ingested and its excretion measured in urine, is now rarely used in the United States. Two studies have shown that elevated levels of Hcy and MMA were detectable in 15% to 33% of patients with normal Schilling tests, indicating the increased specificity of laboratory measurements in diagnosing B12 deficiency.29,30 Magnetic resonance imaging (MRI) is not normally indicated for patients with B12 deficiency, but some reports have noted characteristic V-shaped hyperintensity on T2-imaging when present in the cervical cord and “bumbell” or bilateral nodular shape when present in the thoracic cord in patients with severe myelopathy31,32 (Figure 2).
Figure 2.
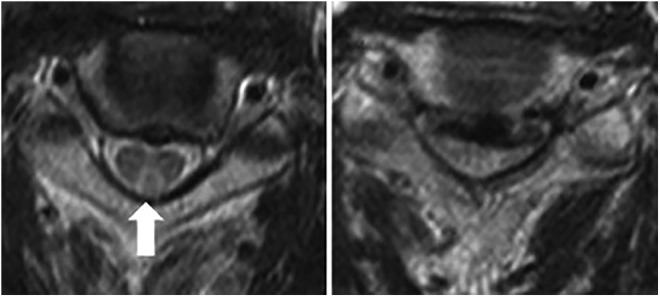
Axial T2-wieghted MRI of a patients with degenerative cervical myelopathy and concomitant B12 deficiency. On the left, a characteristic reverse V-shaped hyperintensity is visible in the posterior column (arrow). On the right significant spinal cord compression is demonstrated. Reprinted with permission from Elsevier: The Spine Journal (Miyazaki et al8).
The clinical presentations of B12 deficiency include megaloblastic anemia and neurological deficits. Megaloblastic anemia is characterized by enlarged red blood cell precursors with asynchronous maturation of the nucleus and cytoplasm. The clinical picture of megaloblastic anemia develops slowly, and symptoms include weakness, palpitations, dyspnea on exertion, fatigue, light-headedness, jaundice, and shortness of breath. These symptoms typically do not arise until the anemia is quite severe, as cardiopulmonary adaptations can alleviate hypoxia.33
Common neurological symptoms include myelopathy, neuropathy, and, less frequently, optic nerve atrophy.12 The best characterized form of myelopathy is known as subacute combined degeneration (SACD). SACD is caused by damage to dorsal and lateral columns and is characterized by symmetric dysesthesia, abnormal proprioception, loss of vibratory sensation, positive Romberg sign, and spastic paraparesis or tetraparesis. Oftentimes, patients initially report sensory loss, presenting as lower limb paresthesia associated with ataxia. In late-stage disease, lateral corticospinal tracts can be involved, leading to impairment of fine motor function and abnormal reflexes.34 Furthermore, a minority of patients present with autonomic disturbances, including bladder and erectile dysfunction.35
Peripheral neuropathy is seen in approximately 25% of patients with B12 deficiency.5 Symptoms include paresthesias, impaired sensation in a “glove and stocking” distribution, painful burning sensations, and muscle wasting.36,37 Occasionally, adult patients with B12 deficiency will present with optic neuropathy, characterized by symmetric, painless, and progressive visual loss. Ophthalmologic findings include central and centrocecal scotomas.35
Although hematologic signs often precede neurological symptoms, neurological symptoms may be the primary manifestation of B12 deficiency in some patients. For example, studies by Lindenbaum et al38 and Healton et al5 showed that up to 28% of patients with neuropsychiatric symptoms of B12 deficiency can present with normal mean corpuscular volume (MCV), hematocrit (HCT), or both. However, although HCT and MCV were normal in these reports, other hematological signs such as neutrophil hypersegmentation were found to be abnormal on inspection of peripheral blood smear.5,38,39 There have been reports of B12-deficient patients with neurological symptoms and normal MCV, HCT, peripheral blood smear, and Hcy levels, although this is quite rare.40
Pathophysiology of DCM
