

Abstract
Colorectal cancer lymph node metastases are common but their genetics and the mechanism whereby these metastases occur is not well understood. Here we present recent data regarding genetic heterogeneity in primary colorectal cancer and their metastasis. In addition, we explain the different potential models of the mechanisms of metastasis and the data supporting them. Multiple studies have also revealed a variety of prognostic molecular markers that are associated with lymph node metastasis in colorectal cancer. Better understanding of genetic heterogeneity and mechanisms of metastasis is critical to predicting clinical response and resistance to targeted therapy.
Keywords: colorectal cancer, cancer genetics, lymph node metastasis, intratumor heterogeneity
Introduction
Colorectal cancer (CRC) the third most common cause of cancer and of cancer related death in males and females in the United States [1]. The 5-year survival for colorectal cancer with distant metastasis is only around 20%. The first sign of advanced disease is traditionally thought to be the presence of malignant spread to nearby lymph nodes (LN) but the timing and mechanism whereby this occurs is not clear. Though LN metastasis is not the direct cause of mortality in patients, these patients reap improved survival when they receive systemic chemotherapy and thus, this appears to me a marker of systemic disease. It is important to fully elucidate the biology of LN spread in colorectal cancer. Through this review, we hope to shed light on recent developments in the understanding of LN spread in colorectal cancer.
The traditional model of linear progression from primary tumor to LN to distant metastasis informs most decision-making, dictates cancer staging, and guides treatment selection. Surgical lymphadenectomy is accepted to improve local tumor control in some cancer types but is only thought of as a staging method in others. A scientific understanding of whether LNs harbor the source of metastatic clones is critical to tailoring the extent (and therefore the morbidity) of lymphadenectomy, especially in the metastatic setting. Physicians and surgeons treating cancer recognize LNs as a poor prognostic marker, but whether their total resection results in improved survival or only accurate staging is a debate that can be appreciated across surgical disciplines when we consider the controversy surrounding extended lymphadenectomy in gastric cancer, total mesocolic excision, high versus low ligation of the inferior mesenteric artery, and the value of lateral pelvic node dissection. In addition, increased appreciation of genetic heterogeneity in primary tumors and metastases has important ramifications for personalized medicine utilizing targeted therapy. In this setting, LN biology may be critical as many colorectal cancer patients have LNs as their only known site of tumor dissemination.
A word of caution about methods
An accurate understanding of cancer’s molecular evolution is dependent on the method of genetic analysis, tumor sampling strategy, and an understanding of the technological limitations and baseline assumptions underlying the research strategy. Type and depth of genetic analysis are critical in determination of mutational divergence between tumor samples. Genetic analysis may be performed by using gene panels, assessing a type of somatic alteration (such as small insertions/deletions [indels]), or employ whole exome sequencing (WES) or whole genomic sequencing (WGS). The number of genetic markers employed in the study has a critical impact on the estimate of molecular similarity estimated by the analysis. Studies employing gene panels are often biased to known driver genes, most of which play an important role in early tumorigenesis/malignant transformation and are therefore more likely to be shared across sub-clones. WES is more cost-effective than WGS, but only 1.5% of the genome is included in the exome, so molecular evolution events effecting introns, some of which have critical roles in gene expression, are not examined. In short, the more markers employed by the study, the more divergent the tumor and metastasis will appear, but NGS has its own flaws compared to traditional Sanger sequencing in terms of variation in coverage in individual genetic regions. In addition, even when WES or WGS is used, if the depth of coverage used to assess for mutations is low, then low frequency mutations will not be identified and heterogeneity and sub-clonality cannot be assessed. In reality, it may be best to perform WES or WGS followed by deep custom panels. Estimates of normal tissues in the specimen must be incorporated into comparisons between samples as differing amounts of normal DNA between samples can falsely increase or decrease estimates of heterogeneity. Furthermore, non-mutational alterations to the genetic code, such as methylation, histone modification, copy number variations, and micro-RNA binding may create a selective advantage for tumoral sub-clones in ways not analyzed by sequencing. Similarly, sampling strategy is critical for analysis. If the primary tumor is not exhaustively sampled and analyzed, heterogeneous primary sub-clones are not identified and mutations in metastases are falsely identified as private metastatic mutations. Indentation of rare sub-clones may require high sequencing depth and assessment of multiple geographic regions. Analysis of metastatic lesions poses a particular challenge as most metastases are not sampled or surgically resected, and those which are made available for genetic analysis have undergone pre-treatment with chemotherapeutics. An exception to this rule, is the LN metastasis which is typically resected at the same time as the primary tumor and therefore undergoes similar treatment within a distinct environment. Lastly, one must take into account copy number variation (CNV) where large or small segments of the genome, often including many genes at once, are amplified or deleted which can be important in and of themselves but can also alter the effects of mutations. CNV can be assessed using WES or single nucleotide polymorphism arrays.
