

Abstract
The unfolded protein response (UPR) plays a vital role in maintaining cell homeostasis as a consequence of endoplasmic reticulum (ER) stress. However, prolonged UPR activity leads to cell death. This time-dependent dual functionality of the UPR represents the adaptive and cytotoxic pathways that result from ER stress. Chronic UPR activation in systemic and neurodegenerative diseases has been identified as an early sign of cellular dyshomeostasis.
The Protein Kinase R-like ER Kinase (PERK) pathway is one of three major branches in the UPR, and it is the only one to modulate protein synthesis as an adaptive response. The specific identification of prolonged PERK activity has been correlated with the progression of disorders such as diabetes, Alzheimer’s disease, and cancer, suggesting that PERK plays a role in the pathology of these disorders. For the first time, the term “PERK-opathies” is used to group these diseases in which PERK mediates detriment to the cell culminating in chronic disorders. This article reviews the literature documenting links between systemic disorders with the UPR, but with a specific emphasis on the PERK pathway. Then, articles reporting links between the UPR, and more specifically PERK, and neurodegenerative disorders are presented. Finally, a therapeutic perspective is discussed, where PERK interventions could be potential remedies for cellular dysfunction in chronic neurodegenerative disorders.
Keywords: eIF2α, EIF2AK3, endoplasmic reticulum, neurodegeneration, PERK, tau, unfolded protein response
INTRODUCTION
The endoplasmic reticulum (ER) provides an optimal environment to host fundamental biological processes such as protein folding, calcium storage, and lipid metabolism, making it an indispensable organelle for cell survival [1–3]. As such, evolutionarily conserved mechanisms regulate ER dynamics to ensure maintenance of these functions. Over a third of nascent proteins are folded in the ER lumen, where molecular chaperones triage misfolded proteins for refolding or degradation [4]. If they cannot be refolded, chaperone substrates (or clients) are targeted for degradation either by a proteasome-dependent pathway called ER-associated degradation (ERAD) or autophagy [5]. Effective chaperone function and clearance of clients out of the ER prevents secretion of unfolded or misfolded proteins. Unfolded and misfolded proteins have exposed hydrophobic signatures, and if they are released into the cytoplasm, the exposed unfolded sequence would trigger a heat shock protein response [5].
In neurological tissues, the aggregation of unfolded or misfolded proteins appears as the hallmark of several proteinopathies such as tauopathies (e.g. Alzheimer’s disease, fronto-temporal dementia, and progressive supranuclear palsy), synucleinopathies (e.g. Lewy body disease and Parkinson’s disease), aggregation of TDP-43 (amyotrophic lateral sclerosis, frontotemporal dementia, and hippocampal sclerosis), and diseases of poly-glutamine aggregation (e.g. Huntington’s disease and ataxias), among many others.
Increasing evidence suggests that several neurodegenerative disorders are rooted in aberrant ER function. For instance, amyloid precursor protein (APP) is processed by secretases on the ER membrane. Under pathogenic conditions, APP processing yields the amyloid beta (Aβ) peptide that is responsible for neurotoxicity and amyloid plaque formation [5]. In AD, cleavage of APP favors production of Aβ, which is released from the ER membrane and initiates neurotoxic cascades. On the other hand, soluble and pathogenic tau, the aggregation of which leads to formation of tangles in AD and nineteen other tauopathies, impairs ER-associated degradation (ERAD) leading to chronic activation of the UPR [6]. In Parkinson’s and Lewy Body Disease, α-synuclein is internalized in the ER, where it impairs protein transport between the ER and the Golgi network [7]. The mechanisms with which the ER combats proteinopathic insults are grouped into the unfolded protein response (UPR).
ER STRESS AND THE UNFOLDED PROTEIN RESPONSE
ER stress results from abnormalities that overwhelm normal ER performance. ER stress can be elicited by viral infection [5], blockage of ER protein clearance pathways such as ERAD [8], calcium disruptors, hypoglycemia, exposing cells to compounds such as tunicamycin, thapsigargin, and dithiothreitol, and hypoxia [9]. In response to ER stress, the cell activates the UPR [2]. The overall goal of this response is to restore ER function by decreasing input of nascent proteins and increasing output of folded proteins. In consequence, the UPR regulates size, shape, and abundance of luminal and transmembrane proteins [10], all of which contribute to the reestablishment of homeostasis.
