

Summary
Synaptic vesicle and active zone proteins are required for synaptogenesis. The molecular mechanisms for coordinated synthesis of these proteins are not understood. Using forward genetic screens, we identified the conserved THO nuclear export Complex (THOC) as an important regulator of presynapse development in C. elegans dopaminergic neurons. In THOC mutants, synaptic messenger RNAs are retained in the nucleus, resulting in dramatic decrease of synaptic protein expression, near complete loss of synapses and compromised dopamine function. CRE binding protein (CREB) interacts with THOC to mark synaptic transcripts for efficient nuclear export. Deletion of Thoc5, a THOC subunit, in mouse dopaminergic neurons causes severe defects in synapse maintenance and subsequent neuronal death in the Substantia Nigra Compacta. These cellular defects lead to abrogated dopamine release, ataxia and animal death. Together, our results argue that nuclear export mechanisms can select specific mRNAs and be a rate-limiting step for neuronal differentiation and survival.
Keywords: THO complex, nuclear export, presynapse assembly, CREB, dopamine neurons, neurodegeneration
Introduction
Neurons communicate with each other through the synapse, a highly specialized structure for signal transmission and reception. Synapses are assembled during neuron development. They consist of a pre- and postsynapse, which are built from hundreds of proteins (Laßek et al., 2015). While the molecular composition and architecture of pre- and postsynapses has been widely explored (Frank and Grant, 2017; Laßek et al., 2015; Van Vactor and Sigrist, 2017), much less is known, how synaptogenesis is regulated at the level of gene expression. Is there a mechanism that coordinates the expression of functionally related proteins such that they are ready to assemble into higher order structures concomitantly?
Regulation of gene expression has been shown to be important for neuronal development and function under many circumstances. Gene expression is a multi-step process starting with the transcription of messenger RNA (mRNA), followed by mRNA maturation, nuclear export and translation in the cytoplasm. At the level of gene transcription, terminal selector transcription factors (TFs) initiate and maintain the differentiation program of neuronal subtypes by controlling the expression of neuron-type specific gene batteries (Hobert, 2016). Furthermore, the CREB (CRE binding protein) TF has been shown to mediate many neuronal functions. During neuron development, it is involved in neuron survival, synapse number as well as dendritic growth (Aguado et al., 2009; Mantamadiotis et al., 2002; Redmond et al., 2002). In adult neurons, CREB couples neuronal activity to long-term changes in synaptic plasticity and memory formation by inducing the transcription of immediate early genes (IEGs) (Sakamoto et al., 2011). mRNA splicing adds another layer of gene expression regulation. Alternative splicing of cell adhesion molecules such as the mammalian neurexins and the drosophila Dscam was shown to regulate neuronal cell recognition and the establishment of specific neuronal circuits (Raj and Blencowe, 2015). At the level of protein translation, local translation has emerged as a way to spatially regulate protein synthesis within a neuron thereby facilitating proper axon pathfinding and activity-dependent synaptic changes (Rangaraju et al., 2017). Lastly, gene expression can be modulated at the level of nuclear export of mRNAs to the cytoplasm. Multiple studies demonstrated that defects in nuclear export might be an underlying cause of neurodegenerative disease (Freibaum et al., 2015; Tsoi et al., 2011; Zhang et al., 2015). Little is known about which RNA binding proteins are involved in the regulation of mRNA export in neurons and whether different RBPs might regulate the export of subsets of functionally related mRNAs.
The THO complex (THOC) is a conserved RNA binding complex which has been implicated in mRNA export of subsets of mRNAs (Guria et al., 2011; Saran et al., 2013; Tran et al., 2013, 2014a; Wang et al., 2013). Together with the RNA helicase UAP56 and the export adaptor AlyRef/THOC4, it forms the TREX (TRanscription/EXport) complex that facilitates the formation of export-competent messenger ribonucleoprotein complexes (mRNPs) (Sträßer et al., 2002). Even though THOC’s core function in mRNA maturation and export is conserved from yeast to mammals, it has diverse cellular functions in different cell types. The THOC is involved in embryonic, hematopoietic and intestinal stem cell renewal and survival, testis development and cancer proliferation (Mancini et al., 2010; Pitzonka et al., 2014; Saran et al., 2016, 2013; Tran et al., 2013; Wang et al., 2013, 2009). It appears to regulate different mRNA species in diverse cell types suggesting that its function is tailored to achieve context- or cell type specific transcriptional programs. Only little is known about THOC’s function in neurodevelopment and neurodegenerative disease. Missense mutations in the THOC2 and THOC6 genes were shown to be linked to intellectual disability (Beaulieu et al., 2013; Kumar et al., 2015). In primary neurons, THOC is highly enriched and mis-localized to cytoplasmic protein aggregates, a hallmark of neurodegenerative disease (Woerner et al., 2016). These data suggest that impairment in nucleocytoplasmic transport in part facilitated by THOC may contribute to the cellular pathology of neurodegenerative disease.
In this study we identified the THOC as an important regulator of presynaptic gene expression in C. elegans. THOC mutants have strong defects in dopaminergic synaptogenesis resulting in impaired dopamine (DA) signaling and corresponding behavioral deficits. We show that presynaptic transcripts are retained in the nucleus of DA neurons, leading to a depletion of their mRNAs in the cytoplasm and hence reduced protein synthesis. We demonstrate that THOC’s function in presynaptic gene expression is especially pivotal under neuronal activity. Based on our genetic as well as biochemical data we propose a model, whereby the activity-dependent TF CREB recruits THOC to presynaptic transcripts, hence facilitating their nuclear export, allowing for concerted translation, which ultimately enables a temporally coordinated assembly of DA presynapses.
Lastly, using conditional Thoc5 knock out mice, we demonstrate that THOC’s function in neurons is conserved from worms to mammals. Mice homozygous mutant for Thoc5 just in DA neurons start to lose DA synapses at 3 weeks of life. At 6 weeks, most DA neurons die leading to sever impairment of DA release and locomotion defects. Taken together, our data demonstrate that the evolutionarily conserved THOC plays an essential role in DA synaptogenesis.
Results
Dopaminergic presynapses are severely impaired in THOC mutants
To identify novel master regulators of presynaptic gene expression, we performed a visual forward genetic screen using the C. elegans PDE dopamine (DA) neurons as model systems. These neurons have their cell bodies in the posterior part of the animal, they extend sensory, ciliated dendrites into the cuticle and project their axons ventrally, where they bifurcate and run along the entire ventral nerve cord (Figure 1A and (White et al., 1986)). They form evenly spaced en-passant synapses along the axon, which can be visualized by the expression of fluorescently tagged presynaptic components under a DA specific promoter (Figure 1A, B).
Figure 1: DA synapses are severely impaired in THOC mutant animals.
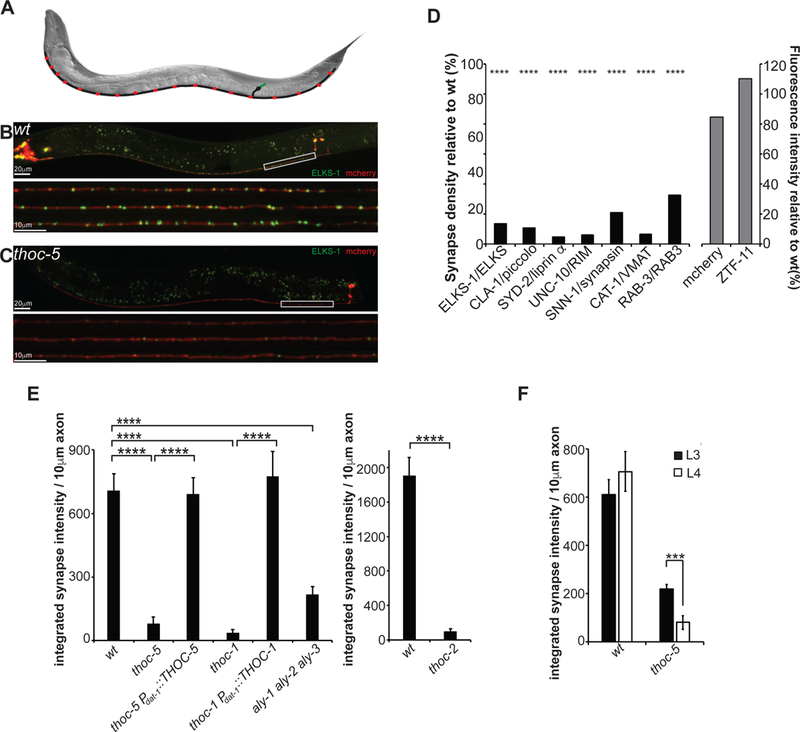
(A) Schematic of the dopaminergic PDE neuron in C. elegans (green: dendrite; black: cellbody and axon; red: presynapses)
(B) Image of wt PDE neuron at larval stage L4. Lower panel: Line scans of 3 PDE axons.
(C) Image of thoc-5 PDE neuron at larval stage L4. Lower panel: Line scans of 3 PDE axons.
(D) Presynaptic markers (black bars) are downregulated in thoc-5 mutants, however control proteins (grey bars) are unaffected.
(E) Quantification of thoc-1, thoc-2, thoc-5 single mutant and aly-1 aly-2 aly-3 triple mutant phenotypes. Cell-specific expression of THOC-1 and THOC-5 rescues the impairment of DA presynapses.
N=10–20 animals at L4; ****, p<0.0001, one-way Anova with Tukey post-hoc test for (D) & (E)
(F) Synapse defect aggravates with age. Dopaminergic presynapses were quantified at L3 and L4 in wt and thoc-5. N=10–20; ***, p<0.001, student t-test.
Averages and SEM are plotted.
We isolated two mutations wy817 and wy822 with dramatic defects in presynapses, however intact axonal morphology (as assayed by GFP::ELKS-1 and cytoplasmic mCherry, Figure 1B, C). In both mutants, only 10% of synapses were retained, which were smaller and dimmer than wildtype (wt) (Figure 1C, D, E and Figure S1A, B, C). To understand whether these mutations affected the expression of other presynaptic components, we crossed wy822 to an array of strains expressing fluorescently tagged active zone molecules (AZ), synaptic vesicle (SV) proteins or control proteins. While all presynaptic markers showed a dramatic decrease in synaptic density and size in the mutant (Figure 1D), control proteins such as cytosolic mCherry or the pro-neurogenic TF ZTF-11 were unaffected (Figure 1D).
Using whole genome sequencing, we identified wy817 as a splice acceptor site mutation in the 11th intron of THOC-1, which led to a 70% reduction in thoc-1 mRNA (Figures S1G). The wy822 mutation introduced an early stop codon in THOC-5 (R288stop). THOC-1 and THOC-5 are part of a nuclear, multi-protein complex conserved from yeast to mammals, which has been implicated to work at the interface of transcription and mRNA export (Sträßer et al., 2002). A deletion mutant of THOC-2, another core member of the THOC, showed similar presynaptic defects as thoc-1 and thoc-5 mutants (Figure 1E). Using cell-specific expression of THOC-1 or THOC-5 cDNA in the corresponding mutant strain background, we show that THOC functions cell-autonomously in DA neurons (Figure 1E and Figure S1A, B, C).
To rule out that loss of DA synapses is a secondary phenotype due to pleiotropic defects caused by the thoc mutation, we examined different fluorescent organelle markers in PDE. We did not detect any defect in the abundance, shape and localization of mitochondria, ribosomes, ER, Golgi, endosome and sensory cilia (Figure S2). These data suggest that THOC is not required for many developmental and functional aspects of the PDE neurons, however is indispensable for the expression of synaptic proteins.
To address whether THOC functions in initial DA synapse development or synapse maintenance, we quantified DA synapses at different larval stages. We found that thoc-5 mutants at L3 contained more abundant and brighter synapses as compared to L4 (Figure 1F and Figure S1D, E, F). In contrast, synapse density did not change with age in wt and synapse intensity showed a slight trend of growth (Figure 1F and Figure S1D, E, F).
Taken together, our forward genetic screen identified the THOC as an important regulator of presynaptic gene expression in DA neurons.
THOC also affects other types of presynapses, however to a lesser extent
Next we wanted to explore whether THOC plays a role in presynaptic gene expression of all neurons or whether it only affects DA neurons. First, we assayed endogenous presynaptic protein levels by quantitative western blot. We examined five presynaptic proteins including SNB-1/synaptobrevin, SNG-1/synaptogarin, SNT-1/synaptotagmin, UNC-64/syntaxin and ELKS-1/ELKS. All these proteins except SNT-1 showed a mild reduction in thoc-5 mutant animals (Figure 2A). For ELKS-1, we examined the expression level in a transgenic strain, so that both the endogenous pan-neuronal ELKS-1 as well as the DA-specific GFP::ELKS-1 could be assayed simultaneously. Endogenous ELKS-1 was affected much less than DA neuron specific GFP::ELKS-1 (Figure 2A).
Figure 2: THOC most severely affects DA synapses.
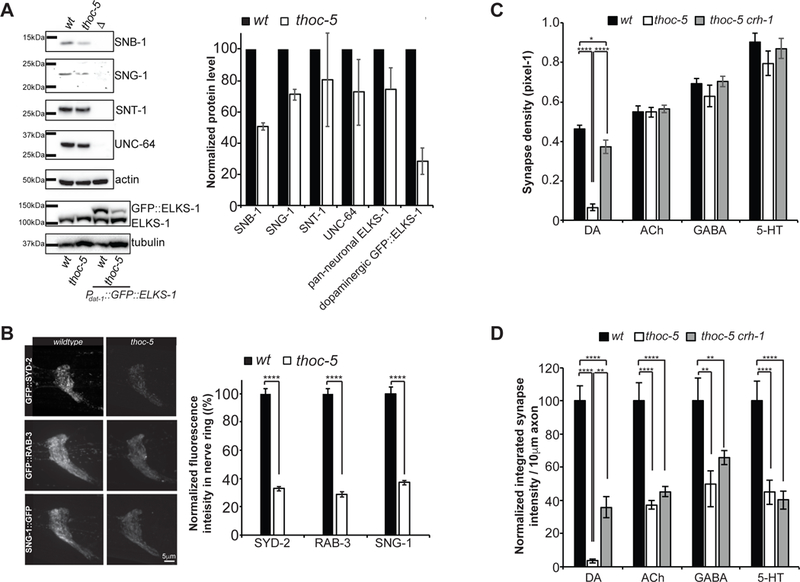
(A) Quantitative western blotting of endogenous, pan-neuronally expressed presynaptic proteins. Representative blot images and quantification of 3 independent experiments is shown.
(B) Endogenous GFP knock-in strains for SYD-2, RAB-3 and SNG-1. Representative images of the nerve ring is shown for wt and thoc-5. Quantification of total nerve ring fluorescence is plotted. N=10–20. ****, p<0.0001, student t-test.
(C) Synapse density quantification for different neuron types in wt, thoc-5 and thoc-5 crh-1.
(D) Quantification of normalized integrated synaptic intensity for different neuron types in wt, thoc-5 and thoc-5 crh-1.
N=10–20 animals at L4; ****, p<0.0001; **, p<0.01, one-way Anova with Tukey post-hoc test for (C) & (D)
DA: Dopamine, Ach: Acetylcholine, GABA: γ-aminobutyric acid, 5-HT: SerotoninAverages and SEM are plotted.