There are a number of pathophysiological factors that result in DCM: (1) static compression of the spinal cord, (2) dynamic injury resulting from mobile degenerative cervical spine elements compressing the cord, and (3) tethering of the cord or altered cord tension due to changes in the cervical spine alignment or cord compression.1,41 These various mechanisms contribute to spinal cord dysfunction by causing reversible and irreversible injury to neuronal tissue. Reversible tissue injury includes demyelination, Wallerian degeneration, edema, and inflammatory changes. Whereas irreversible injury manifests after frank loss of neuronal tissue has occurred.42 The underlying pathobiological mechanisms causing neuronal death are multifold. Mechanical compression initiates an inflammatory process that can be further exacerbated by disruption of blood flow and the blood-spinal cord barrier. Disruption of blood supply may result in variable degrees of cellular injury. This may be caused by direct blood vessel compression, as well as increased spinal cord tension, which may not only cause stretching of nerve fibers but also flattening of blood vessels.1,41,43 The degree of injury is highly variable and is affected by the degree of cord compression, the number of levels involved, and whether the compression is static or dynamic. Consequently, the natural history and clinical manifestations of DCM are highly variable. Clinically, patients typically present with problems using items with their hands and/or problems with their gait.3 In more severe cases, urine incontinence may also manifest. While diagnosis of DCM is based on clinical examination, imaging evidence of spinal cord compression or cord tethering on MRI is required to confirm the diagnosis (Table 2).
Table 2.
Clinical Findings That May Appear on Examination in Patients With DCM.
| Clinical Symptoms | Clinical Signs | MRI Findings |
|---|---|---|
| • Corticospinal motor deficits | • Hoffmann sign | • Cord compression |
| • Numbness of hands | • L’Hermitte’s phenomenon | • Cord flattening |
| • Atrophy of hand muscles | • Ankle clonus | • Cord torsion |
| • Hyperreflexia and spasticity | • Babinski sign | • T2WI Cord hyperintensity |
| • Gait disturbances (broad based) | • Romberg sign | • T1WI cord hypointensity |
| • Clumsy hands | ||
| • Weakness | ||
| • Paraesthesia | ||
| • Urinary incontinence (in severe cases) |
On MRI, patients typically present with one or more levels of cord compression. The direction of the compressive force typically originates from the anterior or anterior and posterior (pincer effect). In most, but not all patients, T2-weighted hyperintensity will approximate the site of cord compression, representing nonspecific inflammatory changes ranging from edema to cavitation depending on the signal intensity and appearance.42 T1-weighted hypointensity changes can occur in approximately one fifth of DCM patients at the site of T2-weighted hyperintensity, indicating cavitation and that frank neuronal tissue loss has occurred.42
Rationale for Investigating B12 Deficiency in DCM
On the Basis of Epidemiology
Both B12 deficiency and DCM are most prevalent in the elderly, and with estimates of 20% of B12 deficiency, even a proportional prevalence among DCM patients would indicate a high rate of potential deficiency among the DCM population. Clinically, reports of patients with known B12 deficiency and superimposed DCM have shown that patients appear with a degree of neurological impairment out of proportion of what would be expected based on imaging, and that treatment with B12 can optimize neurological recovery.7-9 In other case reports, patients with suspected diagnosis of DCM, but underlying SACD, experienced a resolution of symptoms after B12 administration.44-46 These findings have the following implications: patients with definitive DCM and concomitant B12 deficiency require treatment for both conditions to optimize neurological recovery, but care should be taken for patients with mild cord compression and possible B12 deficiency prior to surgical treatment, as cord compression may be a false positive finding and treatment with B12 may resolve symptoms.
A high index of suspicion for B12 deficiency among DCM patients should be placed among patient with history of gastrointestinal resection or comorbidities, such as atrophic gastritis and irritable bowel disease, which may be an underlying cause for unrecognized B12 deficiency.47 When suspected, laboratory findings of megaloblastic anemia, low B12 levels, and high levels of homocysteine may be helpful.
