

Abstract
Silicon oxynitride (Si–O–N) is a new biomaterial in which its O/N ratio is tunable for variable Si release and its subsequent endocytotic incorporation into native hydroxyapatite for enhanced bone healing. However, the effect of nitrogen and hydrogen bonding on the formation and structure of hydroxyapatite is unclear. This study aims to uncover the roles of H and N in tuning Si–O–N surface bioactivity for hydroxyapatite formation. Conformal Si–O–N films were fabricated by plasma-enhanced chemical vapor deposition (PECVD) onto Ti/Si substrates. Fourier transform infrared spectroscopy (FTIR) and Rutherford backscattering spectrometry (RBS) analysis indicated increased Si–H and N–H bonding with increased N content. Surface energy decreased with increased N content. X-ray absorbance near edge structure (XANES) analysis showed tetrahedral coordination in O-rich films and trigonal coordination in N-rich films. O-rich films exhibited a 1:1 ratio of 2p3/2 to 2p1/2 electron absorbance, while this ratio was 1.73:1 for N-rich films. Both Si and N had a reduced partial charge for both O- and N-rich films, whereas O maintained its partial charge for either film. O-rich films were found to exhibit random bonding SizOxNy, while N-rich films exhibited random mixing: [Si–Si]–[Si–O]–[Si–N]. Thus, hydrogen bonding limits random nitrogen bonding in Si–O–N films via surface Si–H and N–H bonding. Moreover, increased nitrogen content reduces the partial charge of constituent elements and changes the bonding structure from random bonding to random mixing.
Graphical Abstract
1. INTRODUCTION
Amorphous silicon oxynitride (Si–O–N) thin films have been extensively studied as a result of their use in microelectronic and optical devices. Plasma enhanced chemical vapor deposition (PECVD) has been established for fabricating these types of films because of its tunability and reproducibility.1 These features of Si–O–N films make them a potential attractive coating for biomedical devices used in fracture healing. In particular, these materials can be used for sustained release of ionic Si (Si4+) which enhances the mineralization process and up-regulates several important physiological processes that occur in bone mineralization.2–7 While Si–O–N films used in optoelectronics require process optimization to reduce hydrogen bonding (N–H and Si–H) and subsequent hydrolysis, hydrogen bonding may prove favorable for hydrolysis within physiological environments. Thus, PECVD films for applications in improving bone-implant bonding may actually benefit from significant levels of N–H and Si–H bonding when local delivery of Si4+ ions is desired.
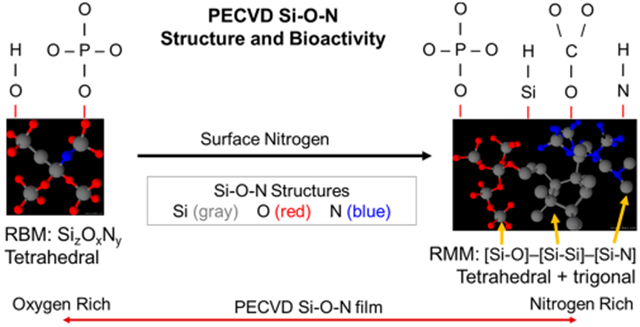
The long-term stability of PECVD films for bone healing applications prompted an investigation into the chemical structure of such films, and two distinct theoretical models have been proposed: the random mixing model (RMM) and the random bonding model (RBM) represented in Figure 1. The RMM states that a nonstoichiometric silicon oxynitride (Si–O–N) can be treated as a simple mixture of Si–O, Si–N, and Si–Si polyhedra.8–14 In the RBM, silicon, oxygen, and nitrogen incorporate into the basic silicon dioxide tetrahedral network to form SizOxNy, where x + y = 4,15 and account for multiple ringed structures composed of three to eight tetrahedra.16 The basic structure of Si3N4 (Si–N bonding) is a trigonal planar space group in which a N atom bonds with three Si atoms to form a 3D network while Si–Si bonding is constrained to either 4-fold or 6-fold coordination and is denoted for the excess Si structure within the RMM.
Figure 1.

Schematic representation of the random bonding model (RBM, left) and random mixing model (RMM) of the amorphous Si–O–N elemental system.
Both models have been used to examine SiO2, Si3N4, and Si–O–N thin films produced using PECVD, LPCVD (low pressure CVD), and other deposition methods under a variety of reaction conditions15,17–19. ‘ On one hand, these films have been generally described by random bonding in which Si bonds to O or N as a substituted species (i.e., SizOxNy).15,18 On the other hand, these films have also been described by random mixing in which Si–Si, Si–O, and Si–N bonds exist in a simple mixture with no clustering of these structures.20 These research groups have used a variety of methods including X-ray photoelectron spectroscopy (XPS), XANES, FTIR, and ellipsometry to verify the composition and model fit. XANES analysis is optimized to detect singly or multiply coordinated surface structures. For example, for PECVD-based Si–O–N material Si K-edge analysis, researchers have found the SiO4 tetrahedron to describe the structure of Si–O–N.21 However, unless there is fabrication of Si nanostructures within a silicon nitride or Si–O–N materials, such as those fabricated by Wilson et al.,21 the Si K-edge cannot discern such clusters within PECVD Si–O–N film. Si–Si structures in Si K-edge analysis may be observed when the beam energy shifts to lower energies.21 However, follow-up of these spectra with the Si L-edge is still needed to discern the multiply coordinated structures more definitively. Such examination has been lacking from previous research into examining PECVD amorphous silicon oxynitride surface structures.
This need for more detailed XANES analysis is further indicated by our previous work on biomineralization using Si–O–N thin films. In that work, it was found that trigonally coordinated carbonate species, tetrahedrally coordinated phosphate species, and calcium species formed on the surface within a few hours of in vitro immersion.22 This was determined using X-ray absorbance near edge structure (XANES) spectroscopy (Ca L-edge, P L-edge, O K-edge). This “configurational match” of the Si–O–N surface would be desired to elicit similarly epitaxially grown biomineral of like coordination to maximize the density of grown film.23 Thus, this combination of our previous results and the presence of enhanced hydrogen bonding in these films along with their bioactive effects warranted an examination of the surface chemical structure in terms of the applicability of either the RMM or the RBM. An improved understanding of these models with respect to the structure of silicon oxynitride may aid in production of osteogenic materials having enhanced and reproducible bone healing capabilities.
In the present study, Si–O–N films with various oxygen and nitrogen ratios were fabricated via a PECVD process designed to yield a potentially bioactive coating for biomedical devices. The model material surface was commercially pure Ti. It was applied to a Si wafer using electron-beam physical vapor deposition (EBPVD). Process validity and repeatability measures were obtained via transmission electron microscopy (TEM), XPS, ellipsometry, and refractometry. Scanning electron microscopy (SEM) with energy dispersive spectroscopy (EDS) was used to evaluate coating composition. Fourier transform infrared spectroscopy (FTIR) was used to investigate N–H and Si–H bonding. Nuclear reaction analysis (NRA) was used to measure the absolute concentration of H, C, N, and O (atoms/cm2) in these films, and Rutherford backscattering spectrometry (RBS) was used to measure the Si content. XANES spectroscopy evaluated the chemical structure of the Si–O–N films for their fit with the RBM or RMM. The low energy Si L2,3 absorption edge resolved with a high resolution third generation synchrotron provided insight into the coordination of Si and its binding with either oxygen or nitrogen. We concluded that surface hydrogen bonding with nitrogen (N–H) influenced the substitution of N in amorphous Si–O–N films prepared by PECVD. The presence of a high concentration of Si–H bonding combined with N–H bonding contributed to the formation of a random mixture of Si–Si, Si–O, and Si–N bonds within high-N-content films. The presence of N–H surface bonding contributed to the formation of randomly bonded N-substitution within the amorphous silica network (i.e., SizOxNy) for high-O-content films.
2. EXPERIMENTAL AND/OR THEORETICAL METHODS
2.1. Device Fabrication.
To create the Ti/TiO2–SiOx devices and Si–O–N overlays, processes were tailored around rapid prototyping of various deposition and analysis techniques. The procedure described below was modified from previous work.22 The fabrication process started with standard cleaning of single crystal Si wafers; these were used as the base substrate. Ti layers were deposited using electron-beam physical vapor deposition (EB-PVD). Thermal oxidation was then employed to create a sufficient layer of TiO2 to anchor overlays. PECVD of amorphous silica-based overlays was then conducted to build SiOx/Si–O–N onto structured Ti/TiO2 surfaces. All materials were fabricated at the University of Texas in the Arlington Nanotechnology Center.
