

Abstract
Osteopenia is observed in some patients affected by phenylalanine hydroxylase (PAH) deficient phenylketonuria (PKU). Bone density studies, in diverse PKU patient cohorts, have demonstrated bone disease is neither fully penetrant nor uniform in bone density loss. Biochemical assessment has generated a muddled perspective regarding mechanisms of the PKU bone phenotype where the participation of hyperphenylalaninemia remains unresolved. Osteopenia is realized in the Pahenu2 mouse model of classical PKU; although, characterization is incomplete. We characterized the Pahenu2 bone phenotype and assessed the effect of hyperphenylalaninemia on bone differentiation. Employing Pahenu2 and control animals, cytology, static and dynamic histomorphometry, and biochemistry were applied to further characterize the bone phenotype. These investigations demonstrate Pahenu2 bone density is decreased 33% relative to C57BL/6; bone volume/total volume was similarly decreased; trabecular thickness was unchanged while increased trabecular spacing was observed. Dynamic histomorphometry demonstrated a 25% decrease in mineral apposition. Biochemically, control and PKU animals have similar plasma cortisol, adrenocorticotropic hormone, and 25-hydroxyvitamin D. PKU animals show moderately increased plasma parathyroid hormone while plasma calcium and phosphate are reduced. These data are consistent with a mineralization defect. The effect of hyperphenylalaninemia on bone maturation was assessed in vitro employing bone-derived mesenchymal stem cells (MSCs) and their differentiation into bone. Using standard culture conditions, PAH deficient MSCs differentiate into bone as assessed by in situ alkaline phosphatase activity and mineral staining. However, PAH deficient MSCs cultured in 1200 μM PHE (metric defining classical PKU) show significantly reduced mineralization. These data are the first biological evidence demonstrating a negative impact of hyperphenylalaninemia upon bone maturation. In PAH deficient MSCs, expression of Col1A1 and Rankl are suppressed by hyperphenylalaninemia consistent with reduced bone formation and bone turnover. Osteopenia is intrinsic to PKU pathology in untreated Pahenu2 animals and our data suggests PHE toxicity participates by inhibiting mineralization in the course of MSC bone differentiation.
1. Introduction
PAH deficient PKU is the paradigm for a treatable inborn error of metabolism [1–4]. Dietary PHE restriction continues to serve as the primary means of PKU intervention [4–7]; however, contemporary approaches include cofactor therapy (Kuvan) and phenylalanine ammonia lyase enzyme therapy (Pegvaliase) [8–12]. Newborn screening facilitates early intervention which counters the most severe elements of the neurologic phenotype but cognitive deficit, executive function deficit, neuropsychiatric phenotypes, and bone disease remain common among both therapy compliant and therapy noncompliant patients.
In 1962 Feinberg and Fisch described osteopenia in PKU affected children [13], which was confirmed by Murdoch and Holman [14], and subsequently reported many times thereafter [15–21]. Lumbar spine bone mineral density Z scores of −2.0 are observed among early-identified, continuously treated patients [22]. Similar reduction in total body bone mineral density has also been observed [23]. Equivalent degrees of low bone mineral density have also been observed in therapy noncompliant patients [22].
Pathophysiological mechanisms of PKU bone disease remain ambiguous [22]. The bone phenotype was originally attributed to secondary manifestations of diet therapy where by an undefined mechanism, bioavailability of calcium, phosphorous, and other bone forming material were reduced; however, this is poorly supported as bone disease is observed in patients that never received diet therapy and very young patients that received only short term therapy [22]. Several studies find no correlation [18,21,24–27] between hyperphe-nylalaninemia and bone disease; others show a negative correlation between hyperphenylalaninemia and bone disease [28,29]. Biochemical ambiguity does not end with PHE toxicity as representation of bone formation markers [28,29], bone resorption markers [30,31], and other biochemical metrics related to bone, while investigated, remain less than fully resolved [28,30,32].
PKU bone disease has significant knowledge gaps; essentially all patient investigations are descriptive and pathophysiology is largely uninvestigated. Central to effective intervention is defined pathophysiology. While the bone phenotype is not fully penetrant in humans, the Pahenu2 mouse model of classical PKU reliably displays abnormal bone phenotypes [33]. We investigated the bone phenotype in Pahenu2 to further define its characteristics and to investigate potential pathophysiological mechanisms based upon the bone phenotype. In untreated Pahenu2 animals, the bone phenotype and biochemical assessment gives evidence for a mineralization defect. Employing PAH deficient and PAH replete mesenchymal stem cells (MSCs) and differentiating these into bone, we demonstrate PAH deficient cells retain bone forming capacity (albeit lower than controls); however, hyper-phenylalaninemia leads to a significant loss of mineralization. These investigations are the first to assess developmental aspects in the pathology of the PKU bone phenotype. While a genetic component is evident as PAH deficient MSCs have reduced capacity for bone development, hyperphenylalaninemia further reduces mineralization. These studies demonstrate a complex pathology to the PKU bone phenotype involving contribution of both hyperphenylalaninemia and PAH deficiency.