Models of metastasis
LN metastasis are rarely accounted for in models of tumor progression or appear only as a stopping point on the way to distant metastasis. Specifically, as it relates to LN metastasis, 3 potential models can be imagined (Figure 1). In the first model, which is clearly not in agreement with recent studies, the tumor is made up of a genetically uniform population and move to the LNs. In the second model, the primary tumor is made up of many genetically related but distinct sub-clones and a single, especially fit, sub-clone metastasizes to the LNs sequentially. The third model, again presents a heterogeneous primary tumor but here the LN metastases are poly-clonal with variability in the specific distribution between different nodes. This third model is explained through multiple waves of metastasis of single or polyclonal clumps of cells. We now know from multiple studies that solid tumors are genetically heterogeneous with multiple sub-clones [2–5]. Current data supports Model 3. Many open questions still exist with regard to the sub-clonality of nodal and distant metastasis including when metastasis occurs and whether certain mutations make it more likely and whether sub-clones cooperate to metastasize successfully [6]. What is becoming clear, is that the process of metastasis, including regional metastasis, is far more complex than previously envisioned.
Figure 1.
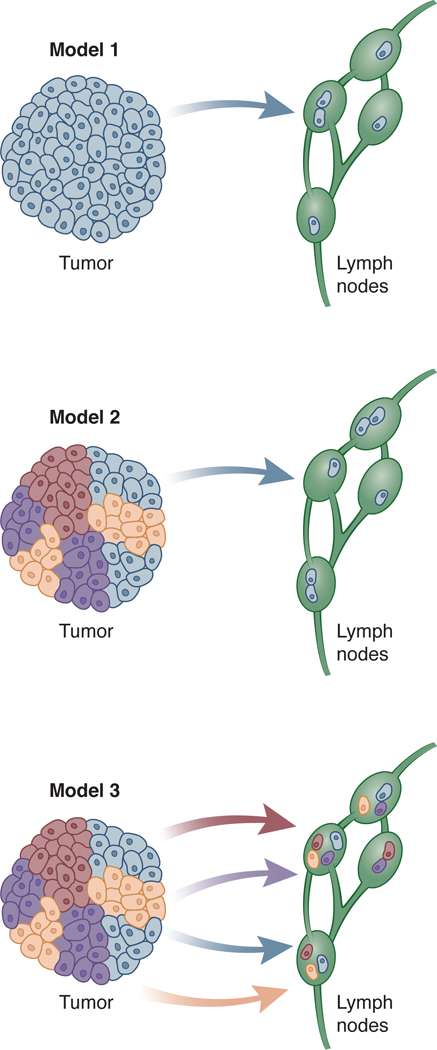
Three models of lymph node metastasis.
The two general over-arching systems of tumor progression describing the evolution of primary tumors and their paired distant metastasis are the linear and parallel models. In the traditional linear progression model of cancer, the bulk of molecular evolution progresses within the primary tumor it becomes a cancer and before metastasis. Selective pressures favor the fittest sub-clone which dominates through clonal sweeps as the primary tumor progresses. This model was put forth for colorectal cancer progression by Vogelstein and Fearon in 1990 [7]. This genetically uniform tumor then colonizes regional, followed by distant metastatic sites. In this model, the ability to metastasize is gained within the primary tumor and mutational divergence between primary and metastatic lesions is predicted to be small, reflecting the role of metastasis as daughter clones. Furthermore, genetic analyses in this model are expected to produce a hierarchical phylogeny from primary tumor to LN to metastasis. The linear progression model has dominated the cancer paradigm. It is reflected by the TNM staging system and informs the decision to provide chemotherapy to LN-positive patients and withhold it from those with negative nodes. For the surgeon, it provides a logical rationale for performing extensive lymphadenectomy, both for staging and disease-control purposes.