2.2. Electron Beam Physical Vapor Deposition (EBPVD) of Ti Films.
P-type Si wafers with 〈100〉 orientation (Nova Electronic Materials, Flower Mound, TX) were employed as substrates for deposition of a uniform layer of Ti using electron beam physical vapor deposition (EB-PVD). Columnar Ti grains were deposited to a depth of 300 nm at a rate of 1 Å/s using an emission current of 3.1 mA and a chamber pressure of 5 × 10−8 Torr.
2.3. Plasma-Enhanced Chemical Vapor Deposition (PECVD) of Si–O–N Films.
A TRION ORION II PECVD/LPECVD system (Trion Technology, Clearwater, FL) was used to deposit a uniform (nonuniformity < 1%) 100 nm SiOx layer followed by 100–1000 nm deposition of a Si–O–N amorphous film. All coatings were fabricated at a substrate temperature of 400 °C, a chamber pressure of 900 mTorr, an ICP power of 30 W, and an applied excitation frequency of 13.56 MHz. Source gases included silane (SiH4) carried by argon (Ar) (15%/85%), nitrous oxide (N2O), nitrogen (N2), and ammonia (NH3). The silane flow rate was kept low at 24 standard cubic centimeters per minute (sccm) to prevent undesirable gas-phase reactions. The nitrogen and ammonia flow rates were kept high at 225 and 50 sccm, respectively, to increase N–H as well as Si–H bonding within the thin films. Five different types of films were prepared by varying the N2O flow rate, as shown in Table 1. The refractive indices and the thickness of the films were measured using ellipsometry at a wavelength of 632.8 nm (Gaertner LS300). The results were confirmed through the use of a reflectometer (Ocean Optics NC-UV–vis TF Reflectometer) and a scanning electron microscope (Hitachi S-3000N VP SEM). Deposition rates were determined from thickness measurements and plasma-on times.
Table 1.
Gas Flow Rates for Silicon Oxynitride (Si–O–N) Layer Deposition as They Relate to Their Measured Index of Refraction by Ellipsometry
| gas flow rates (sccm) |
|||||||
|---|---|---|---|---|---|---|---|
| sample | refractive index | deposition rate (angstroms/min) | N2O/SiH4 | 15% SiH4/Ar | N2O | N2 | NH3 |
| 1 | 2.0 | 320 | 0 | 24 | 0 | 225 | 50 |
| 2 | 1.82 | 360 | 0.13 | 24 | 3 | 225 | 50 |
| 3 | 1.65 | 390 | 0.67 | 24 | 16 | 225 | 50 |
| 4 | 1.57 | 520 | 6.46 | 24 | 155 | 225 | 50 |
| 5 | 1.45 | 550 | 6.67 | 24 | 160 | 225 | 50 |
2.4. Transmission Electron Microscopy (TEM).
TEM facilities were provided by the Center for Nanophase Materials Sciences (CNMS) at Oak Ridge National Laboratory, Oak Ridge, TN (ORNL). After fabrication, samples were sectioned using a Hitachi NB5000 dual-beam scanning electron microscope focused ion beam (SEM-FIB) onto Cu grids prior to imaging by TEM. TEM imaging was carried out using a Hitachi HF-3300 300kV FEG TEM/STEM with electron energy loss spectroscopy (EELS).
2.5. X-ray Photoelectron Spectroscopy (XPS).
XPS facilities were also provided by CNMS at ORNL. XPS analysis was conducted using a Thermo Scientific K-Alpha XPS with capabilities for sample through-thickness milling. A monochromatic Al Kα source with an energy of 1486.6 eV and Ar+ beam sputtering operated at an energy of 4.2 keV was used for XPS analysis. This type of analysis aids in measuring surface and interfacial elementals.
2.6. X-ray Diffraction (XRD).
X-ray diffraction was used to investigate films for the formation of crystalline phases. A Bruker D8 Advance diffractometer was used to collect θ–2θ (Bragg–Brentano) scans using Cu Kα radiation (λ = 1.5418 Å) at room temperature. Data were recorded over the 2θ range of 20–80° with a 0.02° step size and a dwell time of 1 s. XRD was carried out at the Center for Characterization in Materials and Biology at the University of Texas at Arlington.
2.7. Fourier Transform Infrared (FTIR) Spectroscopy.
A FTIR spectrometer (Nicolet 6700, Thermo-Nicolet Corp., Madison, WI) in attenuated total reflectance (ATR) mode was used to collect FTIR spectra. Each spectrum was recorded using 32 scans from 650 to 4000 cm−1 with a resolution of 4 cm−1. An uncoated Si wafer was used to perform background subtraction before each measurement. Quantitation of N–H bond concentration was developed by Lanford and Rand,11,12,24,25 and they determined the absorption cross section of the N–H bond to be 5.3 × 10−18 cm2.11 The FTIR-ATR results should be held in relative context and represent semiquantitative analysis. Therefore, the area under the curve was normalized to an oxide wafer reference to estimate the relative N–H bond concentration. A similar process was used to evaluate normalized Si–H bonding. Its reported absorption cross section is 7.4 × 10−18 cm2.11 The FTIR-ATR results were normalized relative to pure amorphous silica (SiOx), since this compound’s FTIR-ATR results correspond well with FTIR transmission results. The ATR technique accounts only for the relative shifts in band intensity and absolute shifts in frequency. The relative intensity shift is well described and can be easily corrected, whereby the absolute shift in frequency domain is more difficult to correct. Therefore, the frequency shift is often neglected. The ATR correction algorithm in the FTIR software corrects the relative shifts. ATR correction also accounts for the variations in effective path-length by scaling the ATR spectrum accordingly.
2.8. Contact Angle Measurement and Surface Energy Evaluation.
A sessile drop technique was used to determine contact angles of distilled, deionized water and diiodomethane (>99%, Sigma-Aldrich, St. Louis, MO, USA) on the coated wafers. For each liquid, nine repeat drops were measured at 25 °C. Each coating was tested in triplicate. The surface tension of the testing fluids, including the polar and dispersive components, was taken from literature values under the same experimental conditions. The surface energy (γLV) and its corresponding dispersive (γd) and polar (γp) component values for water are 72.8, 21.8, and 51.0 mJ/m2, respectively, and 50.8, 50.4, and 0.4 mJ/m2, respectively, for diiodomethane.26,27 Surface hydrophilicities were inferred from contact angle measurements; surface energy values were obtained by relating the probing liquid contact angles (θ) to solid surface energy dispersive and polar components using the Owens–Wendt–Kaeble relation. Wetting characteristics of a solid strongly depend on the roughness of its surface, and preparation of a smooth solid surface is essential for obtaining accurate and reproducible contact angle measurement.28–30 Surface roughness measured via scanning probe microscopy (SPM) imaging indicated that the as-deposited roughness for all sample groups was very small (Ra < 1 nm), so the sample surface roughness will have no effect on the measurement of equilibrium contact angles.12–14
2.9. Scanning Electron Microscopy (SEM) and Electron Dispersive Spectroscopy (EDS).
Surface morphology, film composition, and film thickness were investigated using a scanning electron microscope (Hitachi S-3000N Variable Pressure SEM) equipped with an energy dispersive X-ray spectroscopy system (EDS). SEM micrographs were captured at an acceleration voltage of 20 keV and a working distance of 15 mm.
2.10. X-ray Absorbance Near Edge Structure (XANES) Spectroscopy.
XANES spectroscopy was carried out on SiO2 and Si3N4 model compounds and four Si–O–N samples–two high-nitrogen-content samples (1 and 2) and two high-oxygen-content samples (4 and 5). The acquired XANES spectra were compared with the model compounds for silicon (Si), oxygen (O), and nitrogen (N). The Si L2,3-edge and Si, O, and N K-edges were used to characterize the chemical nature and structural environment in the amorphous films. The spectra were obtained at the Canadian Light Source (CLS, Saskatoon, Canada) using the Variable Line Spacing Plane Grating Monochromator (VLS-PGM, 11ID-2) for Si L-edge and the High Resolution Spherical Grating Monochromator (SGM, 11ID-1) beam lines for Si K-edge, O K-edge, and N K-edge. The total electron yield (TEY) data for these edges is considered to be collected from the near-surface (~25 nm for O K-edge, ~70 nm for Si K-edge, and ~20 nm for N K-edge), while the fluorescence yield (FY) data contains information from a relatively higher depth of penetration (~150 nm for O K-edge, ~70 nm for Si L-edge, and ~125 nm for N K-edge).31,32
Sample sections of 12 mm × 12 mm × 0.5 mm were attached to the stage with carbon tape and examined under a vacuum with the beam normal to the sample surface. For the Si L2,3-edge acquired on the VLS-PGM beam line, the fluorescence yield (FY) signal was recorded. For the Si K-, O K-, and N K-edges acquired on the SGM beam line, total electron yield (TEY) and partial fluorescence yield (PFY) data was recorded. All sample edges were acquired with a 1 s dwell time and a step size of 0.5/0.1/0.25 eV for the pre-edge/edge/post-edge spectral features. At least two spectra were acquired per sample per edge. Spectra were energy shifted using reference compounds, collected during the same session using established peak positions, Si3N4 nanopowder (Nanostructured & Amorphous Materials Inc., Houston, TX, USA), and SiO2 (>99.5%, Sigma-Aldrich, St. Louis, MO, USA). Linear background subtraction was performed using the spectral pre-edge for peak area calculations; however, spectra without background subtraction are presented for clarity.