2. Methods and materials
2.1. PKU Animal model and controls
Pahenu2 mice were propagated under an approved protocol in the Rangos Research Center animal facility at Children’s Hospital of Pittsburgh. To generate experimental and control animals, matings paired heterozygous females and heterozygous males. Genotyping of offspring was performed as previously described [34]. Alternative homozygous genotypes (experimental enu2/enu2, control wt/wt) were selected for experimentation. After weaning, animals were provided a continuous diet of standard mouse chow to maintain hyperphenylala-ninemia (~2200 μM PHE females; ~2000 μM PHE males) among homozygous experimental animals. Control wild type litter mates, provide an identical diet, have plasma PHE of ~40–70 μM. All experiments used animals in the fed state. Animals were sacrificed by cervical dislocation at 3–5 months of age.
2.2. Cytology and microcomputed tomography
Formalin fixed spinal tissue were utilized for hematoxylin and eosin staining as previously described [37]. Histomorphometric analysis of trabecular bone was performed using microcomputed tomography [35,36] in a randomized and blinded manner. The lumbar vertebrae, originally fixed in 4% formalin, were scanned in 70% ethanol at 6 μm resolution using a Skyscan 1272 microCT (Bruker, Billerica, MA) with 0.25 mm aluminum filter; source voltage and current were set at 60 kV and 166 μA. Images were reconstructed using NRecon and InstaRecon software (Bruker). Cross sectional images were obtained using Data-viewer software and three dimensional images using CTvox software (Bruker). For quantitative histomorphometric analysis the wild type group consisted of five animals (3 female, 2 male) and the PKU group consisted of six animals (3 male, 3 female). We analyzed a 1.2 mm segment centered at the midpoint of the fourth lumbar vertebral body. Using CTan software (Bruker), we determined bone volume/total volume (percent bone), trabecular thickness, trabecular separation and trabecular number. Bone mineral density was also determined using CTan software with calibration standards provided by the manufacturer scanned under the conditions described above. Quantitative data are generated from the entire animal cohort (five control animals, six PKU) and not parsed by sex. Dynamic histomorphometry was performed by intra-peritoneal injection of calcein (25 μg/g body weight), seven and two days prior to sacrifice. Both the PKU and control groups consisted of four animals where two are male and two are female. Measurement of calcein-labeled surface and bone formation rate were performed as described [37].
2.3. Blood chemistry analyses
Terminal blood draw from the heart was performed with four wild type control animals (2 male, 2 female) and 12 homozygous Pahenu2 animals (5 male, 7 female) with serum separated by centrifugation. Analysis of cortisol, calcium, and phosphate utilized the Beckman Coulter AU System applying clinical assay standards and procedures. Quantification of 25-hydroxyvitamin D utilized a Waters MS/MS system and clinical assay standards. Mouse parathyroid hormone (PTH) and adrenocorticotropic hormone were assessed with a commercial enzyme linked immunosorbent assay using goat primary antibodies (MyBioSource) according to the manufacturer.
2.4. Mesenchymal stem cell preparation and osteoblast differentiation
From experimental and control animals, the femur and tibia were dissected, epiphysis removed, and bone marrow flushed with RPMI-1640 plus 10% fetal calf serum using an insulin syringe. For both PKU and control cells were prepared from four animals (2 male, 2 female). Erythrocytes were removed with red cell lysis buffer. Cells were plated and incubated for 2 h at 37 °C to remove rapidly adhering fibroblast-like cells. Remaining cells were plated into a new cell culture plate at 1 × 106 cells/cm2. After 72 h, non-adherent cells were discarded and adherent cells were washed with phosphate-buffered saline and changed into proliferation medium (MesenCult Proliferation Kit (mouse), Stemcell Technologies). Cells were passaged at ~60% confluence to avoid differentiation and re-plated at 2 × 105/cm2. Osteogenic differentiation of MSCs utilized culture for 21 days in growth medium supplemented with 35 μg/ml L-ascorbic acid and 10 mM β-glycerophosphate, 10pM ACTH, 10 nM 1α,25-dihydrox-yvitamin D3, and 0.5 mM CaCl2. During the course of differentiation, both PAH replete cells and PAH deficient cells were cultured at either standard cell culture condition (193.7 μM PHE) or in the context of 1200 μM PHE being the minimum concentration defining classical PKU [8]. Culture media were replaced at ~72h intervals. Bone differentiation was assessed by in situ alkaline phosphatase activity and mineralization. In situ alkaline phosphatase activity used 0.05% napthol AS-MX substrate and fast blue product visualization as previously described [37]. Visualizing mineralization used von Kossa silver staining as described [37]. Densitometry of stained culture plates was performed as described [37,38].
2.5. Gene expression studies
PAH deficient and PAH replete MSC’s were cultured and differentiated down the bone pathway as described above. During the differentiation process, cells were cultured at either a standard cell culture condition or at a hyperphenylalaninemic condition (1200 μM PHE). Cells were harvested and RNA prepared with RNeasy Mini Kit (Qiagen). Reverse transcription and quantitative PCR employed the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and Brilliant III SYBR Green QPCR Mastermix (Agillent) respectively. Supplemental Table 1 provides primer sequences to amplify Gapdh, Col1a1, and Rankl. Amplification conditions and conversion of realtime PCR data to relative expression was as described [37]. All expression studies were performed in triplicate.
2.6. Statistical assessment
Student’s t-test was employed in comparisons with data shown asmean +/− standard deviation. P values ≤.05 is considered significant. Analysis employed GraphPad Prism software [37].
