

Abstract
Polybrominated diphenyl ethers (PBDEs) have been used as flame retardants in household materials. Their environmental persistence has led to continuous human exposure and significant tissue levels. Three PBDE congeners (BDE-47, BDE-100, and BDE-153) have been frequently detected in human serum. Although these compounds appear to possess endocrine disrupting activity, studies are largely missing to determine the biological mechanisms of PBDEs in breast cancer cells. Here, we assessed PBDE bioactivities with three complementary strategies: receptor binding/activity assays; nonbiased RNA-sequencing analysis using an estrogen-dependent breast cancer cell line MCF-7aroERE; and in vivo assessments using patient-derived xenograft (PDX) models of human breast cancer. According to the results from in vitro experiments, the PBDE congeners regulate distinct nuclear receptor signaling pathways. BDE-47 acts as a weak agonist of both estrogen receptor α (ERα) and estrogen-related receptor α (ERRα); it could stimulate proliferation of MCF-7aroERE and induced expression of ER-regulated genes (including cell cycle genes). BDE-153 was found to act as a weak antagonist of ERα. BDE-100 could act as (1) an agonist of aryl hydrocarbon receptor (AhR), inducing expression of CYP1A1 and CYP1B1 and (2) as a very weak agonist/antagonist of ERα. In vivo, a mixture of the three congeners with ratios detected in human serum was tested in an ER+ PDX model. The mixture exhibited estrogenic activity through apoptosis/cell cycle regulation and increased the expression of a proliferation marker, Ki-67. These results advance our understanding of the mechanisms of PBDE exposure in breast cancer cells.
Keywords: breast cancer, PBDE, ER, PR, ERR, AhR, ER+, PDX
Polybrominated diphenyl ethers (PBDEs) have been used as flame retardants in plastics, furniture, and other household products (EPA, 2006). Widespread use of these brominated hydrocarbons began in the United States in the 1970s. Because PBDEs resist degradation, environmental contamination has been observed in the soil, air, and water (McGrath et al., 2017). Although PBDE production and use have been significantly curtailed in the United States, recent epidemiological studies have still detected them with high frequency in human specimens across the globe. These results suggest continued worldwide exposure even today. PBDEs have been associated with a variety of adverse health effects. They include neurodevelopmental, immune, reproductive toxicity, and other health effects. Additionally, PBDEs are considered endocrine disrupting chemicals (EDCs) (EPA, 2006; Gore et al., 2015). Their adverse effects are intensified by their high lipid solubility. Like polychlorinated biphenyls (PCBs), the tissue concentrations of PBDEs increase at higher levels of the food chain (biomagnification) and bioaccumulate in fat tissues in humans, including mammary glands (Pohl et al., 2017).
Although PBDEs were gradually discontinued in the United States from 2004 to 2013, recent studies were motivated by environmental persistence of residual PBDEs and bioaccumulation in the human body (Pohl et al., 2017; Sjodin et al., 2008). According to earlier studies (Guo et al., 2016; Zota et al., 2013), PBDE exposure declined when their use was discontinued. More recently, assessments by Hurley et al. suggest that the decline in exposure may have plateaued; according to their analysis of biomonitoring data from the California Teachers Study, levels of some PBDEs congeners may be even on the rise (ie, mean PBDE concentrations in human blood: BDE-47, 25.56 ng/g lipid; BDE-100, 5.08 ng/g lipid; and BDE-153, 12.03 ng/g lipid) (Hurley et al., 2017; Petreas et al., 2003). Although many biomonitoring studies have been performed on PBDEs, only few have focused on the mechanisms by which PBDEs can interfere with the human endocrine system. Moreover, animal cancer bioassays of BDEs are restricted to deca-BDE (Pohl et al., 2017). Likewise, studies are largely missing in assessing how such exposure may relate to cancer incidence. The nuclear receptor reporter assays used to characterize these endocrine-disrupting activities include estrogen receptor (ER), progesterone receptor (PR), thyroid receptor (TR), androgen receptor (AR), and aryl hydrocarbon receptor (AhR) (Hamers et al., 2006; Kojima et al., 2009; Meerts et al., 2001). These receptors play important roles in various cancer types and are particularly important for endocrine-related cancers. One important and well-recognized instance is the hormone-dependent growth of breast cancer. Approximately 70% of all breast cancers are ER positive; their growth is stimulated by estrogen. Because roughly 85% of breast cancers occur in women who have no family history of breast cancer (Haber et al., 2012), other factors besides genetic background appear to increase the risk of breast cancer, such as exposure to estrogen-like EDCs. Moreover, ER is hypersensitive during menopausal transition, the activity of estrogen-like EDCs potentially could enhance through exposure during this critical window of susceptibility.
Mechanistic studies may further inform whether additional environmental remediation of PBDEs is necessary. Although PBDEs appear to have endocrine-disrupting activities, studies are largely missing to determine the relationship between PBDE exposure and molecular changes in breast cancer cells/tissues. Moreover, different types of PBDEs (ie, congeners) appear to have different biological activities and affect different nuclear receptors. One important yet unmet need is to systematically study how individual PBDE congeners impact genes that are important in breast cancer. Toward this, here we focused on the three most widely detected PBDE congeners: BDE-47, BDE-100, and BDE-153 (Figure 1). PBDEs consist of a central biphenyl ether structure that is functionalized with up to 10 bromine atoms. Each congener varies in terms of the number and position of bromine atoms on the 2 aromatic rings. Although this class of compounds has undergone structure-activity analyses (Yang et al., 2010), the precise relationship between structure and function remains poorly understood. Therefore, we first characterized the effects of these PBDE congeners on 3 important nuclear receptors related to breast cancer: ERα, PR, and estrogen-related receptor (ERRα). For a nonbiased assessment of the complex biological activities of PBDEs, RNA-sequencing (RNA-Seq) analysis was used to identify their gene targets in MCF7aroERE cells. They are an aromatase-transfected ER-positive breast cancer cell line. We validated our in vitro findings in vivo using a clinically relevant, ER+ patient-derived xenograft (PDX) model; we exposed it to a mixture of PBDEs with concentration ratios as detected in human serum. The results provide fundamental insight into the biological activity of three PBDEs, individually or in combination, and lay the foundation for understanding their mechanisms of action in breast cancer cells.
Figure 1.

Effects of PBDEs on ERα. A, Structure of PBDEs. B, To examine the binding of PBDEs to ERα, a ligand competition assay was performed using recombinant ERα protein. Competitive ligand binding to the ERα was detected by displacement of a tracer (fluorescent E2) from ERα; this resulted in a loss of FRET signal between the antibody and the tracer. 10-point titrations ranging from 5 nM to 100 µM were used to generate a dose-response curve for each PBDE congener per assay. All PBDEs showed weak binding to ER. 17β-estradiol (E2) was used as an internal control to determine IC50 values. The 0% displacement control was defined as the well that did not contain E2 in the reaction. The well with the highest concentration of E2 was defined as 100% displacement. Data are expressed as mean ± SD of the mean using duplicate assays. C, Three PBDEs (BDE-47, BDE-100, and BDE-153) were tested in the C4-12 ERα assay for their estrogenic and/or anti-estrogenic properties. Cells were treated with each PBDE for 24 h. The agonist mode had PBDE only; the antagonist mode had PPT (ERα-specific agonist) + PBDE. Activity was measured using a luciferase reporter system. For the agonist mode, luciferase activity of the control (DMSO treatment) was defined as 0. The activity was compared with the positive control, PPT (10 nM), which was defined as 100% for the antagonist mode. Data are expressed as mean ± SD of the mean using triplicate assays. D, Three PBDEs (BDE-47, BDE-100, and BDE-153) were tested in MCF7aroERE for their cell proliferation activity. 5 days after treatment, cell proliferation was measured by MTT assay. E2 (0.5 nM), ICI (100 nM), PPT (10 nM), and MPP (1 μM). Data are expressed as mean ± SD of the mean using triplicate assays.
MATERIALS AND METHODS
Chemicals
BDE-47 [bromine substitution pattern 2,2′,4, 4′], BDE-100 [2,2′,4,4′,6], and BDE-153 [2,2′,4,4′,5,5′] (Figure 1A) were purchased from AccuStandard, Inc. (New Haven, Connecticut). 4,4′,4″’-(4-propyl-1H-pyrazole-1,3,5-triyl)tris[phenol] (PPT) (Tocris Bioscience, Bristol, UK), 17β-estradiol (E2) (Sigma-Aldrich, St. Louis, Missouri), 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride (MPP) (Tocris Bioscience), Mifepristone (RU486) (Tocris Bioscience), ICI 182, 780 (ICI) (Sigma-Aldrich) were purchased. For all chemicals, stock solutions were prepared by dissolving them in dimethyl sulfoxide (DMSO, 99.7%; Sigma-Aldrich).
High-resolution gas chromatography/mass spectrometry
Using commercially available Br-dioxin/furan calibration standards (Cambridge Isotope Laboratories, Inc., Tewksbury, Massachusetts), PBDE standard solutions were further tested for Br-dioxin/furan impurities on gas chromatography-high-resolution mass spectrometry (GC-HRMS) (dual focusing sector, ThermoFisher, Bremen, Germany). The PBDE standard solution in DMSO was exchanged to tetradecane before injection. The analysis was performed at the Environmental Chemistry Laboratory of the California Environmental Protection Agency’s Department of Toxic Substances Control.
Cell culture
AroER Tri-screen cells (Chen et al., 2014), ie, MCF-7aro ERE, were maintained in Eagle’s minimal essential medium (MEM; Genesee Scientific, El Cajon, California) supplemented with 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, California), 2 mM l-glutamine (Fisher Scientific, Chino, California), 1 mM sodium pyruvate (Fisher Scientific, Chino, California), 100 U/ml penicillin-streptomycin (Fisher Scientific, Chino, California), 100 μg/ml of G418 (Omega Scientific, Tarzana, California), and 50 μg/ml hygromycin (Invitrogen, Waltham, Massachusetts). MCF7aro cells (Zhou et al., 1990) were maintained in MEM supplemented with 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin-streptomycin, and 100 μg/ml of G418. C4-12 ERαERE cells (Kanaya et al., 2015) were maintained in phenol red-free high-glucose DMEM supplemented with 10% charcoal-dextran stripped FBS (Omega Scientific, Tarzana, California), 2 mM l-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin-streptomycin, and 100 μg/ml of hygromycin. T47D cells were maintained in RPMI-1640 supplemented with 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, and 100 U/ml penicillin-streptomycin. We obtained ERR reporter cells (Lynch et al., 2018) from Dr Christina Teng at the National Institute of Environmental Health Sciences (NIEHS). The ERR reporter cells were cultured in high-glucose DMEM medium supplemented with 10% FBS, 4 mM l-glutamine, 1 mM sodium pyruvate, and 100 U/ml penicillin-streptomycin. All cell lines were cultured at 37°C with 5% CO2.