Activation of the UPR begins by the dissociation of glucose-regulating proteins (GRPs) from three types of ER transmembrane anchors, namely IRE1 (Inositol-Requiring Protein 1 or Serine/Threonine-Protein Kinase/Endoribonuclease), ATF6 (Activating Transcription Factor α or Cyclic AMP-Dependent Transcription Factor), or PERK. GRPs are ER chaperones that facilitate refolding of nascent proteins [4]. Once detached from the membrane, GRPs associate with nascent proteins to facilitate their folding and secretion from the ER. Therefore, GRPs are ER-resident chaperones, and their dysfunction alone can lead to conditions such as juvenile onset glaucoma and blindness [6]. The most abundant GRPs are Grp78 (binding immunoglobulin protein or BiP) and Grp94 [11]. Meanwhile, each anchor, IRE1, ATF6, and PERK is free to initiate its own signaling pathways (Fig. 1).
Fig. (1). Adaptive and pro-apoptotic pathways of the UPR.
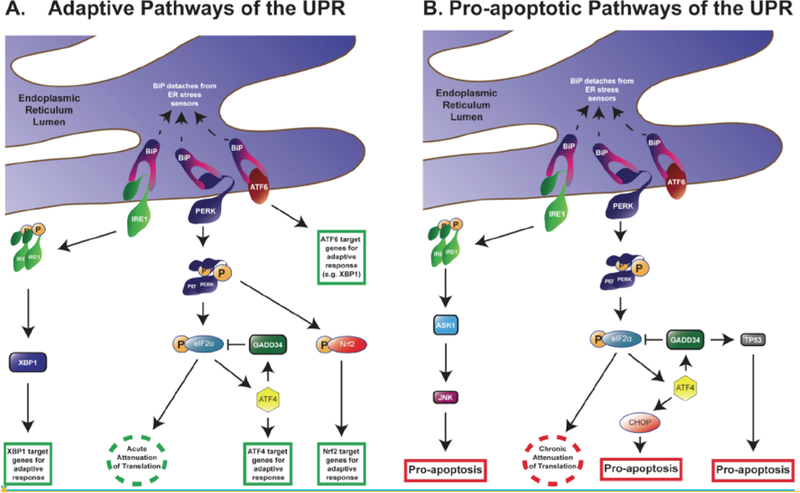
Under homeostatic conditions, IRE1, PERK, and ATF6 are anchored to the ER membrane by association with BiP. Upon activation of the UPR, BiP releases the effectors of the three branches of the UPR. (A) In the pro-survival, adaptive response, IRE1 dimerizes and autophosphorylates. Phosphorylated IRE1 activates XBP1, which in turn is translocated into the nucleus to upregulate the transcription of other adaptive UPR genes. Meanwhile, PERK dimerizes, autophosphorylates, and targets eiF2α and Nrf2. Once phosphorylated, eIF2α cannot contribute to activation of eIF2 thereby halting the initiation of translation. This provides relief to the ER by reducing input. Nonetheless, uORF-containing transcripts, such as ATF4, elude the attenuation of translation and become enriched. ATF4 can transactivate other adaptive UPR genes as well as trigger GADD34 activity. In turn, GADD34 dephosphorylates eIF2α, which restarts initiation of translation. Once phosphorylated, Nrf2 also serves as a transactivator for pro-survival genes. ATF6, the third branch of the UPR, translocates into the nucleus to enhance transcription of UPR genes such as XBP1. (B) Chronic activation of the UPR leads to pro-apoptotic signals. IRE1 induces apoptosis by activating ASK1 and consequently JNK. In the PERK pathway, sustained attenuation of translation by p-eIF2α suppresses protein synthesis. Neurons are particularly susceptible to the impact of prolonged reduction of nascent proteins. In the pro-apoptotic phase of ATF4, CHOP induces cell death cascades; long-term activation of the ATF4-GADD34 pathway leads to activation of the pro-apoptotic protein TP53. Abbreviations: UPR: unfolded protein response; IRE1: inositol-requiring enzyme 1; PERK: protein kinase R-like ER kinase; ATF6: cyclic AMP-dependent transcription factor-6; XBP1: X box-binding protein 1; eIF2α: eukaryotic initiation factor 2α; Nrf2: nuclear factor erythroid 2-related factor 2; ATF4: cyclic AMP-dependent transcription factor-4; uORF: upstream open reading frame; GADD34: growth arrest DNA damage-inducible GADD34; ASK1: apoptosis signal-regulated kinase 1; JNK: c-Jun N-terminal kinase; CHOP: C-EBP-homologous protein; TP53: cellular tumor antigen p53.