3. Results
3.1. Pahenu2 bone phenotyping identifies a mineralization defect
Gross bone morphology was assessed by hematoxylin and eosin staining, followed by μCT assessment to provide higher resolution and enable quantification of bone metrics. Fig. 1A displays hematoxylin and eosin staining of lumbar spine vertebrae four from Pahenu2 and control animals. The Pahenu2 animal displays lower bone density and fewer trabeculae compared to the wild type control. Further, the control animal displays a more highly developed end plate than observed in Pa-henu2. Fig. 1B displays μCT reconstruction of lumbar spine vertebrae 4 from Pahenu2 and control animals. These data are consistent with hematoxylin and eosin staining (Fig. 1A) as reduced bone density, fewer trabeculae, and less developed end plates are evident. Quantitative static histomorphometry parameters were assessed in the lumbar spine from six PKU animals (three male, three female) and five control animals (2 male, three female). Data were analyzed together and not parsed by sex. In the PKU mouse, all static histomorphometry parameters (Fig. 1C, graphs A-C) show statistically significant differences compared to controls with reduced bone mineralization (P = .04), increased trabecular spacing (P = .05), and reduced bone volume to total bone (P = .025). Dynamic histomorphometry (Fig. 1C, graph D) on undecalcified spine vertebrae sections from four PKU animals (two male, two female) and four control animals (two male, two female) display similar labeled surface (data not shown); however mineral ap-positional in PKU animals was decreased by 25% (P = .001). These data are consistent with a mineralization defect.
Fig. 1.
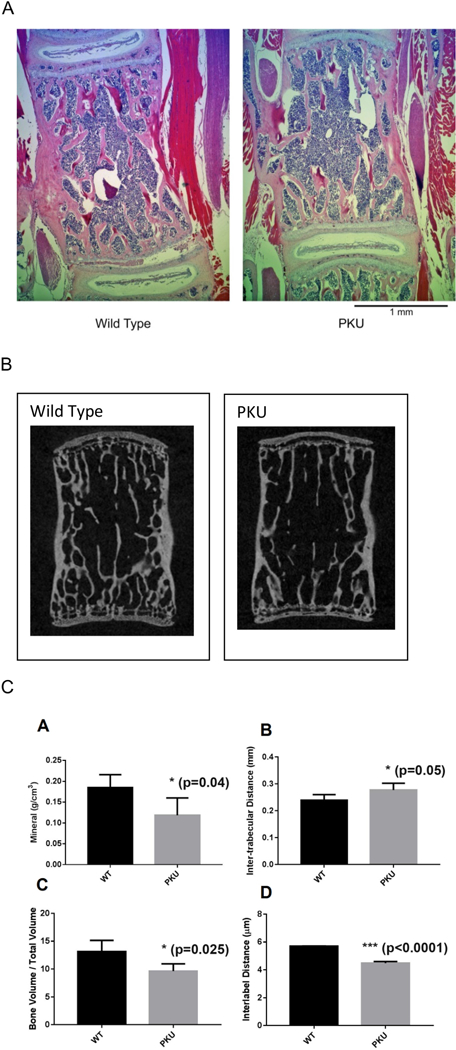
Reduced Bone Formation in Pahenu2. 1A. Hematoxylin and eosin staining of lumbar spine vertebrae four from wild type (left) and Pahenu2 (right). H&E staining demonstrates Pahenu2 has reduced bone mass, fewer trabecula and thinner trabecula compared to a wild type litter mate. 1B. Micro-CT reconstructed images of lumbar spine vertebrae four from wild type (left) and Pahenu2 (right). Micro-CT demonstrates Pahenu2 has reduced bone mass, fewer trabecula and thinner trabecula compared to a wild type litter mate. Micro-CT data and H&E data (1A) are in agreement. 1C. Static and dynamic histo-morphometry parameters. Panels A-C Static parameters A-C were generated from lumbar spine Micro-CT data of Pahenu2 (six animals, 3 male, 3 female) and wild type litter mates (five animals, two male, three female). Micro-CT demonstrates in Pahenu2 animals approximately 35% reduced mineral content, p = .04 (A) with increased trabeculae spacing, p = .05 (B) and increased bone volume/total volume, p = .025 (C). Panel D dynamic histomporphometry. Calcein labeling of Pahenu2 (four animals, two male, two female) and wild type litter mates (four animals, two male, two female) demonstrates appositional bone growth was decreased 25%, p < .001.
3.2. Biochemical parameters
Serum concentrations of cortisol, PTH, adrenocorticotropic hormone, Ca++, phosphate, and 25-hydroxyvitamin D were assessed between Pahenu2 (12 animals five male, seven female) and control animals (four animals, two male, two female). Fig. 2 displays serum values for Ca++, phosphate, and PTH where statistically significant differences were observed between experimental and control animals. Ca++ and phosphate were both low in PKU animals (P = .003 and P = .01 respectively). Imbalance between calcium and phosphate is a recognized osteoporosis risk factor. Moreover, PTH was elevated (P = .02) in PKU animals indicating a chronic paucity of Ca++ and disruption of bone homeostasis. Cortisol, adrenocorticotropic hormone, and 25-hydro-xyvitamin D were similarly represented between PKU and control animals (data not shown).
Fig. 2.