Competitive binding analysis of PBDEs to ERα and PR
To measure the binding affinities of BDE-47, BDE-100, and BDE-153 to ERα and PR, we used Invitrogen’s SelectScreen Nuclear Receptor Profiling Service in the Competitive Binding Assay and Coactivator Assay formats, respectively. Ten-point titrations ranging from 5 nM to 100 µM were applied to generate a dose response curve for each PBDE congener per assay. A more detailed protocol can be found on the Invitrogen product website (https://www.thermofisher.com/us/en/home/products-and-services/services/custom-services/screening-and-profiling-services.html. Last accessed March 6, 2019).
Cell proliferation assay
Prior to the assay, MCF7aroERE cells were hormone deprived for 2 days and then seeded at 5 × 103 cells/100 μl in a 96 well plate. Twenty-four hour post-seeding, the cells were treated with varying concentrations of PBDE. After 5 days of treatment, cell proliferation was measured by adding MTT (thiazolyl blue tetrazolium bromide; Sigma-Aldrich) reagent and recording absorbance at 570 nm on a microplate reader.
C4-12 ERα reporter assay
C4-12ERα ERE cells were seeded in tissue culture-treated white 96-well plates (Falcon, Durham, North Carolina) at 5.0 × 104 cells per well. After overnight incubation, cells were treated with varying concentrations of the compounds of interest. Both an ERα-specific agonist (PPT, 10 nM) and ER degrader (ICI, 100 nM) were used as positive and negative controls. After 24 h incubation, the luciferase signal was measured using the One-Glo Luciferase Assay System (Promega Corporation, Madison, Wisconsin). The luciferase signal was read and measured using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, California) and normalized by protein concentration against the DMSO control. C4-12ERα ERE cells are valuable for examining chemicals for ERα-specific activity, but their proliferation does not depend on estrogen (Petrossian et al., 2018).
Construction of pGL4.26 (PRE)2
Two tandem copies of the PRE consensus sequence insert were synthesized, annealed, and subcloned into the pGL4.26 luciferase reporter vector (Promega Corporation, Madison, Wisconsin). The nucleotide sequences of two strands of the PRE insert are 5′-AGAACAAACTGTTCT AGAACAAACTGTTCT-3′ and 5′-AGAACAGTTTGTTCTAGAACAGTTTGTTCT-3′.
The orientation and sequences of the PRE insert were verified by DNA sequencing. The resulting reporter vector was named pGL4.26 (PRE)2.
PR reporter assay
Prior to the assay, T47D cells were cultured for 3 days in phenol red-free RPMI-1640 containing 10% charcoal-dextran-treated FBS. Then they were seeded into 96-well plates at 3.0 × 104 cells/100 μl/well in tissue culture-treated 96-well white assay plates (Falcon). On the following day, pGL4.26-(PRE)2 and Renilla plasmids (QIAGEN, Germantown, Maryland) were transfected into the cells using X-tremeGENE HP Reagent (Roche, Basel, Switzerland), according to manufacturer’s protocol. Twenty-four hour post-transfection, the cells were treated with varying concentrations of the chemicals of interest. Both a PR agonist (R5020) and antagonist (RU486) were used as controls. After 24 h of incubation, the luciferase signal was measured using the Dual-Glo Luciferase System (Promega Corporation) according to the manufacturer’s instructions. The luminescence was read on a SpectraMax M5 microplate reader (Molecular Devices). Firefly luciferase activity was normalized to Renilla luciferase signals.
ERRα reporter assay
ERRα reporter cells were developed to screen ligands of ERRα and were kindly provided by Dr Christine T. Teng (Lynch et al., 2018). ERRα reporter cells were suspended in culture medium without antibiotics and were dispensed at 4.0 × 104 cells/100 μl/well in tissue culture-treated 96-well white assay plates (Falcon). After the cells were incubated for 24 h, they were treated with the compounds at varying concentrations. The assay plates were incubated at 37°C for 24 h; then luciferase reagent from the One-Glo Luciferase Assay System (Promega Corporation) was added following the manufacturer’s protocol. The luminescence was quantified using a SpectraMaxM5 plate reader (Molecular Devices) and normalized by protein concentration against the DMSO control.
RNA extraction and quantitative PCR (qPCR)
Total RNA was extracted from cultured cells using RNeasy Mini kit (QIAGEN, Germantown, Maryland) and then used to synthesize complementary DNA (cDNA) with SuperScript IV VILO Master Mix (Invitrogen). qPCR was performed using CFX Connect Real-Time System (Bio-Rad, Hercules, California) with PerfeCTa SYBR Green FastMix (Quantabio, Beverly, Massachusetts). The primers used in the qPCR are shown in Supplementary Table 1 (Kanaya et al., 2019). The target gene messenger RNA (mRNA) level was normalized to GAPDH expression.
In vivo mouse studies
Surgical resections (2 × 2 mm2) from consented ER+ breast cancer patients were orthotopically engrafted into the mammary fat pad of 6- to 8-week-old female NOD-scid/IL2Rγ-/- (NSG) mice to derive parental tumors. COH-SC31 is a ER+ tumor model in which estrogen-mediated ER activation is the major driving force of growth (Hsu et al., 2018). For the in vivo mouse studies of PBDEs, tumor slices (2–3 mm) from the parental tumors were engrafted into the intact NSG mice. Once tumors were established to be 100–200 mm3 in size, mice were randomized and then orally treated for 1 week with either control (DMSO) or PBDEs (n = 3 for each group). PBDEs are present at high levels in diets, air, and house dust, suggesting the main human exposure route is oral/dermal. Therefore, PBDEs were added to the diet to mimic human exposure. Diets were designed as soy-free and generated as pellets (matches macronutrient composition of Teklad 2018) by Research Diets, Inc. (New Brunswick, New Jersey). PBDE concentration was decided based on our preliminary dosage testing experiments (below).
For preliminary dosage testing experiments, mice were fed the diet with three different dosages (high, medium, and low: Supplementary Table 3) of three PBDE congeners (BDE47, BDE-100, and BDE-153). Blood was collected to measure levels of each PBDEs after 1 week of oral exposure (Kanaya et al., 2019). The PBDE serum levels were measured using triplicate samples from each treatment group.
The City of Hope Institutional Review Board approved this study and all patients provided written informed consent prior to tissue collection. All animal experiments were done under a protocol approved by the Institutional Animal Care and Use Committee. Facilities are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and operated according to NIH guidelines.
PBDE measurement using mouse serum
Single native PBDE standards and 13C-labeled internal standards were purchased from Wellington Laboratories Inc. (Guelph, Ontario, Canada). The recovery standard, 13C12-labeled PCB-209, used to calculate the 13C-labeled internal standard recoveries, was purchased from Cambridge Isotope Laboratories, Inc. Oasis HLB columns (Waters Corp, Milford, Massachusetts), silica gel (Sigma Aldrich, St. Louis, Missouri), sodium sulfate anhydrous (EMD, Billerica, Massachusetts), and sulfuric acid (EMD) were used for serum extraction and clean-up. Bovine serum (Thermo Scientific HyClone Fetal Bovine Serum) was used for method blanks and internal QC samples to test method precision. NIST Standard Reference Material “Organic Contaminants in Fortified Human Serum” (SRM 1958, National Institute of Standards and Technology) and Arctic Monitoring and Assessment Programme (AMAP) samples were used to test method accuracy.
Preparation, extraction, and instrumental analysis of serum samples were conducted following the method used in a previous study (Guo et al., 2016). Briefly, the serums were transferred to DTSC laboratory in frozen state and stored in –20°C freezer until analysis. Serum samples were thawed completely and then mixed by vortex (5–10 s at 2500 rpm). Between 350 and 450 µl of serum were pipetted into prelabeled 16 × 100 mm test tubes with Teflon-lined screw caps. A total of 75 µl sera from each sample was aliquoted and sent to Boston Children’s Hospital for total cholesterol and triglycerides measurements. Total lipid content was calculated based on the total cholesterol and triglycerides by using Phillips’ formula. Each serum sample was spiked with a panel of 13C12 labeled PBDE surrogate standard, denatured with formic acid, extracted using Waters Oasis HLB SPE cartridges, and cleaned up with 33% sulfuric acid silica using the Biotage RapidTrace system. The extracts were concentrated in a Biotage TurboVap LV system. After the addition of a recovery standard (13C12-PCB209) the samples were analyzed by GC/HRMS. In-house laboratory control sample (prespiked bovine serum with known amount of target PBDE) and SRM 1958 were used as QC samples.
Analysis of three PBDE congeners, BDE-47, BDE-100, and BDE-153, was performed on a Thermo Scientific DFS Magnetic Sector Gas Chromatography High-Resolution Mass Spectrometer. The analysis was performed on a 15-m DB-5ms column (Agilent Technologies, Inc.). The GC oven program was initiated at 130°C, held for 2 min, ramped at 15°C/min to 200°C, ramped at 5°C/min to 280°C, ramped at 10°C/min to 320°C, and finally held for 6 min. The transfer line temperature is 280°C. A resolution setting of 10 000 was used for the analysis. Sample concentrations were calculated from external calibration standard’s average response factors. TargetQuan (version 3.2, Thermo Scientific) was used for sample quantitation.
The recoveries of BDE-47, BDE-100, and BDE-153 in SRMs were 99 ± 10%, 81 ± 6%, and 101 ± 5%, respectively. The bovine serum matrix spike showed similar recoveries: BDE-47 = 99 ± 10%; BDE-100 = 90 ± 1%; and BDE-153 = 101 ± 3%.
RNA-Seq library preparation and sequencing on the illumina Hiseq2500
MCF7aroERE cells were treated for 24 h with the one of following: DMSO, PPT (10 nM), PPT (10 nM) + MPP (1 μM), BDE-47 (20 μM), BDE-100 (20 μM) + PPT (10 nM), or BDE-153 (20 μM) + PPT (10 nM). For the in vivo analysis, mice implanted with COH-SC31 PDX tumors (Hsu et al., 2018) were treated for 7 days with either control diet or a diet that contained a mixture of the 3 PBDEs. Total RNA was extracted from cultured cells or tumors using RNeasy Mini kit. From 250 ng of total RNA from each sample (all with RIN > 8), RNA-Seq libraries were prepared with the Kapa Stranded mRNA-Seq kit (Kapa Biosystems, Cat KK8421) according to the manufacturer’s protocol. The sequencing libraries were validated with the Agilent Bioanalyzer by running DNA high-sensitivity Chip and quantified with Qubit. Sequencing of 51 bp reads was performed in the single read mode on an Illumina HiSeq 2500 with HiSeq SBS V4 Kits (Illumina, FC-401-4002). Real-time analysis (RTA) 2.2.38 software was used to process the image analysis and base calling was performed using bcl2fastq (version2-2.18).