2.11. Nuclear Reaction Analysis and Rutherford Backscattering Spectroscopy.
Nuclear reaction analysis (NRA) and Rutherford backscattering spectroscopy (RBS) were used to measure the elemental content of the films. Films were prepared as described above using PECVD, with the exception that they were deposited onto Si wafers (001, p-type) for the NRA and RBS analysis. These MeV ion beam analysis techniques have the major advantage that they give absolute concentration in atoms/cm2 without the need for measurements relative to “standard samples.” These methods are described in detail in refs 25 and 33. Briefly, the H content is determined by bombarding the films with ~6.5 MeV 15N ions, which can induce a nuclear reaction with protons (hydrogen) in the target emitting a characteristic gamma-ray. By varying the bombarding energy and measuring the number of characteristic gamma-rays emitted, the H content vs depth can be determined. The other light elements in the target (C, N, and O) were determined by bombarding with a 1.2 MeV deuteron beam and measuring the counts from nuclear reactions. The C, N, and O contents were determined by the yields from the 12C(d,po)13C, 14N(d,α1)12C, and 16O-(d,po)17O, respectively.25,33 The Si content was determined by conventional 2 MeV 4He RBS. All three techniques rely on nuclear reaction or scattering cross sections. They are independent of chemical bonding, resulting in determination of absolute elemental concentrations (atoms/cm2). Once a film composition was determined as outlined above, it was used to predict the complete RBS energy spectrum with no adjustable parameters. The comparison of its predicted spectrum with its measured spectrum provided a robust check and ensured that there were no large errors or omission in the film’s analysis.
2.12. RBM, RMM, and Partial Charge Theory.
The RMM calculations were based on the theory that the model is a simple mixture of Si3N4, Si–Si, and SiO2 phases. The Bruggeman approximation equation helps associate the refractive index to the volume fractions of the phases.16,34 It is given by
| (1) |
where vx is the volume fraction of that phase, nx is the refractive index of the phase, and n is refractive index of the film.
The Bruggeman effective medium approximation (EMA) method assumes that the grains of each phase in the composite are randomly mixed, and that their dimensions are much smaller than the wavelength of light but large enough for each grain to be characterized by its bulk optical response.35 For our model, we considered excess Si, Si3N4, and SiO2 (i.e., Si–Si, Si–N, and Si–O bonds) phases.
We assumed that all of the oxygen and nitrogen was found in the form of bonded Si3N4 and SiO2 states. Thus, using mass balance equations
one can derive
| (2) |
The factor of 1.3 stems from the ratio of the atomic concentration of oxygen in SiO2 (4.64 × 1022 cm−3) and that of nitrogen in Si3N4 (5.88 × 1022 cm−3).34
The volume fractions of all of the phases should sum to 1
| (3) |
Using eqs 1, 2, and 3, we modeled the expected phase mixture ratios of SiO2, Si3N4, and Si–Si phases. This was denoted as the RMM fit, and the results are shown in Table 3.
Table 3.
Calculations for the RMM and RBM Mathematical Models for Si–O–N Filmsa
| RMM, [Si–Si]–[Si–O]–[Si–N] |
RBM, SizOxNy |
partial charge (Pi) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| sample | a:b | Si–Si | Si–O | Si–N | z | x | y | Si | O | N | ∑Pi |
| Si3N4 | 1.73 | 0.02 | 0.04 | 0.94 | 1.00 | 0.00 | 1.33 | 0.24 | 0.00 | −0.18 | 0.00 |
| 1 | 1.72 | 0.05 | 0.11 | 0.83 | 1.00 | 0.14 | 3.86 | 0.36 | −0.21 | −0.09 | 0.00 |
| 2 | 1.7 | 0.01 | 0.35 | 0.64 | 1.00 | 0.49 | 3.51 | 0.38 | −0.20 | −0.08 | 0.00 |
| 3 | 0.01 | 0.63 | 0.36 | 1.00 | 1.24 | 2.76 | 0.40 | −0.18 | −0.06 | 0.00 | |
| 4 | 1.2 | 0.08 | 0.90 | 0.02 | 1.00 | 3.65 | 0.35 | 0.49 | −0.13 | −0.01 | 0.00 |
| 5 | 1.15 | 0.00 | 1.00 | 0.00 | 1.00 | 4.00 | 0.00 | 0.51 | −0.13 | 0.00 | 0.00 |
| SiO2 | 1.02 | 0.01 | 0.99 | 0.00 | 1.00 | 2.00 | 0.00 | 0.40 | −0.20 | 0.00 | 0.00 |
a:b represents the peak area ratio based on XANES analysis of the silicon L-edge. Partial charge calculations assert the validity of the RBM model (∑Pi = 0).
For the RBM, there are five distinct stoichiometric tetrahedrons possible: SizOxNy where x + y = 4. By convention, these tetrahedrons are referred to as Siz where z is equal to the number of silicon atoms in a given tetrahedron. A relation for the distribution of these Siz tetrahedrons as a function of oxygen (x) and nitrogen (y) contents has been developed by Phillips and is given in eq 417
| (4) |
where W(x, y) is the work function associated with the random bonding model. The ratios of O/Si (x) and N/Si (y) are used along with the coordination number (v) for finding the random distribution of the five potential tetrahedrons that represent the SiOvN4−v models.
Partial charge calculations were conducted to observe the trend in the shift of the Si 2p3/2 peaks as compared to the partial charge on Si. In addition, when the net charge (summation of partial charges on Si, O, and N) was considered equal to zero, the mathematical model used to find the different O and N stoichiometry in each film composition was completely satisfied.9 The equations used for partial charge calculations were as follows
| (5) |
where SSiON = (SSikSOmSNn)1/(k+m+n)
| (6) |
| (7) |
| (8) |
3. RESULTS
3.1. Device Fabrication and Characterization.
Three regions were fabricated on top of the Si-wafer (Figure 2a and b). Region C is the EB-PVD Ti layer, region B is the thermally own TiO2 layer, and region A is the PECVD-based SiOx layer. Ti layers were found to have a columnar structure, and within ie Ti layer, columnar planes of Ti atoms were clearly observed. The thermally grown TiO2 was then overlaid with a PECVD silica layer. PECVD overlays were found to be conformal with the underlying columnar Ti layer surface. The interface between the PECVD overlay and underlying Ti/TiO2 was observed to have a gradual transition with structural features distinct for each region and a less distinctive structure between regions (Figure 2b). EDS analysis showed low levels of contaminant elements for high-nitrogen and high-oxygen samples (Figure 2c and d).
Figure 2.

Transmission electron micrographs of (a) the fabricated Ti–TiO2–SiOx based biomedical device and (b) the magnified view of the Ti–O–Si interface. EDS spectra showing low contaminant element levels for a (c) high-nitrogen and (d) high-oxygen Si–O–N sample.
3.2. XPS Analysis.
Results from the XPS analysis (Figure 3a) were obtained by milling through the thickness of the device. These data present the changes in elemental composition for the entire coating (overlays) as a function of deposition depth, including regions A, B, and C. The binding energy of individual elements observed in these regions is given in Figure 3b for each region. The XPS results confirmed the coating thickness to be approximately 100 nm of PECVD SiOx, 300 nm of Ti layer, and a graded interface between the SiOx overlay and TiO2 layers. This graded interface exhibits near stoichiometric concentrations of SiO2 and TiO2 at the interface, indicating that the silica and titania layers formed direct ionic bonding between the two layers. The TiO2 layer was distinct and had a thickness of approximately 10 nm. However, data for oxygen within the Ti layer (O–Ti) showed oxygen diffused into the Ti layer, likely along the columnar grain boundaries.36
Figure 3.

XPS analysis of elemental composition through the thickness of the device from Figure 2a. (a) Depth scale determined by a sputter rate of 15 nm/min. Sputter cycles were 30 s. Shaded areas show particular regions that had different chemical signals, i.e., N was present in the SiO2 layer in e chemical form and was present in a different chemical form in the Ti layer. (b) Individual spectra from these regions are compared for Si, O, Ti, id N. Note that the N (NHx and nitride) signals are all plotted magnified by a factor of 10.