In contrast, the parallel progression model proposes that tumor sub-clones start to metastasize early in tumorigenesis, with multiple waves of distinct, early and late metastatic monoclonal and polyclonal cell clusters exiting the tumor in parallel with primary tumor growth. These sub-clones colonize LNs and distant sites and evolve temporally in parallel to the primary tumor, but under a unique set of environmental selective pressures. Therefore, mutational divergence between primary tumor and metastasis is expected to be high and the generated phylogeny complex and inter-related. Parallel progression has been empirically demonstrated in nodal spread of colorectal cancer with genomic analyses revealing contributions of spatially and temporally distinct polyclonal primary tumor sub-clones to individual LNs [8]. Branched evolution has also been demonstrated in colorectal cancer distant metastasis [4]. In some diseases, the parallel progression model also informs clinical and surgical decision-making. For example, in breast cancer when considering omission of axillary node dissection in favor of systemic treatment in the presence of a positive sentinel LN, the parallel progression model is invoked. Importantly, parallel and linear models are not mutually exclusive, and both types of progression may occur within the same cancer type. Studying somatic variants in hypermutable DNA regions in 213 primary, nodal, and distant metastases from 17 colorectal cancer patients, researchers identified common clonal origin of LNs and metastases in 35% of cases and an independent origin in 65% [9].
Intra-tumor heterogeneity
Knowledge of intratumor heterogeneity in cancer has increased substantially over the last several years with improved sequencing tools but the implications of tumor sub-clones for metastasis and treatment was hypothesized by Nowell in 1976 [10]. Intra-tumoral heterogeneity occurs as neoplastic cells divide, acquire mutations and separate spatially [5]. As a result, spatially separated cells in a large tumor have been shown to be more different from each other than close neighbors [11]. Mutations that increase mutation frequency (MSH2, MSH6, MLH1, and PMS2) and those that allow the tumor to better tolerate genomic instability (TP53, ATM) increase the mutational burden and the heterogeneity. Genetic intra-tumoral heterogeneity (ITH) has been demonstrated across a variety of solid tumors including rectal cancer and is now a generally accepted feature of all cancers [2]. Increased ITH in the primary tumor for multiple tumor types has been shown to correlate with worse outcome [12].
Intra-tumor heterogeneity in driver mutations was found in colon cancer by Baldus et al. who performed mutation analysis from tumor tissues that were harvested from primary sites, LNs, and distal sites[13]. They found the KRAS, BRAF, and PIK3CA mutations to be heterogeneous between regions of primary tumors. When comparing the tumor center to the invasion fronts, heterogeneity was present in 20% of the tumors with a KRAS mutation, 14% of the tumors with a BRAF mutation, and 24% of the tumors with a PIK3CA mutation. Kosmidou et al. compared the KRAS mutations from the tumor center to the tumor periphery as well. They found the heterogeneity of KRAS mutations between these two sites to be 44% [14]. They subsequently compared the mutations at a different tumor center site as well and found the heterogeneity to be present in about 34.8% of the samples [14]. A similar analysis was done by Jeantet et al. to further examine the intratumor heterogeneity in colon cancer. By examining KRAS and NRAS mutations in 18 tissues samples from patients with CRC, they found that there was about 33% intratumor heterogeneity among the samples [15]. Intratumor heterogeneity is clinically significant because the heterogeneity of key mutations, such as KRAS, can dictate response to therapy, and even survival [16,17]. Sottoriva et al. also found substantial heterogeneity across colon adenocarcinoma and adenomas through assessment of CNV and through modeling hypothesized that this heterogeneity originates very early in tumor evolution [3].