Processing of RNA-Seq data and differential expression analysis
Reads were aligned against the human genome (hg19) using TopHat2 (Kim et al., 2013). Read counts were tabulated using htseq-count (Anders et al., 2015), with UCSC known gene annotations (TxDb.Hsapiens.UCSC.hg19.knownGene, Hsu et al., 2006). Fold-change values were calculated from Fragments Per Kilobase per Million reads (FPKM, Mortazavi et al., 2008) normalized expression values, which were also used for visualization (following a log2 transformation). Aligned reads were counted using GenomicRanges (Lawrence et al., 2013). Log2 (FPKM + 0.1) expression values were also used for creating the principal component analysis (PCA) (using R-3.3 base functions). False discovery rate (FDR) values were calculated using the method of Benjamini and Hochberg (Benjamini and Hochberg, 1995), based upon the p-value distribution (using the method specific to each experiment, as described below). Prior to p-value calculation, genes were filtered to only include transcripts with an FPKM expression level of 0.1 (after a rounded log2 transformation) in at least 50% of samples (Warden et al., 2013) as well as genes greater than 150 bp in size. For the separate PBDE treatments, p-values were calculated from raw counts using edgeR, and genes were defined as differentially expressed if they had a |fold-change| > 1.5 and FDR < 0.05. For the PDX combined PBDE treatments, p-values were calculated from raw counts using DESeq2 (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8. Last accessed March 6, 2019), and genes were defined as differentially expressed if they had a |fold-change| > 1.2 and FDR < 0.05. Heatmaps for differentially expressed genes were creating using heatmap.3.R (https://github.com/obigriffith/biostar-tutorials/blob/master/Heatmaps/heatmap.3.R. Last accessed March 6, 2019). Raw data have been deposited in NCBI Gene Expression Omnibus with an accession number of GSE119588.
Ingenuity pathway analysis
Ingenuity pathway analysis (IPA, Ingenuity Systems, www.ingenuity.com. Last accessed March 6, 2019) was used to identify the biological functions, pathways, and mechanisms for the filtered genes (fold change > 1.5, FDR < 0.05).
Gene set enrichment analysis
Estrogen-upregulated gene set (Frasor et al., 2003) was used for enrichment analysis. Two types of gene set enrichment analysis (GSEA) were performed for the PDX experiment. (1) GSEA (Subramanian et al., 2005) was run to compare the PBDE samples versus the control DMSO samples. A gene set permutation (instead of a sample permutation) was used. The minimum number of genes in a gene set was set to 3, and the random seed was set to 0. Otherwise, default parameters were used for this analysis. (2) Single-sample GSEA (ssGSEA, as implemented in GenePattern [Reich et al., 2006]) was used to calculate scores for each sample. Scores were then exported, with ANOVA p-values calculated in R using the aov function. Box plots with jittered points also were created in R.
Statistical analysis
For reporter assays and mRNA analysis of PBDE mixture, t tests were performed of each treatment compared with control. For the luciferase assays cotreated with XCT790 and mRNA expression analysis and cell proliferation assay, 1-way ANOVA analysis of variance with post hoc Dunnett’s analysis was performed. For the PR mRNA expression levels cotreated with ICI data, t tests were performed of each treatment compared with DMSO, and with ICI and without ICI treatment. The statistical significance was set at p values of .01 (**) and .05 (*).
RESULTS
Purity Analysis of Commercial PBDEs
PBDEs were purchased from AccuStandard, Inc. (New Haven, Connecticut). These PBDE products were indicated as being 100% pure and free from bromodioxins/furans. Nevertheless, we further tested all PBDE solutions for bromodioxin/furan impurities. None of the three PBDE solutions contained bromodioxins/furans (Supplementary Table 3 [Kanaya et al., 2019]). An important example of this class of compounds is the most potent known AhR receptor agonist, 2,3,7,8-tetrabromodioxin. We did not detect this compound or bromodioxins/furans in any of the three PBDE solutions.
Effects of BDE-47, BDE-100, and BDE-153 on ERα Through Cell-Based Assays
We assessed binding between ERα and each of the PBDE congeners: BDE-47, BDE-100, and BDE-153. Binding was determined using a ligand competition assay with recombinant ERα protein. All three PBDEs bound to ERα at the following IC50 values (from most to least potent): BDE-153 = 12.6 μM, BDE-100 = 50.6 μM, and BDE-47 > 100 μM (Figure 1B). We further evaluated the functional impact of each PBDE on ER’s transcriptional regulation. Using a luciferase reporter system, each PBDE was tested in a C4-12ERα ERE assay for estrogenic and/or anti-estrogenic properties. The C4-12ERα ERE cells were established in our laboratory from C4-12 cells, derived from MCF-7 breast cancer cells, that are deficient in endogenous ER expression; we previously confirmed that the reporter signal is highly specific for ERα (not ERβ) (Kanaya et al., 2015). In the agonist mode of this assay, cells were treated only with the individual PBDE (no cotreatment). In this mode BDE-47 and BDE-100 exhibited luciferase induction in a dose-dependent manner (Figure 1C). In the antagonist mode, cells were treated with one PBDE plus an ERα-specific agonist (ie, PPT). BDE-100 and BDE-153 exhibited dose-dependent inhibition of PPT-induced luciferase activity (Figure 1C). Although BDE-47 had the weakest binding affinity for ERα, it was the most effective agonist among the three PBDEs. Additionally, a cell proliferation analysis was performed to evaluate potential PBDE-mediated biological effects on ER+ breast cancer cells; MCF7aroERE cells were treated with each PBDE and then an MTT assay was performed. BDE-47 and BDE-100 increased the cell proliferation in a dose dependent manner. BDE-47- and BDE-100-induced cell proliferation was inhibited by ICI (an ER degrader), suggesting that the increased cell proliferation was ERα dependent (Figure 1D).
Effects of BDE-47, BDE-100, and BDE-153 on PR
Although PR is important in breast cancer development, very few reports describe how PR is affected by EDCs. Here, we evaluated whether each PBDE congener acts as an agonist or antagonist of PR. Because PR-binding assay systems are not currently available, we tested PBDE activities with PR coregulator assays. The results indicate that only BDE-153 inhibited the coactivator recruitment to PR. The inhibitory effects of BDE-153 were approximately 700 times weaker than the known PR antagonist RU486 (Figure 2A).
Figure 2.

Effects of PBDEs on PR. A, A PR co-regulator assay was performed to test the functional ability of a compound to affect co-activator recruitment. BDE-153 inhibited the co-activator recruitment of progesterone (a PR agonist). Progesterone was used as a positive control for the agonist assay and Mifepristone (a PR antagonist) was used for the antagonist assay. For the agonist mode, 0% activation was defined as the DMSO-treated well; the highest concentration of progesterone was defined as 100% activation. In the antagonist mode, the DMSO-treated well was used to determine 100% inhibition. Data are expressed as mean ± SD of the mean using duplicate assays. B, Three PBDEs (BDE-47, BDE-100, and BDE-153) were tested in T47D PR assay for their progesterone and/or antiprogesterone-like properties. Cells were treated with each PBDE for 24 h (agonist mode: PBDE only and antagonist mode: R5020 [PR-specific agonist] + PBDE) and activity was measured using luciferase reporter system. For the agonist mode, luciferase activity of the control (DMSO treatment) was defined as 0. The efficacies were compared with the positive control, R5020 (50 nM) as 100% for antagonist mode. Data are expressed as mean ± SD of the mean using duplicate assays.
Commercially available PR reporter systems currently do not attain high signal induction and low background. To address this, we developed a PR activity assay; it comprises a luciferase system that is transiently transfected into T47D cells. This breast cancer cell line is often used to examine PR function because of its robust expression levels of endogenous PR. We constructed a PRE-luciferase reporter that achieves both low luciferase background and high reporter induction by using the pGL4.26 vector reporter (Chen et al., 2014). To assess the sensitivity and specificity of the assay, cells were first transiently transfected with pGL4.26 (PRE)2 plasmid; then they were treated with either R5020 (a PR-specific agonist) or a combination of R5020 and RU486 (a PR-specific antagonist). The agonist R5020 treatment produced significant luciferase induction with approximately 20 times increased signal compared with baseline. R5020-induced activity was inhibited back down to baseline by RU486 (Supplementary Figure 1 [Kanaya et al., 2019]). The results confirm the robustness of this PR reporter system. The system was then used to evaluate the three PBDEs. In the agonist mode of this assay, cells were treated only with the individual PBDEs (no cotreatment). In the antagonist mode, cells were treated with one PBDE plus a PR-specific agonist (R5020). No significant effect was observed in the cells treated with any of the congeners in the agonist and antagonist modes (Figure 2B).
Effects of BDE-47, BDE-100, and BDE-153 on ERRα
The estrogen-related receptor, ERRα, binds to an extended estrogen response element (ERE) half site. However, estrogen does not bind to this orphan nuclear receptor. In breast cancer, increased ERRα gene expression is associated with poor prognosis (Park et al., 2016). Each PBDE congener was tested for its effects on ERRα transcriptional activity using ERRα cells (Lynch et al., 2018). BDE-47 exhibited luciferase induction in a dose-dependent manner. In contrast, both BDE-100 and BDE-153 had no effect (Figure 3A). To confirm that the BDE-47-induced luciferase activity was through the ERRα pathway, cells were treated with BDE-47 and an ERRα-specific inverse agonist called XCT (Busch et al., 2004); XCT treatment inhibited BDE-47-induced activity (Figure 3B). These results demonstrate that BDE-47 has an ERRα agonistic activity.
Figure 3.

Effects of PBDEs on ERRα. A, Three PBDEs (BDE-47, BDE-100, and BDE-153) were tested in the ERRα assay. Cells were treated with each PBDE for 24 h and activity was measured using a luciferase reporter system. The efficacies were compared with the positive control genistein (10 μM), which was defined as 100% activity. Data are expressed as mean ± SD of the mean using triplicate assays. BDE-47 exhibited luciferase induction in a dose-dependent manner, but BDE-100 and BDE-153 did not. B, To confirm ERRα activation of BDE-47 (20 μM), it was co-administered in the ERRα cells with XCT790 (20 μM), which is a known inverse agonist of ERRα. The luminescence was quantified, normalized by protein concentration, and shown as relative luciferase unit (RLU). BDE-47-induced ERRα activity was significantly decreased by 10 μM of XCT790.