3.3. Refractive Index of the Films.
From this point forward, the study of the various film compositions was conducted using a Si wafer as the substrate for PECVD-based Si–O–N films. This was done to allow for further characterization without interference from the Ti surface. Moreover, the methods used below were calibrated to the Si wafer substrate which was subtracted from all analyses presented herein.
The refractive indices and deposition rates for each set of gas flow conditions was determined using ellipsometry (Table 1). The refractive index inhomogeneity was determined to be less than 0.005, and the thickness nonuniformity as measured at 15 points on each 4″ wafer was in the range of 1.2–2.4%. The refractive indices ranged from near that of silicon nitride (Si3N4, n ≈ 2.0)37,38 to near that of silicon dioxide (SiO2, n ≈ 1.46).39 Deposition rates decreased and refractive indices increased as the flow of nitrous oxide decreased. This was expected, as N2O was the primary source of oxygen incorporated into the film. Table 1 also shows the refractive index of the samples as a function of the ratio of the flow rates for the primary reactive gases, N2O and SiH4.
3.4. X-ray Diffraction (XRD) Analysis.
X-ray diffraction was used to confirm the amorphous nature of the films. Figure 4a shows representative XRD spectra from the highest-nitrogen- and highest-oxygen-content samples. Although X-ray spectra do not show the two broad 2θ peaks typical to amorphous Si–O–N films40 (i.e., 20–30 and 60–80°), it can be assumed that the SiOx sublayer dominates the spectra, resulting in one amorphous silica peak. All collected XRD spectra were found to have a similar broad peak between 20 and 40°.
Figure 4.

Surface characterization of PECVD Si–O–N films. (a) X-ray diffraction results indicating the presence of amorphous silica-based films. (b) FTIR spectra of Si–O–N films with the Si–H and N–H spectral contribution magnified. (c) Plot of normalized N–H and Si–H bond concentration as a function of refractive index. The concentrations of N–H (0.128 × 1022 atoms cm−3) and Si–H (1.8 × 1022 atoms cm−3) on amorphous silica surfaces (n = 1.45) evaluated by FTIR-ATR in this work were used to illustrate the relative change in these bond concentrations for higher refractive index samples. (d) Sessile drop contact angle of water and diiodomethane. (e) Calculated surface energy of samples, broken into polar γp and dispersive γd components. The dispersive component is relatively constant, while the polar component decreases with increasing nitrogen content.
3.5. FTIR Characterization and Nuclear Reaction Analysis (NRA).
A thicker coating (1.0 μm) of Si–O–N was deposited for FTIR and NRA characterization. Figure 4b shows the absorption spectra of the Si–O–N films. Strong Si–O bending and Si–O stretching peaks near 790 and 1140 cm−1, respectively, were seen in all spectra, indicating that information from the oxide sublayer was most likely also being observed. It should be noted that the region from 2100 to 4000 cm−1 was magnified 5-fold to highlight the contribution of both the 2270 cm−1 Si–H and 3385 cm−1 N–H stretching bands. The position of the Si–H band shifts from about 2270 cm−1 in the high refractive index samples to about 2252 cm−1 in the lower index samples, while the N–H peak position remains constant. The hydrogen concentration was calculated by deconvolution of the Si–H and N–H bands and fitting them to Gaussian distributions. The observed H concentration from Si–H bonds gradually increased as a function of refractive index, ranging from 1.32 to 1.65 × 1021 atoms/cm3, while the N–H bond concentration showed only a modest increase from the low to the high refractive index samples. The Si–H and N–H bond concentrations (normalized to pure amorphous silica, SiOx) are shown in Figure 4c.
Table 2 shows the results of nuclear reaction analysis of the as-deposited PECVD overlays. Samples are arranged from the highest N-rich overlays (sample 1) to the highest O-rich overlays (sample 5). The increased presence of H with increasing N content appears to limit the random mixing of N in the Si–N structure. This is indicated by the lower concentration of Si and N in the high N-rich structure as compared to their stoichiometric values given in Table 2 for Si3N4. This is likely due to under-coordinated Si and N within the Si–N overlay.
Table 2.
Atomic Concentration of Elements in Si–O–N Overlays Measured by Nuclear Reaction Analysisa
| element surface concentration (1015 atoms/cm2) |
||||||
|---|---|---|---|---|---|---|
| sample | C | N | O | Si | H | areal density (mg/cm2) |
| 1 | 8.337 | 562.7 | 29.65 | 410.7 | 238.0 | 0.03372 |
| (at. %) | 0.6673 | 45.04 | 2.373 | 32.88 | 19.05 | |
| 2 | 8.187 | 589.0 | 122.7 | 420.1 | 256.0 | 0.03729 |
| (at. %) | 0.5865 | 42.19 | 8.811 | 30.09 | 18.33 | |
| 3 | 7.159 | 333.3 | 225.2 | 356.0 | 175.0 | 0.03089 |
| (at. %) | 0.6526 | 30.38 | 20.51 | 32.45 | 15.99 | |
| 4 | 6.031 | 44.95 | 710.7 | 424.7 | 44.9 | 0.04009 |
| (at. %) | 0.4899 | 3.652 | 57.76 | 34.50 | 3.646 | |
| 5 | 5.726 | 0 | 814.7 | 408.8 | 22.10 | 0.04104 |
| (at. %) | 0.4577 | 0 | 65.15 | 32.68 | 1.767 | |
The areal density was determined from the sum of the elemental concentration and multiplying by the average molecular weight. This method is very sensitive to carbon, and the carbon results above are likely a surface contamination (almost universally present on surfaces).
3.6. Surface Energy Evaluation.
Water contact angles (Figure 4d) increased with increasing nitrogen content. High-oxygen-content samples (4 and 5) were more hydrophilic than the high-nitrogen-content samples (1 and 2). For this sample set, the diiodomethane contact angle was relatively constant, indicating that the dispersive component was relatively constant for these surfaces. Differences observed in the contact angle with water can be primarily attributed to a change in the polar component of the surface energy. Figure 4e shows the polar (γp), dispersive (γd), and total surface energy (γ) values calculated from the Owens–Wendt–Kaeble equation in mJ/m2. The dispersive component for all samples ranged from 23.1 to 24.9 mJ/m2, while the polar component varied with nitrogen content. The highest-oxygen-content sample (5) had a polar component of 32.1 mJ/m2, while the highest-nitrogen-content sample (1) was nearly half at around 16.1 mJ/m2. The variation of oxygen and nitrogen in these coatings allowed for tunability of the polar component of the surface energy. This may provide a limited method for control over protein adhesion and cell attachment.41 The tunability of the surface energy of these coatings makes them attractive for use in biomedical applications. This stems from the fact that the O/N ratio in these films influences the contact angle, the surface energy, and in turn the attachment, migration, and growth of cells on the Si–O‒N surface.41
3.7. XANES Analysis.
The Si L2,3-edge42,43 and Si K42-edge can be used both to investigate the chemical species that give rise to changes in binding energies and to distinguish between 4-fold and 6-fold coordination of Si, while the O K-edge44 and N K-edge can be used to investigate local oxygen and nitrogen structure.
3.7.1. Silicon K-Edge.
Figure 5b shows the Si K-edge TEY data for four sample chemistries and the SiO2 and Si3N4 standards. There are two main peaks of interest in these spectra: peak a, the main Si K-edge peak around 1847 ± 0.3 eV, and a post-edge peak b feature common to all spectra around 1864.8 ± 0.2 eV. Peak a can be thought of as the sum total contribution of Si–Si bonding (~1841 eV), Si–N bonding (~1844.5 eV), and Si–O bonding (1847 eV) resonances.21 In the Si3N4 standard, some oxygen contamination was observed and the peak was split between Si–N and Si–O resonant energies. On the other hand, the high-N-content samples showed evidence of all three bonding forms with a leading edge around 1841 eV and a broad absorption peak encompassing both Si–N and Si–O resonant energies. The high-O-content samples primarily showed Si–O bonding, with a small spectral contribution around 1841 eV, suggesting Si–Si bonding. In contrast, the SiO2 standard peak a was centered at 1847 ± 0.3 eV and had a leading edge at 1843 ± 0.3 eV, which was well above the energy level suggesting Si–Si bonding.
Figure 5.

(a) Silicon L2,3-edge XANES spectra and (b) silicon K-edge XANES spectra of four Si–O–N coating chemistries with Si3N4 and SiO2 standards for comparison.