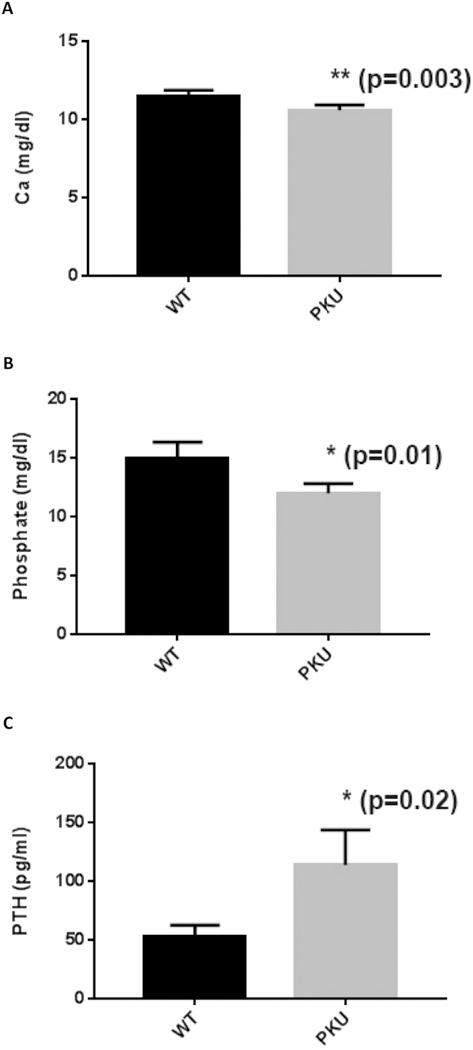
Statistically significant biochemical metrics differing between Pahenu2 and Controls. 2A. A moderate yet statistically significant reduction (p = .03) in plasma calcium is observed between PKU and control animals. 2B. A statistically significant reduction (p = .03) in plasma phosphate (p = .01) is observed between PKU and control animals. 2C. Moderate hyperparathyroidism is observed in Pahenu2 animals (p = .02) indicating low Ca++ and disruption of bone homeostasis. Cortisol, adrenocorticotropic hormone, and 25-hydro-xyvitamine D were assessed but no statistical differences in their representation were observed (data not shown). All measurements were made with four control animals (2 male, 2 female) and twelve PKU animals (5 male, 7 female).
3.3. Hyperphenylalaninemia augments reduced MSC bone differentiation in PAH deficient cells
The role of hyperphenylalaninemia in the PKU bone phenotype is controversial as data in human studies has been inconsistent [18,21,24–29]. Morphologic, histomorphometry, and biochemical data demonstrate a mineralization defect; therefore, we investigated bone differentiation in PAH deficient and PAH replete MSCs in the context of standard culture conditions and hyperphenylalaninemic conditions (1200 μM PHE, low-end metric defining classical PKU). Fig. 3 demonstrates both PAH deficiency and hyperphenylalaninemia negatively impacts MSC differentiation into bone. Overall, PAH replete MSCs display more robust bone development in comparison to PAH deficient MSCs. In situ alkaline phosphatase activity is similar in PAH replete MSCs under standard and hyperphenylalaninemic conditions; however, a modest yet statistically significant decline in mineralization is observed in the context of hyperphenylalaninemia (Fig. 3B). PAH deficient MSC’s retain bone forming capacity as in situ alkaline phosphatase activity and mineralization demonstrate (Fig. 3A). However, the formation of bone is less robust as statistically significant decreases in both in situ alkaline phosphatase (p < .01) activity and mineralization (p < .001) are observed. At hyperphenylalaninemic conditions, the already diminished mineralization capacity of PAH deficient MSCs display a profound further loss of mineralization capacity (p < .001). These data are the first to demonstrate that both PAH deficiency and PHE toxicity contribute to decreased MSC differentiation into bone. Moreover, this evidence provides a potential disease mechanism for the PKU bone phenotype being an intrinsic defect in stem cell differentiation which is further augmented by hyperphenylalaninemia.
Fig. 3.