RNA-Seq Analysis as a Non-Biased Measure of the Biological Activities of PBDEs
Our results and those of others (Hamers et al., 2006; Meerts et al., 2001) demonstrate that PBDEs interact with multiple nuclear receptors to regulate their pathways. RNA-Seq analysis was performed to identify additional genes targeted by PBDEs in an estrogen-dependent breast cancer cell line, MCF7aroERE. Considering the importance of ERα in breast cancer development, the experimental design was based on our ERα reporter assay results: BDE-47 was estrogenic; BDE-100 was estrogenic in the agonist mode but antiestrogenic in the presence of PPT, an ERα-specific agonist; BDE-153 had antiestrogenic activity. When the 3 PBDEs were assessed using RNA-Seq, we chose to analyze BDE-100 in the antagonist mode. Although previously reported results indicate BDE-100 has estrogenic activity (Meerts et al., 2001), to the best of our knowledge its antiestrogenic activity has not been reported; thus our analysis was focused on the antiestrogenic activity to reveal novel functions. BDE-153 was also examined in the antagonist mode because it was found to be an ERα antagonist through the reporter assay. Thus, RNA-Seq analysis was performed on 6 treatment groups: control (DMSO), PPT, PPT+MPP, BDE-47, BDE-100+PPT, and BDE-153+PPT. PPT and MPP were used as controls for ER agonist and ER antagonist gene signatures, respectively. To ensure that each treatment produced definitive changes in RNA expression, MCF7aroERE cells were treated with each PBDE at a concentration of 20 μM; the concentration was chosen because it produced significant signal for ERα reporter assay.
The effects of the 6 treatments were compared by PCA to visually assess global differences between the sample groups. As expected, expression patterns for PPT (ER agonist) were different from the vehicle control; this difference was not seen in PPT+MPP (ER agonist + antagonist) compared with the vehicle control because the agonist effects on gene expression were counteracted by the antagonist effects. A different gene expression pattern was observed with BDE-47 compared with the vehicle control; not surprisingly the difference was less pronounced than PPT versus the vehicle control. These results suggest that BDE-47 acts as a weak agonist of ERα. The BDE-100 and BDE-153 antagonist treatments exhibited similar expression patterns to PPT alone. Their ERα antagonistic effects appear to be weak (Figure 4A).
Figure 4.

RNA-Sequencing (RNA-Seq) analysis to identify the genes targeted by PBDEs in MCF7aroERE cells. RNA-Seq was performed on MCF7aroERE cells that had been treated with PBDEs for 24 h. A, Principal component analysis (PCA) indicating 6 treatment groups in triplicates: control (DMSO), BDE-47, PPT, PPT + MPP, BDE-100 + PPT, and BDE-153+PPT. PPT is an ERα agonist; MPP is an ERα antagonist. B, Unsupervised hierarchical clustering of samples (triplicate, 1–3) using all genes of the RNA-Seq. C, The top canonical pathway (cell cycle control of chromosomal replication) in BDE-47-treatred cells. D, mRNA expression levels of selected genes were evaluated using qPCR analysis in cells treated with each PBDE for 24 h. Selected genes were the ER-regulated genes PGR, PDZK1, and PS2; and the ER-regulated cell cycle genes AURKA, E2F1, CCNB1, and FOXM1. Each gene was expressed relative to the levels of GAPDH. Each bar represents the mean ± standard deviation (n = 3). E, PGR mRNA expression levels were measured using qPCR analysis in the cells treated with each PBDEs (20 μM) with or without ICI 182 780 (ICI at 100 nM). Each bar represents the mean ± standard deviation (n = 3). *p < .05 between each treatment with ICI versus without ICI.
Figure 4.

Continued.
Figure 4.

Continued.
Clustering of differentially expressed genes were visualized using heatmaps (Figure 4B). Our comparison between BDE-47 and PPT revealed similar expression profiles. The results suggest that the main activity of BDE-47 may be driven by its ER agonistic effects. In contrast, the BDE-100 heatmap exhibited 2 clusters: ER antagonistic (yellow) and other (white). The expression patterns of genes in the yellow cluster were different from those regulated by PPT; however, they exhibited a similar pattern to those regulated by PPT plus an ERα antagonist (ie, PPT+MPP). These results suggest that BDE-100 can act as a weak ERα antagonist and are consistent with findings from the luciferase reporter assays (Figure 1D). In addition, we identified unique gene sets that were not affected by PPT but specifically regulated by BDE-100 (white). These results suggest that BDE-100 has other effects in addition to its ERα antagonistic activity. Likewise, the BDE-153 heatmap exhibited 2 clusters. Because gene expression clusters of BDE-100 and BDE-153 were not identical (Figure 4B), there are likely unique genes regulated by BDE-100 and BDE-153, respectively. To identify the key pathways regulated by each PBDE, IPA was performed using the filtered genes (fold change > 1.5, FDR < 0.05, called as differentially expressed genes) (Tables 1 and 2). IPA generated a Tox list using differentiated genes to understand molecular toxicity. Canonical pathway analysis was used to predict the most significant pathways affected based on gene expression. For BDE-47, the most significantly upregulated pathways were cell cycle and DNA damage. Relative to the effects of PPT alone, the following fold changes in gene expression were observed for the top 3 genes of BDE-100 and BDE-153 treatment groups, respectively: (1) PPT+BDE-100—CYP1A1 = +16.1, CYP1B1 = +6.2, BFSP2 = +3.5; (2) PPT+BDE-153—CYP1A1 = +3.8, EGR1 = +3.3, and CYP1B1 = +2.6 (ie, BDE-100 upregulated them in AhR signaling, and BDE-153 did so in a cytochrome P450 panel). Interestingly, BDE-100 downregulated genes associated with immune functions related to the pattern recognition of bacteria and viruses. BDE-153 downregulated immune pathways such as interferon signaling (Table 2).
Table 1.
Number of Differentially Expressed Genes
| Comparison | Upregulated | Downregulated |
|---|---|---|
| PPT versus control | 968 | 1570 |
| PPT+MPP versus control | 299 | 336 |
| BDE-47 versus control | 796 | 523 |
| BDE-100 + PPT versus control | 979 | 1360 |
| BDE-153 + PPT versus control | 1038 | 1511 |
| BDE-100 + PPT versus PPT | 91 | 54 |
| BDE-153 + PPT versus PPT | 78 | 72 |
Table 2.
Summary of Ingenuity Pathway Analysis
| p-Value | Overlap | ||
|---|---|---|---|
| Tox list | |||
| BDE-47 versus control | |||
| Cell cycle: G2/M DNA damage checkpoint regulation | 2.82E-09 | 42% | 21/50 |
| BDE-100 + PPT versus PPT | |||
| Aryl hydrocarbon receptor signaling | 2.11E-05 | 7% | 8/122 |
| BDE-153 + PPT versus PPT | |||
| Cytochrome P450 panel | 5.08E-03 | 18% | 2/11 |
| Canonical pathway | |||
| BDE-47 versus control | |||
| Cell cycle control of chromosomal replication | 2.49E-19 | 71% | 27/38 |
| Role of BRCA1 in DNA damage response | 1.86E-13 | 42% | 32/76 |
| BDE-100 + PPT versus PPT | |||
| Role of pattern recognition receptors in recognition of bacteria and viruses | 2.82E-06 | 8% | 8/93 |
| Interferon signaling | 1.01E-05 | 16% | 5/31 |
| BDE-153 + PPT versus PPT | |||
| Interferon signaling | 5.18E-07 | 19% | 6/31 |
| Role of pattern recognition receptors in recognition of bacteria and viruses | 3.24E-04 | 7% | 6/93 |
Cell Cycle Regulation of BDE-47
Based on the RNA-Seq data, BDE-47 appears to upregulate genes in a cell-cycle-controlled pathway. According to the IPA network analysis (Figure 4C), multiple genes were upregulated in cell cycle control of chromosomal replication. The genes are involved in the G1-S transition; their upregulation indicates rapid replication of DNA, which results in increased cell proliferation. Using a proliferation assay, we confirmed that BDE-47 can increase cell proliferation (Figure 1D). Additionally, we used qPCR to further assess how cell cycle control is affected by the potential estrogenic effects of BDE-47. qPCR was performed on BDE-47-treated cells and used to measure expression of ER-regulated cell cycle genes (eg, Petrossian et al., 2018). Compared with the control (DMSO), BDE-47 increased the expression of 4 selected cell cycle-related genes: AURKA, E2F1, CCNB1, and FOXM1. It also increased other known ER-regulated genes: PGR, PDZK1, and PS2 (Figure 4D). A validation experiment was performed by combining BDE-47 with ICI. ICI decreased the BDE-47-induced expression of PGR. Thus, the effects of BDE-47 appear to involve ER regulation (Figure 4E). These results confirm that BDE-47 acts like an estrogen-mimic and increases cell proliferation through cell cycle regulation.
Effects of BDE-100 and BDE-153 on AhR
As demonstrated by RNA-Seq analyses, BDE-100 and BDE-153 can (1) upregulate 2 key genes, CYP1A1 and CYP1B1 and (2) affect AhR signaling. We examined how each of the 2 PBDEs individually affected expression of CYP1A1 and CYP1B1. Using qPCR, changes in expression were assessed after MCF7aroERE cells were treated with either BDE-100 or BDE-153 (without PPT). Both CYP1A1 and CYP1B1 were significantly induced by BDE-100 and BDE-153. However, the magnitude of induction by BDE-153 was approximately 5 times lower than BDE-100 (Figure 5A). To validate the findings, we measured the enzymatic activity of CYP1A1: MCF7aroERE cells were treated with an individual congener for 24 h; then enzymatic activity was measured with a P450-Glo CYP1A1 Assay System. Although no significant activity was observed for BDE-47 and BDE-153, the results confirmed that BDE-100 treatment increased CYP1A1 activity significantly (Supplementary Figure 2 [Kanaya et al., 2019]).
Figure 5.

Effects of PBDE on the expression of ER and AhR targets genes. A, mRNA expression levels of selected genes (the AhR-regulated genes CYP1A1 and CYP1B1) were evaluated using qPCR analysis in cells treated with each PBDE for 24 h. Each gene was expressed relative to the levels of β-actin. Each bar represents the mean ± standard deviation (n = 3). B, Interaction network generated by IPA of the selected genes changed in the groups treated with BDE-100 + PPT or BDE-153+ PPT compared with PPT. Node color indicates upregulated genes (red) and downregulated genes (green). Node shape represents function: diamond = enzyme; square = growth factor; double circle = group/complex; oval = transmembrane receptor; trapezoid = transporter; circle = other. Edges (lines and arrows between nodes) represent direct (solid lines) and indirect (dashed lines) interactions between molecules as supported by information in the Ingenuity knowledge base.