To further test if synaptically localized SV and AZ proteins are affected by thoc-5 in vivo, we generated knocked-in strains, where GFP was inserted in the endogenous genetic loci of syd-2/liprinα, rab-3/RAB3 and sng-1/synaptogyrin with CRISPR/Cas9. We quantified the total fluorescence intensity of these proteins in the nerve ring, the C. elegans brain, in wt and thoc-5. Again, we found a global decrease in fluorescence intensity in thoc5 mutants for all three synaptic proteins (Figure 2B).
Lastly, to directly study THOC’s function in different neuron types, we utilized a cell-type specific endogenous GFP knock-in strain (Schwartz and Jorgensen, 2016), in which the tagging of the SV protein RAB-3 with GFP was activated by the expression of flippase under different neuron-type specific promoters. We found that cholinergic, GABAergic and serotonergic synapses did not show a dramatic decrease in synaptic density as compared to DA synapses (Figure 2C). However, the integrated synapse intensity was reduced in all neuron types, with the most significant decrease in DA neurons (Figure 2D).
Thus, our data suggest that THOC affects overall synaptic protein levels in all neurons. However, its function is more essential in DA neurons.
THOC mutants display impaired DA signaling and behavioral deficits
To understand whether the morphological synaptic defect leads to a deficit in DA signaling, we analyzed the basal slowing response, a DA dependent behavior (Sawin et al., 2000). Well-fed wt animals slow down in the presence of food (Figure S3A) and this behavior depends on DA neurons. In contrast, cat-2 mutants that cannot synthesize DA, do not slow down in the presence of food (Figure S3A). Interestingly, both thoc-2 and thoc-5 mutants are impaired in the basal slowing response (Figure S3A). Low concentrations of exogenous DA restored the behavior back to wt for thoc-2 and thoc-5 mutant animals (Figure S3B). Taken together, THOC mutants are highly impaired in DA signaling resulting in a corresponding behavioral deficit.
To rule out that the decreased DA signaling in THOC mutant animals arose from sensory defects of the DA neurons, we analyzed the activity of PDE neurons by imaging their calcium transients in feely-behaving worms. Animals expressing the calcium sensor Gcamp6 in DA neurons were imaged during the basal slowing response (Hardaway et al., 2015; Piggott et al., 2011). Upon entry into the bacterial food, wt PDE neurons were activated (Figure S3C) and this activation was the same in thoc-5 mutant animals. Thus, defects in DA signaling in THOC mutant animals arise from impaired presynaptic rather than sensory function.
THOC regulates mRNA export of presynaptic genes
Previous studies implicated THOC in multiple steps of mRNA maturation, mRNP formation and nuclear export (Luna et al., 2012; Tran et al., 2014a; Wang et al., 2013). In order to elucidate THOC’s role in presynaptic gene expression we performed single molecule in situ hybridization (smFISH) experiments to specifically visualize and determine the intracellular localization of presynaptic as well as control transcripts in PDE DA neurons. Under wt conditions, all transcripts predominantly showed cytoplasmic localization (Figure 3A, B and Figures S4A). However, in thoc-5 mutants, presynaptic transcripts displayed 1.5- to 5-fold increase in transcripts retained in the nucleus, while the subcellular localization of control transcripts, including TBA-1/tubulin, UNC-33/CRMP and UNC-44/ankyrin, was unperturbed (Figure 3A, B and Figure S4A). Interestingly, the total number of transcripts as approximated by the number of smFISH puncta, was unchanged between wt and thoc-5 mutants (Figure S4B).
Figure 3: Presynaptic transcripts are retained in the nucleus in thoc-5 mutants.
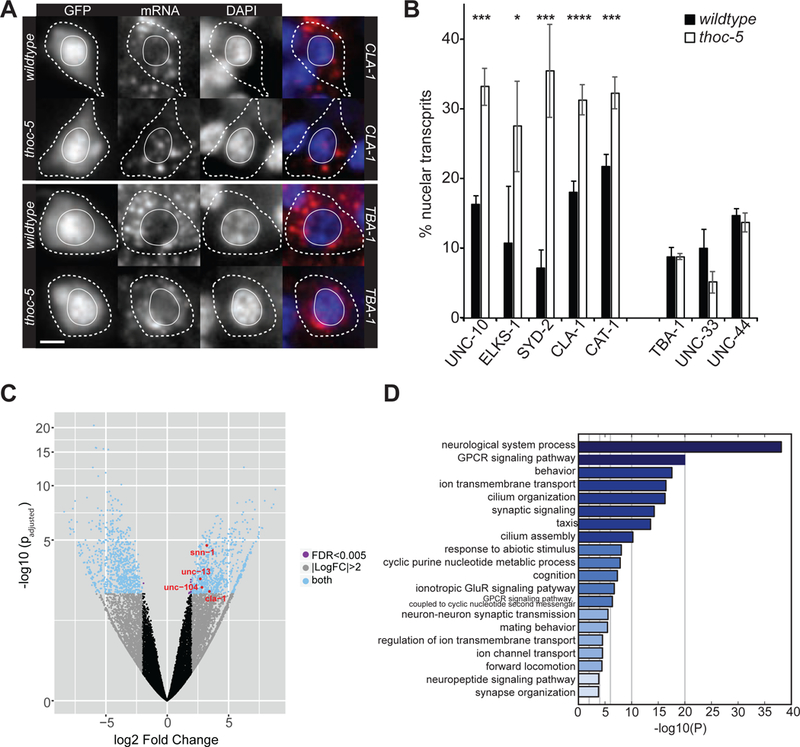
(A) smFISH for presynaptic (upper panel) and control (lower panel) transcripts in wt (upper row) and thoc-5 (lower row). GFP: PDE cellbody; red: signal for FISH probes; blue: DAPI staining of nuclei. Scale bar represents 2μm.
(B) Quantification of nuclear transcripts per PDE neuron in wt and thoc-5. N=10–50. Averages and SEM are plotted.
(C) Volcano plot of differentially expressed genes between wt and thoc-5. Red genes correspond to significantly up-regulated presynaptic genes further examined throughout this paper. Biological replicates: N=2 for wt and N=3 for thoc-5.
(D) Gene enrichment analysis for up-regulated genes.
See also Figure S4.
Next, we wanted to obtain more comprehensive insight into THOC’s function on neuronal gene expression. To this end, we performed whole transcriptome analysis of FACS-sorted wt and thoc-5 mutant neurons. We found that 763 genes are up-regulated (fold change ≥ 2 and p < 0.005) and 938 genes are down-regulated (fold change ≤ 2 and p < 0.005) (Figure 3C and Supplemental Table1). Gene enrichment analysis revealed that the up-regulated genes are highly enriched in synaptic genes (Figure 3D). Down-regulated genes do not show a strong neuron-specific functional enrichment, they rather associate with a wide variety of biological functions (Figure S4C). Using qPCR, we confirmed the up-regulation of a few presynaptic transcripts in thoc-5 mutants (Figure S4D). Interestingly, while we see a decrease in presynaptic proteins, we detect an increase in the corresponding transcripts. We speculate that this increase in mRNA might be due to a feedback loop, through which the neurons try to boost presynaptic protein production. Alternatively, mRNAs might be trapped in a cellular compartment, where they are inaccessible to translation, however protected from degradation.
Collectively, these observations suggest that THOC plays an important role in the nuclear export of synaptic transcripts.
Thoc5 conditional knock-out mice exhibit DA synapse loss at 3 weeks of age
To investigate whether the function of Thoc5 is evolutionarily conserved in mice DA neurons, we crossed Thoc5 floxed (Thoc5fl/fl) mice with DAT-IRES-Cre mice to produce a conditional knockout (cKO) of Thoc5 in DA neurons (DAT-Cre;Thoc5fl/fl). Thoc5 cKO mice exhibited normal body size and locomotive behavior during the first 3 postnatal weeks. They started to loose body weight from 4 weeks of age (Figure 4A, B). and became ataxic by 6 weeks. Surprisingly, the compromised body growth and locomotion was accompanied by premature mortality at around 6 to 9 weeks of age (Figure 4C).
Figure 4. THOC5 cKO mice exhibit developmental defects, reduced DAT expression, but normal DA release at 3 weeks of age.
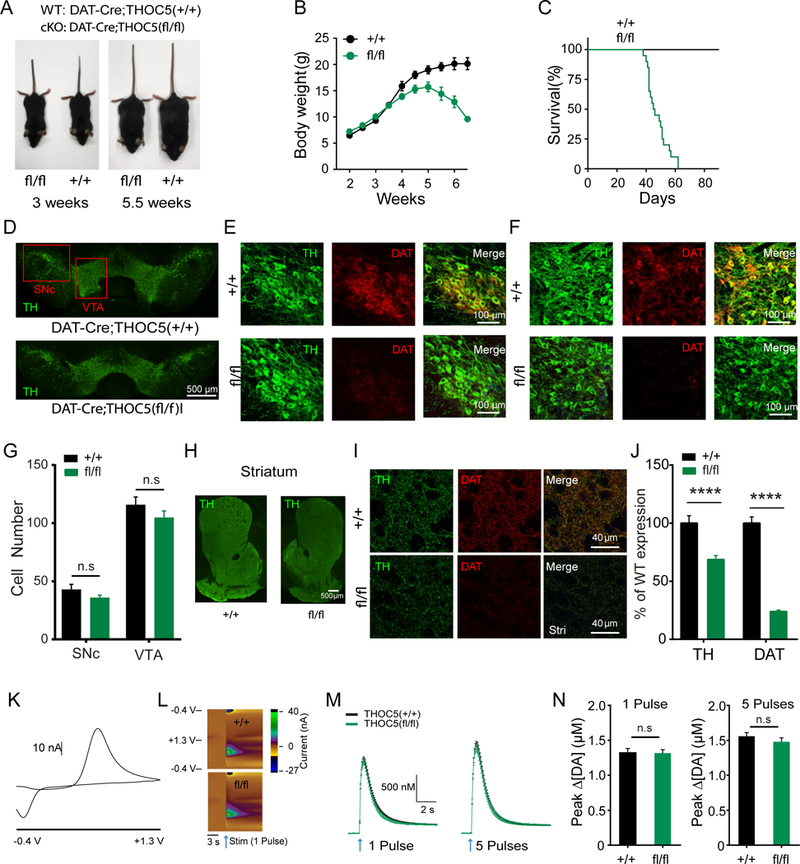
(A) Representative body size in 3 week old (left) and 5.5 week old (right) DAT-cre;Thoc5+/+ (wt) and DAT-cre;Thoc5fl/fl (cKO).
(B) Summary of wt and cKO body weight (N=10 for wt, and N=10 for cKO)
(C) Survival curve (N=13 for wt, and N=21 for cKO)
(D) Representative images of midbrain TH immufluorescence in 3 weeks old wt and cKO mice.
(E) Representative images of TH and DAT immunofluorescence in SNc from 3 weeks old wt (top) and cKO (bottom) mice.
(F) Representative images of TH and DAT immunofluorescence in VTA from wt (top) and cKO (bottom).
(G) Quantification of DA cell number in midbrain (p>0.05, two-way ANOVA with Bonferroni post-hoc test, wt: N = 12, cKO: N = 18)
(H) Representative images of striatum showing TH immufluorescence in wt (left) and cKO (right) mice.
(I) Representative high magnification images of TH and DAT immunofluorescence in dorsal striatum from wt (top) and cKO (bottom).
(J) Quantification of TH and DAT immunofluorescence in striatum (****, p<0.0001, two-way ANOVA with Bonferroni post-hoc test, wt: N = 24, cKO: N = 36).
(K) 2D voltammogram showing oxidation and reduction peaks of DA.
(L) Representative 3D voltammograms from 1 pulse. x-axis: recording time, y-axis: applied potential, color map: recorded current. Arrows represent time of stimulation.
(M) Summary of fast-scan cyclic voltammetry (FSCV) recordings evoked by 1 pulse (left) and 5 pulses (right) stimulations (wt: N=24, cKO: N=29).
(N) Quantification of peak DA release evoked by 1 pulse (left) and 5 pulses (right) stimulations (p>0.05, Mann-Whitney, wt: N=24, cKO: N=29).
Next we asked whether cKO of Thoc5 has deleterious effects on midbrain DA neurons. To this end, we checked the number of DA neurons in 3 weeks old mice by double immunostaining for tyrosine hydroxylase (TH) and DA transporter (DAT) in substantia nigra pars compacta (SNc) and ventral tegmental area (VTA). We found no difference in DA cell numbers in neither SNc nor VTA (Figure 4D-G). However, 3 week old Thoc5 cKO mice exhibited less TH, and much less DAT immunoreactivity in the axons in the striatum, the major target area of midbrain DA neurons (Figure 4H-J).
To assess the physiological consequence of DA synapse loss in Thoc5 cKO mice, we performed fast-scan cyclic voltammetry (FSCV) to measure DA release from DA axons in the dorsal striatum. We prepared brain slices from these mice and evoked DA release by local electrical stimulation. Consistent with an equal number of TH+ cell bodies in the midbrain, we did not find a difference in DA release between wt and mutant (Figure 4L-N). These data are in agreement with previous studies, where it was shown that depletion of up to 80% of DA neurons was necessary to evoke a defect in DA release (Zigmond et al., 1984).
Taken together, our data suggest that DA neurons of Thoc5 cKO mice display a substantial loss of synapses at 3 weeks of age, phenocopying THOC mutant worms.
DA neurons degenerate in Thoc5 cKO mice at 6 weeks of age
To explore whether the synaptic defect of Thoc5 mutant DA neurons aggravated with age as seen for worms (Figure 1F), we conducted the same double immunostaining experiment in ~6 week old mice. We found a significant loss of DA neurons in Thoc5 cKO mice, especially in SNc (Figures 5A-D). This phenotype was accompanied by a massive decrease of axon terminals in the striatum (Figure 5E-G). The loss of TH immunoreactivity in dorsal striatum was more pronounced than in ventral striatum, consistent with a greater loss of TH+ cells in SNc compared to VTA.
Figure 5. THOC5 cKO mice exhibit severe loss of DA synapses and diminished DA release at 6 weeks of age.
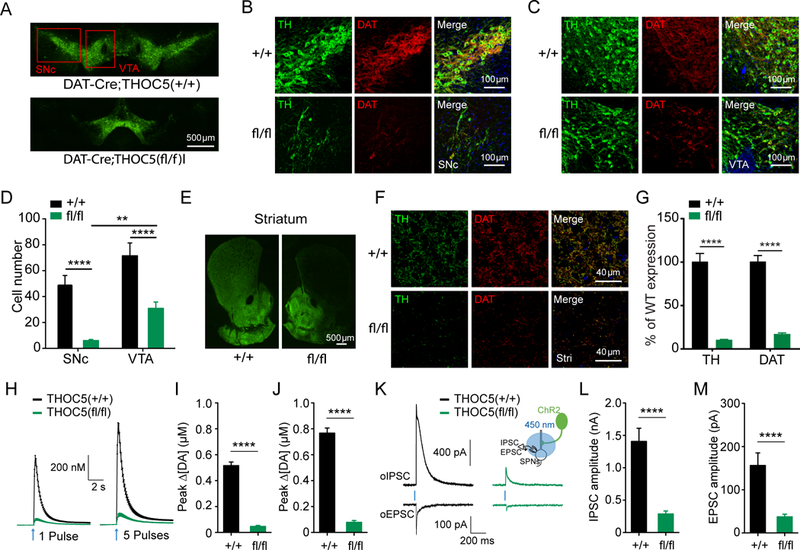
(A) Representative images of midbrain TH immufluorescence in ~6 week-old DAT-cre;Thoc5+/+ (wt) and DAT-cre;Thoc5fl/fl (cKO) mice.