From the Si K-edge data above, [4]Si and [6]Si coordination in O- and N-containing samples was difficult to resolve. The main 1847 eV Si–O peak is attributed to [4]Si coordination, [6]Si coordination peaks occur around 1848.5 eV (main line) and 1853 eV (post-edge resonance). Whereas the higher-O-content samples and SiO2 standard show no evidence of either peak, these spectral features overlap with the broad high-N-content main edge peak, which both have a post-edge shoulder that extends to energies higher than 1853 eV, making it difficult to draw a conclusion about any [6]Si content in these samples. Also of note is the shift toward lower energy of peak a as the N content increases in the samples. This was also observed by Wilson et al.,21 in which it was suggested to manifest as a result of multiple coordinated structures. Further investigation of the Si L-edge was therefore needed to ascertain if such coordination structures could be observed.
3.7.2. Silicon L2,3-Edge.
Figure 5a shows the FY data for four sample chemistries and the SiO2 and Si3N4 standards. Six peaks of interest were identified and labeled a–f. Peaks a and b at 105.5 ± 0.1 and 106.1 ± 0.1 eV, respectively, arise from the transition of 2p electrons to unoccupied 3d orbitals that have been split by ~0.6 eV by spin–orbital splitting.42,43 Table 3 shows the calculated a:b peak area ratio for the two model compounds and the four sample films investigated. The a:b peak area ratio data suggest that, while the silica tetrahedron-based RBM may provide a useful approximation of the Si–O–N bonding in the higher-oxygen-containing samples, the high-nitrogen-content samples may form a structure better approximated by that of the β-Si3N4 standard. The ratio of these peaks has been used to differentiate the contribution from disparately substituted SiO4 tetrahedra.31 That is, the a:b integrated peak area ratio can be used as a semiquantitative measure of nitrogen substitution into a SiO4 tetrahedron, which aligns well with the RBM view of Si–O–N structure.
The highest intensity peak c that was found in both standards and all samples is the main Si L2,3-edge peak. Its position can be used to distinguish between 4-fold ([4]Si) and 6-fold ([6]Si) coordinated silicon in silicate glasses.42,45 For [4]Si, the main peak is centered at 107.9 ± 0.2 eV, while in [6]Si an additional main edge peak is observed at 106.7 ± 0.2 eV.43 In Figure 5a, the peak c line was centered at 108.0 ± 0.2 eV, indicating that [4]Si was in all samples and standards. However, second order derivative fitting of samples 1 and 2 indicates that there may be a peak centered near 106.8 eV, which may be an indication that a mix of [4]Si and [6]Si was present in these chemistries. Peak d centered at 115.3 ± 0.2 eV was a resonance Si-peak found in both Si3N4 and SiO2 and therefore cannot be used to distinguish between the chemical fingerprints of either structure. Peak e is characteristic of Si–N resonance bonding21 and was found centered at 103.2 ± 0.2 eV for the Si3N4 standard. This peak was observed to shift to higher energies in high-N-content samples 1 (104.0 ± 0.2 eV) and 2 (104.7 ± 0.2 eV), which indicates a distortion of the Si–N bond in a nitride matrix.21 Peak f at 99.7 ± 0.2 eV was found in all samples, though the intensity of the main edge peak makes it difficult to ascertain in samples 4 and 5. This peak has previously been observed in other Si–O–N films with excess Si and is attributed to Si–Si bonding in silicon nanoclusters (Si-nc) found within the coating.21
Silicon L-edge data support the theory that the high-oxygen-content Si–O–N sample coating chemistries can be described as N-substituted silica tetrahedrons ([4]Si) with small amounts of Si-nc to account for excess Si according to the RBM view of the structure. However, the high-nitrogen-content samples show inconclusive evidence of both [4]Si and [6]Si coordination with oxygen substituted into a β-Si3N4-like matrix that becomes increasingly distorted with additional oxygen content.
3.7.3. Oxygen K-Edge.
Figure 6 shows the O K-edge TEY and FY data for four sample chemistries and the SiO2 standard. All data have been scaled to the signal intensity of the SiO2 standard. There were three main peaks observed in these spectra: peak a, a pre-edge peak around 529.9 ± 0.2 eV, the main O K-edge peak b around 535.2 ± 0.2 eV, and a post-edge peak c feature around 558.5 ± 0.2 eV. Peak a, which was observed as a distinct peak in all sample chemistries in the TEY data and as a pre-edge shoulder in the SiO2 standard in both the TEY and FY data, suggests the presence of silanol (Si–OH) bonding at the near surface, while the absence of this peak in the FY data suggests that the primary oxygen bonding in the bulk of the films for all samples can be attributed to Si–O–Si bridges, as evidenced by the main peak b and post-edge resonance peak c.15
Figure 6.

Oxygen K-edge XANES TEY and FY spectra of four Si–O–N coating chemistries with an SiO2 standard for comparison.
3.7.4. Nitrogen K-Edge.
Figure 7 shows the N K-edge TEY and FY data for four sample chemistries and the Si3N4 standard. All data have been scaled to the signal intensity of the Si3N4 standard. There were three main peaks observed in these spectra: peak a, around 398.6 ± 0.2 eV, the main peak b around 402.6 ± 0.2 eV, and a post-edge peak c feature around 421.4 ± 0.2 eV. Peak a has been attributed to the presence of under-coordinated (i.e., 2-fold coordinated) or hydrogen-bound nitrogen which indicates either the presence of N–H bonding or the substitution of a nitrogen atom for an oxygen atom into a Si–O–Si bridge.15,46
Figure 7.

Nitrogen K-edge XANES TEY and FY spectra of four Si–O–N coating chemistries with an Si3N4 standard.
While this peak was particularly prominent in the TEY spectra of the lowest-N-containing sample, it was also observed as a pre-edge shoulder in the other TEY spectra. The apparent increased intensity of the peak in the low-N sample 5 chemistry suggests that a proportionately larger amount of N in this sample was 2-fold coordinated, or, conversely, that the Si–O–Si bridge network remains relatively undisturbed by the limited amount of N incorporation into the coating. The main edge peak was similar for all samples and the Si3N4 standard, suggesting that the bulk of the spectral contribution was due to Si–N bonding. However, the post-edge peak c resonance appears distorted in the lower-N-content sample FY data. The peaks observed between 425 and 435 eV in these spectra may be due to a higher order spectral contribution from silicon, which overwhelmed the N signal in these relatively Si-rich and N-poor films. Also of note was the apparent shift of the N K-edge spectra (TEY) as a function of increasing N content, which was similarly found in the Si K-edge spectra and associated with a change in bond coordination as explained above.
3.8. RMM and RBM Calculations.
Calculations for the RMM and RBM are presented in Table 3 and Figures 8 and 9. For the random mixing model, the calculations were performed using the RBS compositional data and eqs 1–3. The calculated RMM values are given in Table 3. For the RMM, the calculated values for the O-rich samples (refractive index of 1.45 and 1.57) are not in agreement with the XANES spectra, because the Si Land K-edges do not indicate the presence of Si–Si or Si3N4 bonding. Thus, the RMM fit for the O-rich films is not applicable. For the RBM based on calculation from eq 4 and represented in Figure 8, it can be observed that the SiO4 tetrahedron has a higher ratio (in the higher-O-content samples), indicating tetrahedral coordination for Si. This was further confirmed by the XANES silicon L-edge results showing the a:b peak ratio closer to unity for the higher-O-content (1.45 and 1.57 refractive index) samples. Figure 8 shows the calculated work function based on the RBM view of the structure using the measured RBS data in which several types of silica tetrahedra were compared for fit. According to the RBM, it was found that even the high-N-content samples have Si in tetrahedral coordination as opposed to the trigonal coordination observed from the XANES data. Therefore, the RBM is applicable only for high-O-content samples.
Figure 8.

Results of calculations using the data from RBS for the Si–O–N samples in Table 2. For the O-rich samples (n = 1.45, 1.57), W(x, y) was estimated to be close to unity, indicating their fit with the SiO4 tetrahedron, while the samples with increased N content (n = 1.65, 1.82, 2.0) exhibited coordination that is not reflected in the tetrahedral model.
Figure 9.

Calculated partial charge of silicon P(Si), oxygen P(O), and nitrogen P(N) as a function of Si–O–N film refractive index. Also plotted in this figure are the experimentally determined O/Si ratio (x) and N/Si ratio (y) and the a:b peak area ratio based on XANES analysis of the silicon L-edge.
The results of partial charge calculations are given in Figure 9. Reflecting on these results, decreased N content results in an increase in the partial charge on each atom, an increase in the electronegativity between the Si–N and Si–O bonds, and a decrease in the a:b peak ratio resulting in higher surface polarity. This was also observed in surface energy evaluation, where lowering N content was shown to have increased surface energy, and decreased contact angle, indicating the surface was more wettable. Improved biological activity, protein adhesion, and cell-attachment have been reported for relatively more hydrophilic (lower contact angle) surfaces.22,47,48 Thus, the lower a:b peak ratio observed in the XANES data can be related to the lower contact angle measurements; these results help predict the potential ability of a material to facilitate cell growth and migration.