To understand the significant biological networks of BDE-100 and BDE-153, IPA network analysis was performed using differentially expressed genes in our analysis (Figure 5B). Network analysis showed that BDE-100 interacts with AhR and TR based on the expression of the genes in the network. These two receptors formed a major hub in the main network. For BDE-153, ER and TR were identified as hubs to interact with other genes. Interestingly, both BDE-100 and BDE-153 downregulated interferon networks; the most downregulated gene was the Interferon-simulated gene (IFIT1). Compared with PPT alone, PPT+BDE-100 downregulated IFIT1 by −3.5; PPT+BDE-153 downregulated IFIT1 by −4.0. Using IPA upstream regulator analysis, we identified the cascade of transcriptional regulators that can potentially explain these observed changes in gene expression. Activation z-score was calculated based on relationships in the molecular network that (1) represent experimentally observed gene expression and (2) are associated with a published literature-derived regulation direction which can be either activating or inhibiting. We identified the top 3 upstream regulators for BDE-100. (1) Interferon alpha (IFNA2) had activation z-scores of −4.1 (BDE-100) and −4.5 (BDE-153). (2) Prolactin (PRL) had activation z-scores of −3.9 (BDE-100) and −3.4 (BDE-153). (3) Interferon lambda1 (IFNL1) had activation z-scores of −3.9 (BDE-100) and −4.2 (BDE-153). Taken altogether, both BDE-100 and BDE-153 appear to have immune suppression effects through interferon regulation.
Effects of a PBDE Mixture
To assess how a mixture of three PBDEs affects ER and AhR, we treated the MCF7aroERE cells with a combination of BDE-47, BDE-100, and BDE-153. We chose a ratio for the 3 PBDEs that mimics the ratio observed in serum of women from the California Teachers Study (Hurley et al., 2017): The median concentration of BDE-47 was 13.34 ng/g lipid; BDE-100 was 2.31 ng/g lipid; and BDE-153 was 4.89 ng/g lipid. To replicate these proportions, the ratio of the mixture we used was 1 BDE-47:0.2 BDE-100:0.4 BDE-153. This ratio was maintained as we varied the concentrations of the mixture. At the highest concentration, the mixture comprised 10 µM of BDE-47, 2 µM of BDE-100, and 4 µM of BDE-153. In the agonist mode, the mixture caused induction of 2 ER-regulated genes, PR and PDZK1. These results suggest that the mixture primarily elicits estrogenic effects. The mixture also showed significant AhR agonistic effects as determined by CYP1A1 induction; most of this activity can be attributed to BDE-100 (Figure 6A). Moreover, mixture increased cell proliferation of MCF7aroERE and ICI treatment reversed the effects (Figure 6B).
Figure 6.

Effects of PBDE mixtures. A, mRNA expression levels of the ER and AhR-regulated genes were measured using qPCR analysis in cells treated with mixture of three PBDEs with the ratio observed in the human serum: BDE-47 = 20 μM, BDE-100 = 4 μM, and BDE-153 = 8 μM. B, Mixture was tested in MCF7aroERE for their cell proliferation activity. After 5 days of treatment, cell proliferation was measured by MTT assay. E2 (0.5 nM) and ICI (100 nM). Data are expressed as mean ± SD of the mean using triplicate assays.
Lipid-Adjusted Steady-State Concentrations of PBDEs in Mice
In order to design a mouse diet for in vivo study to evaluate the effects of PBDE on PDX tumor model, preliminary study was conducted to reproduce physiologically relevant ratios of PBDEs. The majority of in vivo studies on PBDE exposure in rodents have involved slightly higher dosage regimens than environmentally relevant (Dunnick et al., 2018; Stoker et al., 2004; Zhou et al., 2002); for example, in some in vivo studies (Ceccatelli et al., 2006; Costa et al., 2015; Liu et al., 2011; McIntyre et al., 2015) the dose of BDE-47 was equal to or greater than 1 mg/kg. Based on a toxicokinetics study of BDE-47 (Staskal et al., 2005), a mouse diet was designed to comprise three different dosages of BDE-47: low = 0.1 mg/kg, medium = 1 mg/kg, and high = 10 mg/kg. In human serum, a typically observed ratio for the congeners is 1 BDE-47:0.2 BDE-100:0.4 BDE-153 (Hurley et al., 2017); this ratio was applied to BDE-47 (low−high) to calculate corresponding mouse dosages for BDE-100 and BDE-153 (Supplementary Table 3) (Kanaya et al., 2019). Food intake averages for the different treatment groups were 3.5 ± 0.5 g (control), 3.4 ± 0.5 g (low), 3.2 ± 0.3 g (medium), and 3.5 ± 0.7 g (high). Thus, no significant differences in intake were observed between the groups. At the end of the dosing regimen, blood was drawn and serum was used to measure PBDE levels. Using data from the 3 dosage groups (low−high), a linear regression model was generated. The results suggest that mouse sera concentrations (ng/ml) correlated with administered dosages (mg/kg bw/day, Supplementary Figure 3) (Kanaya et al., 2019). Based on linear regression model, diet was modified to generate the ratio similar to human exposure and experimental diet was generated (BDE-47 = 1 mg/kg bw/day, BDE-100 = 0.056 mg/kg bw/day, and BDE-153 = 0.126 mg/kg bw/day [Supplementary Table 4] [Kanaya et al., 2019]). For levels of exposure, in order to correlate molecular characteristics to a definitive PDX changes, we decided to use BDE-47 = 1 mg/kg bw/day (medium), BDE-100 = 0.056 mg/kg bw/day, and BDE-153 = 0.126 mg/kg bw/day (Supplementary Table 5). This diet resulted in generating approximately 10–30 times (BDE-47; 32 times, BDE-100; 9.6 times, and BDE-153; 25 times) higher PBDE serum levels in our experimental mice (BDE-47; 24336 ng/g lipid, BDE-100; 1790 ng/g lipid, and BDE-153; 9479 ng/g lipid) (Supplementary Table 5) (Kanaya et al., 2019) compared with the highest human exposure levels (BDE-47; 749 ng/g lipid, BDE-100; 186 ng/g lipid, and BDE-153; 379 ng/g lipid) (Hurley et al., 2017).
PBDE Effects on ER+ PDX Model
The most common type of breast cancer is ER+ and is stimulated by estrogen. ER+ breast tumors can be robustly modeled using PDXs. In these biologically relevant systems, characteristics of human cancer tumors are recapitulated by engrafting mice with fresh tumors from patients. For example, COH-SC31 is an ER+ tumor model in which estrogen-mediated ER activation is the major driving force of growth (Hsu et al., 2018). Here, we used COH-SC31 to further assess the estrogenic activity of the PBDE mixture in vivo. Mice harboring COH-SC31 were treated with PBDE mixtures via diet. After 1 week of treatment, both the body weight and food intake of PBDE-exposed mice were similar to control mice.
At the end of study, PDX tumors were collected, RNA was extracted, and RNA-Seq was performed to assess potential estrogenic activity of the PBDE mixture. Compared with tumors from control diet (DMSO)-fed mice, PBDE treatment upregulated 234 genes and downregulated 227 genes (fold change > 1.2, FDR < 0.05, p-value < .05, referred to as differentially expressed genes) in tumors. Agreeing with our in vitro results in MCF7aroERE cells, we observed significant upregulation of the same estrogen-regulated genes in vivo: PDZK1 = +1.6, AURKA = +1.3, CCNB1 = +1.3, and FOXM1 = +1.3. Results from heatmap analysis demonstrated (1) clear differences between PBDE and control and (2) definitive changes to gene expression patterns in ER+ tumors exposed to PBDEs (Figure 7A). To identify the key pathways regulated by the PBDEs, IPA was performed using the differentially expressed genes. Top network pathway showed that PBDE exposure upregulated the ER pathway. IPA upstream regulator analysis was used to identify any transcriptional regulators potentially associated with the observed gene expression changes. Results based on the prolife of the ER-regulated gene set suggested that ESR1 activity was increased (activation z-score = 4.543, p-value of overlap = 6.85E-11). We then compared the in vivo PBDE-specific gene set to published data on estrogen upregulated gene expression for biological networks and pathways in human breast cancer cells (Frasor et al., 2003). We tested whether the PBDE mixture increased estrogen-regulated genes in 4 important biological networks: (1) estrogen regulated-cell cycle and apoptosis; (2) growth factors, cytokines, and hormones; (3) signal transduction proteins; and (4) transcription factors and transcriptional coregulators (Frasor et al., 2003). Among these biological networks, in vivo administration of the PBDE mixture appears to most upregulate estrogen regulated-cell cycle and apoptosis pathways (Figure 7B). These in vivo results are consistent with our results in vitro. Additionally, the proliferation marker Ki-67 was significantly upregulated in PBDE-exposed tumors (fold change +1.4, p-value 9.18E-05) (Figure 7C). To the best of our knowledge, this is the first report demonstrating estrogenic activity of PBDE mixtures in vivo using a clinically relevant ER+ PDX model and promotion of tumor growth as indicated by the increase of Ki-67.
Figure 7.

RNA-Seq analysis to identify the genes targeted by PBDEs in COH-SC31 PDX. A, Unsupervised hierarchical clustering of samples (triplicate, 1–3) using all genes of the RNA-Sequencing. B, GSEA (left) and ssGSEA (right) plots for custom gene signatures of estrogen-regulated pathways. C, Dot plot for Ki67 in PBDE-exposed ER+ PDX (COH-SC31).
DISCUSSION
PBDEs have been associated with adverse health effects and are thought to be EDCs (EPA, 2006; Gore et al., 2015). Yet, the PBDE bioactivity in endocrine-related breast cancer has not been fully demonstrated. The unique findings of our study was our comprehensive application of a battery of twenty-first century in vitro and in vivo toxicity testing approaches (ie, RNA-Seq analysis as a nonbiased measure of the biological activities of PBDEs and testing effects using clinically relevant models [PDX]) beyond classical reporter assay system used in past toxicological studies (Hamers et al., 2006; Kojima et al., 2009; Liu et al., 2011; Meerts et al., 2001). Furthermore, PBDEs, such as BDE-47, were shown to be able to act as a weak agonist of orphan receptor ERRα. Moreover, we investigated the mechanisms of BDEs using suitable breast cancer model systems and discussed the potential risk during menopausal transition based on breast cancer biology as well as ER hypersensitivity during this window of susceptibility. This manuscript highlighted the importance of a re-evaluation of PBDE breast cancer risk because recent epidemiological studies have still detected them with high frequency in human specimens across the globe. For example, in 2017 alone, there were 41 biomonitoring studies measuring PBDE levels in humans and 81 studies for examining the presence of PBDEs in environmental samples such as water and soil. These studies indicate a continued attention in the effects of PBDEs even though its use has already been phased out in the United States, due to the continuous existence of residual PBDE in our environment and bioaccumulation in the human body.