Under homeostatic conditions, IRE1 is constitutively attached to Grp78 under homeostatic conditions. Once detached, IRE1 dimerizes and autophosphorylates, which in turn activates RNase domains. Phosphorylated and active IRE1 targets and cleaves X box-binding protein 1 (XBP1), which is a transcriptional activator of UPR target genes such as ERAD proteins and ER chaperones [12, 13] (Fig. 1). Other downstream transcription factors that are activated by XBP1 facilitate production of phospholipids that allow the ER membranes to expand under stress [12, 13]. If ER stress is maintained, sustained IRE1 function mediates activation of signaling cascades involved in cell death. Specifically, this apoptotic cascade activates the apoptosis signal-regulated kinase 1 (ASK1) and c-Jun N-terminal kinase (JNK) [14, 15] (Fig. 1).
The ATF6 pathway also begins by its dissociation from Grp78, which provides S1 and S2 proteases to release ATF6 from the membrane. ATF6 translocates to the nucleus, where it promotes expression of Grp78, XBP1, C-EBP-Homologous Protein / Growth Arrest DNA Damage-Inducible transcript 3 (CHOP/GADD153), and Grp94. These effectors participate in protein folding, protein secretion, and ERAD [9, 16–19]. Together, this response facilitates protein output from the ER thereby offering relief and restoration of other functions.
PERK
The third branch of the UPR is initiated by PERK. The immediate objective of this cascade is to reduce translation of RNA in order to limit the input of nascent proteins in the ER. PERK has two regions: a luminal region that changes conformation when in contact with unfolded proteins and a cytoplasmic region containing a kinase domain that is necessary for activation [9]. Upon dissociation from Grp78, PERK dimerizes and autophosphorylates in a manner that is similar to the activation of IRE1. In turn, phosphorylation of PERK activates the kinase domain, which then targets substrates to activate the cascade. The best characterized PERK target is the eukaryotic initiation factor 2a (eIF2α) [5]. Phosphorylation of eIF2α prevents it from forming a complex with GTP, a step that is necessary for reloading eIF2 to initiate translation of mRNA. Therefore, p-eIF2α reduces protein synthesis [2, 20, 21]. Nonetheless, proteins that participate in UPR such as ERAD components, ER chaperones, UPR transcription factors, and mediators of the three branches of the UPR elude this mechanism (reviewed in [22]). The mRNA for these proteins contains pseudo-open or upstream open reading frames (uORFs) that avoid the p-eIF2α-mediated attenuation of translation. As a result of increased and selective expression of uORF-containing transcripts, the UPR pathways become robust permitting a return to homeostasis or activation of apoptosis [21].
Activating Transcription Factor-4 (ATF4) is another downstream effector of the PERK cascade. ATF4 is selectively enriched during ER stress by eluding PERK-mediated suppression of translation [20, 21, 23]. ATF4 plays a critical role in both relieving the ER for cell adaptation and activating apoptosis. As in the case of IRE1 and ASK1, the mechanisms responsible to this dual ATF4 function are crucial to understand the role of the UPR in cell survival versus death decisions. A key factor regulating the switch from activating pro-survival to favoring cell death pathways is the extent of time in which the UPR is active [24, 25].
In the case of acute PERK activity, ATF4 stimulates expression of proteins involved in cell recovery and adaptation such as ER chaperones. However, when PERK activity is chronic, sustained ATF4 levels upregulate pro-apoptotic proteins such as CHOP and growth arrest and DNA damage-inducible 34 (GADD34) [1, 2, 26]. Therefore, increased ATF4 decreases cell survival suggesting that prolonged translation of ATF4 is a primary signal for apoptosis under ER stress [3, 20]. ATf4 facilitates expression of CHOP, which in turn promotes apoptosis by enhancing expression of DR5 and tribbles-related protein 3. In parallel, ATF4 also participates in the inhibition of peroxisome proliferator-activated receptor γ, which triggers pro-apoptotic signals [4, 26–28].
Besides CHOP, ATF4 also induces the phosphatase activity of GADD34, which targets p-eIF2α. Initial expression of GADD34 negatively feeds back on the PERK pathway by dephosphorylating eIF2α to restore protein synthesis to its normal rate [29, 30]. However, extended GADD34 activity is associated with apoptosis via phosphorylation of (cellular tumor antigen p53) Tp53 [31] (Fig. 1).