(B) Representative images of TH and DAT immunofluorescence in SNc from ~6 week-old wt (top) and cKO (bottom) mice.
(C) Representative images of TH and DAT immunofluorescence in VTA from the same wt (top) and cKO (bottom) mice.
(D) Quantification of DA cell number in midbrain (**, p<0.01, ****, p<0.0001, two-way ANOVA with Bonferroni post-hoc test, wt: N = 10, cKO: N = 18)
(E) Representative images of striatum showing TH immufluorescence in wt (left) and cKO (right) mice.
(F) Representative high magnification images of TH and DAT immunofluorescence in dorsal striatum from wt (top) and cKO (bottom) mice.
(G) Quantification of TH and DAT immunofluorescence in striatum (****, p<0.0001, two-way ANOVA with Bonferroni post-hoc test, wt: N = 12, cKO: N = 18).
(H) Summary of fast-scan cyclic voltammetry (FSCV) recordings evoked by 1 pulse (left) and 5 pulses (right) stimulations.
(I) Quantification of peak DA release evoked by 1 pulse stimulation (****, p<0.0001, unpaired t-test, wt: N = 13 samples, cKO: N = 12).
(J) Quantification of peak DA release evoked by 5 pulse stimulation (****, p<0.0001, unpaired t-test, wt: N = 13 samples, cKO: N = 12).
(K) Representative traces for oIPSC and oEPSC recorded from wt (left) and cKO (right) SPNs. Insert: experimental configuration.
(L) Quantification of peak oIPSC (****, p<0.0001, unpaired t-test, wt: N = 9 samples, cKO: N = 14).
(M) Quantification of peak oEPSC (****, p<0.0001, unpaired t-test, wt: N = 9 samples, cKO: N = 13).
To assess the physiological consequence of DA neuron loss in 6–9 weeks old Thoc5 cKO mice, we performed FSCV experiments. To selectively activate dopaminergic terminals, DAT-Cre;Thoc5fl/fl mice were further bred with Ai32 mice containing a conditional floxed allele of H134R variant of channelrhodopsin2 (ChR2) in the Rosa26 locus. We prepared brain slices from these mice and evoked DA release with 450 nm blue light (Figure 5H-J). Within the dorsal striatum, DA release from 1 or 5 light pulses was severely impaired in Thoc5 cKO mice (Figure 5H, I & J), demonstrating that DA neuron axons are severely degenerated.
There is accumulating evidence that γ-aminobutyric acid (GABA) and glutamate are co-released along with DA from dopaminergic terminals in the striatum (Hnasko et al., 2010). These co-released neurotransmitters induce inhibitory postsynaptic currents (IPSC) in the nano-ampere range (GABA) and much smaller, but reproducible, excitatory postsynaptic currents (EPSC). To test whether the release of these two neurotransmitters is also affected in Thoc5 cKO mice, we performed whole-cell patch clamp recordings in postsynaptic striatal spiny projection neurons (SPNs). Optogenetically evoked IPSC (oIPSC) and EPSC (oEPSC) were dramatically diminished in Thoc5 cKO mice (Figure 5K-M).
Together, our data support the conclusion that deletion of Thoc5 from mouse DA neurons leads to rapid synapse loss followed by DA neurodegeneration and animal death.
A human thoc1 mutation found in ALS patients renders THOC-1 dysfunctional
Defects in nuclear export have become an emerging pathogenic mechanism in many neurodegenerative diseases. As our mouse data implicated a potential role for THOC in neurodegeneration, we searched public databases from large human genome-wide association studies for missense and loss-of-function mutations that are found in patients with neurodegenerative diseases such as Parkinson’s Disease (PD) and Amyotrophic Lateral Sclerosis (ALS) (Lill et al., 2011, 2012). Multiple missense mutations in THOC1, THOC2 and THOC5 were reported in these databases, however their statistical significance for disease was rather low. To enrich for putative disease causing mutation, we focused on mutations that occurred on conserved amino acid residues and were absent from a much larger dataset of the control human population. Specifically, we studied the thoc1 R138H mutation that was found in the ALS database (Figure S5A). We introduced this mutation in the corresponding location of the worm THOC-1 gene, expressed it under the DA specific promoter and assayed, whether it was able to rescue the thoc associated DA synapse loss. We found that this mutant allele of THOC-1 was unable to rescue the synapse defect (Figure S5B). However the disease-unrelated mutation V338I found in multiple control individuals (Lek et al., 2016) did not affect the function of THOC-1 (Figure S5B). The R138H mutation lies within the N-terminal domain of the protein, which has been implicated for protein interactions with other THOC components and hence might impact the function of the entire complex (Peña et al., 2012).
THOC’s function in presynaptic gene expression is pivotal under neuronal activity
Recent studies in mouse and drosophila suggest that THOC is not generally needed for bulk mRNA export, rather it facilitates the export of subsets of mRNAs (Guria et al., 2011; Katahira et al., 2009; Rehwinkel et al., 2004; Wang et al., 2013). Furthermore, THOC5 has been shown to regulate the transcription, processing and export of immediate early genes (IEGs) in response to different stimuli in various non-neuronal tissues (Tran et al., 2013, 2014b, 2014a). In neurons, IEGs play a prominent role in long-term synaptic changes and memory formation (Okuno, 2011). The expression of IEGs is mediated by the TFs CREB (CRE binding protein) and SRF (serum response element).
In order to explore whether neuronal activity modulates THOC-dependent presynaptic protein expression, we silenced PDE activity by a histamine gated chloride channel (HisCl) (Pokala et al., 2014). Acute silencing of DA neurons by cell-type specific expression of HisCl in thoc-5 mutant animals partially but significantly restored DA synapses (Figure 6B). This result suggests that THOC-5 is particularly important for presynaptic protein production when neurons are active.
Figure 6: THOC-dependent presynaptic gene expression is pivotal under neuronal activity.
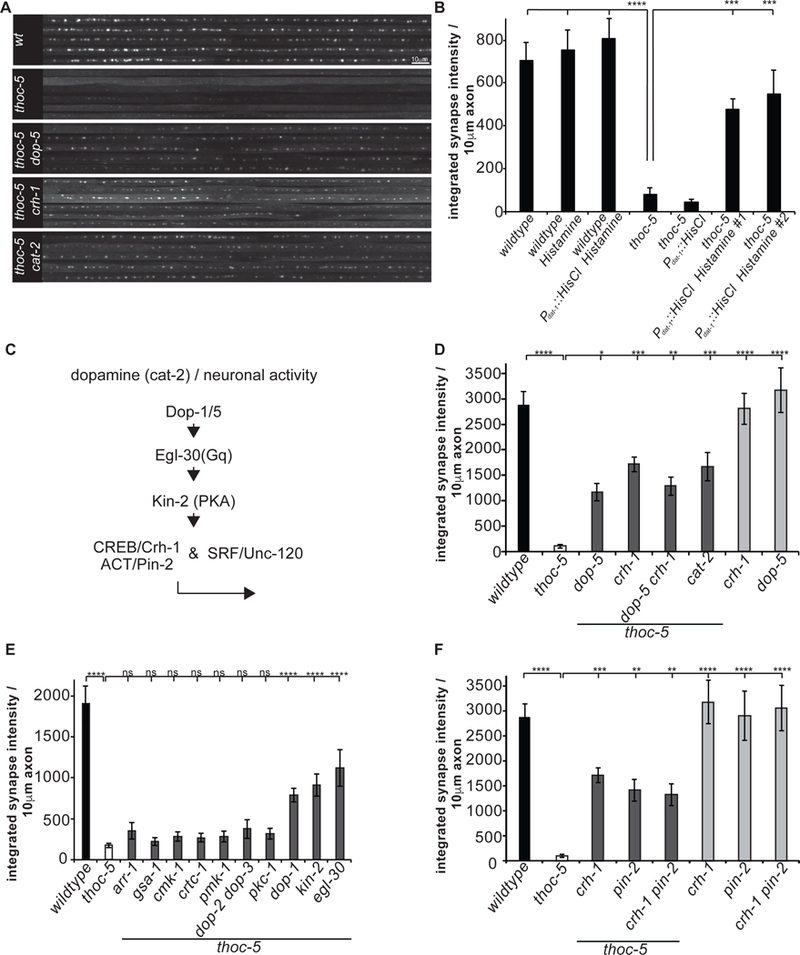
(A) PDE axonal line scans of endogenous GFP::RAB-3 in wt, thoc-5 and thoc-5 DA signaling double mutant animals.
(B) Silencing of DA neurons suppresses THOC-dependent synapse defect. Integrated synaptic intensity is quantified.
(C) Schematic of DA signaling cascade feeding into THOC-dependent presynaptic transcription.
(D) Integrated synaptic intensity is quantified for DA signaling mutants.
(E) Extensive double mutant analysis identifies signaling components down-stream of neuronal activity.
(F) CREB functions with the nuclear factor PIN-2, a LIM-domain containing protein. Integrated synaptic intensity is quantified.
Averages and SEM are plotted in all graphs.
N=10–20 animals; ****, p<0.0001, ***, p<0.001, one-way Anova with Tukey post-hoc test.
See also Figure S6.
To further understand the molecular mechanism underlying this activity-dependent regulation, we created double mutants for thoc-5 and crh-1 (C. elegans homologue of CREB). thoc-5 crh-1 mutants also showed partial but significant rescue of DA synapses. (Figure 6A, C,D). Loss of crh-1 in DA neurons alone was sufficient to partially restore PDE synapses in thoc-5 mutants, suggestive of a cell autonomous function of CREB in PDE neurons (Figure S6A.). Interestingly, in other neuronal celltypes, thoc-5 crh-1 double mutants did not show any rescue of presynaptic defects (Figure 2C, D).
As the presynaptic defect was partially restored in thoc-5 crh-1 mutants, we wondered whether it was also the case for the nuclear export defect of presynaptic transcripts. Indeed, smFISH experiments in thoc-5 crh-1 mutants revealed that nuclear export was restored to wt (Figure S6B).
In mammals it has been shown that synaptic gene expression is often regulated by multiple TFs including SRF and MEF2 (Rodríguez-Tornos et al., 2013). To explore whether these TFs might function similarly as CREB in the context of THOC, we built double mutants between thoc-5 and unc-120, the worm homologue of SRF. Indeed, DA synapses were partially rescued in thoc-5 unc-120 mutants (Figure S6C). Thus, neuronal activity in PDE might induce both CREB and SRF-mediated transcripts, which are particularly dependent on THOC for nuclear export.
Next, we asked how PDE is activated by examining genetic interactions between thoc-5 and mutants that affect PDE activity. The thoc-5 synaptic phenotype could be partially rescued in a cat-2 thoc-5 double mutant background, which is defective in DA synthesis (Figure 6A, C, D). Consistent with this notion, mutations in DA receptors dop-1 and dop-5, when combined with thoc-5, also mitigated presynaptic loss in PDE (Figure 6A, C, D & E). In contrast, combining dop-2 and/or dop-3 mutations with thoc-5 did not lead to increased DA synapses, arguing for receptor specificity (Figure 6E).
To further unravel the signaling cascade involved in linking neuronal activity to CREB in THOC-dependent synapse formation, we performed an extensive double mutant analysis with known upstream signaling molecules of CREB. CREB can be phosphorylated and activated by several kinases, such as PKA, MAP kinases and Ca2+/Calmodulin-dependent kinases (CaMKs) (De Cesare et al., 1999). While double mutants between thoc-5 and cmk-1, pmk-1 and pkc-1 did not show any suppression of DA synapses (Figure 6C&E), double mutants between thoc-5 and kin-2, the regulatory subunit of PKA, partially restored them (Figure 6C&E). Furthermore, we identified the heterotrimeric G protein alpha subunit Gq egl-30, but not the Gs gsa-1 and β−arrestin arr-1 to be involved in this signaling cascade (Figure 6C&E). CREB often functions in concert with co-activators to promote gene transcription. It is thought that various external stimuli and signaling cascades might lead to recruitment of different co-activators and hence drive expression of specific sets of target genes (Altarejos and Montminy, 2011; Fimia et al., 1999). Double mutant animals between thoc-5 and pin-2, the C. elegans homologue of LIM-only protein ACT, but not between thoc-5 and crtc-1 showed suppression of synapse loss of the thoc-5 animals (Figure 6C&F).
Lastly, we were interested in identifying additional RNA binding proteins, which might work together with THOC to facilitate the export of synaptic transcripts. PolyA binding proteins (PABs) have been previously implicated in mRNA quality control and mRNA nuclear export (Brune et al., 2005). We built double mutants between thoc-5 and the pab-2 and examined synapses in PDE neuron. We found that thoc-5 pab-2 mutants showed significant rescue of DA synapses (Figure S6D). We speculate that PAB-2 acts as a quality control factor retaining synaptic transcripts in the nucleus in THOC mutant animals. However, in the absence of PAB-2, synaptic transcripts undergo less stringent quality assessment hence being released into the cytoplasm. To test this model, we performed smFISH experiments in thoc-5 pab-2 mutants. Indeed, we found that synaptic mRNA export was restored to wt levels in these animals (Figure S6E).
CREB interacts with THOC to mark transcripts for efficient export
CREB and THOC localize to the nucleus, which corresponds well with their function in transcription and mRNA maturation, respectively (Cha-Molstad et al., 2004; Tran et al., 2014b; Wang et al., 2013). We studied their localization in C. elegans DA neurons by expressing fluorescently tagged CREB and THOC proteins. THOC-2 displayed diffuse localization in the nucleus and cytoplasm of DA neurons, while THOC-5 was strictly nuclear excluded (Figure 7A). The three AlyRef/THOC-4 homologues ALY-1,−2,−3, THOC-1 and CREB showed predominant nuclear localization (Figure 7A, B). Co-expression of THOC-1 and CREB in PDE neurons revealed perfect co-localization of the two proteins (Figure 7B). We tested whether the localization of THOC and CREB depended on each other. We did not observe any change in localization for neither THOC components nor CREB in crh-1 and thoc-5 mutant background, respectively (Figure S7A). Collectively, our localization data supports our genetic link between THOC and CREB and raises the question whether they might work together through physical interaction to facilitate presynaptic gene expression.
Figure 7: CREB physically interacts with THOC to recruit it onto activity-dependent transcripts.