On the Basis of Pathophysiology
The average patient receiving surgical treatment for DCM has moderate to severe neurological impairment at presentation,48 and nonoperative management has been shown to result in neurological deterioration in 20% to 62% of patients at 3 to 6 years of follow-up.49 When surgical treatment is undertaken, the average patient experiences meaningful neurological recovery.48 However, not all patients experience significant improvement, others maintain their preoperative levels of function, and less commonly, patients experience neurological deterioration. The occurrence of suboptimal recovery can be expected since it is known that DCM can have elements of reversible and irreversible neuroanatomic changes—the balance of which influences the degree of functional recovery.50 Approximately 80% of patients with DCM present with either no significant changes or only T2-weighted hyperintensity signal on conventional MRI,50 and these MRI findings suggest that most patients have a large component of nonspecific inflammatory changes, including Wallerian degeneration, that are potentially reversible. Reversible neurological function, however, is partly attributable to remyelination, which requires B12.10 While there have been no direct clinical studies looking at B12 and DCM, basic science research has shown that peripheral nerve crush injury alter the levels of B12 at the nerve,51 and it has been suggested that B12 with dexamethasone or nonsteroidal anti-inflammatory drugs can be used to treat peripheral nerve crush injury and reduce Wallerian degeneration.52,53 Sun et al52 suggested that upregulation of brain-derived neurotrophic factor (BDNF) may be a mechanism of action for this improvement.
Vitamin B12 may also have a role in attenuating neurological deterioration after surgery for DCM. While the occurrence of deterioration is infrequent, not clearly understood, and difficult to anticipate, it has been suggested that reperfusion injury and subsequent glutamate excitotoxicity after cord decompression may be responsible for this phenomenon.54,55 It has been shown that treatment with a combination of B-vitamins (including B1, B6, B12) can reduce neuronal injury,56 and it has been suggested that B12 potentially depresses glutamate-induced neurotoxicity.57 These studies suggest that in addition to raising B12 levels in those with suboptimal levels, higher levels may also provide a therapeutic benefit to patients receiving surgery for DCM. This however remains speculative.
On the Basis of Preoperative Planning
An intriguing and clinical relationship between B12 and myelopathy that may be highly relevant for patients with DCM is also interaction of nitrous oxide (N2O) during anesthesia with perioperative myelopathy development.47 N2O irreversibly oxides the cobalt ion at the center of B12, and impedes its crucial cofactor function for methionine synthetase. This enzyme is required for the formation of tetrahydrofolate (THF) and methionine. THF is involved in thymidine synthesis and DNA production, while methionine is required for the methylation of myelin sheath phospholipids.58 Consequently, patients with already low levels of B12 or methylene-tetrahydrofolate-reductase deficiency are particularly at risk for perioperative myelopathy due to N2O administration.58 Although rare and an underrecognized phenomenon, there have been numerous case reports describing the development of SACD after anesthesia with N2O administration.47,59-62 Given that N2O can be used during spine surgery, this points to the necessity of routinely monitoring B12 levels in patients with DCM to optimize surgical outcomes and prevent perioperative or postoperative neurological deficit development. Recognition of this phenomenon is important, as intramuscular injection of B12 has been shown to rapidly reverse SACD symptoms.47
Conclusion
It is clear that B12 is necessary for maintaining spinal cord function, and deficiency can result in SACD. Given the high prevalence of clinical and subclinical B12 deficiency in the elderly, the role of B12 in myelination, and B12 deficiency as a differential diagnosis of DCM, there is considerable rationale to conduct routine assessment of B12 levels in patients with DCM. Going forward, it will be necessary to assess additional aspects of this relationship, including (1) whether DCM patients with B12 deficiency present differently on clinical exam, (2) whether patients with B12 deficiency and DCM have suboptimal surgical outcomes, (3) whether patients with deficiency who are supplemented with B12 achieve optimal outcomes, and (4) whether increasing B12 levels in patients with no deficiency improves surgical outcomes more than otherwise expected. Since preoperative assessment includes routine blood work, this additional diagnostic measurement would not unnecessarily burden the patient or substantially increase costs. In the event that a patient appears to have suboptimal levels or deficiency, treatment with B12 would not be costly, is unlikely to adversely affect the patient, and may optimize surgical outcome. Further studies in this area are needed and would be highly feasible given the fact that B12 is an essential vitamin, cheap, and readily accessible. We intend to investigate this relationship and will seek to report on how to incorporate B12 assessment into the clinical management of patients with DCM.