Nrf2 (nuclear factor erythroid 2-related factor 2) is another direct substrate of PERK [32]. This second PERK substrate is a transcriptional activator that promotes expression of proteins involved in adaptation to oxidative stress [33]. Nrf2 exists ubiquitously in the cytoplasm attached to Keap 1 (Kelch-like ECH-associated protein 1). Upon activation of the UPR, PERK-directed phosphorylation of Nrf2 dissociates the Keap1/Nrf2 complex. Consequently, Nrf2 is translocated to the nucleus where it activates transcription of proteins with antioxidant activity.
In addition to these direct interactions between PERK and eIF2α or PERK and Nrf2, the PERK pathway plays a central role in regulating the entire UPR. Expression of dominant-negative PERK facilitates activation of ATF6 and its downstream effector XBP1 [28]. The inability of cells to activate the PERK pathway under conditions of ER stress (e.g. exposure to tunicamycin) results in attenuated phosphorylation of eIF2α and delayed suppression of translation. In response, the ATF6 pathway is further enhanced compared to control cells [28]. These data suggest that UPR activity is exquisitely tuned in three different yet integrated branches. In addition, PERK facilitates ATF6 function by enhancing transport of ATF6 from the ER and Golgi [34]. These studies suggest that PERK is a central integrator of the UPR, and specific modulation of PERK activity could lead to overall UPR-directed cell survival/death pathways.
SYSTEMIC IMPLICATIONS
Diabetes Mellitus
Diabetes mellitus is caused by impaired insulin signals and decreased insulin secretion [35]. In both Type I and Type II diabetes, apoptosis of β-cells is the primary mechanism of cell death [35]. According to the American Diabetes Association, diabetes mellitus symptomatology includes frequent urination, feeling excessively thirsty or hungry, extreme fatigue, blurry vision, cuts and bruises that heal slowly, and tingling or numbness in the hands or feet. In the United States, 29.1 million people have diabetes, with approximately 27.8% of those people being undiagnosed. According to the Centers for Disease Control and Prevention’s 2014 “National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States”, 1.7 million Americans aged 20 or older were diagnosed with the disease in 2012.
PERK-mediated cell death, is implicated in the pathogenic process leading to diabetes mellitus [35]. Secretory cells, such as islet β-cells that are integral for insulin production, produce high levels of PERK [35, 36]. Demand for insulin requires these β-cells to produce upwards of one million molecules per minute, meaning the ER must modify and package the proteins for proper use [37]. PERK signaling is important for the normal function and survival of secretory cells [36]. The pancreatic β-cells are particularly susceptible to ER stress and therefore depend on PERK for proper function [35]; however, PERK might be responsible for this susceptibility. Just like in other cells, PERK can be protective and harmful in the pancreas. Under normal cellular conditions, the survival properties of PERK dominate; but under extreme stress, such as what is associated with diabetes, induction of cell death pathways becomes more prevalent [36].
PERK abnormalities are directly associated with diabetes. In Scandinavians, the PERK gene maps to a locus that is implicated in the development of Type I diabetes [36]. Under more severe conditions, a mutation in the PERK gene, EIF2AK3, causes Wolcott-Rallison Syndrome (WRS), a rare form of monogenic diabetes that manifests as infantile-onset, insulin requiring diabetes [38]. WRS is characterized by early destruction of pancreatic beta cells [36]. This disease is very rare, with under 60 cases noted as of 2010 [39, 40], although it may be underdiagnosed due to death occurring before patients exhibit the typical signs and symptoms [41]. Alongside neonatal diabetes, the main features of this WRS include multiple epiphyseal dysplasia (impaired ability of bones to elongate and grow normally) and hepatic dysfunction [39, 41, 42]. More recent studies have reported 39 distinct mutations in the PERK gene in WRS patients, which result in early termination of the protein or missense mutations in the kinase domains [41]. As of yet, there is no link between any specific mutation and WRS symptoms or onset.
As a key mediator of the UPR, PERK activity is necessary to protect cells from the apoptotic signals inherent in prolonged UPR activation. Studies using in vitro and in vivo models show that PERK-deficient pancreatic β-cells are more sensitive to apoptosis induced by the ER. In addition, PERK-deficient mice develop neonatal hyperglycemia caused by islet proliferation defects and increased apoptosis [43]. In fact, embryonic development of β-cells requires eIF2α phosphorylation, and peIF2α maintains full function of differentiated β-cells [43]. Although there are other enzymes that can also phosphorylate eIF2α, these data suggest that the PERK pathway is a primary mediator of β-cell development.