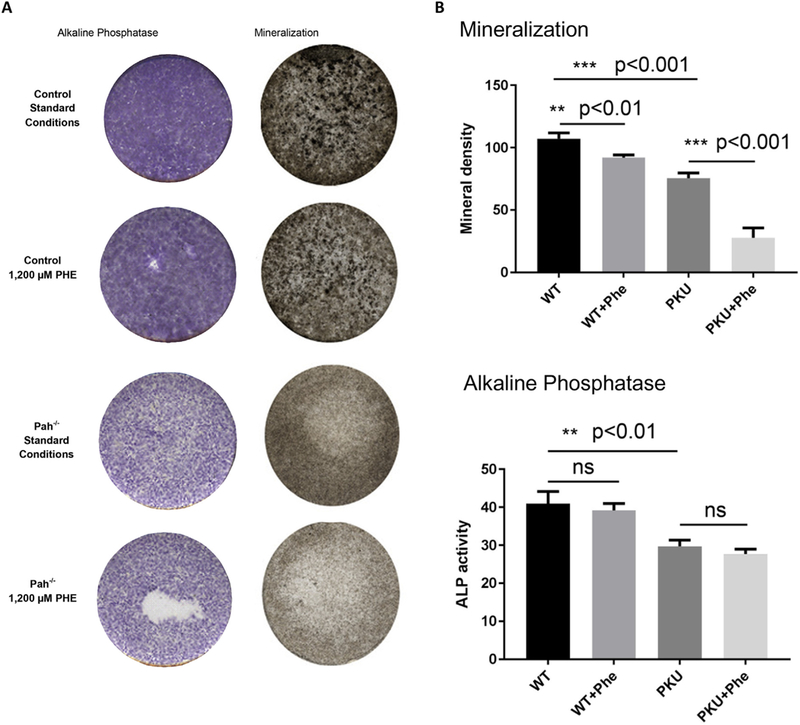
Effect of hyperphenylalaninemia on Pahenu2 and wild type MSC osteoblast differentiation. 3A. In situ alkaline phosphatase activity and von Kossa silver stain for mineralization in wild type control cells at standard culture conditions, wild type control cells in the context of 1200 μM PHE, PAH deficient cells at standard culture conditions, and PAH deficient cells in the context of 1200 μM PHE. Wild type cells (Pah replete) display abundant alkaline phosphatase activity and mineralization under standard conditions; however, mineralization is moderately reduced under hyperphenylalaninemic conditions. PAH deficient cells retain alkaline phosphatase activity and mineralization capacity under standard conditions; albeit reduced in comparison to PAH replete cells. PAH deficient cells under hyperphenylalaninemic conditions experience no loss of alkaline phosphatase activity while mineralization is reduced. 3B. Densitometry enables statistical analysis of alkaline phosphatase activity and mineralization. Alkaline phosphatase activity in PAH replete cells is not influenced by hyperphenylalaninemia. In PAH deficient cells alkaline phosphatase activity is significantly reduced compared to PAH replete cells (P < .01) but hyperphenylalaninemia does not cause further reduction of alkaline phosphatase activity in PAH deficient cells. PAH replete cells generate more robust mineralization than PAH deficient cells (P < .001). Hyperphenylalaninemia causes modest yet significant down-regulation of mineralization in PAH replete cells (P < .01) while in PAH deficient cells, hyperphenylalaninemia has greater impact to down-regulate mineralization (p < .001).
3.4. PAH deficient and PAH replete MSCs display altered gene expression in response to hyperphenylalaninemia
Fig. 4 displays relative expression of Col1a1 (collagen type 1 alpha 1 chain) and Rankl (TNF superfamily member 11) in PAH deficient and PAH replete MSCs following bone differentiation under standard or hyperphenylalaninemic conditions. Expression is normalized to Gapdh expression with all studies representing a minimum of 4 replicates. Col1a1 is an indicator of bone formation. Both PAH deficient and PAH replete cells show Col1a1 down-regulation under hyperphenylalani-nemic conditions. Control cells under hyperphenylalaninemic conditions have similar Collai expression to PAH deficient cells under physiological conditions supporting culture studies (Fig. 3) as both display bone formation. Collai expression is exceedingly low in PAH deficient cells under hyperphenylalaninemic conditions reflecting low bone formation (Fig. 3). Rankl is a marker of bone turn-over. PAH replete cells generate far more bone than PAH deficient cells (Fig. 3) reflected in higher Rankl representation. Expression of Rankl is significantly suppressed in PAH deficient cells. Under hyperphenylalani-nemic conditions far less bone is generate by PAH deficient cells underlying Rankl’s near total absence.
Fig. 4.
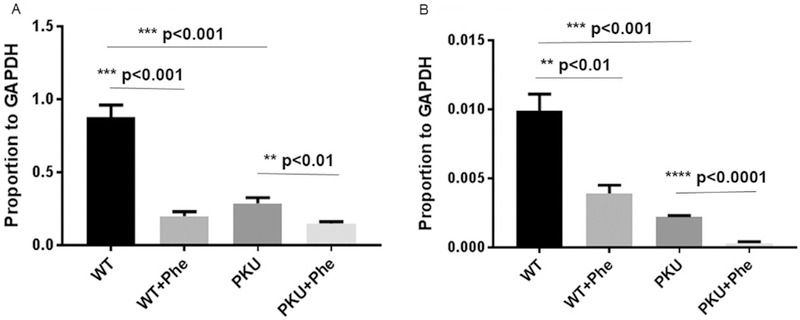
Effect of hyperphenylalaninemia on expression of Col1A1 and Rankl during Pahenu2 and wild type MSC osteoblast differentiation (A-B). Expression data is calculated relative to Gapdh. All expression studies were performed in triplicate. 4A. Expression of Collai in wild type cells (Pah replete) is abundant during osteoblast differentiation reflecting robust bone formation. Wild type cells under hyperphenylalaninemic conditions display lower Collai expression. Pah deficient cells express Collai at levels similarly to wild type cells under hyperphenylalaninemic conditions reflecting residual bone forming capacity. Pah deficient cells under hyperphenylalaninemic conditions display the lowest Collai expression. 4B. Expression of Rankl in wild type cells (Pah replete) is abundant as greater bone turn-over occurs as a consequence of high level bone formation. Under hyperphenylalaninemic conditions wild type cells express less Rankl as lower bone turn-over will occur owing to lower bone formation. In Pah deficient cells under standard culture conditions Rankl expression is reduced owing to lower bone formation with subsequent lower bone turn-over. Pah deficient cells under hyperphenylalaninemic conditions form little bone, exceedingly little Rankl expression is observed as bone turn-over is not required.