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Sciubba is a consultant for Medtronic, Depuy-Synthes, Stryker, Nuvasive, and K2M. The other authors have no conflicts of interest to declare.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Aria Nouri, MD, MSc  https://orcid.org/0000-0002-4965-3059
https://orcid.org/0000-0002-4965-3059
References
- 1. Nouri A, Tetreault L, Singh A, Karadimas SK, Fehlings MG. Degenerative cervical myelopathy: epidemiology, genetics and pathogenesis. Spine (Phila Pa 1976). 2015;40:E675–E693. [DOI] [PubMed] [Google Scholar]
- 2. Harrop JS, Naroji S, Maltenfort M, et al. Cervical myelopathy: a clinical and radiographic evaluation and correlation to cervical spondylotic myelopathy. Spine (Phila Pa 1976). 2010;35:620–624. [DOI] [PubMed] [Google Scholar]
- 3. Kalsi-Ryan S, Karadimas SK, Fehlings MG. Cervical spondylotic myelopathy: the clinical phenomenon and the current pathobiology of an increasingly prevalent and devastating disorder. Neuroscientist. 2013;19:409–421. [DOI] [PubMed] [Google Scholar]
- 4. Andres E, Loukili NH, Noel E, et al. Vitamin B12 (cobalamin) deficiency in elderly patients. CMAJ. 2004;171:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Healton EB, Savage DG, Brust JC, Garrett TJ, Lindenbaum J. Neurologic aspects of cobalamin deficiency. Medicine (Baltimore). 1991;70:229–245. [DOI] [PubMed] [Google Scholar]
- 6. Carmel R. Subclinical cobalamin deficiency. Curr Opin Gastroenterol. 2012;28:151–158. [DOI] [PubMed] [Google Scholar]
- 7. Xu Y, Chen W, Jiang J. Cervical spondylotic myelopathy with vitamin B deficiency: two case reports. Exp Ther Med. 2013;6:943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyazaki T, Sudo H, Hiratsuka S, Iwasaki N. Cervical spondylotic myelopathy with subacute combined degeneration. Spine J. 2014;14:381–382. [DOI] [PubMed] [Google Scholar]
- 9. Haghighi SS, Zhang R, Stein D. Cervical myelopathy due to chronic vitamin B12 deficiency or herniated cervical disc or both. Electromyogr Clin Neurophysiol. 2003;43:443–447. [PubMed] [Google Scholar]
- 10. Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368:2041–2042. [DOI] [PubMed] [Google Scholar]
- 11. Nielsen MJ, Rasmussen MR, Andersen CB, Nexo E, Moestrup SK. Vitamin B12 transport from food to the body’s cells—a sophisticated, multistep pathway. Nat Rev Gastroenterol Hepatol. 2012;9:345–354. [DOI] [PubMed] [Google Scholar]
- 12. Kumar N. Neurologic aspects of cobalamin (B12) deficiency. Handb Clin Neurol. 2014;120:915–926. [DOI] [PubMed] [Google Scholar]
- 13. Black MM. Effects of vitamin B12 and folate deficiency on brain development in children. Food Nutr Bull. 2008;29(2 suppl):S126–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Lau LM, Smith AD, Refsum H, Johnston C, Breteler MM. Plasma vitamin B12 status and cerebral white-matter lesions. J Neurol Neurosurg Psychiatry. 2009;80:149–157. [DOI] [PubMed] [Google Scholar]
- 15. Lovblad K, Ramelli G, Remonda L, Nirkko AC, Ozdoba C, Schroth G. Retardation of myelination due to dietary vitamin B12 deficiency: cranial MRI findings. Pediatr Radiol. 1997;27:155–158. [DOI] [PubMed] [Google Scholar]
- 16. Yamatsu K, Kaneko T, Kitahara A, Ohkawa I. Pharmacological studies on degeneration and regeneration of peripheral nerves. (1) Effects of methylcobalamin and cobamide on EMG patterns and loss of muscle weight in rats with crushed sciatic nerve [in Japanese]. Nihon Yakurigaku Zasshi. 1976;72:259–268. [PubMed] [Google Scholar]