In order to clearly define the action of 3 PBDEs, we carefully chose cell lines with selected/preferential nuclear activities for our studies. Here, key bioactivities of 3 PBDEs were determined and are summarized in Figure 8. (1) BDE-47 was assessed in ERα-positive breast cancer cells (MCF7aroERE) and acted as a weak estrogen-mimic (in ERα-expressing C4-12 cells). It increased cell proliferation through ER-regulated cell cycle genes. In addition, BDE-47 acted as a weak agonist of ERRα. (2) BDE-100 acted as an agonist of AhR; the compound caused induction of CYP1A1 and CYP1B1. BDE-100 also acted as a weak agonist of ERα. It upregulated PR expression and led to increased proliferation of MCF7aroERE cells. Conversely, BDE-100 can also block activity of ERα agonists and thereby act as a weak ERα antagonist when estrogen is present. (3) BDE-153 act as a weak antagonist of ERα. BDE-153 inhibited coactivator recruitment to PR. However, the anti-PR effects were not conclusively demonstrated in a PR reporter assay. Furthermore, these 3 PBDEs did not modulate the activity nor the expression of aromatase in our aromatase activity assays (Chen et al., 2015) and promoter/exon 1-specific q-PCR analyses (Imir et al., 2007), respectively (data not shown). Therefore, 3 PBDEs can act as ligands of ERα, a key driver in estrogen-dependent breast cancers.
Figure 8.
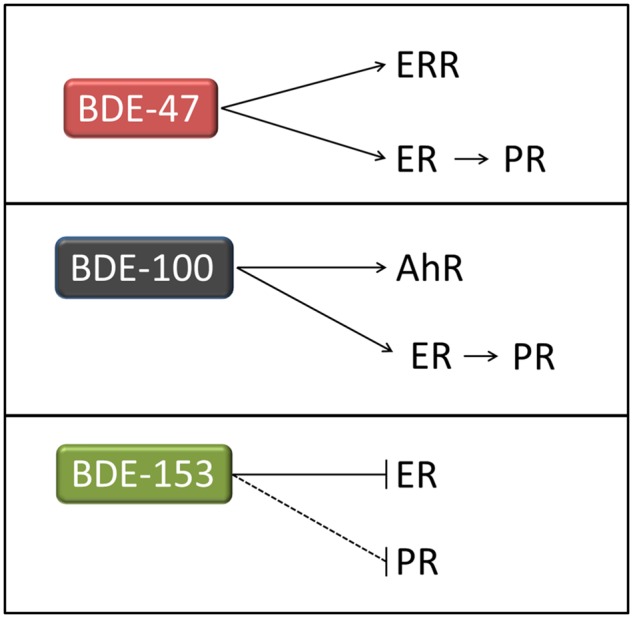
Schematic summary of the effects of each PBDE on key nuclear receptors (ERR, ER, AhR, and PR).
For the first time, the impact of PBDEs on ERRα was assessed in our study. Unlike the ER, ERRα is classified as an orphan receptor. It does not bind natural estrogen but rather the estrogen-related response element (ERRE = TCAAGGTCA). Although ERRE is similar to the half-sites of ERE, the target genes of ER and ERRα overlap only to a small extent. ERRα preferentially binds to metabolic genes involved in glucose/glutamine metabolism, mitochondrial activity, lipid metabolism, and energy sensing (Audet-Walsh and Giguere, 2015). These functions and ERRα expression have been associated with a number of diseases. For example, high ERRα expression in breast cancer is associated with poor prognosis (Park et al., 2016). Congruently, ERRα activation enhances a central metabolic pathway in breast cancer—glutamine metabolism (McGuirk et al., 2013). Several studies have examined the relationship between PBDE exposure, mitochondrial function, and neurotoxicity (Shao et al., 2008; Wong and Giulivi, 2016). Our data indicates that BDE-47 can act as a weak ERRα agonist. However, 20 µM of BDE-47 was necessary to elicit ERRα activation relative to genistein (10 µM). Therefore, other ERRα ligands present in the environment could have greater effects than BDE-47. In order to define BDE-47’s weak ERRα agonistic effects, future studies need to potentially provide insight into the ERRα-mediated action of PBDEs on metabolic control and related diseases using environmentally relevant exposures levels.
Using RNA sequencing, we performed a nonbiased assessment of the biological activities of PBDEs. As opposed to our and others focused studies on a subset of nuclear receptors using reporter assay systems, RNA sequencing can capture total transcriptome changes. RNA sequencing results showed that BDE-47 can act like estrogen, which increases cell proliferation through cell cycle regulation. In addition to be weak ligands of ERα, BDE-100 and BDE-153 acted like AhR agonists and increased the expression of CYP1A1 and CYP1B1. AhR is a ligand-activated transcriptional factor. It forms a complex with the aryl hydrocarbon receptor nuclear translocator (ARNT). The complex binds to xenobiotic response elements (XREs) and induces the expression of downstream genes including cytochrome P450 (CYP) 1 family members: CYP1A1 and CYP1B1. Studies by others have assessed whether PBDE congeners act as AhR receptor agonists or antagonists (Chen and Bunce, 2003). Although their binding is much weaker than TCDD, PBDEs can still act as ligands of AhR (Chen et al., 2001). According to our results, BDE-100 and BDE-153 increase mRNA expression of CYP1A1 and CYP1B1 in MCF7aroERE cells. However, the effect of BDE-153 on AhR-regulated genes was approximately 5 times weaker than BDE-100. Moreover, enzymatic assay results of BDE-153 did not show any significant effects. These results suggest that BDE-100 clearly activates the AhR pathway whereas the effects of BDE-153 were too weak to be significant. Results of our RNA-seq analysis support our findings from nuclear receptor assays. Importantly, these results indicate that each of the 3 PBDEs has weak but preferential interactions with distinct sets of receptors. We currently do not have sufficient data to attribute these distinct interactions to structural differences of the PBDEs. Further studies can be used to systematically assess the impact of structural features, such as bromine substitution patterns (Figure 1A), on PBDE binding patterns to ERα, ERRα, and AhR. Several studies have demonstrated that PBDEs modulate immune functions. Here, IPA analysis predicted that immune function was suppressed by BDE-100 and BDE-153. Moreover, treatment with these PBDEs significantly decreased the expression of interferon-stimulated genes in cells. Likewise, reports have demonstrated that PBDEs suppress host immune defense mechanisms (Fair et al., 2012; Feng et al., 2016). Beyond these reports, the effects of PBDEs on the immune system have been largely overlooked. Importantly, AhR has been suggested to modulate immune function (Cella and Colonna, 2015). Thus, continued investigation is warranted in each disease phenotype (ie, breast cancer) to elucidate how PBDEs may modulate the immune system and whether AhR is involved.
Treatment with a mixture of the 3 PBDEs in vitro primarily elicited estrogenic effects in MCF7aroERE cells. To evaluate the estrogenic activity of PBDE mixtures in a biologically relevant in vivo model, mice were engrafted with an ER+ PDX, COHSC-31. This is an ER+ tumor model in which estrogen-mediated ER activation became the major driving force of growth (Hsu et al., 2018). We observed that treatment with PBDE mixtures in vivo, just for 1 week, upregulated an estrogen-regulated cell cycle pathway. The results from the PDX tumor model confirm with the in vitro results. Moreover, the PBDE-treated group showed increased expression of a proliferation marker Ki67. Post-treatment changes to Ki67 have been used as a prognostic factor for estrogen treatment response of COHSC-31(Hsu et al., 2018). Because the PBDE exposure was only 1 week, the tumor volume and weight were not significantly different between control and PBDE-treated mice (Supplementary Figure 4 [Kanaya et al., 2019]).
PBDEs have been systematically screened with systems for ER, PR, and AhR by several groups (Hamers et al., 2006; Kojima et al., 2009; Meerts et al., 2001); previously reported ER assays involved either T47D cells (ER+ breast cancer) (Hamers et al., 2006; Meerts et al., 2001) or CHO-K1 (Chinese hamster ovary cells) (Kojima et al., 2009). Results from these assessments are consistent with our results using MCF7aroERE cells. Likewise, previous reports on the antiestrogenic activity of BDE-153 are consistent with the weak activity we observed. Before, the PR-antagonistic effects of BDE-100 and BDE-153 were reported using U-2 OS (human osteoblast) cells (Hamers et al., 2006). Although our PR-competitive binding analysis accordingly showed anti-PR effects of BDE-153, we did not observe significant changes in our PR reporter assay using a T47D cell system. Additionally, BDE-153 was previously reported to act as a weak AhR agonist (Meerts et al., 2001). Importantly, our results suggest that BDE-100 is a more potent AhR agonist than BDE-153. The difference in potency was based on differences in downstream induction (CYP1A1 and CYP1B1) and CYP1A1 enzymatic activity. In general, our results are consistent with previous reports describing the effects of PBDEs on nuclear receptors. However, we recognize that such assessments of EDC activities can depend on the cellular context. Thus, we also have defined PBDE activities with (1) gene expression profile analysis and (2) proliferation studies in an estrogen-dependent breast cancer cell line, MCF-7aroERE. Moreover, we have assessed PBDE activities in vivo using mice carrying an estrogen-responsive breast cancer PDX model, COH SC-31. This model was derived and extensively characterized in our laboratory (Hsu et al., 2018; Kanaya et al., 2017).