PERK knockout mice are born with seemingly normal islets of Langerhans, but there is progressive destruction of β-cells over the first few weeks of their lives [36]. This is likely the result of inability to cope with the high demand for insulin. These data suggest that PERK cycling is critical for adaptation to ER function under extreme conditions of protein secretion. PERK knockdown in cells shows similar results, where reduced PERK activity keeps eIF2α in a dephosphorylated state, which also inhibits the GTP cycling and obstructs translation [21]. As a result, cells become sensitive to ER dysfunction and accumulation of misfolded proteins in the ER [44]. PERK loci variants are associated with the risk of prediabetes, such as the minor C allele of rs867529 (a PERK SNP), which has been linked with a 1.3-fold increased risk [38].
Obesity, a leading cause of Type II Diabetes Mellitus, is associated with initiation of cellular stress signaling and inflammatory pathways, including ER stress [45]. Deprivation of nutrients and glucose or increased synthesis of secretory proteins is a major change in response to obesity; compounded over time, these factors induce ER stress [45]. UPR proteins such as Grp78 are also increased in obese mice, indicating that the ER is undergoing stress and the UPR is active during obesity [45]. In addition, PERK, peIF2α, and JNK are increased in liver extracts of obese mice when compared to lean control mice [45]. These data suggest that obesity is directly linked to chronic induction of ER stress and the UPR (Fig. 1B). These data also suggest that the duration of the UPR could be suspended by removing the need for increased metabolic demands and protein synthesis.
A widely used model for diabetes, the Akita mouse, has a spontaneous mutation that causes early-onset non-obese diabetes [46]. In the Akita mouse, the ER of secretory β-cells distends and contains increased levels of BiP indicating that the ER has become stressed [23, 36]. In addition, progressive hyperglycemia is accompanied by CHOP induction, which occurs downstream of PERK and leads to apoptosis [23, 35]. Conversely, CHOP knockout mice have a delayed onset of islet cell destruction and hyperglycemia as well as reduced cell death by any kind of ER stress [23]. These studies highlight the important role of CHOP as a downstream effector of the PERK pathway and mediator of cell death.
Tumor Growth and Cancer
ER stress also plays a role in various cancers and tumor growth. Cancer cells become highly dependent on glycolysis due to increased metabolic demands for growth and propagation [47]. Increased demand of the glycolytic pathway causes glucose deprivation inducing ER stress [47]. In response, the UPR pathway is an effective early sensor of cellular stress associated with tumorigenesis [48]. Further research into the UPR’s relationship with cancer and tumor growth could provide useful information for early detection. In addition, targeting the UPR with pro-apoptotic compounds (e.g. PERK enhancers) could serve as therapeutic strategies.
PERK activity regulates tumor growth in several tissues. Tumors from MMTV-Neu transgenic mice were transduced with a retrovirus that excised PERK [48]. When transplanted into mammary fat pads of SCID mice, the PERK-deficient tumors had a reduced volume compared to PERK-positive cells [48]. This function extrapolates to other cell types throughout the body, where partial blocking of the UPR via PERK/ATF4 knockdown reduces the production of angio-genic mediators [20]. Knockdown of PERK also leads to reduced cell migration, which in turn reduces metastasis [49]. Further, PERK-deficient tumor cells have markedly less volume compared to PERK-positive cells [48]. Therefore, PERK knockdown leads to attenuation of tumor cell growth. Tests in vivo show that PERK silencing slows tumor growth and decreases the blood vessel density in tumors.
Due to their increased growth rate, cancer cells require aberrantly amplified ER activity in order to handle the ER membrane swelling demands in protein folding [47]. This increase in ER activity and cellular demand causes tumor cells to outgrow their blood supply. As a result, solid tumors, and in particular malignant tumors, present a toxic tumor microenvironment, which is characterized by sustained hypoxic conditions [4]. In response, the cell undergoes ER stress and activates the UPR. Various types of cancers have increased levels of BiP that correlates with tumor growth and proliferation [47]. In fact, the level of BiP expression directly correlates with stage of malignancy [4]. Therefore, hypoxia-mediated activation of the UPR, and its angiogenic properties become beneficial for tumor cells [4, 50]. Indeed, specific PERK activation helps upregulate angiogenic genes [50].