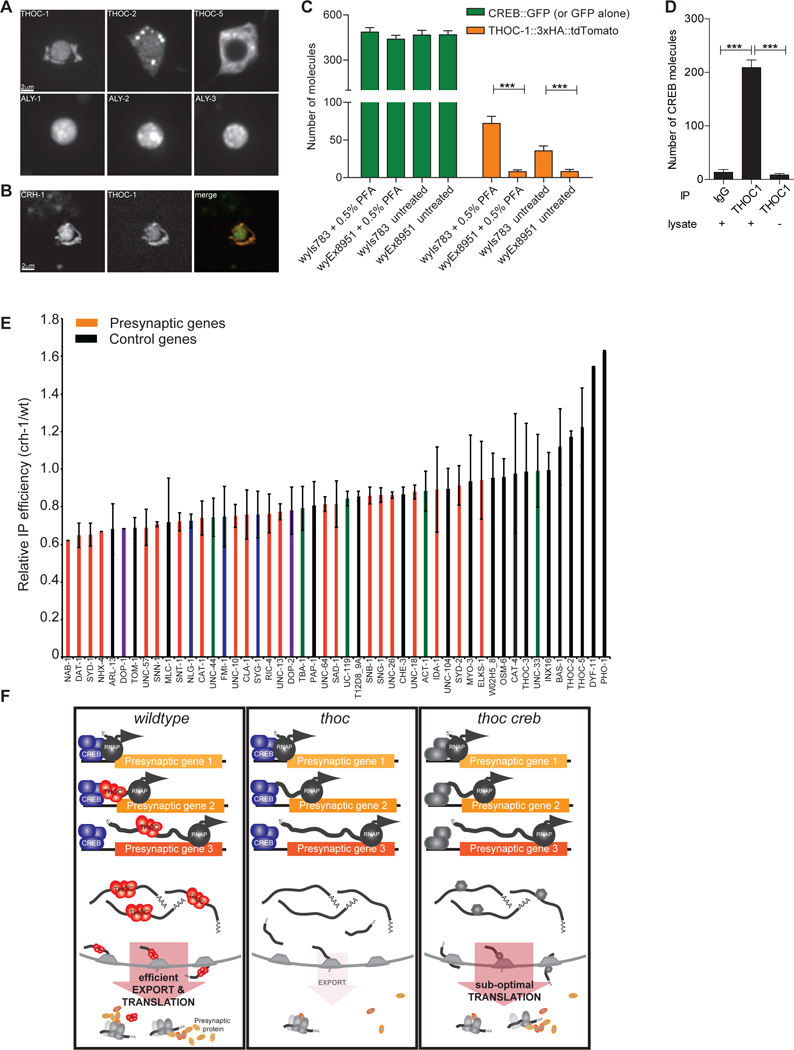
(A) Localization of GFP-tagged THOC components in PDE cell body.
(B) Co-expression analysis of fluorescently tagged THOC-1::tdTomato and CREB::GFP in PDE neurons.
(C) Quantification of simPULL assay showing that THOC-1 and CREB transiently interact in C. elegans. GFP serves as negative control. ****, p<0.0001 student t-test.
(D) Quantification of simPULL assay showing that THOC1 and CREB transiently interact in mouse striatal brain lysate. No lysate and non-specific IgG antibodies served as negative control. ***, p<0.0001 one-way Anova
(E) Immunoprecipitation of THOC-1-bound RNAs from wt and crh-1 mutant neurons. Red: synaptic genes, purple: postsynaptic genes, blue: cell adhesion molecules, green: cytoskeletal genes, black: other pan-neuronal and control genes.
(F) Current model of how THOC functions to regulate presynaptic gene expression in DA neurons. In wt, CREB recruits THOC onto presynaptic transcripts to facilitate their efficient nuclear export for concerted translation. In THOC mutant background, these transcripts are retained in the nucleus, hence leading to decreased presynaptic proteins and defective synapse assembly. This defect can be alleviated by removal of CREB, thereby allowing for a constitutive backup export pathway to kick in and to re-establish efficient export of presynaptic transcripts.
Averages and SEM are plotted in all graphs.
See also Figure S7.
To address whether THOC and CREB interact with each other, we constructed worm strains expressing THOC-1::3xHA::tdTomato with either CREB::GFP (wyIs783) or GFP (wyEx8951) and performed single molecule pull-down (SiMPull) experiments (Jain et al., 2011). While THOC-1 did not bind to GFP alone, we detected a small, but significant number of THOC-1 molecules interacting with CREB under native condition (Figure 7C, Figure S7B). Furthermore, the detected interaction was more prominent when samples were crosslinked with paraformaldehyde suggestive of a weak, but specific interaction between these two molecules. To explore whether the physical interaction between THOC and CREB is conserved in mice, we performed simPULL experiments with mouse striatal brain lysate. Again, we readily detected co-immunoprecipitation between THOC1 and CREB (Figure 7D). Taken together our data support a model, where transient interaction between CREB and THOC-1 might mark presynaptic transcript for efficient nuclear export.
To directly test this model, we performed RNA immunoprecipitation (IP) experiments. Specifically, we IPed pan-neuronally expressed THOC-1::3xHA::tdTomato under wt and CREB mutant conditions and quantified the THOC-1-bound and co-IPed mRNAs by qPCR. We hypothesized that in a CREB mutant strain background, THOC would be less efficiently loaded onto synaptic transcripts and hence, less synaptic mRNAs would be precipitated. In line with our hypothesis, we found that THOC-1 bound considerable amounts of presynaptic transcripts under wt conditions (Figure S7C). In CREB mutants, roughly 2-fold less mRNA was precipitated by THOC-1 as compared to wt. Precipitation of synaptic transcripts was strongly dependent on the presence of CREB, while other neuronal and housekeeping transcripts were less affected (Figure 7E). This change in IP efficiency was not due to overall changes in gene expression between the two genotypes, as there was no correlation between total mRNA level and IPed mRNA quantities for synaptic and control genes. (Figure S7D). In conclusion we propose a model, where activity-dependent transcripts get efficiently exported out of the nucleus due to the CREB-dependent loading of THOC onto these transcripts (Figure 7F).
Discussion
During neuronal differentiation, the majority of synapses of a neuron form in a relative short time window, during which the expression of synaptic proteins needs to be up-regulated in a concerted manner. How the production of synaptic proteins is coordinated is not well understood. We identified the THOC as an important regulator of presynaptic gene expression in DA neurons. Loss of THOC leads to dramatic synapse development deficit in both worms and mouse. THOC is recruited onto activity-dependent mRNAs by physical interaction with the CREB TF, thereby, with the help of additional RNA binding proteins such as PAB-2, facilitating efficient nuclear export and translation of synaptic transcripts (Figure 7F). These results reveal another layer of neuronal gene expression regulation and demonstrate that nuclear export is an important regulatory mechanism for neurodevelopment.
Transcription factor-dependent loading of THOC onto mRNAs as a novel mechanism for target selectivity
THOC was shown to be essential during early mouse development, as thoc5 and thoc1 full knock-out animals die during embryogenesis (Mancini et al., 2010; Wang et al., 2006). Furthermore, this protein complex is also indispensable for survival of adult mice, as full knock-out of thoc5 after birth leads to rapid death. Interestingly, even though THOC seems to be an important RNA metabolism factor, it does not affect mRNA on a global level, rather it was shown to regulate subsets of transcripts in different tissues and under diverse conditions (Guria et al., 2011; Katahira et al., 2013; Rehwinkel et al., 2004; Tran et al., 2013; Wang et al., 2013). How THOC selects its targets is still an open question in the field. In yeast, a studies has shown that THOC is required for the transcription of long and/or GC rich transcripts (Chávez et al., 2001). While some mammalian THOC targets contain higher GC content, this feature is not sufficient to specify THOC targets. In mammalian cell culture, THOC5 was shown to play a role in 3’ end processing (Katahira et al., 2013; Tran et al., 2014a). As most genes in metazoans contain introns and multiple polyadenylation signals, these mechanisms cannot explain THOC’s target selectivity satisfyingly. Here we provide evidence for a novel target selectivity mechanism for THOC. Our data supports a model, whereby physical interaction between a TF and THOC recruits this complex directly onto nascent transcripts, thereby selectively marking them for efficient downstream processing and nuclear export. This model offers an explanation on how THOC with thus far unknown and “unspecific” RNA binding affinities can still selectively bind to a specific group of target mRNAs. Furthermore, this model provides an organism with an elegant mechanism to harness a single RNA binding complex to facilitate distinct gene expression profiles by coupling different TFs to THOC and thereby providing an express lane for certain mRNAs out of the nucleus for efficient protein translation.
THOC and CREB work together to efficiently export activity-induced transcripts
Here we describe a novel function of THOC in terminally differentiated neurons. Our understanding of THOCs function in the brain so far only based on human genetic studies (Beaulieu et al., 2013; Kumar et al., 2015), however how THOC affects neurons at the cellular or molecular level is completely unknown. We show that THOC does not affect neuronal proliferation and fate determination, but has a conserved role in synapse formation and maintenance. In THOC mutant worms and mice, DA neurons proliferate and develop normally, however their synapses are dramatically reduced. Our mRNA localization studies in THOC mutants revealed a nuclear export defect specifically for presynaptic, but not control transcripts. How does THOC affect a specific set of genes including presynaptic genes? First clues came from studies in non-neuronal cells demonstrating that THOC has a prominent role in the processing and export of IEGs in LPS-stimulated macrophages and serum-stimulated MEFs (Tran et al., 2013, 2014a). In neurons, IEGs are induced by neuronal activity. Interestingly, some synaptic genes can be classified as IEGs in neurons (Okuno, 2011). In this study we propose a model, whereby the activity-dependent TF CREB loads THOC specifically onto activity-induced transcripts (Figure 7E). We show that CREB interacts with THOC (Figure 6A, D and Figure 7C, D) and that CREB is necessary to efficiently recruit THOC onto synaptic transcripts (Figure 7E).
A conserved role for THOC in dopaminergic synaptogenesis
As THOC and its molecular function in mRNA maturation and export is conserved from yeast to mammals, and as multiple human genetic studies implicated THOC in normal brain function, we explored whether THOC’s role in presynaptic gene expression, as discovered in C. elegans, was conserved in mice. Indeed, cKO of Thoc5 in mouse DA neurons led to a significant synapse loss at 3 weeks of age, while DA cell number was unchanged. These data nicely recapitulate the worm thoc-5 mutant phenotype. Furthermore, they underscore THOC’s new function in terminally differentiated cells. Thoc5 cKO mice at 6 weeks of age displayed an even more dramatic synapse loss due to DA neuron degeneration, which resulted in ataxia and eventual animal death. While we could not detect DA neuron degeneration and cell death in C. elegans, we indeed found an age-dependent decline of DA synapses, mirroring the progressive synapse loss in mice (Figure 1F and Figure 4 & 5).
Might loss of THOC play a role in neurodegenerative disease? While defects in nuclear export has emerged as a common cause of multiple neurodegenerative diseases (Freibaum et al., 2015; Tsoi et al., 2011; Zhang et al., 2015), and components of the THOC were shown to mislocalize in these diseases (Woerner et al., 2016), the causative link between THOC mislocalization and neurodegeneration still awaits further investigation. It is interesting to note that large human genome-wide association studies identified multiple mis-sense and loss-of-function mutations in different THOC components that show association with neurodegenerative diseases, in particular with ALS and to a lesser extent with PD (Lill et al., 2011, 2012). Interestingly, introduction of the disease specific R138H thoc1 mutation in the corresponding amino acid of the worm THOC-1 protein rendered the mutant protein dysfunctional, such that it was unable to rescue the synapse defect of worm DA neurons (Figure S5). These data might offer a first glimpse into THOC’s potential role in neurodegeneration. In the future, further massive sequencing efforts of human genomes, together with continuing studies in mouse and C. elegans will be needed to shed more light onto THOC’s role in neurodegeneration.
STAR methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kang Shen (kangshen@stanford.edu) and Jun B. Ding (dingjun@stanford.edu).
Experimental Model and Subject Details
C. elegans worm strains were grown on NGM plates seeded with OP50 bacteria. To make NGM + Histamine plates and NGM + dopamine plates, Histamine (Sigma Aldrich histamine-dihydrochloride, 1 M stock in water) or Dopamine (Sigma Aldrich Dopamine-hydrochloride, 1 M stock in M9 buffer) was added to NGM agar at ~65°C before pouring the plates. Worms were grown at 16° to 23°C. Worm strains used in this study are listed above in the Key Resources Table.
Mouse work: Mice were housed in pathogen-free and light- and temperature-controlled conditions. Food and water were available ad libitum. 3 week or 6–9 week old mice (male and female) were used for immunostaining and electrophysiological experiments. Thoc5fl/fl mice were provided from Teruko Tamura’s lab in Germany (Saran et al., 2013). The Thoc5fl/+ mice were crossed to generate Thoc5+/+, Thoc5fl/+ and Thoc5fl/fl. Then Thoc5fl/+ was crossed with DAT-IRES-Cre (B6.SJL-Slc6a3tm1.1(cre)Bkmn/J, JAX stock number 006660) mice to produce DAT-Cre;Thoc5fl/+ mice. DAT-Cre;Thoc5fl/+ mice were further bred with Ai32 mice (B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J, JAX stock number 012569) to generate DAT-Cre;Ai32;Thoc5fl/+ mice. Finally, these mice were bred with Thoc5 fl/fl or Thoc5 +/+ mice from the F2 generation of the same founding parents. This mating created DAT-Cre;Ai32;Thoc5fl/fl and DAT-Cre;Ai32;Thoc5+/+ mice for immunostaining, FSCV and electrophysiological studies. All the mice used for experiments were heterozygous for alleles encoding Cre recombinase under control of the DAT promoter (DAT-IRES-Cre) and ChR2-EYFP (Ai32). All experimental procedures were conducted in accordance with protocols approved by Stanford University’s Administrative Panel on Laboratory Animal Care.
Method Details
Imgaging:
Worms were mounted on 2% agarose pads in 5 mM levamisol + 0.05 Micron Polysterene Polybeads (Polysciences) diluted in M9 buffer. Imaging was performed using a Zeiss Axio Observer.Z1 microscope equipped with 488 and 561 laser lines, a Yokagawa spinning disk head, a Hamamatsu C9100–13 EM-CCD camera and a Zeiss 40X/1.3 objective. Metamorph Imaging software Version 7.7.9.0 (Molecular Devices) was used to acquire images. Imaging settings (e.g. exposure time, laser power) were identical for all genotypes. To image the entire PDE neuron, 3–4 z-stacks of 30um range (1um step size) were acquired, and then stitched together using Photoshop software.
For smFISH experiments, the wide-field set-up on the same microscope was used. Specifically, samples were illuminated through a Zeiss 100X/1.2 objective with the HXP120C light source, and the signal was detected with the QuantEM:512SC camera. Z-stacks of 5um (0.5 um step size) were acquired for three colors, GFP, DAPI and CAL Fluor Red or Quasar 670.
Image analysis was performed with ImageJ software. Briefly, image stacks were z-projected using maximum projection. PDE axons were straightened using the plugin “straighten to line”. Synapses were then thresholded and analyzed using “analyze particles”. Parameters such as axon length, synapse size, average synapse intensity as well as integrated synapse intensity were measured. These parameters were then used to calculate integrated synapse intensity / 10um axon.
Whole worm lysate and western blotting:
Worms from 5 60mm plates were harvested into a single 1.5ml tube. Worm pellets were resuspended in 2x NuPAGE LDS sample buffer + 50mM DTT and flash frozen in liquid nitrogen. Samples were then thawed on ice, sonicated 3x for 60 seconds (100% Amplitude) in a Cup Horn sonicator with intermittent chilling on ice. Samples were boiled for 5min at 95 degree followed by a 5min spin at 13’000rpm.
Proteins were separated on NuPAGE® Novex® 4–12% Bis-Tris Protein Gels and transferred onto nitrocellulose membranes through conventional tank electrotransfer for 2 to 4 hours. Membranes were then blocked with 5% milk in PBS followed by primary and secondary antibody incubation (antibody dilutions: anti SNB-1 1:20000; anti-SNG-1 1:1000; anti-SNT-1 1:5000; anti-UNC-64 1:5000; anti-ELKS-1 1:2000; anti-actin 1:10000; anti-tubulin 1:5000; anti-HA 1:2000; goat-anti-chicken HRP 1:2000; mouse-anti-rabbit HRP 1:2000). Membranes were washed extensively with PBS+0.1% Tween, before the signal was detected by chemiluminescence with the ChemiDoc XRS+ system.