4. Discussion
The PKU bone phenotype is under-investigated with a majority of studies being patient-based and descriptive. The role of hyperphenylalaninemia in the PKU bone phenotype has been controversial with some reports suggesting a correlation between hyperphenylalaninemia and bone mineral loss while other studies find no relationship. Inconsistencies may be attributable to diverse cohorts, lack of standardization in assessment regimens, and widely differing levels of treatment compliance between patients. An intuitive route to clarify these inconsistencies is assessment of animal models where all metrics regarding study cohorts and their assessment are readily standardized. These investigations sought to further characterize the bone phenotype in the Pahenu2 mouse; followed with assessment of possible disease mechanisms while addressing the role of hyperphenylalaninemia.
Osteopenia is observed in the Pahenu2 mouse. Heterozygote crosses generated experimental and control animals from common litters, thus both cohorts experienced a highly similar in utero environment and were subsequently propagated under identical conditions (chow, light/ dark cycle, housing, etc.); largely eliminating differences owing to developmental or environmental influences. Cytology and quantitative μCT metrics demonstrate significant bone loss (low mineralization, increased trabecula spacing, reduced bone volume to total bone). Dynamic histomorphometry demonstrated reduced mineral apposition. Biochemical metrics (low Ca++, low phosphate, elevated PTH) indicate a disruption of bone homeostasis. These combined phenotypes demonstrate a mineralization defect in the PKU mouse that is not realized in the C57bl/6 background strain. PKU is primarily defined by its neurological phenotype which is poorly reflected in Pahenu2 [39]; however, for the purposes of investigating the bone phenotype, Pahenu2 is better suited to model human disease.
Leveraging MSCs and directing their differentiation down the bone pathway is a novel approach to investigate developmental bone processes within the context of PAH deficiency and hyperphenylalani-nemia. Fig. 3 cell culture studies and Fig. 4 gene expression studies are complementary as both support defective MSC differentiation into bone and demonstrate a role of hyperphenylalaninemia therein. While PAH replete MSCs demonstrate robust bone formation, hyperphenylalani-nemia (1200 μM PHE) leads to a modest yet significant loss of mineralization. The impact of hyperphenylalaninemia on PAH replete MSCs is more pronounced in gene expression studies demonstrated by down-regulated Collal and Rankl expression. Bone differentiation in PAH deficient MSCs is more complex. A genetic component is evident as overall, PAH deficient MSC display far less robust bone differentiation (Fig. 3). Decreased bone differentiation is further evidenced by lower Collal and Rankl expression (Fig. 4). The influence of hyperphenyla-laninemia on mineralization in PAH deficient MSC reflects the observation in PAH replete cells; however, the intrinsic mineralization capacity of PAH deficient cells is lower and impact of hyperphenyla-laninemia upon mineralization is more profound (~50% decreased mineralization). Current dogma dictates PAH gene expression and oxidation of PHE as restricted to liver and kidney. Based upon PAH expression, the effect of hyperphenylalaninemia upon bone formation must owe to either or both a toxic effect of PHE and/or interference of amino acid transport (i.e. SLC7A5). Our MSC data indicate two things: 1. PAH deficiency causes a statistically significant loss of bone forming capacity; and 2. Hyperphenylalaninemia acts upon a PAH deficient background to cause a large loss of mineralization. If loss of bone forming capacity were purely toxic or purely involved aberrant amino acid transport, it would be anticipated that PAH deficient and PAH replete MSCs would have similar bone forming capacity and respond similarly to hyperphenylalaninemia, which is clearly not observed. The magnitude of hyperphenylalaninemia induced mineralization loss in PAH deficient MSCs far exceeds that in PAH replete cells (Fig. 3B). While seemingly unlikely, these data may suggest PAH has a role in MSC differentiation involving either PHE oxidation or an unrecognized PAH moonlighting role. While in vitro MSC differentiation is a well-established tool to assess bone pathophysiology; ultimately in vivo confirmation of these results will be necessary.
The role of hyperphenylalaninemia in the PKU bone phenotype remains controversial. We contend that PKU phenotypes are viewed through the lens of neurologic manifestations where high-level PHE toxicity is clearly causal. It is possible that MSCs, in the course of bone differentiation, may have heightened sensitivity to PHE toxicity. That osteopenia is observed in well controlled as well as poorly controlled patients may suggest this to be the case. Previous Pahenu2 bone studies [33,40] assessed treated animals where hyperphenylalaninemia was in excess well controlled PKU. Our standard condition for MSC differentiation had PHE far lower than treatment levels in previous Pahenu2 studies [33,40]. To fully define the influence of hyperphenylalaninemia on the Pahenu2 bone phenotype, it will be necessary to assess animals over a broad range of PHE homeostasis. Continued studies in PAH deficient MSCs is focused upon identifying a PHE toxicity threshold triggering reduced MSC bone differentiation.
Supplementary Material
Acknowledgements
Supported in part by the Department of Veterans Affairs grant I01BX002490 and by the National Institutes of Health grant AR065407 to H.B.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymgme.2018.08.010.