4. DISCUSSION
Five distinct Si–O–N chemistries were fabricated via a repeatable PECVD process designed to create potentially bioactive Si–O–N films on commercially pure Ti surfaces. These films ranged from nitrogen-rich Si–O–N (for films produced at low N2O flow rates between 0 and 16 sccm) to oxygen-rich Si–O–N (for films produced at high N2O flow rates of 155 and 160 sccm). XRD spectra indicated that the films were amorphous. XPS results demonstrated formation of a Ti–O–Si interface between the PECVD films and the underlying Ti/TiO2.
The relatively high N–H and Si–H bond concentrations calculated from the FTIR spectra suggest that these films may be partially soluble under physiological conditions because readily available surface silanols by the PECVD process49,50 and low nitrogen content (high N–H and Si–H bonding) allow immediate dissolution/degradation in vitro as explained in our previous work.5,22 The highest concentration of Si–N and Si–H bonding was observed in the highest-nitrogen-content samples. Comparing the N–H bond concentration to the nitrogen concentration in the samples revealed that a greater percentage of the nitrogen in the low-nitrogen-content samples was bound to hydrogen. Thus, hydrogen bonding may limit nitrogen substitution within the Si–O network as a byproduct of the PECVD process, and it prevents alterations of the Si–O tetrahedral structure. This change in relative under-coordinated nitrogen in the films may have implications for in vitro dissolution.51
On the basis of surface energy and contact angle measurement, the high-O-containing samples were generally more wettable than the high-N-containing samples. Reduced wettability has been linked to the decreased O–H density with increased N doping.50,52 Our results further indicate that poorer wettability (higher contact angle) and decreased surface energy were associated with increased surface H bonding with increasing N doping. In previous work, these trends were observed to promote more osteoblast migration on Si–O surfaces as compared to Si–O–N or Si–N surfaces.22 However, Si–O–N surfaces promoted rapid human periosteum cell formation of collagen-like ECM and enhanced their osteogenic response as compared with Si–O or Si–N surfaces.22 These results and discussion suggest that surface polarization, contact angle, and surface energy associated with hydrogen bonding are likely reliable predictors of cell migration, whereas other factors, such as surface chemical coordination associated with surface nitrogen bonding, surface N–H bonding, and ion dissolution, play more of a role during osteoblast differentiation, collagen ECM bonding, and mineralization.
From the XANES spectral features in these elemental edges, it is apparent that increased N content alters the bonding environment of the Si–O–N films. For the higher-nitrogen-content films, the Si L-edge a:b peak ratio remains close to that of Si3N4 for these samples which suggests that the Si–N bonding environment is similar to the nitride standard, which would be expected in a random mixture of silica and silicon nitride. The broad main peak a in the Si K-edge spectra for these samples also suggests that Si–O and Si–N bonding both exist in forms similar to the silica Si–O bonding and the nitride Si–N bonding structures simultaneously, since the broad peak overlaps with both of the standards. For the higher-oxygen-content films, the Si L-edge a:b peak ratio remains close to that of SiO2 for these samples which suggests that the Si–O bonding environment is similar to that of silica. The narrow main peak a in the Si K-edge spectra for these samples also suggests that silica-like Si–O bonding dominates the structure. The presence of the sharp resonance peak in the N K-edge data also suggests that nitrogen is substituted into the place of a bridging oxygen atom, which would occur in systems where the silica tetrahedron is the basic structural unit.
The a:b peak ratio has been used to differentiate the contribution from disparately substituted SiO4 tetrahedra.31 As applied in this work, this ratio could be used as a semiquantitative measure of nitrogen substitution into a SiO4 tetrahedron and would align with the RBM view of Si–O–N structure. However, this association does not hold for higher N-rich Si–O–N films. On one hand, Criado et al.15 showed that increasing N content did not induce a change in the absorption edge for Si–N as compared to Si–O. They found them to be of the random bonding model, which aligns more with the topological constraint theory that generally associates the coordination structure of amorphous films to the average coordination number of the constitutive atoms. On the other hand, Rahman et al.20 viewed the structure from a random mixing model in which the individual structures of Si–Si, Si–O, and Si–N simply mix and do not phase segregate or cluster based on compositional analysis. These 2 opposing viewpoints have been shared fairly equally among a number of research groups, hence, added computational analysis was needed to ascertain the appropriate model for the XANES, RBS, and refractive index data.
Computational analysis of the compositions coupled with surface characterization found that the high-O-containing films conformed to the RBM evidence by the work function (W(x, y)) showing a value of unity for the higher-O-content sample (n = 1.45). Meanwhile, partial charge calculations showed that O maintained a partial charge close to that of the pure oxide, which indicated that Si–O bonding governed O incorporation in high-N-containing films. Coupled with this was a loss in the partial charge of N with increasing N content. These results indicate Si–O, Si–N, and Si–Si bonding when coupled with XANES analysis. This illustrates that the RMM is suitable for high-N-containing films. Table 3 was revised to reflect the application of RBM and RMM models to the samples based on composition, XANES a:b peak ratios, and our calculations using the RBM view of the electronic structure.
The change in the a:b peak ratio with a change in the nitrogen content in the films was of note. Pure silicon will always have an a:b peak area ratio equal to 2, since this represents 4 2p electrons with 3/2 energy (2p3/2) vs 2 2p electrons with 1/2 energy (2p1/2). These 2p core shell electron binding energies can shift depending on the amount of N and O added to Si. This is due to a charge transfer that occurs between Si and O or N.53 In the case of oxygen, this highly electronegative atom shifts the 2p core electrons toward higher energy, as indicated by a shift in the a:b peak ratio from 2:1 to 1:1. As the nitrogen concentration increased, the binding energy of the Si 2p core shell electrons shifts toward a lower value because of the higher surrounding electron density.51 This effect of increasing N content is accompanied by a shift of the N 1s line (N K-edge spectra), which is connected with the increased number of next-nearest and further far-neighbor N atoms (induction effect).53 Thus, the decrease in partial charge on Si with increasing N content suggests that the 2p core shell electron shifts to lower energy, thereby resulting in an increased a:b peak ratio.
In previous work,22 we found that these films promote different types of biominerals. High-O-containing Si–O–N and pure Si–O films promoted the formation of calcium phosphate, while a mixture of phosphate and carbonate was observed for higher-N-containing Si–O–N and pure Si–N samples, as shown by the O K-edge XANES analysis of Si–O–N samples (Figure 10a). The coupled formation of carbonate and phosphate near edge structures indicates the presence of hydroxycarbonate apatite with increasing carbonate to phosphate ratio as the N/O ratio increases in the Si–O–N film. Given that calcium carbonate and calcium phosphate can exhibit trigonal and tetrahedral bonding structures, respectively, it may be likely that formation of these different mineral types depends on random bonding or random mixing within the Si–O–N films. Random bonding within tetrahedrally coordinated Si–O–N likely promoted the surface formation of tetrahedrally coordinated phosphate-containing mineral, while the random mixing of Si–O (tetrahedral) and Si–N (trigonal) likely promoted the formation of phosphate- and carbonate-containing (trigonal) biomineral. This carbonate to phosphate ratio of resultant biomineral is vital to the healing rate of different physiological biomineral structures. Thus, the ability for Si–O–N films to be tuned to elicit varying resultant carbonate-to-phosphate ratios allows for tailored design of osseointegratable implants for a range of bone implant applications with predictable healing rates.
Figure 10.

(a) XANES analysis of O K-edge data showing that, as the N content increases, the presence of O peaks coordinated in a carbonate structure appears with increasing intensity relative to the peak showing O coordinated into a phosphate structure. (b) Raman spectroscopy showed the initial layers of silica on Si–O–N surfaces. (c) Similarly, the optical micrograph showed no minerals present on the surfaces initially. (d) After 4 weeks of in vitro cell culture, Si–O–N surfaces exhibited the formation of carbonate, phosphate, and amide peaks associated with mineralized tissue to indicate the presence of collagenous matrixes. (e) Optical micrograph showing the presence of hydroxycarbonate apatite biomineral on the Si–O–N surface, indicated by the formation of white nodules on the surface. Reprinted with permission from American Chemical Society: ACS Applied Materials and Interfaces,22 copyright (2015).