Heterogeneity in metastasis
The heterogeneity among tumor sub-clones is not limited to primary tumors alone but persists throughout LNs and distant sites. Few studies have been performed in colon cancer LN metastasis. Baldus et al. showed heterogeneity in driver mutations KRAS, BRAF, and PIK3CA between LNs and matched primary tumors for some colorectal cancers [13]. The authors found that 31% of the LNs had discordant KRAS mutations when compared to primary tissue. They also found 9% of LNs to have discordant BRAF mutations and 13% of LNs to have discordant PIK3CA mutations. When examining the distant metastatic tissues, the discordant mutations were 10% for KRAS, 0% for BRAF, and 5% for PIK3CA. These findings show that heterogeneity persists throughout disease progression [13].
Another example of using mutational analysis to determine heterogeneity between primary tumor and metastasis is the study done by Jeantet et al [15]. These authors analyzed patient tissue samples for KRAS, NRAS, and BRAF mutations. They found 36% of intertumoral heterogeneity between primary tumor samples and LNs or distant metastatic samples. In these samples, the primary tumors harbored a RAS mutation that was a wild type in the LNs or distant metastatic lesions. They also found that there were different forms of the RAS mutations between the samples as well indicating convergent evolution.
Mutation analysis is not the only way that heterogeneity is evident. Kim et al. established heterogeneity by measuring copy number alterations in addition to mutation analysis [18]. In terms of mutation analysis, the authors performed 35 multiregional biopsies of primary and metastatic CRC lesions among five different CRCs. They performed WES to further characterize the mutations into five spatial categories: universal, metastasis-clonal, metastasis-private, primary-private, and unclassified. They found that, depending upon which tumor was sampled, only 19.8% to 53.9% of the mutations were universal, meaning that they were present in both the primary tumor and the metastatic lesion. Among the rest of the mutations that were sub-clonal, 13.8% to 56.0% were primary-private. These were defined as mutations present in the primary tumor, but not in the metastatic lesion. Metastatic-private mutations, those that were clonally present in metastatic lesions but were not present in the primary tumor, comprised of 2.4–40.7% of the sub-clonal mutations. Interestingly, about 1.4–37.2% of mutations were biopsy-specific and not found elsewhere [18]. The authors also utilized copy number alterations in combination with mutation analysis to develop phylogeny trees to further examine the linearity of evolution of the cancers. Analysis of these phylogeny patterns revealed that four of the five cancers experienced non-linear or branched evolution.
Using mathematical modelling tumor sub-clones can be inferred from in depth data on heterogeneity. Findings that perhaps represent the most significant departure from the monoclonal model of cancer metastasis are those described by Ulintz et al [8]. For this study, seven colon cancers were used to collect 33 samples. These included spatially disparate regions in each primary tumor and also one to four matched LNs. These samples were then analyzed using WES, deep targeted sequencing, CNV analysis, and Pyclone analysis to distinguish one sub-clone from another. The results of these analyses were then used to develop phylogenic trees. Ulintz et al. showed the presence of heterogeneity in mutations and CNV between primary tumor samples and their matched LN metastases. Interestingly, they also showed heterogeneity between metastatic lesions. Furthermore, they showed that LN metastases were polyclonal and established that the LN metastases originated from different geographic regions of the primary tumor leading them to propose a model of LN metastasis where there are multiple waves over time. This spatial-temporal relationship is a novel finding that expands our understanding of LN metastasis (consistent with Model 3 in Figure 1). These findings question common clinical practice of selecting targeted therapeutics based on the mutation status of the primary tumor.
Drivers and predictors of LN metastasis
Driver genes of colorectal tumorigenesis, such as APC, TP53, KRAS, MAPK, MEK, BRAF, PTEN, etc. are well known [19]. However, despite intensive research effort, there is no genetic signature which can distinguish a localized from a metastatic colorectal tumor. Drivers of metastasis may be more difficult to discover, as metastatic lesions are less routinely biopsied and resected. However, some authors feel that the majority of meaningful cancer driver genes have been discovered [20] or that genetic drivers of metastasis do not exist and metastasis can be explained as a stochastic process alone [21]. As such, a LN metastasis could be explained as chance event as the tumor randomly sheds malignant cells. The ability of non-neoplastic cells to colonize LNs and form functioning organoids has been cited by some authors as a potential support of this theory [22]. However, some gene mutations and somatic alterations have been associated with increased LN metastasis in colorectal cancer. Driver gene mutations in KRAS and TP53 but not APC or PIK3CA have been found to be associated with LN and distant metastasis [23]. Less well-known transcription factors, FOXM1 and eIF3e have also been associated with LN metastasis and poor prognosis in colorectal cancer [24][25]. Certain copy number variations have also been associated with colorectal LN metastasis. For example, copy number deletion of JK-1 was found to be more frequent in colorectal adenocarcinomas than adenomas and conferred an increased risk of nodal disease [26].