References
- [1].Guthrie R, Sussi A, A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants, Pediatrics 32 (1963) 318–322. [PubMed] [Google Scholar]
- [2].Følling A, Uber Ausscheidung von Phenylbrenztraubensaure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillitat, Hoppe-Seylers Z Physiol Chem 277 (1934) 169. [Google Scholar]
- [3].Jervis GA, Phenylpyruvic oligophrenia: Deficiency of phenylalanine oxidizing system, Proc. Soc. Exp. Biol. Med. 82 (1953) 514. [PubMed] [Google Scholar]
- [4].Bickel H, et al. , Influence of phenylalanine intake on phenylketonuria, Lancet 265 (1953) 812. [DOI] [PubMed] [Google Scholar]
- [5].García MI, Araya G, Coo S, Waisbren SE, de la Parra A, Treatment adherence during childhood in individuals with phenylketonuria: early signs of treatment discontinuation, Mol. Genet. Metab. Rep. 28 (11) (2017) 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pena MJ, Almeida MF, van Dam E, Ahring K, Bélanger-Quintana A, Dokoupil K, Gokmen-Ozel H, Lammardo AM, MacDonald A, Robert M, Rocha JC, Special low protein foods for phenylketonuria: availability in Europe and an examination of their nutritional profile, Orphanet J. Rare Dis. 22 (10) (2015) 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Al Hafid N, Christodoulou J, Phenylketonuria: a review of current and future treatments, Transl Pediatr. 4 (4) (2015) 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Al Hafid N, Christodoulou J, Phenylketonuria: a review of current and future treatments, Transl Pediatr. 4 (4) (2015) 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bell SM, Wendt DJ, Zhang Y, Taylor TW, Long S, Tsuruda L, Zhao B, Laipis P, Fitzpatrick PA, Formulation and PEGylation optimization of the therapeutic PEGylated phenylalanine ammonia lyase for the treatment of phenylketonuria, PLoS One 10 (12(3)) (2017) e0173269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Longo N, Harding CO, Burton BK, Grange DK, Vockley J, Wasserstein M, Rice GM, Dorenbaum A, Neuenburg JK, Musson DG, Gu Z, Sile S, Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, multicentre, phase 1 dose-escalation trial, Lancet 384 (2014) 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tansek MZ, Groselj U, Kelvisar M, Kobe H, Lampret BR, Battelino T, Long-term BH4 (sapropterin) treatment of children with hyperphenylalaninemia - effect on median Phe/Tyr ratios, J. Pediatr. Endocrinol. Metab. 1 (29(5)) (2016) 561–566. [DOI] [PubMed] [Google Scholar]
- [12].Longo N, Arnold GL, Pridjian G, Enns GM, Ficicioglu C, Parker S, CohenPfeffer JL, Phenylketonuria demographics, outcomes and safety registry. Long-term safety and efficacy of sapropterin: the PKUDOS registry experience, Mol. Genet. Metab. 114 (4) (2015) 557–563. [DOI] [PubMed] [Google Scholar]
- [13].Feinberg SB, Fisch RO, Roentgenologic findings in growing long bones in phenylketonuria. Preliminary study, Radiology 78 (1962) 394–398. [DOI] [PubMed] [Google Scholar]
- [14].Murdoch MM, Holman GH, Roentgenologic bone changes in phenylketonuria. Relationship to dietary phenylalanine and serum alkaline phosphatase, Am. J. Dis. Child. 107 (1964) 523–532. [PubMed] [Google Scholar]
- [15].Porta F, Spada M, Lala R, Mussa A, Phalangeal quantitative ultrasound in children with phenylketonuria: a pilot study, Ultrasound Med. Biol. 34 (7) (2008) 1049–1052. [DOI] [PubMed] [Google Scholar]
- [16].Schwahn B, Mokov E, Scheidhauer K, Lettgen B, Schönau E, Decreased trabecular bone mineral density in patients with phenylketonuria measured by peripheral quantitative computed tomography, Acta Paediatr. 87 (1) (1998) 61–63. [DOI] [PubMed] [Google Scholar]
- [17].Zeman J, Bayer M, Stepán J, Bone mineral density in patients with phenylketonuria, Acta Paediatr. 88 (12) (1999) 1348–1351. [DOI] [PubMed] [Google Scholar]
- [18].Allen JR, Humphries IR, Waters DL, Roberts DC, Lipson AH, Howman-Giles RG, Gaskin KJ, Decreased bone mineral density in children with phenylketonuria, Am. J. Clin. Nutr. 59 (2) (1994) 419–422. [DOI] [PubMed] [Google Scholar]
- [19].Coakley KE, Douglas TD, Goodman M, Ramakrishnan U, Dobrowolski SF, Singh RH, Modeling correlates of low bone mineral density in patients with phenylalanine hydroxylase deficiency, J. Inherit. Metab. Dis. 39 (3) (2016) 363–372. [DOI] [PubMed] [Google Scholar]
- [20].Pérez-Dueñas B, Cambra FJ, Vilaseca MA, Lambruschini N, Campistol J, Camacho JA, New approach to osteopenia in phenylketonuric patients, Acta Paediatr. 91 (2002) 899–904. [DOI] [PubMed] [Google Scholar]
- [21].Modan-Moses D, Vered I, Schwartz G, Anikster Y, Abraham S, Segev R, Efrati O, Peak bone mass in patients with phenylketonuria, J. Inherit. Metab. Dis. 30 (2007) 202–208. [DOI] [PubMed] [Google Scholar]