The effect of the a:b peak ratio on the biomineral carbonate to phosphate peak ratio may be explained as follows. In our previous work, a shift in the O K-edge to lower binding energy was observed with increased N content, resulting in formation of the carbonate structure for mineralized tissue that formed by human periosteum cells on the surface of Si–O–N films (Figure 10). The shift toward lower energy in the 2p core electron shell of Si in Si–O–N films coincides with a similar shift in the O K-edge in the biomineral. This associated effect may induce the formation of calcium carbonate via the lower binding energy of electrons in the oxygen within the biomineral bound to the Si atoms in the Si–O–N films. The incorporation of N into the silica network promoted the rapid formation of collagen ECM by human periosteum cells. This could be due to the increased surface N–H groups (Figure 4c) that can bind and substitute for amide groups within collagen (Figure 10d) as it forms prior to mineralization. From an application point of view, this means that fabrication of biomaterials for targeted bone formation and bone chemistry can be influenced by the fabrication of the material by targeting the a:b peak ratio. This suggests that the a:b peak ratio could be potentially used as a predictive tool for targeting various types of bone formation; in turn, this a:b peak ratio can be targeted on the basis of the input conditions of the PECVD process.
5. CONCLUSIONS
The aim of this study was to investigate the role that nitrogen and hydrogen play on the surface chemical structure of PECVD-based amorphous Si–O–N films for future bone implant applications. A range of Si–O–N compositions was used for these studies. XANES analysis revealed that high-O-containing Si–O–N films and pure silica exhibited Si–O bonding with 2p3/2:2p1/2 (a:b peak ratio is Si L-edge) to be close to unity, while high-N-containing Si–O–N films and pure silicon nitride films exhibited Si–Si, Si–O, and Si–N bonding with 2p3/2:2p1/2, which increased with increasing N content. Ellipsometry and RBS analysis combined with computational analysis showed that high-O-containing Si–O–N and pure silica films are described by the RBM, hence denoting their tetrahedral structure as SizOxNy. High-N-containing Si–O–N and pure silicon nitride films are described by the RMM, hence denoting their structure as [Si–Si]–[Si–O]–[Si–N]. The change in the electron configuration and bonding structure of Si–O–N films with increasing N content was attributed to changes in the partial charge on Si, N, and O based on the combined analyses. FTIR analysis and NRA-RBS data showed that Si–H and N–H bonding increased with increasing N content. In low-N-containing Si–O–N samples, the small amount of N that was present was likely bound to surface hydrogen as a consequence of the PECVD process. Thus, the density of hydrogen to nitrogen bonding influences the coordination of N in amorphous Si–O–N prepared by PECVD.
ACKNOWLEDGMENTS
The authors would like to thank Drs. Nickolay Lavrick, Harry Meyer, and Karen Moore for their assistance with characterization of materials in the HTML at ORNL via the CNMS and SHARE program. The authors would like to thank Drs. Lucia Zuin, Tom Regier, and Youngfeng Hu at the Canadian Light Source Inc. for their assistance with XANES analysis. The XANES experiments were performed at the Canadian Light Source, which is supported by NSERC, NRC, CIHR, and the University of Saskatchewan. We also thank Kamal Awad for his help with data analysis. Si–O–N thin films were deposited at the Nanofabrication Facility at UT Arlington. FTIR experiments were conducted at Center for Characterizations for Materials and Biology at The University of Texas at Arlington. The work was supported by a grant from National Institutes of Health/National Institute for Dental and Craniofacial Research (1R03DE023872-01A1, Varanasi PI).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Ay F; Aydinli A Comparative Investigation of Hydrogen Bonding in Silicon Based Pecvd Grown Dielectrics for Optical Waveguides. Opt. Mater 2004, 26, 33–46. [Google Scholar]
- (2).Tousi NS; Velten MF; Bishop TJ; Leong KK; Barkhordar NS; Marshall GW; Loomer PM; Aswath PB; Varanasi VG Combinatorial Effect of Si 4+, Ca 2+, and Mg 2+ Released from Bioactive Glasses on Osteoblast Osteocalcin Expression and Biomineralization. Mater. Sci. Eng. C 2013, 33, 2757–2765. [DOI] [PubMed] [Google Scholar]
- (3).Varanasi VG; Owyoung JB; Saiz E; Marshall SJ; Marshall GW; Loomer PM The Ionic Products of Bioactive Glass Particle Dissolution Enhance Periodontal Ligament Fibroblast Osteocalcin Expression and Enhance Early Mineralized Tissue Development. J. Biomed. Mater. Res. Part A 2011, 98A, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Varanasi V; Saiz E; Loomer P; Ancheta B; Uritani N; Ho S; Tomsia A; Marshall S; Marshall G Enhanced Osteocalcin Expression by Osteoblast-Like Cells (Mc3t3-E1) Exposed to Bioactive Coating Glass (Sio 2–Cao–P 2 O 5–Mgo–K 2 O–Na 2 O System) Ions. Acta Biomater. 2009, 5, 3536–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ilyas A; Odatsu T; Shah A; Monte F; Kim HK; Kramer P; Aswath PB; Varanasi VG Amorphous Silica: A New Antioxidant Role for Rapid Critical-Sized Bone Defect Healing. Adv. Healthcare Mater. 2016, 5, 2199–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Varanasi V; Leong KK; Jue SM; Dominia LM; Loomer PM; Marshall GW Si and Ca Combinatorially Target and Enhance Early Mc3t3-E1 Osteoblast Expression of Osteocalcin. J. Oral Implantol. 2012, 38, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Varanasi V; Velten M; Odatsu T; Ilyas A; Iqbal S; Aswath P Surface Modifications and Surface Characterization of Biomaterials Used in Bone Healing. Mater. Dev. Bone Disord. 2017, 405. [Google Scholar]
- (8).Sivaram S Chemical Vapor Deposition: Thermal and Plasma Deposition of Electronic Materials; McGraw–Hill: New York, 2013. [Google Scholar]
- (9).Gritsenko V; Kwok R; Wong H; Xu J Short-Range Order in Non-Stoichiometric Amorphous Silicon Oxynitride and Silicon-Rich Nitride. J. Non-Cryst. Solids 2002, 297, 96–101. [Google Scholar]
- (10).Jehanathan N; Liu Y; Walmsley B; Dell J; Saunders M Effect of Oxidation on the Chemical Bonding Structure of Pecvd Sin X Thin Films. J. Appl. Phys 2006, 100, 123516. [Google Scholar]
- (11).Lanford W; Rand M The Hydrogen Content of Plasma-Deposited Silicon Nitride. J. Appl. Phys 1978, 49, 2473–2477. [Google Scholar]
- (12).Feng B; Chen J; Qi S; He L; Zhao J; Zhang X Characterization of Surface Oxide Films on Titanium and Bioactivity. J. Mater. Sci.: Mater. Med 2002, 13, 457–464. [DOI] [PubMed] [Google Scholar]
- (13).Kaelble D Dispersion-Polar Surface Tension Properties of Organic Solids. J. Adhes 1970, 2, 66–81. [Google Scholar]
- (14).Owens DK; Wendt R Estimation of the Surface Free Energy of Polymers. J. Appl. Polym. Sci 1969, 13, 1741–1747. [Google Scholar]
- (15).Criado D; Alayo M; Pereyra I; Fantini M Structural Analysis of Silicon Oxynitride Films Deposited by Pecvd. Mater. Sci. Eng. B 2004, 112, 123–127. [Google Scholar]
- (16).Bruggeman VD Berechnung Verschiedener Physikalischer Konstanten Von Heterogenen Substanzen. I. Dielektrizitatskonstanten Und Leitfahigkeiten Der Mischkorper Aus Isotropen Substanzen. Ann. Phys 1935, 416, 636–664. [Google Scholar]
- (17).Philipp H Optical Properties of Non-Crystalline Si, Sio, Siox and Sio2. J. Phys. Chem. Solids 1971, 32, 1935–1945. [Google Scholar]