Gene-level changes within a tumor’s DNA may ultimately not reflect protein translation due to post-transcriptional changes such as epigenetic alterations and chromatin structural changes. Therefore, several studies have attempted to analyze gene expression patterns between LN-positive and negative tumors. Altered patterns of expression of genes of interest, such as in CDH1 and aquaporins, have been studied in colorectal cancer via immunostaining and found to be associated with LN metastases [27,28]. Similarly, microarray technology has identified differential expression patterns of 41 genes of interest between tumors with and without LN metastasis [29]. Novel methods in proteomics have also identified different levels of novel markers FXYD3, S100A11, and GSTM3 between LN-positive and negative tumors [30]. However, determining causality and clinical relevance in these associative expression studies is challenging.
Similarly, Lan et al. examined 629 patient colorectal cancer samples for genetic factors that can help distinguish LN status [31]. Rather than genes, however, the authors of this study looked for single nucleotide polymorphisms (SNPs) in the genome. Eleven SNPs were found to be significantly different between samples that had LN spread, and those that did not. Of these, eight SNPs were associated with an increased risk of metastasis, whereas three SNPs were associated with a decreased risk of LN spread. In their study, they also found that a patient having seven or more unfavorable genotypes increased their risk of LN spread by 1.97 times as much as those that had less than seven unfavorable genotypes. The major limitation of this study, however, was that all the patients that the samples came from were Asian, which potentially limits the generalizability of the findings [31].
One possible outcome of genomic analysis is development of predictive biomarkers that can identify patients at high risk for LN metastasis, potentially allowing improved selection of patients for neoadjuvant treatment versus up-front surgery. Using a training cohort of 69 patient samples, a gene expression signature using 73 discriminating genes was derived. The model had a positive predictive value of 84.6% and a negative predictive value of 90.7% in detecting LN metastasis [32]. Though this concept would need to be validated in large clinical trials, it does show potential in helping stage patients, thereby helping clinicians provide individualized therapy. Use of molecular marker to risk stratify patients for systemic therapy is on the horizon and being tested in randomized trials.
There are many clinical implications to the findings that have been presented. A deeper understanding of the heterogeneity in the progression of cancer is vital in understanding resistance to therapy. For example, presence of KRAS mutations predicts resistance to therapy epidermal growth factor (EGFR) antibody therapy [33]. Despite this valuable information, it is still difficult to predict clinical response because of the intertumoral heterogeneity of KRAS mutations since a single biopsy may or may not detect the mutation. Intra-metastatic heterogeneity is demonstrated clinically in relapse of metastatic lesions after initial response to chemotherapy. For instance, mathematical modelling of the emergence of resistance to panitumumab in initially-responsive colorectal tumors revealed that resistant sub-clones existed prior to the initiation of targeted therapy [34]. Any given tumor harbors 100 or more mutations and CNV’s that can vary between geographic locations with the potential for at least some of these to alter response to therapy.
Conclusions
Linear progression of a clonal cancer from primary tumor, to LN, to metastasis is a paradigm that informs much of current cancer diagnosis, staging, treatment, and research. However, as technology has improved to the point where we can interrogate individual LNs and distant metastatic sites, a growing body of evidence suggests that this is an over-simplification. Whether by as-yet obscure molecular mechanisms or by stochastic means alone or a combination, metastases are heterogeneous. Spatially, temporally, and molecularly distinct malignant sub-clones colonize LNs and distant sites. Development of personalized therapy to treat and prevent fatal systemic metastasis requires consideration of the metastatic lesions as heterogeneous collections of sub-clones each harboring unique patterns of resistance. With that in mind, research and treatment strategies aimed at discovering and targeting single mutations in the primary tumor are unlikely to achieve long term, relapse-free success. Therefore, an improved scientific understanding of the heterogeneous nature of LN behavior will be key to tailoring treatment and improving patient care and survival.