- [22].Demirdas S, Coakley KE, Bisschop PH, Hollak CE, Bosch AM, Singh RH, Bone health in phenylketonuria: a systematic review and meta-analysis, Orphanet J. Rare Dis. 15 (10) (2015) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hansen KE, Ney D, A systematic review of bone mineral density and fractures in phenylketonuria, J. Inherit Metab Dis. 37 (6) (2014) 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].de Groot MJ, Hoeksma M, van Rijn M, Slart RH, van Spronsen FJ, Relationships between lumbar bone mineral density and biochemical parameters in phenylketonuria patients, Mol. Genet. Metab. 105 (4) (2012) 566–570. [DOI] [PubMed] [Google Scholar]
- [25].Lage S, Bueno M, Andrade F, Prieto JA, Delgado C, Legarda M, Sanjurjo P, Aldamiz-Echevarria Lj, Fatty acid profile in patients with phenylketonuria and its relationship with bone mineral density, J. Inherit. Metab. Dis. 33 (Suppl. 3) (2010) S363–S371. [DOI] [PubMed] [Google Scholar]
- [26].Zeman J, Bayer M, Stepán J, Bone mineral density in patients with phenylketonuria, Acta Paediatr. 88 (12) (1999) 1348–1351. [DOI] [PubMed] [Google Scholar]
- [27].Hillman L, Schlotzhauer C, Lee D, Grasela J, Witter S, Allen S, Hillman R, Decreased bone mineralization in children with phenylketonuria under treatment, Eur. J. Pediatr. 155 (Suppl. 1) (1996) S148–S152. [DOI] [PubMed] [Google Scholar]
- [28].P1 Adamczyk, Morawiec-Knysak A, Piudowski P, Banaszak B, Karpe J, Pluskiewicz W, Bone metabolism and the muscle-bone relationship in children, adolescents and young adults with phenylketonuria, J. Bone Miner. Metab. 29 (2) (2011) 236–244. [DOI] [PubMed] [Google Scholar]
- [29].P1 Barat, Barthe N, Redonnet-Vernhet I, Parrot F, The impact of the control of serum phenylalanine levels on osteopenia in patients with phenylketonuria, Eur. J. Pediatr. 161 (12) (2002) 687–688. [DOI] [PubMed] [Google Scholar]
- [30].Nagasaka H, Tsukahara H, Takatani T, Sanayama Y, Takayanagi M, Ohura T, Sakamoto O, Ito T, Wada M, Yoshino M, Ohtake A, Yorifuji T, Hirayama S, Miida T, Fujimoto H, Mochizuki H, Hattori T, Okano Y, Cross-sectional study of bone metabolism with nutrition in adult classical phenylketonuric patients diagnosed by neonatal screening, J. Bone Miner. Metab. 29 (6) (2011) 737–743. [DOI] [PubMed] [Google Scholar]
- [31].Porta F, Roato I, Mussa A, Repici M, Gorassini E, Spada M, Ferracini R, Increased spontaneous osteoclastogenesis from peripheral blood mononuclear cells in phenylketonuria, J. Inherit. Metab. Dis. 31 (Suppl. 2) (2008) S339–S342. [DOI] [PubMed] [Google Scholar]
- [32].Hillman L, Schlotzhauer C, Lee D, Grasela J, Witter S, Allen S, Hillman R, Decreased bone mineralization in children with phenylketonuria under treatment, Eur. J. Pediatr. 155 (Suppl. 1) (1996) S148–S152. [DOI] [PubMed] [Google Scholar]
- [33].Yannicelli S, Medeiros DM, Elevated plasma phenylalanine concentrations may adversely affect bone status of phenylketonuric mice, J. Inherit. Metab. Dis. 25 (5) (2002. September) 347–361. [DOI] [PubMed] [Google Scholar]
- [34].Dobrowolski SF, Vockley J, Spridik K, Lyons-Weiler J, Epigenomics in Neuropathology of PAH Deficient PKU in the Pahenu2 Model, Mol. Genet. Metab. 117 (3) (2016) 254. [Google Scholar]
- [35].Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R, Guidelines for assessment of bone microstructure in rodents using micro-computed tomography, J. Bone Miner. Res. 25 (7) (2010. July) 1468–1486. [DOI] [PubMed] [Google Scholar]
- [36].Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM, Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee, J. Bone Miner. Res. 28 (1) (2013. January) 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Blair HC, Kalyvioti E, Papachristou NI, Tourkova IL, Syggelos SA, Deligianni D, Orkoula MG, Kontoyannis CG, Karavia EA, Kypreos KE, Papachristou DJ, Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice, Lab. Investig. 96 (7) (2016. July) 763–772. [DOI] [PubMed] [Google Scholar]
- [38].Rodenacker K, Bengtsson E, A feature set for cytometry on digitized microscopic images, Anal. Cell. Pathol. 25 (1) (2003) 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sarkissian CN, Boulais DM, McDonald JD, Scriver CR, A heteroallelic mutant mouse model: a new orthologue for human hyperphenylalaninemia, Mol. Genet. Metab. 69 (3) (2000. March) 188–194. [DOI] [PubMed] [Google Scholar]
- [40].Solverson P, Murali SG, Litscher SJ, Blank RD, Ney DM, Low bone strength is a manifestation of phenylketonuria in mice and is attenuated by a glycomacropeptide diet, PLoS One 7 (9) (2012) e45165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.