One limitation of these studies is the use of in vitro cell line models to simulate environmentally relevant exposures to PBDEs. Real-life exposures will typically occur at lower dosages and for longer periods of time (the duration of exposure for most of cell culture experiments was 24 h.). Although the concentration range we used was similar to the ranges used by others in in vitro systems (Hamers et al., 2006; He et al., 2008a,b; Kojima et al., 2009; Lefevre et al., 2016; Meerts et al., 2001), we performed a series of experiments to clarify the impact of exposure dosages. We compared the published human serum/tissue concentrations with the dosages we used to treat cell cultures. In pilot experiments, cells were treated with a mixture of all 3 PBDEs at either a nM or pM concentration range. One mixture was 5 nM BDE-47 + 1 nM BDE-100 + 2 nM BDE-153. Another was 100 pM BDE-47 + 20 pM BDE-100 + 40 pM BDE-153. These low concentration ranges did not significantly change target gene expression (Supplementary Figure 5 [Kanaya et al., 2019]). Therefore, we compared with in vitro cell culture treatments at concentrations in the range of 10 μM (the lowest concentration at which we see reporter signal increase) to human serum concentration. Hurley et al. reported that the mean serum concentration of BDE-47 in California women (2011–2015) was 25.56 ng/g lipid; the maximum observed concentration was 746 ng/g lipid (Hurley et al., 2017). Lipid-adjusted BDE-biomonitored serum concentrations are converted to µg/l serum by using mean adult human serum total lipid concentration (approximately 5 mg lipid/ml) (Bernert et al., 2007). This maximum observed concentration was approximately 1300 times lower than the minimum concentration at which we observed in vitro signals (10 µM). We also considered the impact of tissue accumulation, because PBDEs are lipophilic and can deposit in fat tissues. Adipose tissue is particularly abundant in the breast and surrounds the mammary glands. Johnson-Restrepo et al. reported that the median concentration of BDE-47 in adipose tissue of New York populations was 132 ng/g lipid and maximum was 2720 ng/g lipid (Johnson-Restrepo et al., 2005). Still, the maximum observed concentration was approximately 350 times lower than 10 µM. For in vivo study, our diet regime resulted in generating approximately 300–1000 times (BDE-47, 952 times; BDE-100, 352 times; and BDE-153, 787 times) higher PBDE serum levels in our experimental mice (BDE-47, 24336 ng/g lipid; BDE-100, 1790 ng/g lipid; and BDE-153, 9479 ng/g lipid) compared with mean human levels (BDE-47, 25.56 ng/g lipid; BDE-100, 5.08 ng/g lipid; and BDE-153, 12.03 ng/g lipid) (Hurley et al., 2017). Also compared with highest concentration scenario in human exposure (ie, maximum observed serum concentration, BDE-47, 749 ng/g lipid; BDE-100, 186 ng/g lipid; and BDE-153, 379 ng/g lipid) (Hurley et al., 2017), mouse serum concentration was still 10–30 times higher than human exposure. It is important to recognize this dosimetry concern and propose future approaches to advance understanding PBDEs affects. It is known that ER increases its sensitivity to E2 as an adaptive mechanism to estrogen deprivation. Masumura et al. reported that after 1–6 months estrogen deprivation on MCF7 cells, only 10−14–10−15 mol/l E2 could stimulate the estrogen-deprived MCF7 cell to grow maximally. In contrast, parental MCF7 cell required 10−10 mol/l E2, which is 4–5 orders of magnitude different, to grow at the same rate (Masamura et al., 1995). We speculated based on these observations that the risk of PBDE increases when ER is hypersensitive, such as menopausal transition (estrogen deprivation). During menopausal transition (perimenopause), endogenous levels of estrogen start to decline. Therapy with these hormones eases symptoms of both natural and surgically induced (oophorectomy) menopause. When hormone therapy (together with progesterone) is applied shortly after menopause, it may be associated with several health risks. According to studies by the Women’s Health Initiative (WHI), intervention at this juncture may be linked to increased breast cancer incidence and mortality (Prentice et al., 2009). When estrogen levels are low (during perimenopause), mammary gland cells become hypersensitive to estrogen. Therefore, future study will be needed to test using environmentally relevant dosage of PBDEs to answer the critical question, whether PBDEs can be a risk of human breast cancer, or only a low-priority health concern using in vitro and in vivo models with hypersensitive ER (focusing on “the window of sensitivity”). Furthermore, other naturally occurring ER agonists that are substantially more potent than PBDEs can be also present in the blood at significantly higher concentrations/activities than PBDEs, as shown in this study. Therefore, it is critical to identify the PBDE’s contribution to the cancer with or without those stronger ER activators because overall PBDE’s effects shown in this study was relatively weak. During menopausal transition, women are exposed to multiple chemicals which can modulate estrogen- and/or progesterone-mediated mechanisms. Although it remains to be challenging to define the combined action of chemical mixtures in breast cancer promotion, our findings on PBDEs will be valuable in designing experiments to examine mixtures that include PBDEs.
In summary, we tested 3 PBDEs that are widely detected in human samples and determined that these compounds possess distinct biological activities using unique nuclear receptor-expressing cell lines. Together with a nonbiased RNA-seq analysis, we provided the fundamental understandings of BDE-47, BDE-100, and BDE-153 effects in breast cancer through modulation of ERα, ERRα, PR, and/or AhR pathways. Furthermore, we confirm the estrogenic activity of PBDE mixtures in vivo using a clinically relevant ER+ PDX model. However, the higher concentration of PBDE exposure compared with the levels detected in human were required to observe activities, therefore the results may or may not indicate a role of PBDE in breast cancer when they are present alone. By assessing the activities of individual PBDEs and a mixture of PBDEs with three different approaches, we have advanced our understanding of potential mechanisms of PBDE exposure that may or may not influence breast cancer.
DATA AVAILABILITY
Supplementary data are available at https://doi.org/10.5061/dryad.c115v23.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
As part of the Breast Cancer and Environmental Research Program, this work was supported by NIH (U01ES026137-01) (MPIs Chen and Neuhausen). The City of Hope Core Facilities are supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572.
ACKNOWLEDGMENTS
We thank the City of Hope Core Facilities, including the Integrative Genomics Core, for the excellent technical support. The authors thank Ian Talisman, PhD for editing the manuscript, and Joginder Dhaliwal, Roshni Sarala, and Dr Reber Brown from California Department of Toxic Substances Control for analytical support.
REFERENCES
- Anders S., Pyl P. T., Huber W. (2015). HTSeq – A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet-Walsh E., Giguere V. (2015). The multiple universes of estrogen-related receptor alpha and gamma in metabolic control and related diseases. Acta Pharmacol. Sin. 36, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300. [Google Scholar]
- Bernert J. T., Turner W. E., Patterson D. G. Jr, Needham L. L. (2007). Calculation of serum “total lipid” concentrations for the adjustment of persistent organohalogen toxicant measurements in human samples. Chemosphere 68, 824–831. [DOI] [PubMed] [Google Scholar]
- Busch B. B., Stevens W. C. Jr, Martin R., Ordentlich P., Zhou S., Sapp D. W., Horlick R. A., Mohan R. (2004). Identification of a selective inverse agonist for the orphan nuclear receptor estrogen-related receptor alpha. J. Med. Chem. 47, 5593–5596. [DOI] [PubMed] [Google Scholar]
- Ceccatelli R., Faass O., Schlumpf M., Lichtensteiger W. (2006). Gene expression and estrogen sensitivity in rat uterus after developmental exposure to the polybrominated diphenylether PBDE 99 and PCB. Toxicology 220, 104–116. [DOI] [PubMed] [Google Scholar]
- Cella M., Colonna M. (2015). Aryl hydrocarbon receptor: Linking environment to immunity. Semin. Immunol. 27, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Bunce N. J. (2003). Polybrominated diphenyl ethers as Ah receptor agonists and antagonists. Toxicol. Sci. 76, 310–320. [DOI] [PubMed] [Google Scholar]
- Chen G., Konstantinov A. D., Chittim B. G., Joyce E. M., Bols N. C., Bunce N. J. (2001). Synthesis of polybrominated diphenyl ethers and their capacity to induce CYP1A by the Ah receptor mediated pathway. Environ. Sci. Technol. 35, 3749–3756. [DOI] [PubMed] [Google Scholar]
- Chen S., Hsieh J. H., Huang R., Sakamuru S., Hsin L. Y., Xia M., Shockley K. R., Auerbach S., Kanaya N., Lu H., et al. (2015). Cell-based high-throughput screening for aromatase inhibitors in the Tox21 10K library. Toxicol. Sci. 147, 446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou D., Hsin L. Y., Kanaya N., Wong C., Yip R., Sakamuru S., Xia M., Yuan Y. C., Witt K., et al. (2014). AroER tri-screen is a biologically relevant assay for endocrine disrupting chemicals modulating the activity of aromatase and/or the estrogen receptor. Toxicol. Sci. 139, 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa L. G., Pellacani C., Dao K., Kavanagh T. J., Roque P. J. (2015). The brominated flame retardant BDE-47 causes oxidative stress and apoptotic cell death in vitro and in vivo in mice. Neurotoxicology 48, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick J. K., Pandiri A. R., Merrick B. A., Kissling G. E., Cunny H., Mutlu E., Waidyanatha S., Sills R., Hong H. L., Ton T. V., et al. (2018). Carcinogenic activity of pentabrominated diphenyl ether mixture (DE-71) in rats and mice. Toxicol. Rep. 5, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA (2006). Polybrominated diphenyl ethers (PBDEs) project plan. Report Number: 740R06002. Washington, DC 20460.