- 17. Watanabe T, Kaji R, Oka N, Bara W, Kimura J. Ultra-high dose methylcobalamin promotes nerve regeneration in experimental acrylamide neuropathy. J Neurol Sci. 1994;122:140–143. [DOI] [PubMed] [Google Scholar]
- 18. Nishimoto S, Tanaka H, Okamoto M, Okada K, Murase T, Yoshikawa H. Methylcobalamin promotes the differentiation of Schwann cells and remyelination in lysophosphatidylcholine-induced demyelination of the rat sciatic nerve. Front Cell Neurosci. 2015;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scalabrino G. The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. Prog Neurobiol. 2009;88:203–220. [DOI] [PubMed] [Google Scholar]
- 20. Gan L, Qian M, Shi K, et al. Restorative effect and mechanism of mecobalamin on sciatic nerve crush injury in mice. Neural Regen Res. 2014;9:1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scalabrino G, Veber D. Normal prions as a new target of cobalamin (vitamin B12) in rat central nervous system. Clin Chem Lab Med. 2013;51:601–606. [DOI] [PubMed] [Google Scholar]
- 22. Allen LH. How common is vitamin B-12 deficiency? Am J Clin Nutr. 2009;89:693S–696S. [DOI] [PubMed] [Google Scholar]
- 23. Campbell AK, Miller JW, Green R, Haan MN, Allen LH. Plasma vitamin B-12 concentrations in an elderly Latino population are predicted by serum gastrin concentrations and crystalline vitamin B-12 intake. J Nutr. 2003;133:2770–2776. [DOI] [PubMed] [Google Scholar]
- 24. Lindenbaum J, Rosenberg IH, Wilson PW, Stabler SP, Allen RH. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr. 1994;60:2–11. [DOI] [PubMed] [Google Scholar]
- 25. Hannibal L, Lysne V, Bjorke-Monsen AL, et al. Biomarkers and algorithms for the diagnosis of vitamin B12 deficiency. Front Mol Biosci. 2016;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aparicio-Ugarriza R, Palacios G, Alder M, Gonzalez-Gross M. A review of the cut-off points for the diagnosis of vitamin B12 deficiency in the general population. Clin Chem Lab Med. 2015;53:1149–1159. [DOI] [PubMed] [Google Scholar]
- 27. Iqbal N, Azar D, Yun YM, Ghausi O, Ix J, Fitzgerald RL. Serum methylmalonic acid and holotranscobalamin-II as markers for vitamin B12 deficiency in end-stage renal disease patients. Ann Clin Lab Sci. 2013;43:243–249. [PubMed] [Google Scholar]
- 28. Valente E, Scott JM, Ueland PM, Cunningham C, Casey M, Molloy AM. Diagnostic accuracy of holotranscobalamin, methylmalonic acid, serum cobalamin, and other indicators of tissue vitamin B12 status in the elderly. Clin Chem. 2011;57:856–863. [DOI] [PubMed] [Google Scholar]
- 29. Lindgren A, Swolin B, Nilsson O, Johansson KW, Kilander AF. Serum methylmalonic acid and total homocysteine in patients with suspected cobalamin deficiency: a clinical study based on gastrointestinal histopathological findings. Am J Hematol. 1997;56:230–238. [DOI] [PubMed] [Google Scholar]
- 30. Lindgren A, Bagge E, Cederblad A, Nilsson O, Persson H, Kilander AF. Schilling and protein-bound cobalamin absorption tests are poor instruments for diagnosing cobalamin malabsorption. J Intern Med. 1997;241:477–484. [DOI] [PubMed] [Google Scholar]