Basal slowing response assay:
The basal slowing response assay was performed as described in (Sawin et al., 2000). In brief, on half of the NGM plates, a ring of the E. coli strain HB101 was spread. Empty plates as well as seeded plates were then incubated over night at 37degree. Animals of young adult stage and grown at 20degree were used to perform the assay and all plates were coded so that the experimenter was blinded to the genotype.
Animals were washed in a small droplet of S-basal buffer (5.85 g NaCl, 1 g K2 HPO4, 6 g KH2PO4, 1 ml cholesterol (5 mg/ml in ethanol), H2O to 1 litre), before being placed either on an empty plate or in the middle of a bacterial ring. Five minutes after transfer, the number of body bends within a 20 second interval window was counted sequentially for all the animals transfer to the plate.
To test whether exogenous dopamine could rescue the basal slowing response of cat-2 and THOC mutant animals, L4 animals of each genotype were grown over night on NGM plates containing 2mM dopamine, but were then assayed on plates without dopamine. All behavioral tests were repeated at least three times on different days.
Calcium imaging:
Calcium imaging was performed during the basal slowing response assay using the CARIBN system as previously described (Piggott et al., 2011). Day 1 adult worms were used for imaging under standard laboratory conditions on NGM plates seeded with two thin lines of HB101 bacteria. We imaged calcium transients in PDE soma.
Worm dissociation and FACS sorting
Synchronized day 1 adult worms expressing pan-neuronal GFP were used for neuron isolation. Worms were washed with M9 buffer to remove excess bacteria. Worm pellet (~250 μl) was then washed once with 500 μl lysis buffer (200 mM DTT, 0.25% SDS, 20 mM HEPES pH 8.0, 3% sucrose), resuspended in 750 μl lysis buffer and incubated with gentle rocking for 6.5 min at room temperature. Worms were washed 6x with M9 and resuspended in 250 μl of 20 mg ml−1 pronase from Streptomyces griseus (Sigma-Aldrich). Worms were incubated at room temperature (~20 min) with periodic mechanical disruption (100x pipetting every 5 min). When most worm bodies were dissociated, ice-cold PBS buffer containing 2% fetal bovine serum (Life Technologies) was added to stop the digestion reaction. Prior to sorting, cell suspensions were passed through a 5 μm syringe filter (Millipore). Cell suspension were stained with Hoechst and Propidium Iodide for at least 30min. GFP+, Hoechst+ and Propidium Iodide- cells were sorted on a BD Influx cell sorter at the Stanford Shared FACS Facility. As a control, the corresponding GFP-, Hoechst+, and Propidium Iodide- cells were also collected. Cells were directly sorted into RTL buffer from RNAeasy kit. Sorting gates were determined by comparison to day 1 adult wildtype N2 cell suspension without any labels. At least 2000 to 10000 cells were collected per sample.
RNA isolation, amplification, library preparation and sequencing
RNA isolation was performed using the RNAeasy Plus Mirco Kit according to the manufacturer’s instructions. RNA quality and quantity was assessed by Agilent Bioanalyzer RNA Pico chip. Only samples with RIN > 6 were further processed. 50ng to 500ng of total RNA was used for cDNA generation utilizing the SMART-seq v4 Ultra low input RNA kit from Clontech. From these cDNAs, libraries were generated with the Illumina Nextera XT kit. cDNA and library samples were always analyzed by Agilent Bioanalyzer HS DNA chip for quality control and quantification. Up to 24 libraries were pooled and sequenced paired-ended using an Illumina HiSeq4000.
RNA-seq data processing
Raw sequencing reads were first trimmed using cutadapt (v1.10) to get rid of the low quality bases and adapter sequences. After trimming, paired-end reads with at least 16 nt in length were aligned to C. elegans reference genome (ce10) using STAR (2.4.2a) and quantified by RSEM (v1.2.27). We allowed mismatches up to 10% of reads length and only uniquely mapped reads were used to calculate the expression level of gene. Differential expressed genes (DEGs) were identified using DESeq2 with adjuste p-value ≤ 0.005, fold-change ≥ 2. Metascape was used to identify gene enrichment terms in up/down regulated genes.
Single molecule fluorescence in-situ hybridization (smFISH):
smFISH probes were designed with the Stellaris probe designer software (https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer) and ordered from Biosearch Technologies. Probes were labeled with either Cal Fluor Red 590, 610 or Quasar 670.
Mixed stages of worms were collected and washed multiple times with M9 buffer to rinse away bacteria. Worms were then fixed at room temperature for 45min in 4% PFA in PBS while rotating. Subsequently, worms were washed twice with PBS, resuspended in 70% ethanol and rotated at 4°C for at least 12 hours.
Before hybridization, worms were resuspended in 1ml wash buffer (10% formamide in 2xSCC) for 5 minutes. Then they were resuspended in 100ul of hybridization buffer plus smFISH pobes (1ul of 0.25uM probe stock was added to the hybridization buffer) and incubated in the dark over night at 30°C on a rotating wheel. The next morning samples where washed 3x for 30min with 1ml of wash buffer. In the last wash step, 1.5ul of 5ng/ml DAPI was added to stain for nuclei. After washing, worms were resuspended in a small volume of 2xSCC.
For imaging, small imaging chambers were assembled as described below. 2 to 4 ul of worm suspension were pipetted onto the center of an 8mm round cover glass (Electron Microscopy Sciences, #1.5 thickness). A 22×22mm cover glass was gently taped onto the drop of worms and the cover glass sandwich was flipped immediately. Any excess of buffer was removed with a kimwipe before the cover glass sandwich was adhered onto a square silicon isolator (Grace Biolabs, 20mm Diameter x 0.5 depth) on a regular microscope slide.
Brain slice preparation
Oblique horizontal brain slices (300 μm) containing the dorsal striatum were prepared using standard techniques. We used 300 μm coronal sections for FSCV experiments with 3 week old mice. Mice were anesthetized with isoflurane and decapitated. Then, the brain was quickly removed and exposed to chilled artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 15 mM glucose, 2 mM CaCl2 and 1 mM MgCl2 oxygenated with 95% O2 and 5% CO2 (300~305 mOsm, pH 7.4). Tissue vibratome was used to section chilled brain, producing brain slices containing dorsal striatum. Acute brain slices were first kept in ACSF for 30 min at 34 °C and then maintained for another 30 min at room temperature. After recovery period, slices were moved to a submerged recording chamber perfused with ACSF at a rate of 2~3 ml/min at 30~31 °C and brain slices were recorded within 4 hours after recovery.
Fast-scan cyclic voltammetry (FSCV)
Extracellular dopamine release was monitored by fast-scan cyclic voltammetry recordings performed in dorsal striatum using carbon-fiber microelectrodes (7 μm diameter carbon fiber extending 50~100 μm beyond the tapered glass seal). Cyclic voltammograms were measured with a triangular potential waveform (−0.4 to +1.3 V vs Ag/AgCl reference electrode, 400 V/s scan rate, 8.5 ms waveform width) applied at 100 ms intervals. The carbon fiber microelectrode was held at −0.4 V between scans. Cyclic voltammograms were background-subtracted by averaging 10 background scans. For 6–9 week old mice, ChR2-expressing dopaminergic axon terminals were stimulated by blue laser light (450 nm, 2 ms single pulse for one pulse stimulation, 5 pulses with 2 ms pulse width at 25 Hz for 5 pulses stimulation). For 3 week old mice, dopaminergic axon terminals were stimulated locally (100–200μm from carbon fiber) with an electrode. Optogenetically and electrically evoked dopamine release and subsequent oxidation current was detected and monitored using TarHeel CV system. Changes in dopamine concentration by optical stimulation were quantified by plotting the peak oxidation current of the voltammogram over time. Carbon fiber microelectrode was calibrated at the end of each day of experiments to convert oxidation current to dopamine concentration using 10 μM dopamine in ACSF.
Whole-cell patch clamp recording
Whole-cell patch clamp recordings were made in the dorsal striatum. Spiny projection neurons were visually identified by conventional IR-DIC optics and whole-cell patch clamp recordings were conducted using borosilicate glass pipettes (2.5~3.5 MΩ). Cs+-based low Cl- internal solution containing 126 mM CsMeSO3, 10 mM HEPES, 1 mM EGTA, 2 mM QX-314 chloride, 0.1 mM CaCl2, 4 mM MgATP, 0.3 mM Na3GTP, 8 mM Na2-phosphocreatine (280~290 mOsm, pH 7.3 with CsOH) was used to measure both oIPSC and oEPSC from the same SPN neurons. Membrane potentials were first held at +8 mV (the reversal potential of ionotropic glutamate receptors, liquid junction potential not corrected) to measure oIPSC. After acquiring stable oIPSC, membrane potential was held at –70 mV (the reversal potential of chloride) to measure oEPSC. To selectively activate ChR2-expressing dopaminergic axon terminals, blue laser light (450 nm, 0.5 msec pulses with 60 sec intervals, and saturation power under the objective less than 20 mW) was focused on the back focal plane of the objective to produce wide-field illumination. Access resistance was 10~20 MΩ (no compensation) and only cells in which access resistance changed < 20% were included in the analysis. Whole-cell patch recordings were performed using Multiclamp 700B and signals were filtered at 2 kHz and digitized at 10 kHz. Recording data were acquired online by WinWCP program and analyzed offline using Clampfit 10.0.
Immunohistochemistry
3 week or 6 week old DAT-Cre;Thoc5+/+ and DAT-Cre;Thoc5fl/fl mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) / xylazine (10 mg/kg) solution and perfused transcardially with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. Brains were rapidly removed and maintained in the same fixative at 4°C for overnight. Fixed brain was transferred to 30% sucrose in 0.01M phosphate buffer (PB) for cryoprotection. Serial sections containing striatum and midbrain were made using the frozen sectioning technique. For immunofluorescence staining, sections were incubated with anti-tyrosine hydroxylase antibody (rabbit polyclonal, 1:1000, ab112, abcam) and anti-dopamine transporter antibody (rat monoclonal, 1:500, ab5990, abcam) at 4 °C overnight, followed by secondary antibodies conjugated to Alexa 488 and Alexa 647 fluorophores at room temperature for 1 hour to detect signals. Images were captured using a Leica confocal microscope or a microscope equipped with a CCD camera.
Single molecule pulldown
C. elegans strains wyIs783 (Prab-3::THOC-1::3xHA::tdTomato, Prab-3::CREB::GFP) and wyEx8951 (Prab-3::THOC-1::3xHA::tdTomato & Prgef-1::GFP) were grown on 6-cm dishes, collected and washed, treated with or without 0.5% PFA then dropped in liquid nitrogen to form “worm pearls”. Worm pearls were thawed in a two-fold excess (volume/weight) of lysis buffer (50 mM HEPES pH 7.7, 50 mM KCl, 2 mM MgCl2, 250 mM Sucrose, 1 mM EDTA pH 8.0, with protease inhibitors). After sonication on ice to break cuticle (Fisher Scientific Sonic Dismembrator model 500, amplitude 10%, 5 seconds pulse with 59 seconds pause, 8 cycles), final concentration of 100 mM NaCl and 1% Triton X-100 were added into solution and samples were rotated at 4°C for 1 hour. After centrifugation at 15,000 g 4°C for 30 minutes, supernatants were transferred to new tubes and measured by BCA assay for total protein concentration (11–15 mg/ml). For worm SiMPull experiments, all worm lysates from different samples were then adjusted by lysis buffer to 10 mg/ml total protein concentration. Adjusted lysates were further serially diluted to desired concentrations in order to achieve optimum fluorescent protein density (100–400 molecules in a 2000 um2 imaging area) on SiMPull quartz slides surface which were coated with biotinylated anti-GFP antibodies (Rockland immunochemicals, 600–106-215) for CREB::GFP or GFP pull down, or biotinylated anti-HA antibody (Abcam, ab26228) for THOC-1::3xHA::tdTomato pull down. Briefly, when pulling down GFP, 100x diluted solution for wyIs783 samples and 1000x diluted solution for wyEx8951 samples were used.
Mouse midbrains were homogenized by a glass-Teflon homogenizer in a five-fold excess (volume/weight) of lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM EDTA, 10 mM EGTA, 10 mM Tris-HCl, pH 7.4, with protease inhibitors) and then rotated at 4°C for 30 minutes. Samples were cleared from non-solubilized material by ultracentrifugation (400,000 rpm for 30 minutes) and measured by BCA assay for total protein concentration (1.2–1.5 mg/ml). In order to immobilize THOC1 and visualize its associated CREB from brain lysate, anti-THOC1 (Novus, NB100–174) and normal IgG (Santa Cruz, sc-2025) were dialyzed by Pierce antibody clean-up kit (Thermo Fisher, 44600), labeled with hydrazide-PEG4-biotins (Thermo Fisher, 21360) according to manufacturer’s manual and then purified by Amicon Ultra-4 centrifugal filters (Millipore, UFC8100). Anti-CREB (sigma, PLA0205) was labeled with Alexa Fluor 546 NHS ester dye (Thermo Fisher, A20002) according to manufacturer’s manual and purified by Illustra NAP-5 columns (GE Healthcare 17–0853-02). The concentration of antibodies after labeling was determined by Nanodrop spectrophotometer. For mouse SiMPull experiments, in brief, SiMPull slides were coated with 10 nM biotinylated anti-THOC1 or biotinylated normal IgG which served as a non-specific binding control, brain lysates were incubated then washed away, and captured proteins on slides were stained by 5 nM Alexa Fluor 546 conjugated anti-CREB.
Proteins immobilized on the slides were visualized by a TIRF microscope equipped with excitation laser 488 nm (GFP) and 561 nm (tdTomato or Alexa Fluor 546), and DV2 dichroic 565dcxr dual-view emission filters (520/30 nm and 630/50 nm). Mean spot count per image and standard deviation were calculated from images taken from 10–20 different regions.
RNA-immunoprecipitation:
Worm lysates were prepared from fixed worms as described above. To avoid RNA degradation during sample preparation and immunoprecipitation, we added RNasin RNAse inhibitor and protease inhibitor to the lysis buffer.
Total RNA was extracted from a small aliquot of lysate using the Trizol method, following the manufacturer’s protocol (https://tools.thermofisher.com/content/sfs/manuals/trizol_reagent.pdf). Total RNA concentration was then measured by nanodrop. Worm lysates from different samples were adjusted/diluted to the same RNA concentration before immunoprecipitation. 50ul of Pierce™ Anti-HA Magnetic Beads were added to each sample and incubated for 2 to 3 hours at 4°C on a rotating wheel. Samples were washed 3x for 30min with lysis buffer. Bound RNA was then eluted twice with 125ul elution buffer (50mMTris pH 8, 10mM EDTA, 1.3%SDS, RNasin, preheated to 65°C) for 30min at 65°C. RNA from pooled eluates was then purified using the Trizol extraction method.