1. Acknowledgement
We acknowledge Patricia Beals who created Figure 1.
4. Funding sources: 5P50CA130810, American Surgical Association Foundation Fellowship; 3P30CA046592-S8S3; John S. and Suzanne C. Munn Cancer Research Fund, K08CA190645
Grant Support: KMH: 5P50CA130810, American Surgical Association Foundation Fellowship; 5P30A046592; John S. and Suzanne C. Munn Cancer Research Fund, K08CA190645; SER: National Institute on Aging Grants for Early Medical/Surgical Specialists Transition to Aging Research R03-AG047860, and National Institute on Aging K08-AG047252
Footnotes
Statement of Ethics: The authors have no ethical conflicts to disclose.
Disclosure Statement: The authors have no conflicts of interest to declare.
References:
- 1.Siegel RL, Miller KD, Jemal A: Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Hardiman KM, Ulintz PJ, Kuick RD, Hovelson DH, Gates CM, Bhasi A, Rodrigues Grant A, Liu J, Cani AK, Greenson JK, Tomlins SA, Fearon ER: Intra-tumor genetic heterogeneity in rectal cancer. Lab Invest 2016;96:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, Curtis C: A Big Bang model of human colorectal tumor growth. Nat Genet 2015;47:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim TM, An CH, Rhee JK, Jung SH, Lee SH, Baek IP, Kim MS, Chung YJ: Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget 2015;6:27725–27735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polyak K, Marusyk A: Cancer: Clonal cooperation. Nature 2014;508:52–53. [DOI] [PubMed] [Google Scholar]
- 7.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 8.Ulintz PJ, Greenson JK, Wu R, Fearon ER, Hardiman KM: Lymph Node Metastases in Colon Cancer Are Polyclonal. Clin Cancer Res 2018;24:2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naxerova K, Reiter JG, Brachtel E, Lennerz JK, van de Wetering M, Rowan A, Cai T, Clevers H, Swanton C, Nowak MA, Elledge SJ, Jain RK: Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017;357:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowell PC: The clonal evolution of tumor cell populations. Science 1976;194:23–28. [DOI] [PubMed] [Google Scholar]
- 11.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA: Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467:1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris LG, Riaz N, Desrichard A, Senbabaoglu Y, Hakimi AA, Makarov V, Reis-Filho JS, Chan TA: Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016;7:10051–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE: Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 2010;16:790–799. [DOI] [PubMed] [Google Scholar]
- 14.Kosmidou V, Oikonomou E, Vlassi M, Avlonitis S, Katseli A, Tsipras I, Mourtzoukou D, Kontogeorgos G, Zografos G, Pintzas A: Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum Mutat 2014;35:329–340. [DOI] [PubMed] [Google Scholar]
- 15.Jeantet M, Tougeron D, Tachon G, Cortes U, Archambaut C, Fromont G, Karayan-Tapon L: High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. Int J Mol Sci 2016;17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tougeron D, Lecomte T, Pages JC, Villalva C, Collin C, Ferru A, Tourani JM, Silvain C, Levillain P, Karayan-Tapon L: Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol 2013;24:1267–1273. [DOI] [PubMed] [Google Scholar]
- 17.Joung JG, Oh BY, Hong HK, Al-Khalidi H, Al-Alem F, Lee HO, Bae JS, Kim J, Cha HU, Alotaibi M, Cho YB, Hassanain M, Park WY, Lee WY: Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clin Cancer Res 2017;23:7209–7216. [DOI] [PubMed] [Google Scholar]
- 18.Kim TM, Jung SH, An CH, Lee SH, Baek IP, Kim MS, Park SW, Rhee JK, Lee SH, Chung YJ: Subclonal Genomic Architectures of Primary and Metastatic Colorectal Cancer Based on Intratumoral Genetic Heterogeneity. Clin Cancer Res 2015;21:4461–4472. [DOI] [PubMed] [Google Scholar]