- 31. Pittock SJ, Payne TA, Harper CM. Reversible myelopathy in a 34-year-old man with vitamin B12 deficiency. Mayo Clin Proc. 2002;77:291–294. [DOI] [PubMed] [Google Scholar]
- 32. Sun HY, Lee JW, Park KS, Wi JY, Kang HS. Spine MR imaging features of subacute combined degeneration patients. Eur Spine J. 2014;23:1052–1058. [DOI] [PubMed] [Google Scholar]
- 33. Green R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood. 2017;129:2603–2611. [DOI] [PubMed] [Google Scholar]
- 34. Senol MG, Sonmez G, Ozdag F, Saracoglu M. Reversible myelopathy with vitamin B12 deficiency. Singapore Med J. 2008;49:e330–e332. [PubMed] [Google Scholar]
- 35. Briani C, Torre DC, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5:4521–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ekabe CJ, Kehbila J, Abanda MH, Kadia BM, Sama CB, Monekosso GL. Vitamin B12 deficiency neuropathy; a rare diagnosis in young adults: a case report. BMC Res Notes. 2017;10:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCombe PA, McLeod JG. The peripheral neuropathy of vitamin B12 deficiency. J Neurol Sci. 1984;66:117–126. [DOI] [PubMed] [Google Scholar]
- 38. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318:1720–1728. [DOI] [PubMed] [Google Scholar]
- 39. Thompson WG, Cassino C, Babitz L, et al. Hypersegmented neutrophils and vitamin B12 deficiency. Hypersegmentation in B12 deficiency. Acta Haematol. 1989;81:186–191. [DOI] [PubMed] [Google Scholar]
- 40. Voukelatou P, Vrettos I, Kalliakmanis A. Neurologic symptoms as the only manifestation of B12 deficiency in a young patient with normal hematocrit, MCV, peripheral blood smear and homocysteine levels. Oxf Med Case Reports. 2016;2016:omw091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ames CP, Blondel B, Scheer JK, et al. Cervical radiographical alignment: comprehensive assessment techniques and potential importance in cervical myelopathy. Spine (Phila Pa 1976). 2013;38(22 suppl 1):S149–S160. [DOI] [PubMed] [Google Scholar]
- 42. Nouri A, Martin AR, Mikulis DJ, Fehlings M. Magnetic resonance imaging assessment of degenerative cervical myelopathy: a review of structural changes and measurement techniques. Neurosurg Focus. 2016;40:E5. [DOI] [PubMed] [Google Scholar]
- 43. Karadimas SK, Gatzounis G, Fehlings MG. Pathobiology of cervical spondylotic myelopathy. Eur Spine J. 2015;24(suppl 2):132–138. [DOI] [PubMed] [Google Scholar]
- 44. Yokoyama K, Kawanishi M, Sugie A, et al. A case of subacute combined degeneration caused by vitamin B12 deficiency in a cervical spondylosis surgery referral [in Japanese]. No Shinkei Geka. 2016;44:1059–1063. [DOI] [PubMed] [Google Scholar]
- 45. Li J, Zhang L, Zhang Y, Deng B, Bi X. Misdiagnosis of spinal subacute combined degeneration in a patient with elevated serum B12 concentration and sensory deficit level. Neurol Sci. 2016;37:1577–1578. [DOI] [PubMed] [Google Scholar]
- 46. Alonso F, Rustagi T, Schmidt C, et al. Subacute combined degeneration disguised as compressive myelopathy. Spine Scholar. 2017;1:49–53. [Google Scholar]
- 47. Patel K, Mejia-Munne J, Gunness V, et al. Subacute combined degeneration of the spinal cord following nitrous oxide anesthesia: a systematic review of cases. Global Spine J. 2018;8(1S):172S. [DOI] [PubMed] [Google Scholar]