- (18).Scopel W; Fantini M; Alayo M; Pereyra I Local Structure and Bonds of Amorphous Silicon Oxynitride Thin Films. Thin Solid Films 2002, 413, 59–64. [Google Scholar]
- (19).Criado D; Zuniga A; Pereyra I Structural and Morphological Studies on Sioxny Thin Films. J. Non-Cryst. Solids 2008, 354, 2809–2815. [Google Scholar]
- (20).Rahman HU; Gentle A; Gauja E; Ramer R In Characterisation of Dielectric Properties of Pecvd Silicon Nitride for Rf Mems Applications, Multitopic Conference, 2008. INMIC 2008 IEEE International, IEEE: 2008; pp 91–96. [Google Scholar]
- (21).Wilson PR; Roschuk T; Dunn K; Normand EN; Chelomentsev E; Zalloum OH; Wojcik J; Mascher P Effect of Thermal Treatment on the Growth, Structure and Luminescence of Nitride-Passivated Silicon Nanoclusters. Nanoscale Res. Lett. 2011, 6, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ilyas A.; Lavrik NV; Kim HK; Aswath PB; Varanasi VG. Enhanced Interfacial Adhesion and Osteogenesis for Rapid “Bone-Like” Biomineralization by Pecvd-Based Silicon Oxynitride Overlays. ACS Appl. Mater. Interfaces 2015, 7, 15368–15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pauleau Y Materials and Processes for Surface and Interface Engineering; Springer Sci. Bus. Media: 2012; Vol. 290. [Google Scholar]
- (24).Lanford W Analysis for Hydrogen by Nuclear Reaction and Energy Recoil Detection. Nucl Instrum. Methods Phys. Res. Sect. B 1992, 66, 65–82. [Google Scholar]
- (25).Lanford W; Tesmer J; Nastasi M Handbook of Modern Ion Beam Materials Analysis. Mater. Res. Soc. Symp. Proc 1995, 193. [Google Scholar]
- (26).Amaral M; Lopes M; Santos J; Silva R Wettability and Surface Charge of Si 3 N 4–Bioglass Composites in Contact with Simulated Physiological Liquids. Biomaterials 2002, 23, 4123–4129. [DOI] [PubMed] [Google Scholar]
- (27).Correia NT; Ramos JJM; Saramago BJ; Calado JC Estimation of the Surface Tension of a Solid: Application to a Liquid Crystalline Polymer. J. Colloid Interface Sci. 1997, 189, 361–369. [Google Scholar]
- (28).Wenzel RN. Resistance of Solid Surfaces to Wetting by Water. Ind. Eng. Chem 1936, 28, 988–994. [Google Scholar]
- (29).Bikerman JJ The Surface Roughness and Contact Angle. J. Phys. Colloid Chem. 1950, 54, 653–658. [Google Scholar]
- (30).Morrow NR J. Can. Pet. Technol 1975, 14, 10.2118/75-04-04. [DOI] [Google Scholar]
- (31).Kasrai M; Lennard W; Brunner R; Bancroft G; Bardwell J; Tan K Sampling Depth of Total Electron and Fluorescence Measurements in Si L-and K-Edge Absorption Spectroscopy. Appl. Surf. Sci 1996, 99, 303–312. [Google Scholar]
- (32).Leinweber P; Kruse J; Walley FL; Gillespie A; Eckhardt K-U; Blyth RI; Regier T Nitrogen K-Edge Xanes–an Overview of Reference Compounds Used to Identifyunknown’organic Nitrogen in Environmental Samples. J. Synchrotron Radiat. 2007, 14, 500–511. [DOI] [PubMed] [Google Scholar]
- (33).Wang Y; Pan Z; Ho Y; Xu Y; Du A Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. Nucl. Instrum. Methods Phys. Res. Sect. B 2001, 180, 251–256. [DOI] [PubMed] [Google Scholar]
- (34).Kuiper A; Koo S; Habraken F; Tamminga Y Deposition and Composition of Silicon Oxynitride Films. J. Vac. Sci. Technol., B: Microelectron. Process. Phenom 1983, 1, 62–66. [Google Scholar]
- (35).Xiong Y-M; Snyder PG; Woollam JA; Al-Jumaily G; Gagliardi F; Krosche E R Controlled Index of Refraction Silicon Oxynitride Films Characterized by Variable Angle Spectroscopic Ellipsometry. Thin Solid Films 1991, 206, 248–253. [Google Scholar]
- (36).Golightly F; Stott F; Wood G The Influence of Yttrium Additions on the Oxide-Scale Adhesion to an Iron-Chromium-Aluminum Alloy. Oxid. Met 1976, 10, 163–187. [Google Scholar]
- (37).Kuo Y Plasma Enhanced Chemical Vapor Deposited Silicon Nitride as a Gate Dielectric Film for Amorphous Silicon Thin Film Transistors—a Critical Review. Vacuum 1998, 51, 741–745. [Google Scholar]
- (38).Subramanian A; et al. Low-Loss Singlemode Pecvd Silicon Nitride Photonic Wire Waveguides for 532–900 Nm Wavelength Window Fabricated within a Cmos Pilot Line. IEEE Photonics J. 2013, 5, 2202809–2202809. [Google Scholar]
- (39).Idris I; Sugiura O Film Characteristics of Low-Temperature Plasma-Enhanced Chemical Vapor Deposition Silicon Dioxide Using Tetraisocyanatesilane and Oxygen. Jpn. J. Appl. Phys 1998, 37, 6562. [Google Scholar]
- (40).Gaind A; Hearn E Physicochemical Properties of Chemical Vapor-Deposited Silicon Oxynitride from a Sih4-Co 2-Nh 3-H 2 System. J. Electrochem. Soc 1978, 125, 139–145. [Google Scholar]
- (41).Redey S; Razzouk S; Rey C; Bernache-Assollant D; Leroy G; Nardin M; Cournot G Osteoclast Adhesion and Activity on Synthetic Hydroxyapatite, Carbonated Hydroxyapatite, and Natural Calcium Carbonate: Relationship to Surface Energies. J. Biomed. Mater. Res 1999, 45, 140–147. [DOI] [PubMed] [Google Scholar]
- (42).Li D; Bancroft G; Kasrai M; Fleet M; Secco R; Feng X; Tan K; Yang B X-Ray Absorption Spectroscopy of Silicon Dioxide (Sio 2) Polymorphs; the Structural Characterization of Opal. Am. Mineral. 1994, 79, 622–632. [Google Scholar]
- (43).Demirkiran H; Hu Y; Zuin L; Appathurai N; Aswath PB Xanes Analysis of Calcium and Sodium Phosphates and Silicates and Hydroxyapatite–Bioglass® 45s5 Co-Sintered Bioceramics. Mater. Sci. Eng., C 2011, 31, 134–143. [Google Scholar]
- (44).Rajendran J; Gialanella S; Aswath PB Xanes Analysis of Dried and Calcined Bones. Mater. Sci. Eng., C 2013, 33, 3968–3979. [DOI] [PubMed] [Google Scholar]
- (45).Li D; Bancroft G; Kasrai M; Fleet M; Feng X; Tan K; Yang B High-Resolution Si K-and L2, 3-Edge Xanes of A-Quartz and Stishovite. Solid State Commun. 1993, 87, 613–617. [Google Scholar]
- (46).Pinakidou F; Katsikini M; Paloura E Xafs Characterization of Buried Si X N Y O Z Samples. Nucl. Instrum. Methods Phys. Res., Sect. B 2003, 200, 66–72. [Google Scholar]
- (47).Grinnell F; Feld M Fibronectin Adsorption on Hydrophilic and Hydrophobic Surfaces Detected by Antibody Binding and Analyzed During Cell Adhesion in Serum-Containing Medium. J. Biol. Chem 1982, 257, 4888–4893. [PubMed] [Google Scholar]
- (48).Giovambattista N; Debenedetti PG; Rossky PJ Effect of Surface Polarity on Water Contact Angle and Interfacial Hydration Structure. J. Phys. Chem. B 2007, 111, 9581–9587. [DOI] [PubMed] [Google Scholar]
- (49).Szili EJ; Kumar S; Smart RSC; Lowe R; Saiz E; Voelcker NH Plasma Enhanced Chemical Vapour Deposition of Silica onto Titanium: Analysis of Surface Chemistry, Morphology and Hydroxylation. Surf. Sci 2008, 602, 2402–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ceiler M; Kohl P; Bidstrup S Plasma-Enhanced Chemical Vapor Deposition of Silicon Dioxide Deposited at Low Temperatures. J. Electrochem. Soc 1995, 142, 2067–2071. [Google Scholar]
- (51).Poon M; Kok C; Wong H; Chan P Bonding Structures of Silicon Oxynitride Prepared by Oxidation of Si-Rich Silicon Nitride. Thin Solid Films 2004, 462–463, 42–45. [Google Scholar]
- (52).Sakai N; Fujishima A; Watanabe T; Hashimoto K Quantitative Evaluation of the Photoinduced Hydrophilic Conversion Properties of Tio2 Thin Film Surfaces by the Reciprocal of Contact Angle. J. Phys. Chem. B 2003, 107, 1028–1035. [Google Scholar]
- (53).Hasegawa S; He L; Inokuma T; Kurata Y Analysis of Photoemission in Amorphous Sio X and Sin X Alloys in Terms of a Charge-Transfer Model. Phys. Rev. B: Condens. Matter Mater. Phys. 1992, 46, 12478. [DOI] [PubMed] [Google Scholar]