Reverse transcription and quantitative real-time PCR
cDNA was synthesized in a 20μl reaction volume using the SuperScript III firststrand synthesis Super Mix (Invitrogen #11752). 2μl of 1:4 dilution of cDNA was used as the template in a 20μl reaction volume from the SsoFast EvaGreen Supermix with Low ROX (Bio-Rad, #172–5211). A list of the real-time PCR primers can be found in the key resource table. Quantitative real-time PCR was performed in duplicates using the Bio-Rad CFX96 Real-Time System. Data were analyzed using the standard curve method. The entire experiments were repeated 3 times on independent RNA preparations.
Quantification and Statistical Analysis
All data are expressed as the mean ± the standard deviation (SD) or mean ± the standard error of the mean (SEM) as stated in the figure legends and results. All statistical tests were performed in GraphPad Prism 7 and specific statistical tests are described in the figure legends. For all experiments with more than two samples we first analyzed the data set for overall statistically significant differences (ANOVA) and subsequently compared the indicated datasets with corrections for multiple comparisons applied to the stated P values. Experiments with sample size of two, data sets were analyzed by unpaired student t-test.
Supplementary Material
Figure S1: Detailed analysis of synaptic defects in THOC mutant animals (Related to Figure 1)
(A) Quantification of synaptic density relative to wt for different THOC mutants and their corresponding cell-specific rescue.
(B) Quantification of synaptic size for different THOC mutants and their corresponding cell-specific rescue.
(C) Quantification of synapse intensity for different THOC mutants and their corresponding cell-specific rescue.
(A-C) N=10–20 animals at L4 stage; ****, p<0.0001, ***, p<0.001**, p<0.01, *, p<0.05, one-way Anova with Tukey post-hoc test.
(D) Quantification of synaptic density for L3 and L4 animals in wt and thoc-5 mutant background.
(E) Quantification of synaptic size for L3 and L4 animals in wt and thoc-5 mutant background.
(F) Quantification of synapse intensity for L3 and L4 animals in wt and thoc-5 mutant background.
(D-F) N=10–20 animals; ***, p<0.001, student t-test.
Averages and SEM are plotted in all graphes.
(G) thoc-1 mRNA quantification in wt and thoc-1 strain backgrounds show a 70% reduction in thoc-1 mRNA level in the mutant.
Figure S2: thoc mutation does not affect PDE neuronal health (Related to Figure 1)
(A) - (C) Visualization and quantification of mitochondria in PDE cell body (A) and axons (B), (C) in wt and thoc-5 mutants.
(D)-(I) thoc-5 mutation does not affect the localization, number and morphology of different organelle markers. (D) ribosomes, (E) ER, (F) Endosome, (G), (H) Golgi and (I) cilia.
Figure S3: THOC mutants are impaired in DA signaling and DA-dependent behavior (Related to Figure 1 & 2)
(A) and (B) Quantitative analysis of basal slowing response in the absence (A) or presence (B) of DA. Locomotion rates in the absence (black bars) or presence (white bars) of bacteria were calculated in a 20 second time window. Wt and cat-2 animals were included as controls. The percentage of slowing is shown on the right. N=30–60 animals; ***, p<0.001, **, p<0.01, one-way Anova with Tukey post-hoc test.
(C) Ca2+ response of PDE neuron during basal slowing response. Calcium trace and quantification of it is shown for wt and thoc-5 mutant animals.
Averages and SEM are plotted in all graphs.
Figure S4: Presynaptic transcript retention in the nucleus is rescued in thoc-5 creb mutant animals. (Related to Figure 3)
(A) Representative images for smFISH for presynaptic and control transcripts in wt (upper row) and thoc-5 (lower row) mutant animals. GFP: PDE cellbody; red: signal for FISH probes; blue: DAPI staining of nuclei. Scale bar represents 2μm.
(B) Total number of smFISH puncta in PDE is not changed by thoc-5 mutation.
(C) Gene enrichment analysis for down-regulated genes in thoc-5 mutant neurons.
(D) qPCR quantification of presynaptic as well control transcripts in wt and thoc-5 mutant strain backgrounds. N = 3 biological replicates for both genotypes. ****, p<0.0001, ***, p<0.001, **, p<0.01; one-way Anova with Tukey post-hoc test. Averages and standard deviations are plotted.
Figure S5: Putative disease-associated thoc-1 R123H mutation renders THOC-1 dysfunctional (Related to Figure 7)
(A) Sequence alignment of human, mouse and C. elegans THOC1. Human, ALS-associated mutations are highlighted. Red: conserved R123H mutation, Blue: other disease associated mutations on un-conserved residues.
(B) Integrated synaptic intensity is quantified for wt, thoc-1, thoc-1 Pdat-1::thoc-1R123H and thoc-1 Pdat-1::thoc-1V338I strain backgrounds. N=20 animals; ****, p<0.0001, one-way ANOVA with Tukey post-hoc test.
Averages and SEM are plotted.
Figure S6: CREB, SRF/UNC-120 and PAB-2 function with THOC to facilitate export of synaptic transcripts (Related to Figure 6)
(A) Integrated synaptic intensity is quantified for two lines, in which crh-1 is cell-specifically knocked-down in DA neuron by CRE-loxP strategy. N=10–20 animals;
(B) Quantification of nuclear transcripts in wt, thoc-5, thoc-5 crh-1 and crh-1 PDE neurons. N=10–50 animals.
(C) Integrated synaptic intensity is quantified for wt, thoc-5, thoc-5 unc-120 and unc-120 mutant animals. N=20–50 animals
(D) Integrated synaptic intensity is quantified for wt, thoc-5, thoc-5 pab-2 and pab-2 mutant animals. N=20–50 animals
(D) Quantification of nuclear transcripts in wt, thoc-5, thoc-5 pab-2 and pab-2 PDE neurons. N=10–50 animals. ***, p<0.001; one-way Anova with Tukey post-hoc test. Averages and SEM are plotted in all graphs.
****, p<0.0001, ***, p<0.001, **, p<0.01, *, p<0.05 one-way Anova with Tukey post-hoc test.
Figure S7: THOC-1 specifically interacts with CREB but not GFP and binds to presynaptic transcripts (Related to Figure 7)
(A) Localization of THOC and CRH-1a does not depend on each other. THOC component are not mis-localized in crh-1 mutant background. CRH-1A localization is not altered by thoc-5 mutation.
(B) Quantification of the interaction between THOC-1 and increasing GFP concentration. No significant interaction between those two proteins was detected even at very high GFP concentration in PFA fixed samples. These result further corroborate the specificity of the direct interaction between CREB and THOC-1.
(C) Relative IP efficiency of synaptic transcripts (orange bars) as well as other pan-neuronal and control genes (black). THOC-1 bound transcripts were pulled-down under wt conditions. Transcripts were then quantified by qPCR and normalized onto total mRNA.
(D) Correlation analysis between total mRNA level and IPed mRNAs for wt and crh-1 mutant animals. No correlation was detected for synaptic transcripts (orange bars) and control transcripts (black bars).
Supplemental Table S1: Summary table of RNAseq data. (Related to Figure 3 and Figure S4)
Supplemental Table S2: qPCR primers used in this study. (Related to STAR methods)
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| C.elegans STRAINS | ||
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV14139 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP] | this study | TV15123 |
| thoc-5(wy822); wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV14937 |
| thoc-5(wy822); wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP] | this study | TV16068 |
| wyIs690[Pdat-1::CLA-1::GFP, Pdat-1::mcherry, Podr-1::RFP] | this study | TV18497 |
| thoc-5(wy822); wyIs690[Pdat-1::CLA-1::GFP, Pdat-1::mcherry, Podr-1::RFP] | this study | TV18706 |
| wyIs692[Pdat-1::3xnovo2GFP::SYD-2, Pdat-1::mcherry, Podr-1::RFP] | this study | TV18524 |
| thoc-5(wy822); wyIs692[Pdat-1::3xnovo2GFP::SYD-2, Pdat-1::mcherry, Podr-1::RFP] | this study | TV18689 |
| wyIs513[Pdat-1::GFP::SNN-1b, Pdat-1::mcherry, Podr-1::RFP] | this study | TV14859 |
| thoc-5(wy822); wyIs513[Pdat-1::GFP::SNN-1b, Pdat-1::mcherry, Podr-1::RFP] | this study | TV19305 |
| wyIs553[Pdat-1::UNC-10::3xGFP, Pdat-1::tdTomato::RAB-3, Podr-1] | this study | TV15119 |
| thoc-5(wy822); wyIs553[Pdat-1::UNC-10::3xGFP, Pdat-1::tdTomato::RAB-3, Podr-1] | this study | TV19407 |
| wyIs521[Pdat-1::CAT-1::GFP, Pdat-1::mcherry, Pord-1::RFP] | this study | TV114867 |
| thoc-5(wy822); wyIs521[Pdat-1::CAT-1::GFP, Pdat-1::mcherry, Pord-1::RFP] | this study | TV19726 |
| ztf-11(wy1077)[ztf-11::zf1::GFPnovo2]; zif-1(gk117) | this study | TV20812 |
| thoc-5(wy822); ztf-11(wy1077)[ztf-11::zf1::GFPnovo2] | this study | TV21659 |
| thoc-5(wy822); wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; wyEx6458[Pdat-1::THOC-5] | this study | TV16056 |
| thoc-2(ok961)/hTs[bli4(e937) let-?(q782) qIs48]; wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP] | this study | TV16277 |
| thoc-1(wy817)/+; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV14850 |
| wyEx7354[Pdat-1::THOC-1::GFP]; thoc-1(wy817)/+; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV17802 |
| aly-1(ok1920) aly-2(wy924) aly-3(wy909); wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV17992 |
| syd-2(wy1073)[GFP::syd-2] | this study | TV20871 |
| syd-2(wy1073)[GFP::syd-2]; thoc-5(wy822) | this study | TV20982 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; Psnt-1::flp | this study | TV22398 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; Psnt-1::flp; thoc-5(wy822) | this study | TV22397 |
| sng-1(ox706)[sng-1::GFP] | Jorgensen lab | EG9408 |
| sng-1(ox706)[sng-1::GFP]; thoc-5(wy822) | this study | TV21323 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP] | this study | TV21223 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822) | this study | TV21801 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822) crh-1(tz2) | this study | TV21529 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8927[Ptph-1::flp; Podr-1::RFP] | this study | TV21944 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8927[Ptph-1::flp; Podr-1::RFP]; thoc-5(wy822) crh-1(tz2) | this study | TV22039 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8929[Punc-47::flp; Podr-1::RFP] | this study | TV21946 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8929[Punc-47::flp; Podr-1::RFP]; thoc-5(wy822) crh-1(tz2) | this study | TV22015 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8929[Punc-47::flp; Podr-1::RFP]; thoc-5(wy822) | this study | TV22020 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyIs821[Pmig-13::flp; Podr-1::RFP]; thoc-5(wy822) | this study | TV22025 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyIs821[Pmig-13::flp; Podr-1::RFP]; thoc-5(wy822) crh-1(tz2) | this study | TV22396 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyIs821[Pmig-13::flp; Podr-1::RFP] | this study | TV22024 |
| cat-2(e1112) | CGC | CB1112 |
| thoc-2(ok961)/hTs[bli4(e937) let-?(q782) qIs48] | CGC | VC673 |
| wyIs700[Pdat-1::GFP; Podr-1::RFP] | this study | TV18678 |
| wyIs700[Pdat-1::GFP; Podr-1::RFP]; thoc-5(wy822) | this study | TV18846 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822); cat-2(e1112) | this study | TV22035 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; dop-5(ok568) | this study | TV22046 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822); dop-5(ok568) | this study | TV22401 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; pin-2(gk435511) | this study | TV22370 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; pin-2(gk435511), chr-1(tz2) | this study | TV22371 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; pin-2(gk435511); thoc-5(wy822) chr-1(tz2) | this study | TV22400 |
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; wyEx8629[Pdat-1::HisCL; Podr-1::GFP] | this study | TV21033 |
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; wyEx8712[Pdat-1::HisCL; Podr-1::GFP]; thoc-5(wy822) | this study | TV21209 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); gsa-1(ce94) | this study | TV21136 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); cmk-1(ok287) | this study | TV20576 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); crtc-1(tm7084) | this study | TV20876 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); pmk-1(km25) | this study | TV21330 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); trp-4(sy695) | this study | TV21791 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); dop-1(vs100) | this study | TV21660 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); egl-30(ad805) | this study | TV21954 |