- 19.Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW: Cancer genome landscapes. Science 2013;339:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernards R, Weinberg RA: A progression puzzle. Nature 2002;418:823. [DOI] [PubMed] [Google Scholar]
- 22.Komori J, Boone L, DeWard A, Hoppo T, Lagasse E: The mouse lymph node as an ectopic transplantation site for multiple tissues. Nat Biotechnol 2012;30:976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang D, Sun W, Zhou Y, Li P, Chen F, Chen H, Xia D, Xu E, Lai M, Wu Y, Zhang H: Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev 2018;37:173–187. [DOI] [PubMed] [Google Scholar]
- 24.Li D, Wei P, Peng Z, Huang C, Tang H, Jia Z, Cui J, Le X, Huang S, Xie K: The critical role of dysregulated FOXM1-PLAUR signaling in human colon cancer progression and metastasis. Clin Cancer Res 2013;19:62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Lin S, Jiang T, Wang J, Lu H, Tang H, Teng M, Fan J: Overexpression of eIF3e is correlated with colon tumor development and poor prognosis. Int J Clin Exp Pathol 2014;7:6462–6474. [PMC free article] [PubMed] [Google Scholar]
- 26.Kasem K, Gopalan V, Salajegheh A, Lu CT, Smith RA, Lam AK: JK1 (FAM134B) gene and colorectal cancer: a pilot study on the gene copy number alterations and correlations with clinicopathological parameters. Exp Mol Pathol 2014;97:31–36. [DOI] [PubMed] [Google Scholar]
- 27.Kang BW, Kim JG, Lee SJ, Chae YS, Jeong JY, Yoon GS, Park SY, Kim HJ, Park JS, Choi GS, Jeong JY: Expression of aquaporin-1, aquaporin-3, and aquaporin-5 correlates with nodal metastasis in colon cancer. Oncology 2015;88:369–376. [DOI] [PubMed] [Google Scholar]
- 28.Kim SA, Inamura K, Yamauchi M, Nishihara R, Mima K, Sukawa Y, Li T, Yasunari M, Morikawa T, Fitzgerald KC, Fuchs CS, Wu K, Chan AT, Zhang X, Ogino S, Qian ZR: Loss of CDH1 (E-cadherin) expression is associated with infiltrative tumour growth and lymph node metastasis. Br J Cancer 2016;114:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertucci F, Salas S, Eysteries S, Nasser V, Finetti P, Ginestier C, Charafe-Jauffret E, Loriod B, Bachelart L, Montfort J, Victorero G, Viret F, Ollendorff V, Fert V, Giovaninni M, Delpero JR, Nguyen C, Viens P, Monges G, Birnbaum D, Houlgatte R: Gene expression profiling of colon cancer by DNA microarrays and correlation with histoclinical parameters. Oncogene 2004;23:1377–1391. [DOI] [PubMed] [Google Scholar]
- 30.Meding S, Balluff B, Elsner M, Schone C, Rauser S, Nitsche U, Maak M, Schafer A, Hauck SM, Ueffing M, Langer R, Hofler H, Friess H, Rosenberg R, Walch A: Tissue-based proteomics reveals FXYD3, S100A11 and GSTM3 as novel markers for regional lymph node metastasis in colon cancer. J Pathol 2012;228:459–470. [DOI] [PubMed] [Google Scholar]
- 31.Lan YT, Yang SH, Lin JK, Lin CC, Wang HS, Chen WS, Lin TC, Jiang JK, Chang SC: Genetic variations are associated with lymph node metastasis in colorectal cancer patients. J Surg Oncol 2014;110:307–312. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe T, Kobunai T, Tanaka T, Ishihara S, Matsuda K, Nagawa H: Gene expression signature and the prediction of lymph node metastasis in colorectal cancer by DNA microarray. Dis Colon Rectum 2009;52:1941–1948. [DOI] [PubMed] [Google Scholar]
- 33.Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P: KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992–3995. [DOI] [PubMed] [Google Scholar]
- 34.Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B: The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]