- 48. Fehlings MG, Wilson JR, Kopjar B, et al. Efficacy and safety of surgical decompression in patients with cervical spondylotic myelopathy: results of the AOSpine North America prospective multi-center study. J Bone Joint Surg Am. 2013;95:1651–1658. [DOI] [PubMed] [Google Scholar]
- 49. Badhiwala JH, Wilson JR. The natural history of degenerative cervical myelopathy. Neurosurg Clin N Am. 2018;29:21–32. [DOI] [PubMed] [Google Scholar]
- 50. Nouri A, Martin AR, Kato S, Reihani-Kermani H, Riehm LE, Fehlings MG. The relationship between MRI signal intensity changes, clinical presentation, and surgical outcome in degenerative cervical myelopathy: analysis of a global cohort. Spine (Phila Pa 1976). 2017;42:1851–1858. [DOI] [PubMed] [Google Scholar]
- 51. Altun I, Kurutas EB. Vitamin B complex and vitamin B12 levels after peripheral nerve injury. Neural Regen Res. 2016;11:842–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun H, Yang T, Li Q, et al. Dexamethasone and vitamin B(12) synergistically promote peripheral nerve regeneration in rats by upregulating the expression of brain-derived neurotrophic factor. Arch Med Sci. 2012;8:924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tamaddonfard E, Farshid AA, Samadi F, Eghdami K. Effect of vitamin B12 on functional recovery and histopathologic changes of tibial nerve-crushed rats. Drug Res (Stuttg). 2014;64:470–475. [DOI] [PubMed] [Google Scholar]
- 54. Vidal PM, Karadimas SK, Ulndreaj A, et al. Delayed decompression exacerbates ischemia-reperfusion injury in cervical compressive myelopathy [published online June 2, 2017]. JCI Insight. doi:10.1172/jci.insight.92512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karadimas SK, Laliberte AM, Tetreault L, et al. Riluzole blocks perioperative ischemia-reperfusion injury and enhances postdecompression outcomes in cervical spondylotic myelopathy. Sci Transl Med. 2015;7:316ra194. [DOI] [PubMed] [Google Scholar]
- 56. Yu CZ, Liu YP, Liu S, Yan M, Hu SJ, Song XJ. Systematic administration of B vitamins attenuates neuropathic hyperalgesia and reduces spinal neuron injury following temporary spinal cord ischaemia in rats. Eur J Pain. 2014;18:76–85. [DOI] [PubMed] [Google Scholar]
- 57. Hung KL, Wang CC, Huang CY, Wang SJ. Cyanocobalamin, vitamin B12, depresses glutamate release through inhibition of voltage-dependent Ca2+ influx in rat cerebrocortical nerve terminals (synaptosomes). Eur J Pharmacol. 2009;602:230–237. [DOI] [PubMed] [Google Scholar]
- 58. Brown S, Sneyd J. Nitrous oxide in modern anaesthetic practice. BJA Education. 2016;16:87–91. [Google Scholar]
- 59. Ahn SC, Brown AW. Cobalamin deficiency and subacute combined degeneration after nitrous oxide anesthesia: a case report. Arch Phys Med Rehabil. 2005;86:150–153. [DOI] [PubMed] [Google Scholar]
- 60. Ilniczky S, Jelencsik I, Kenez J, Szirmai I. MR findings in subacute combined degeneration of the spinal cord caused by nitrous oxide anaesthesia—two cases. Eur J Neurol. 2002;9:101–104. [DOI] [PubMed] [Google Scholar]
- 61. Marie RM, Le Biez E, Busson P, et al. Nitrous oxide anesthesia-associated myelopathy. Arch Neurol. 2000;57:380–382. [DOI] [PubMed] [Google Scholar]
- 62. Beltramello A, Puppini G, Cerini R, et al. Subacute combined degeneration of the spinal cord after nitrous oxide anaesthesia: role of magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 1998;64:563–564. [DOI] [PMC free article] [PubMed] [Google Scholar]