- Fair P. A., Stavros H. C., Mollenhauer M. A., DeWitt J. C., Henry N., Kannan K., Yun S. H., Bossart G. D., Keil D. E., Peden-Adams M. M. (2012). Immune function in female B(6)C(3)F(1) mice is modulated by DE-71, a commercial polybrominated diphenyl ether mixture. J. Immunotoxicol. 9, 96–107. [DOI] [PubMed] [Google Scholar]
- Feng Y., Zeng W., Wang Y., Shen H., Wang Y. (2016). Long-term exposure to high levels of decabrominated diphenyl ether inhibits CD4 T-cell functions in C57Bl/6 mice. J. Appl. Toxicol. 36, 1112–1119. [DOI] [PubMed] [Google Scholar]
- Frasor J., Danes J. M., Komm B., Chang K. C., Lyttle C. R., Katzenellenbogen B. S. (2003). Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 144, 4562–4574. [DOI] [PubMed] [Google Scholar]
- Gore A. C., Chappell V. A., Fenton S. E., Flaws J. A., Nadal A., Prins G. S., Toppari J., Zoeller R. T. (2015). EDC-2: The endocrine society’s second scientific statement on endocrine-disrupting chemicals. Endocr. Rev. 36, E1–E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Holden A., Smith S. C., Gephart R., Petreas M., Park J. S. (2016). PBDE levels in breast milk are decreasing in California. Chemosphere 150, 505–513. [DOI] [PubMed] [Google Scholar]
- Haber G., Ahmed N. U., Pekovic V. (2012). Family history of cancer and its association with breast cancer risk perception and repeat mammography. Am. J. Public Health 102, 2322–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers T., Kamstra J. H., Sonneveld E., Murk A. J., Kester M. H., Andersson P. L., Legler J., Brouwer A. (2006). In vitro profiling of the endocrine-disrupting potency of brominated flame retardants. Toxicol. Sci. 92, 157–173. [DOI] [PubMed] [Google Scholar]
- He P., He W., Wang A., Xia T., Xu B., Zhang M., Chen X. (2008). PBDE-47-induced oxidative stress, DNA damage and apoptosis in primary cultured rat hippocampal neurons. Neurotoxicology 29, 124–129. [DOI] [PubMed] [Google Scholar]
- He Y., Murphy M. B., Yu R. M., Lam M. H., Hecker M., Giesy J. P., Wu R. S., Lam P. K. (2008). Effects of 20 PBDE metabolites on steroidogenesis in the H295R cell line. Toxicol. Lett. 176, 230–238. [DOI] [PubMed] [Google Scholar]
- Hsu F., Kent W. J., Clawson H., Kuhn R. M., Diekhans M., Haussler D. (2006). The UCSC known genes. Bioinformatics 22, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Hsu P. Y., Wu V. S., Kanaya N., Petrossian K., Hsu H. K., Nguyen D., Schmolze D., Kai M., Liu C. Y., Lu H., et al. (2018). Dual mTOR kinase inhibitor MLN0128 sensitizes hr(+)/HER2(+) breast cancer patient-derived xenografts to trastuzumab or fulvestrant. Clin. Cancer Res. 24, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley S., Goldberg D., Nelson D. O., Guo W., Wang Y., Baek H. G., Park J. S., Petreas M., Bernstein L., Anton-Culver H., et al. (2017). Temporal evaluation of polybrominated diphenyl ether (PBDE) serum levels in middle-aged and older California women, 2011-2015. Environ. Sci. Technol. 51, 4697–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imir A. G., Lin Z., Yin P., Deb S., Yilmaz B., Cetin M., Cetin A., Bulun S. E. (2007). Aromatase expression in uterine leiomyomata is regulated primarily by proximal promoters I.3/II. J. Clin. Endocrinol. Metab. 92, 1979–1982. [DOI] [PubMed] [Google Scholar]
- Johnson-Restrepo B., Kannan K., Rapaport D. P., Rodan B. D. (2005). Polybrominated diphenyl ethers and polychlorinated biphenyls in human adipose tissue from New York. Environ. Sci. Technol. 39, 5177–5182. [DOI] [PubMed] [Google Scholar]
- Kanaya N., Bernal L., Chang G., Yamamoto T., Nguyen D., Wang Y., Park J., Warden C., Wang J., Wu X., et al. (2019). Data from: Molecular mechanisms of polybrominated diphenyl ethers (BDE-47, BDE-100, and BDE-153) in human breast cancer cells and patient-derived xenografts. Toxicol. Sci. 169, 380–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya N., Nguyen D. M., Lu H., Wang Y. Z., Hsin L. Y., Petreas M., Nelson D., Guo W., Reynolds P., Synold T., et al. (2015). AroER tri-screen is a novel functional assay to estimate both estrogenic and estrogen precursor activity of chemicals or biological specimens. Breast Cancer Res. Treat. 151, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya N., Somlo G., Wu J., Frankel P., Kai M., Liu X., Wu S. V., Nguyen D., Chan N., Hsieh M. Y., et al. (2017). Characterization of patient-derived tumor xenografts (PDXs) as models for estrogen receptor positive (ER+HER2- and ER+HER2+) breast cancers. J. Steroid. Biochem. Mol. Biol. 170, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H., Takeuchi S., Uramaru N., Sugihara K., Yoshida T., Kitamura S. (2009). Nuclear hormone receptor activity of polybrominated diphenyl ethers and their hydroxylated and methoxylated metabolites in transactivation assays using Chinese hamster ovary cells. Environ. Health .Perspect 117, 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M. T., Carey V. J. (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre P. L., Wade M., Goodyer C., Hales B. F., Robaire B. (2016). A mixture reflecting polybrominated diphenyl ether (PBDE) profiles detected in human follicular fluid significantly affects steroidogenesis and induces oxidative stress in a female human granulosa cell line. Endocrinology 157, 2698–2711. [DOI] [PubMed] [Google Scholar]
- Liu H., Hu W., Sun H., Shen O., Wang X., Lam M. H., Giesy J. P., Zhang X., Yu H. (2011). In vitro profiling of endocrine disrupting potency of 2,2',4,4'-tetrabromodiphenyl ether (BDE47) and related hydroxylated analogs (HO-PBDEs). Mar. Pollut. Bull. 63, 287–296. [DOI] [PubMed] [Google Scholar]
- Lynch C., Zhao J., Huang R., Kanaya N., Bernal L., Hsieh J. H., Auerbach S. S., Witt K. L., Merrick B. A., Chen S., et al. (2018). Identification of estrogen-related receptor alpha agonists in the Tox21 compound library. Endocrinology 159, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamura S., Santner S. J., Heitjan D. F., Santen R. J. (1995). Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J. Clin. Endocrinol. Metab. 80, 2918–2925. [DOI] [PubMed] [Google Scholar]
- McGrath T. J., Ball A. S., Clarke B. O. (2017). Critical review of soil contamination by polybrominated diphenyl ethers (PBDEs) and novel brominated flame retardants (NBFRs); concentrations, sources and congener profiles. Environ. Pollut. 230, 741–757. [DOI] [PubMed] [Google Scholar]
- McGuirk S., Gravel S. P., Deblois G., Papadopoli D. J., Faubert B., Wegner A., Hiller K., Avizonis D., Akavia U. D., Jones R. G., et al. (2013). PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 1, 22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre R. L., Kenerson H. L., Subramanian S., Wang S. A., Kazami M., Stapleton H. M., Yeung R. S. (2015). Polybrominated diphenyl ether congener, BDE-47, impairs insulin sensitivity in mice with liver-specific Pten deficiency. BMC Obes 2, 3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerts I. A., Letcher R. J., Hoving S., Marsh G., Bergman A., Lemmen J. G., van der Burg B., Brouwer A. (2001). In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PDBEs, and polybrominated bisphenol A compounds. Environ. Health Perspect. 109, 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Park S., Chang C. Y., Safi R., Liu X., Baldi R., Jasper J. S., Anderson G. R., Liu T., Rathmell J. C., Dewhirst M. W., et al. (2016). ERRalpha-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep. 15, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreas M., She J., Brown F. R., Winkler J., Windham G., Rogers E., Zhao G., Bhatia R., Charles M. J. (2003). High body burdens of 2,2',4,4'-tetrabromodiphenyl ether (BDE-47) in California women. Environ. Health Perspect. 111, 1175–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrossian K., Kanaya N., Lo C., Hsu P. Y., Nguyen D., Yang L., Yang L., Warden C., Wu X., Pillai R., et al. (2018). ERalpha-mediated cell cycle progression is an important requisite for CDK4/6 inhibitor response in HR+ breast cancer. Oncotarget 9, 27736–27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl H. R., Odin M., McClure P., Zaccaria K., Llados F., Kawa M., Citra M. (2017). Toxicological profile for polybrominated diphenyl ethers (PBDEs), 1–532.
- Prentice R. L., Manson J. E., Langer R. D., Anderson G. L., Pettinger M., Jackson R. D., Johnson K. C., Kuller L. H., Lane D. S., Wactawski-Wende J., et al. (2009). Benefits and risks of postmenopausal hormone therapy when it is initiated soon after menopause. Am. J. Epidemiol. 170, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J. P. (2006). GenePattern 2.0. Nat. Genet. 38, 500–501. [DOI] [PubMed] [Google Scholar]
- Shao J., White C. C., Dabrowski M. J., Kavanagh T. J., Eckert M. L., Gallagher E. P. (2008). The role of mitochondrial and oxidative injury in BDE 47 toxicity to human fetal liver hematopoietic stem cells. Toxicol. Sci. 101, 81–90. [DOI] [PubMed] [Google Scholar]
- Sjodin A., Wong L. Y., Jones R. S., Park A., Zhang Y., Hodge C., Dipietro E., McClure C., Turner W., Needham L. L., et al. (2008). Serum concentrations of polybrominated diphenyl ethers (PBDEs) and polybrominated biphenyl (PBB) in the United States population: 2003–2004. Environ. Sci. Technol. 42, 1377–1384. [DOI] [PubMed] [Google Scholar]
- Staskal D. F., Diliberto J. J., DeVito M. J., Birnbaum L. S. (2005). Toxicokinetics of BDE 47 in female mice: Effect of dose, route of exposure, and time. Toxicol. Sci. 83, 215–223. [DOI] [PubMed] [Google Scholar]
- Stoker T. E., Laws S. C., Crofton K. M., Hedge J. M., Ferrell J. M., Cooper R. L. (2004). Assessment of DE-71, a commercial polybrominated diphenyl ether (PBDE) mixture, in the EDSP male and female pubertal protocols. Toxicol. Sci. 78, 144–155. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden C., Yuan Y., Wu X. (2013). Optimal calculation of RNA-seq fold-change values. Int. J. Comput. Bioinform. In Silico Model. 2, 285–292. [Google Scholar]
- Wong S., Giulivi C. (2016). Autism, mitochondria and polybrominated diphenyl ether exposure. CNS Neurol. Disord. Drug Targets 15, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. H., Wang Z. Y., Liu H. L., Yu H. X. (2010). Exploring the binding features of polybrominated diphenyl ethers as estrogen receptor antagonists: Docking studies. SAR QSAR Environ. Res. 21, 351–367. [DOI] [PubMed] [Google Scholar]
- Zhou D. J., Pompon D., Chen S. A. (1990). Stable expression of human aromatase complementary DNA in mammalian cells: A useful system for aromatase inhibitor screening. Cancer Res. 50, 6949–6954. [PubMed] [Google Scholar]
- Zhou T., Taylor M. M., DeVito M. J., Crofton K. M. (2002). Developmental exposure to brominated diphenyl ethers results in thyroid hormone disruption. Toxicol. Sci. 66, 105–116. [DOI] [PubMed] [Google Scholar]
- Zota A. R., Linderholm L., Park J. S., Petreas M., Guo T., Privalsky M. L., Zoeller R. T., Woodruff T. J. (2013). Temporal comparison of PBDEs, OH-PBDEs, PCBs, and OH-PCBs in the serum of second trimester pregnant women recruited from San Francisco General Hospital, California. Environ. Sci. Technol. 47, 11776–11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supplementary data are available at https://doi.org/10.5061/dryad.c115v23.