| wyIs557[Pdat-1::GFP::ELKS-1, Pdat-1::tdTomato::RAB-3, Podr-1::GFP]; thoc-5(wy822); kin-2(ce179) | this study | TV20140 |
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; thoc-5(wy822); dop-2(vs105); dop-3(vs106) | this study | TV21523 |
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; thoc-5(wy822); pkc-1(ok563) | this study | TV19788 |
| wyEx6497[Pdat-1::THOC-1::GFP; Podr-1::GFP] | this study | TV16143 |
| wyIs724[Pdat-1::THOC-2::GFP; Pdat-1::mcherry; Podr-1::RFP] | this study | TV19593 |
| wyIs737[Pdat-1::THOC-5::GFP, Pdat-1::mcherry; Podr-1::RFP] | this study | TV19785 |
| wyEx8628[Pdat-1::THOC-1::tdTomato; Pdat-1::crh-1a::GFP; Podr-1::RFP] | this study | TV21032 |
| wyEx7120[Pdat-1::aly-1::GFPnovo2; Pdat-1::mcherry, Podr-1::GFP] | this study | TV17347 |
| wyEx7122[Pdat-1::aly-2::GFPnovo2; Pdat-1::mcherry, Podr-1::GFP] | this study | TV17349 |
| wyEx7124[Pdat-1::aly-3::GFPnovo2; Pdat-1::mcherry, Podr-1::GFP] | this study | TV17351 |
| wyIs783[Prab-3::THOC-1::3xHA::tdTomato; Prab-3::crh-1a::GFP, Podr-1::RFP] | this study | TV21307 |
| wyEx8951[Prab-3::THOC-1::3xHA::tdTomato; Prgef-1:: GFP, Podr-1::RFP] | this study | TV21991 |
| wyEx8951[Prab-3::THOC-1::3xHA::tdTomato; Prgef-1:: GFP, Podr-1::RFP]; crh-1(tz2) | this study | TV22380 |
| thoc-1(wy817)/+; wyEx9278[Pdat-1::THOC-1(V338I)::GFP, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV22654 |
| thoc-1(wy817)/+; wyEx9279[Pdat-1::THOC-1(V338I)::GFP, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV22655 |
| thoc-1(wy817)/+; wyEx9338[Pdat-1::THOC-1(P1236T)::GFP, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV22684 |
| thoc-1(wy817)/+; wyEx9337[Pdat-1::THOC-1(P1236T)::GFP, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV22863 |
| thoc-1(wy817)/+; wyEx9353[Pdat-1::THOC-1(P1236T)::GFP, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP] | this study | TV22925 |
| wyEx9292[Pdat-1::CRE, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; thoc-5(wy822) crh-1(wy1152wy1154) | this study | TV22668 |
| wyEx9293[Pdat-1::CRE, Podr-1::GFP]; wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; thoc-5(wy822) crh-1(wy1152wy1154) | this study | TV22669 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822) crh-1(tz2); dop-5(ok568) | this study | TV23052 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822) crh-1(tz2); pin-2(gk435511) | this study | TV22400 |
| edIs6[Punc-119::GFP; rol-6(su1006)] | CGC | DP132 |
| edIs6[Punc-119::GFP; rol-6(su1006)]; thoc-5(wy822) | this study | TV19032 |
| lite-1(xu7); xuEx2165[Pdat-1::GCaMP6f+Pdat-1::sl2::mcherry] | this study | TQ6291 |
| lite-1(xu7); thoc-5(wy822); xuEx2165[Pdat-1::GCaMP6f+Pdat-1::sl2:mcherry] | this study | TQ6340 |
| wyEx6695[Pdat-1::rps-2::3xGFP, Pdat-1::mcherry, Podr-1::GFP] | this study | TV16704 |
| wyEx6695[Pdat-1::rps-2::3xGFP, Pdat-1::mcherry, Podr-1::GFP]; thoc-5(wy822) | this study | TV17593 |
| wyEx6697[Pdat-1::aman-2b::YFP, Pdat-1::mcherry, Podr-1::GFP] | this study | TV16706 |
| wyEx6697[Pdat-1::aman-2b::YFP, Pdat-1::mcherry, Podr-1::GFP]; thoc-5(wy822) | this study | TV17622 |
| wyEx6699[Pdat-1::rab-5::GFP, Pdat-1::mcherry, Podr-1::GFP] | this study | TV16708 |
| wyEx6699[Pdat-1::rab-5::GFP, Pdat-1::mcherry, Podr-1::GFP]; thoc-5(wy822) | this study | TV17594 |
| wyEx6702[Pdat-1::mito::GFP, Pdat-1::mcherry, Podr-1::GFP] | this study | TV16711 |
| wyEx6702[Pdat-1::mito::GFP, Pdat-1::mcherry, Podr-1::GFP]; thoc-5(wy822) | this study | TV17595 |
| wyEx6702[Pdat-1::SP-12::GFP, Pdat-1::mcherry, Podr-1::GFP] | this study | TV16863 |
| wyEx6702[Pdat-1::SP-12::GFP, Pdat-1::mcherry, Podr-1::GFP]; thoc-5(wy822) | this study | TV17592 |
| wyEx6781[Pdat-1::rab-6.2::GFP, Pdat-1::mcherry, Podr-1::GFP] | this study | TV16866 |
| wyEx6781[Pdat-1::rab-6.2::GFP, Pdat-1::mcherry, Podr-1::GFP]; thoc-5(wy822) | this study | TV17616 |
| wyEx6497[Pdat-1::THOC-1::GFP; Podr-1::GFP]; crh-1(tz2) | this study | TV23400 |
| wyIs724[Pdat-1::THOC-2::GFP; Pdat-1::mcherry; Podr-1::RFP]; crh-1(tz2) | this study | TV23437 |
| wyIs737[Pdat-1::THOC-5::GFP, Pdat-1::mcherry; Podr-1::RFP]; crh-1(tz2) | this study | TV23401 |
| wyEx7120[Pdat-1::aly-1::GFPnovo2; Pdat-1::mcherry, Podr-1::GFP]; crh-1(tz2) | this study | TV23402 |
| wyEx8581[Pdat-1::crh-1a::GFP, Podr-1::RFP] | this study | TV20899 |
| wyEx8581[Pdat-1::crh-1a::GFP, Podr-1::RFP]; thoc-5(wy822) | this study | TV20901 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; thoc-5(wy822); pab-2(ok1851) | this study | TV22402 |
| rab-3(ox699)[GFP::FLP-on::rab-3]; wyEx8715[Pdat-1::flp; Podr-1::GFP]; pab-2(ok1851) | this study | TV22044 |
| wyIs700[Pdat-1::GFP; Podr-1::RFP]; pab-2(ok1851) | this study | TV21475 |
| wyIs700[Pdat-1::GFP; Podr-1::RFP]; thoc-5(wy822); pab-2(ok1851) | this study | TV21530 |
| wyIs700[Pdat-1::GFP; Podr-1::RFP]; crh-1(tz2) | this study | TV20417 |
| wyIs700[Pdat-1::GFP; Podr-1::RFP]; thoc-5(wy822) crh-1(tz2) | this study | TV20588 |
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; unc-120(st364) | this study | TV23430 |
| wyIs427[Pdat-1::GFP::ELKS-1, Pdat-1::mcherry, Podr-1::RFP]; unc-120(st364); thoc-5(wy822) | this study | TV23599 |
| Mouse strains | ||
| THOC5(fl/fl) | (Saran et al., 2013) | N/A |
| DAT-IRES-Cre (B6.SJL-Slc6a3tm1.1(cre)Bkmn/J) | JAX | Cat# JAX:006660; RRID:IMSR_JAX:006660 |
| Ai32 (B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J) | JAX | Cat# JAX:012569; RRID:IMSR_JAX:012569 |
| ANTIBODIES | ||
| Rabbit anti SNB-1 | Nonet lab | Ab1092 |
| Rabbit anti SNT-1 | Nonet lab | Ab1095 |
| Rabbit anti UNC-64 | Nonet lab | Ab940 |
| Rabbit anti ELKS-1 | Nonet lab | Ab237 |
| Chicken anti SNG-1 | Nonet lab | Ab1165 |
| Rabbit anti-Chicken IgY (H+L) Secondary Antibody, HRP conjugate | Thermo Scientific | Cat# 31401 |
| Perox-IgG Fraction Monoc Mse Anti-Rab IgG, Lt Chn Spec (min X Bov,Gt,Arm Hms,Hrs,Hu,Ms,Rat,Shp Ig) | Jackson Immuno | Cat# 211–032-171 |
| GFP (goat) antibody biotin conjugated | Rockland inc. | Cat# 600–106-215; RRID:AB_218204 |
| Anti-Tyrosine Hydroxylase antibody | Abcam | Cat# ab112; RRID:AB_297840 |
| Anti-Dopamine Transporter antibody | Abcam | Cat# ab5990; RRID:AB_305226 |
| mono-clonal anti-HA antibody produced in mouse | Sigma | Cat# H3663; RRID:AB_262051 |
| Pierce™ Anti-HA Magnetic Beads | Thermo Scientific | Cat# 88836 |
| Mouse anti-actin, monoclonal (clone: C4) | MP Biomedicals | Cat# 8691001 |
| Monoclonal Anti-α-Tubulin antibody produced in mouse | Sigma | Cat# T6199; RRID:AB_477583 |
| Goat anti rabbit IgG H&L Alexa Fluor 488 | Abcam | Cat# ab150077; RRID:AB_2630356 |
| Goat anti rat IgG H&L Alexa Fluor 647 | Abcam | Cat# ab150167 |
| Mouse anti THOC1, monoclonal | Novus | Cat# NB100–174; RRID:AB_2202241 |
| Rabbit anti CREB | Sigma | Cat# PLA0205 |
| Oligonucleotides | ||
| Oligonucleotides for qPCR | this study | Table S2 |
| Software | ||
| ImageJ, Version 1.45k | NIH | https://imagej.nih.gov/ij/download.html |
| Metamorph, Version 7.7.9.0 | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/15203/~/metamorph®-software-installation-files |
| Prism, Version 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Deposited Data | ||
| Raw and analyzed data | this study | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102300 |
| Critical commercial assay | ||
| ssofast evagreen supermix with low ROX | Biorad | Cat# 172–5211 |
| SuperScript® III First-Strand Synthesis SuperMix for qRT-PC | Invitrogen | Cat# 11752–050 |
| Chemicals | ||
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Scientific | Cat# 34096 |
| NuPAGE® Novex® 4–12% Bis-Tris Protein Gels, 1.0 mm, 12 well | Life Technologies | Cat# NP0322BOX |
| NuPAGE® MOPS SDS Running Buffer (20X) | Life Technologies | Cat# NP0001 |
| NuPAGE® Transfer Buffer (20X) | Life Technologies | Cat# NP0006 |
| NuPAGE® LDS Sample Buffer (4X) | Life Technologies | Cat# NP0007 |
| Methanol | Sigma | Cat# 320390 |
| Blotting grade dry milk | Biorad | Cat# 170–6404 |
| Paraformaldehyde | Sigma | Cat# 158127 |
| Dextran Sulfate 50% Solution, 100 mL | Millipore | Cat# S4030 |
| E.coli tRNA | Roche | Cat# 10109541001 |
| UltraPure™ BSA (50 mg/mL) | Life Technologies | Cat# AM2618 |
| Nuclease-Free Water (not DEPC-Treated) | Life Technologies | Cat# AM9939 |
| 20X SSC | Life Technologies | Cat# AM9770 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dilactate) | Life Technologies | Cat# D3571 |
| Formamide (Deionized) | Life Technologies | Cat# AM9342 |
| Histamine dihydrochloride | Sigma | Cat# H7250 |
| Dopamine hydrochloride | Sigma | Cat# H8502 |
| RNasin Ribonuclease Inhibitor | Promega | Cat# N2115 |
| Complete mini EDTA-free protease inhibitor tablets | Roche | Cat# 11836170001 |
| TRIzol® LS Reagent | Life Technologies | Cat# 10296–010 |
| GlycoBlue™ Coprecipitant (15 mg/mL) | Thermofisher | Cat# AM9515 |
| Levamisol hydrochloride | Sigma | Cat# 31742 |
| RNAeasy Plus Micro Kit | Qiagen | Cat# 74034 |
| SMART-seq v4 Ultra low input RNA kit | Clontech | Cat# 634888 |
| Nextera XT DNA library preparation kit | Illumina | Cat# FC-131–1024 |
| Protease from Streptomyces griseus | Sigma | Cat# P6911 |
| Fetal Bovine Serum, certified, heat inactivated, US origin | Life Technologies | Cat# 10082–139 |
| Hoechst 33258, Pentahydrate (bis-Benzimide) - 10 mg⁄mL Solution in Water | Life Technologies | Cat# H3569 |
| Propidium Iodide - 1.0 mg⁄mL Solution in Water | Life Technologies | Cat# P3566 |
| Isofluorane | Vetone | Cat# 200–070 |
| Ketamine | Vedco | Cat# 50989–161-06 |
| xylazine | Lloyd | Cat# 139–236 |
| NBQX | Tocris | Cat# 1044 |
| (R)-CPP | Tocris | Cat# 0247 |
| SR95531 | Tocris | Cat# 1262 |
| Recombinant DNA | ||
| pCM362: pSM::Pdat-1::tdTomato-rab-3::unc10–3’UTR | this study | N/A |
| pCM367: pSM::Pdat-1::mcherry::unc10–3’UTR | this study | N/A |
| pCM377: pSM::Pdat-1::GFP::ELKS-1::unc-54 3’UTR | this study | N/A |
| pCM378: pSM::Pdat-1::GFP::snn-1::unc-54 3’UTR | this study | N/A |
| pCM413: pSM::Pdat-1::GFP::unc-54 3’UTR | this study | N/A |
| pCM429: pSM::Pdat-1::cat-1::GFP::unc-54 3’UTR | this study | N/A |
| pCM434: pSM::Pdat-1::unc-10::3xGFP::unc-54 3’UTR | this study | N/A |
| pCM483: pSM::Pdat-1::THOC-5::GFP::unc-54 3’UTR | this study | N/A |
| pCM485: pSM::Pdat-1::THOC-5::unc-54 3’UTR | this study | N/A |
| pCM493: pSM::Pdat-1::THOC-2::GFP::unc-54 3’UTR | this study | N/A |
| pCM496: pSM::Pdat-1::THOC-1::GFP::unc-54 3’UTR | this study | N/A |
| pCM526: pSM::Pdat-1::3xGFPnovo2::syd-2::unc-54 3’UTR | this study | N/A |
| pCM530: pSM::Pdat-1::aly-1::GFPnovo2::unc-54 3’UTR | this study | N/A |
| pCM531: pSM::Pdat-1::aly-2::GFPnovo2::unc-54 3’UTR | this study | N/A |
| pCM532: pSM::Pdat-1::aly-3::GFPnovo2::unc-54 3’UTR | this study | N/A |
| pCM562: pSM::Pdat-1::GFP::cla-1::unc-54 3’UTR | this study | N/A |
| pCM698: pSM::Pdat-1::CRH-1a::GFP::unc-54 3’UTR | this study | N/A |
| pCM708: pSM::Pdat-1::THOC-1::tdTomato::unc-54 3’UTR | this study | N/A |
| pCM710: pSM::Prab-3::CRH-1a::GFP::unc-54 3’UTR | this study | N/A |
| pCM711: pSM::Pdat-1::HisCL1::unc-54 3’UTR | this study | N/A |
| pCM712: pSM::Prab-3::THOC-1::3xHA::tdTomato::unc-54 3’UTR | this study | N/A |
| pCM763: pSM::Prgef-1:: GFP::let-858 3’UTR | this study | N/A |
| Other | ||
| EpiShear Probe Sonicator | Active Motif | Cat# 53051 |
| CFX96 Touch™ Real-Time PCR Detection System | Biorad | Cat# 1855196 |
| Zeiss Axio Observer Microscope with Yokagawa spinning disk head | Zeiss/Solamere Technology | N/A |
Acknowledgements
We are grateful to Michael L. Nonet for sharing antibodies. We thank the international Caenorhabditis Genetics Center for strains. We also thank C. Yee and the Stanford FACS facility for help with FACS sorting, C. Gao and K. Vega for technical assistance and members of the Shen lab for valuable comments and discussion. This work is supported by Howard Hughes Medical Institute and NIH (K.S.); NIH NS103037 NS091144, and a GG Technologies gift fund (J.B.D.); R01HL127764 (Y.K.X.); R35GM126917 (X.Z.S.X.)
Footnotes
Data and Software Availability
The RNA-seq data has been deposited in the NCBI GO database with the GEO Accession number GSE102300 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102300].
Declaration of interest
The authors declare no competing interests.
References
- Aguado F, Díaz-Ruiz C, Parlato R, Martínez A, Carmona MA, Bleckmann S, Ureña JM, Burgaya F, Río JA del, Schütz G, et al. (2009). The CREB/CREM Transcription Factors Negatively Regulate Early Synaptogenesis and Spontaneous Network Activity. J. Neurosci. 29, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY, and Montminy M (2011). CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu CL, Huang L, Innes AM, Akimenko M-A, Puffenberger EG, Schwartz C, Jerry P, Ober C, Hegele RA, McLeod DR, et al. (2013). Intellectual disability associated with a homozygous missense mutation in THOC6. Orphanet J. Rare Dis. 8, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune C, Munchel SE, Fischer N, Podtelejnikov AV, and Weis K (2005). Yeast poly(A)-binding protein Pab1 shuttles between the nucleus and the cytoplasm and functions in mRNA export. RNA 11, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H, Keller DM, Yochum GS, Impey S, and Goodman RH (2004). Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc. Natl. Acad. Sci. U. S. A. 101, 13572–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez S, García-Rubio M, Prado F, and Aguilera A (2001). Hpr1 Is Preferentially Required for Transcription of Either Long or G+C-Rich DNA Sequences in Saccharomyces cerevisiae. Mol. Cell. Biol. 21, 7054–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare D, Fimia GM, and Sassone-Corsi P (1999). Signaling routes to CREM and CREB: plasticity in transcriptional activation. Trends Biochem. Sci. 24, 281–285. [DOI] [PubMed] [Google Scholar]
- Fimia GM, De Cesare D, and Sassone-Corsi P (1999). CBP-independent activation of CREM and CREB by the LIM-only protein ACT. Nature 398, 165–169. [DOI] [PubMed] [Google Scholar]
- Frank RA, and Grant SG (2017). Supramolecular organization of NMDA receptors and the postsynaptic density. Curr. Opin. Neurobiol. 45, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders N, Valentine M, Miller BL, Wong PC, et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guria A, Tran DDH, Ramachandran S, Koch A, Bounkari OE, Dutta P, Hauser H, and Tamura T (2011). Identification of mRNAs that are spliced but not exported to the cytoplasm in the absence of THOC5 in mouse embryo fibroblasts. RNA 17, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Sturgeon SM, Snarrenberg CL, Li Z, Xu XZS, Bermingham DP, Odiase P, Spencer WC, Miller DM, Carvelli L, et al. (2015). Glial Expression of the Caenorhabditis elegans Gene swip-10 Supports Glutamate Dependent Control of Extrasynaptic Dopamine Signaling. J. Neurosci. 35, 9409–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, and Edwards RH (2010). Vesicular Glutamate Transport Promotes Dopamine Storage and Glutamate Corelease In Vivo. Neuron 65, 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O (2016). Terminal Selectors of Neuronal Identity. Curr. Top. Dev. Biol. 116, 455–475. [DOI] [PubMed] [Google Scholar]
- Jain A, Liu R, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, Park J, Chen J, Xiang YK, and Ha T (2011). Probing cellular protein complexes using single-molecule pull-down. Nature 473, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Inoue H, Hurt E, and Yoneda Y (2009). Adaptor Aly and co‐adaptor Thoc5 function in the Tap‐p15‐mediated nuclear export of HSP70 mRNA. EMBO J. 28, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Okuzaki D, Inoue H, Yoneda Y, Maehara K, and Ohkawa Y (2013). Human TREX component Thoc5 affects alternative polyadenylation site choice by recruiting mammalian cleavage factor I. Nucleic Acids Res. 41, 7060–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Corbett MA, van Bon BWM, Woenig JA, Weir L, Douglas E, Friend KL, Gardner A, Shaw M, Jolly LA, et al. (2015). THOC2 Mutations Implicate mRNA-Export Pathway in X-Linked Intellectual Disability. Am. J. Hum. Genet. 97, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laßek M, Weingarten J, and Volknandt W (2015). The synaptic proteome. Cell Tissue Res. 359, 255–265. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Abel O, Bertram L, and Al-Chalabi A (2011). Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotroph. Lateral Scler. 12, 238–249. [DOI] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide B-MM, Schjeide LM, Meissner E, Zauft U, Allen NC, et al. (2012). Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson’s Disease Genetics: The PDGene Database. PLOS Genet. 8, e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Hannenhalli S, and Bucan M (2009). Motifs and cis-regulatory modules mediating the expression of genes co-expressed in presynaptic neurons. Genome Biol. 10, R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna R, Rondón AG, and Aguilera A (2012). New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1819, 514–520. [DOI] [PubMed] [Google Scholar]
- Mancini A, Niemann-Seyde SC, Pankow R, Omar EB, Klebba-Färber S, Koch A, Jaworska E, Elaine S, Gruber AD, Whetton AD, et al. (2010). THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Villalba AM, Tronche F, Kellendonk C, Gau D, Kapfhammer J, et al. (2002). Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 31, 47–54. [DOI] [PubMed] [Google Scholar]
- Okuno H (2011). Regulation and function of immediate-early genes in the brain: Beyond neuronal activity markers. Neurosci. Res. 69, 175–186. [DOI] [PubMed] [Google Scholar]
- Peña Á, Gewartowski K, Mroczek S, Cuéllar J, Szykowska A, Prokop A, Czarnocki‐Cieciura M, Piwowarski J, Tous C, Aguilera A, et al. (2012). Architecture and nucleic acids recognition mechanism of the THO complex, an mRNP assembly factor. EMBO J. 31, 1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott BJ, Liu J, Feng Z, Wescott SA, and Xu XZS (2011). The Neural Circuits and Synaptic Mechanisms Underlying Motor Initiation in C. elegans. Cell 147, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzonka L, Ullas S, Chinnam M, Povinelli BJ, Fisher DT, Golding M, Appenheimer MM, Nemeth MJ, Evans S, and Goodrich DW (2014). The Thoc1 Encoded Ribonucleoprotein Is Required for Myeloid Progenitor Cell Homeostasis in the Adult Mouse. PLOS ONE 9, e97628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokala N, Liu Q, Gordus A, and Bargmann CI (2014). Inducible and titratable silencing of Caenorhabditis elegans neurons in vivo with histamine-gated chloride channels. Proc. Natl. Acad. Sci. 111, 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, and Blencowe BJ (2015). Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87, 14–27. [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Dieck S tom, and Schuman EM (2017). Local translation in neuronal compartments: how local is local? EMBO Rep e201744045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, and Ghosh A (2002). Calcium Regulation of Dendritic Growth via CaM Kinase IV and CREB-Mediated Transcription. Neuron 34, 999–1010. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, Herold A, Gari K, Köcher T, Rode M, Ciccarelli FL, Wilm M, and Izaurralde E (2004). Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat. Struct. Mol. Biol. 11, 558–566. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Tornos FM, Aniceto IS, Cubelos B, and Nieto M (2013). Enrichment of Conserved Synaptic Activity-Responsive Element in Neuronal Genes Predicts a Coordinated Response of MEF2, CREB and SRF. PLOS ONE 8, e53848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Karelina K, and Obrietan K (2011). CREB: a multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 116, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saran S, Tran DDH, Ewald F, Koch A, Hoffmann A, Koch M, Nashan B, and Tamura T (2016). Depletion of three combined THOC5 mRNA export protein target genes synergistically induces human hepatocellular carcinoma cell death. Oncogene 35, 3872–3879. [DOI] [PubMed] [Google Scholar]
- Saran Shashank, Tran Doan DH, Klebba-Färber Sabine, Moran-Losada Patricia, Wiehlmann Lutz, Koch Alexandra, Chopra Himpriya, Pabst Oliver, Hoffmann Andrea, Kopfleisch Robert, et al. (2013). THOC5, a member of the mRNA export complex, contributes to processing of a subset of wingless/ integrated (Wnt) target mRNAs and integrity of the gut epithelial barrier. BMC Cell Biol. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, and Horvitz HR (2000). C. elegans Locomotory Rate Is Modulated by the Environment through a Dopaminergic Pathway and by Experience through a Serotonergic Pathway. Neuron 26, 619–631. [DOI] [PubMed] [Google Scholar]
- Schwartz ML, and Jorgensen EM (2016). SapTrap, a Toolkit for High-Throughput CRISPR/Cas9 Gene Modification in Caenorhabditis elegans. Genetics 202, 1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanakis N, Carrera I, and Hobert O (2015). Regulatory Logic of Pan-Neuronal Gene Expression in C. elegans. Neuron 87, 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sträßer K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondón AG, Aguilera A, Struhl K, Reed R, et al. (2002). TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417, 304–308. [DOI] [PubMed] [Google Scholar]
- Tran DD, Saran S, Dittrich-Breiholz O, Williamson AJ, Klebba-Färber S, Koch A, Kracht M, Whetton AD, and Tamura T (2013). Transcriptional regulation of immediate-early gene response by THOC5, a member of mRNA export complex, contributes to the M-CSF-induced macrophage differentiation. Cell Death Dis. 4, e879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran DDH, Saran S, Williamson AJK, Pierce A, Dittrich-Breiholz O, Wiehlmann L, Koch A, Whetton AD, and Tamura T (2014a). THOC5 controls 3′end-processing of immediate early genes via interaction with polyadenylation specific factor 100 (CPSF100). Nucleic Acids Res. 42, 12249–12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran DDH, Koch A, and Tamura T (2014b). THOC5, a member of the mRNA export complex: a novel link between mRNA export machinery and signal transduction pathways in cell proliferation and differentiation. Cell Commun. Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi H, Lau CK, Lau KF, and Chan HYE (2011). Perturbation of U2AF65/NXF1-mediated RNA nuclear export enhances RNA toxicity in polyQ diseases. Hum. Mol. Genet. 20, 3787–3797. [DOI] [PubMed] [Google Scholar]
- Van Vactor D, and Sigrist SJ (2017). Presynaptic morphogenesis, active zone organization and structural plasticity in Drosophila. Curr. Opin. Neurobiol. 43, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Miao Y-L, Zheng X, Lackford B, Zhou B, Han L, Yao C, Ward JM, Burkholder A, Lipchina I, et al. (2013). The THO Complex Regulates Pluripotency Gene mRNA Export and Controls Embryonic Stem Cell Self-Renewal and Somatic Cell Reprogramming. Cell Stem Cell 13, 676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chang Y, Li Y, Zhang X, and Goodrich DW (2006). Thoc1/Hpr1/p84 is essential for early embryonic development in the mouse. Mol. Cell. Biol. 26, 4362–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chinnam M, Wang J, Wang Y, Zhang X, Marcon E, Moens P, and Goodrich DW (2009). Thoc1 Deficiency Compromises Gene Expression Necessary for Normal Testis Development in the Mouse. Mol. Cell. Biol. 29, 2794–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, and Brenner S (1986). The Structure of the Nervous System of the Nematode Caenorhabditis elegans. Philos. Trans. R. Soc. B Biol. Sci. 314, 1–340. [DOI] [PubMed] [Google Scholar]
- Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, et al. (2016). Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176. [DOI] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond MJ, Acheson AL, Stachowiak MK, and Strickerm EM (1984). Neurochemical Compensation After Nigrostriatal Bundle Injury in an Animal Model of Preclinical Parkinsonism. Arch. Neurol. 41, 856–861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Detailed analysis of synaptic defects in THOC mutant animals (Related to Figure 1)
(A) Quantification of synaptic density relative to wt for different THOC mutants and their corresponding cell-specific rescue.
(B) Quantification of synaptic size for different THOC mutants and their corresponding cell-specific rescue.
(C) Quantification of synapse intensity for different THOC mutants and their corresponding cell-specific rescue.
(A-C) N=10–20 animals at L4 stage; ****, p<0.0001, ***, p<0.001**, p<0.01, *, p<0.05, one-way Anova with Tukey post-hoc test.
(D) Quantification of synaptic density for L3 and L4 animals in wt and thoc-5 mutant background.
(E) Quantification of synaptic size for L3 and L4 animals in wt and thoc-5 mutant background.
(F) Quantification of synapse intensity for L3 and L4 animals in wt and thoc-5 mutant background.
(D-F) N=10–20 animals; ***, p<0.001, student t-test.
Averages and SEM are plotted in all graphes.
(G) thoc-1 mRNA quantification in wt and thoc-1 strain backgrounds show a 70% reduction in thoc-1 mRNA level in the mutant.
Figure S2: thoc mutation does not affect PDE neuronal health (Related to Figure 1)
(A) - (C) Visualization and quantification of mitochondria in PDE cell body (A) and axons (B), (C) in wt and thoc-5 mutants.
(D)-(I) thoc-5 mutation does not affect the localization, number and morphology of different organelle markers. (D) ribosomes, (E) ER, (F) Endosome, (G), (H) Golgi and (I) cilia.
Figure S3: THOC mutants are impaired in DA signaling and DA-dependent behavior (Related to Figure 1 & 2)
(A) and (B) Quantitative analysis of basal slowing response in the absence (A) or presence (B) of DA. Locomotion rates in the absence (black bars) or presence (white bars) of bacteria were calculated in a 20 second time window. Wt and cat-2 animals were included as controls. The percentage of slowing is shown on the right. N=30–60 animals; ***, p<0.001, **, p<0.01, one-way Anova with Tukey post-hoc test.
(C) Ca2+ response of PDE neuron during basal slowing response. Calcium trace and quantification of it is shown for wt and thoc-5 mutant animals.
Averages and SEM are plotted in all graphs.
Figure S4: Presynaptic transcript retention in the nucleus is rescued in thoc-5 creb mutant animals. (Related to Figure 3)
(A) Representative images for smFISH for presynaptic and control transcripts in wt (upper row) and thoc-5 (lower row) mutant animals. GFP: PDE cellbody; red: signal for FISH probes; blue: DAPI staining of nuclei. Scale bar represents 2μm.
(B) Total number of smFISH puncta in PDE is not changed by thoc-5 mutation.
(C) Gene enrichment analysis for down-regulated genes in thoc-5 mutant neurons.
(D) qPCR quantification of presynaptic as well control transcripts in wt and thoc-5 mutant strain backgrounds. N = 3 biological replicates for both genotypes. ****, p<0.0001, ***, p<0.001, **, p<0.01; one-way Anova with Tukey post-hoc test. Averages and standard deviations are plotted.
Figure S5: Putative disease-associated thoc-1 R123H mutation renders THOC-1 dysfunctional (Related to Figure 7)
(A) Sequence alignment of human, mouse and C. elegans THOC1. Human, ALS-associated mutations are highlighted. Red: conserved R123H mutation, Blue: other disease associated mutations on un-conserved residues.
(B) Integrated synaptic intensity is quantified for wt, thoc-1, thoc-1 Pdat-1::thoc-1R123H and thoc-1 Pdat-1::thoc-1V338I strain backgrounds. N=20 animals; ****, p<0.0001, one-way ANOVA with Tukey post-hoc test.
Averages and SEM are plotted.
Figure S6: CREB, SRF/UNC-120 and PAB-2 function with THOC to facilitate export of synaptic transcripts (Related to Figure 6)
(A) Integrated synaptic intensity is quantified for two lines, in which crh-1 is cell-specifically knocked-down in DA neuron by CRE-loxP strategy. N=10–20 animals;
(B) Quantification of nuclear transcripts in wt, thoc-5, thoc-5 crh-1 and crh-1 PDE neurons. N=10–50 animals.
(C) Integrated synaptic intensity is quantified for wt, thoc-5, thoc-5 unc-120 and unc-120 mutant animals. N=20–50 animals
(D) Integrated synaptic intensity is quantified for wt, thoc-5, thoc-5 pab-2 and pab-2 mutant animals. N=20–50 animals
(D) Quantification of nuclear transcripts in wt, thoc-5, thoc-5 pab-2 and pab-2 PDE neurons. N=10–50 animals. ***, p<0.001; one-way Anova with Tukey post-hoc test. Averages and SEM are plotted in all graphs.
****, p<0.0001, ***, p<0.001, **, p<0.01, *, p<0.05 one-way Anova with Tukey post-hoc test.
Figure S7: THOC-1 specifically interacts with CREB but not GFP and binds to presynaptic transcripts (Related to Figure 7)
(A) Localization of THOC and CRH-1a does not depend on each other. THOC component are not mis-localized in crh-1 mutant background. CRH-1A localization is not altered by thoc-5 mutation.
(B) Quantification of the interaction between THOC-1 and increasing GFP concentration. No significant interaction between those two proteins was detected even at very high GFP concentration in PFA fixed samples. These result further corroborate the specificity of the direct interaction between CREB and THOC-1.
(C) Relative IP efficiency of synaptic transcripts (orange bars) as well as other pan-neuronal and control genes (black). THOC-1 bound transcripts were pulled-down under wt conditions. Transcripts were then quantified by qPCR and normalized onto total mRNA.
(D) Correlation analysis between total mRNA level and IPed mRNAs for wt and crh-1 mutant animals. No correlation was detected for synaptic transcripts (orange bars) and control transcripts (black bars).
Supplemental Table S1: Summary table of RNAseq data. (Related to Figure 3 and Figure S4)
Supplemental Table S2: qPCR primers used in this study. (Related to STAR methods)