Another complex mechanism within the UPR-PERK pathway is one that impacts autophagy-related proteins. Hypoxia-mediated activation of PERK/ATF4 induces Lysosomal-Associated Membrane Protein 3 (LAMP3), which is necessary for autophagy [49, 51]. The activation kinetics of LAMP3 closely resembles those of the PERK pathway. ATF4 knockdown using siRNA or disrupting the phosphorylation of eIF2α, prevents LAMP3 induction even under hypoxic conditions [51]. For a therapeutic perspective in other stages of cancer progression, the LAMP3 homologs, LAMP1 and LAMP2, are associated with metastasis in breast cancer [49]. While the mechanism by which LAMP3 associates with cancer progression is not yet clear, LAMP expression associates with metastasis in the cervix and it is implicated in breast cancer [49]. Therefore, as an upstream facilitator of LAMP expression, PERK could serve as a therapeutic target or downstream sensor.
NEUROLOGICAL IMPLICATIONS
Many neurodegenerative diseases show upregulation of the UPR including amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), Parkinson’s disease (PD), Alzheimer’s disease (AD), Progressive Supranuclear Palsy (PSP), and Frontotemporal Dementia (FTD). The mechanisms linking these diseases and the UPR are not yet clear; however, an underlying molecular phenomenon of these disorders is the aberrant accrual of unfolded / misfolded proteins such as poly-glutamine, Aβ, tau, synuclein, and TDP-43, among others.
In the context of neurotoxicity, PERK has received special attention. This is in part due to the genetic risk for onset of some tauopathies inherent to SNPs on the gene coding for PERK [52–55], identification of increased pPERK in various neurodegenerative disorders [56–60], and a link between chronic PERK activation and decreased synthesis of synaptic proteins [61]. Neurons require constant protein production for synaptic function, which makes them particularly vulnerable to chronic attenuation of protein translation. Therefore, the PERK pathway hampers neuronal function well before it initiates pro-apoptotic cascades. As a result, PERK is a potent mediator of neuronal dysfunction, which underscores the interest of PERK in the field of neurodegeneration research.
Abrogating the extended activity of other branches of the UPR, that is IRE1 and ATF6, could offer benefits to long-term cell function and survival and ameliorate the long-term consequences of synaptic dysfunction. However, the neuronal dependence on nascent protein production makes the PERK pathway critical for synaptic function. The requirement for nascent protein synthesis extends to proper cellular function and it quickly and potently impacts overall brain function [62].
Amyotrophic Lateral Sclerosis (ALS)
ALS is a motor neuron disease showing neurodegeneration in the spinal ventral horn, most of the nuclei in the brainstem, and cerebral cortex. An estimated 5,000 people are diagnosed with ALS every year [63] and more than 12,000 people meet the standard surveillance definition of ALS [64]. ALS is fatal, and it leads to loss of motor function due to muscle atrophy with death typically occurring less than four years after diagnosis. [2, 65–68]. Phenotypic variation in the onset of this disease contributes to the difficulty in understanding ALS to the fullest extent. Spinal onset, seen in the majority of ALS cases, begins around 60 years of age. Initially, a painless weakness in one limb leads to a clinical examination that reveals further muscle atrophy, hyperreflexia, fasciculation, and hypertonia [69, 70]. The early symptoms are observed at an inconsistent rate, but show contiguous spread in the majority of patients [71]. Bulbar onset is seen in around 20% of all ALS cases [69, 72]. Common symptoms include dysarthria, dysphagia, tongue fasciculation, and jaw jerkiness [69, 72]. Bulbar onset has a worse prognosis than spinal onset, with life expectancy after diagnosis only being 2 years. Respiratory onset is the least common form in ALS, seen in only 3–5% of patients. These patients show orthopnea or dyspnea, with mild or absent spinal or bulbar onset symptoms. This pattern of onset is seen predominantly in males. The short-term life expectancy after diagnosis is 1.4 years with no long-term survival (>10 years) [72, 73]. The majority of cases are sporadic (~90%), but a few cases (~10%) are known to show genetic patterns of inheritance [2]. More than half of familial cases of ALS can be attributed to mutations in SOD1 (superoxide dismutase 1), TARDBP (the gene encoding the protein TDP-43), C9orf72, and the FUS gene [71].
Expression of mutant forms of TDP-43 and SOD1 are associated with PERK activation. TDP-43 is a DNA binding protein found predominantly in the nucleus. However, even under normal circumstances, part of TDP-43 is cytoplasmic, and aberrant localization and sorting of TDP-43 contributes to disease progression [74–76]. Interestingly, TDP-43 can also be a part of co-morbidities in disease when brains are confounded by pathological tau [77]. Previous studies have shown that stress granule formation correlates with expression of ALS-associated mutations [78]. Stress granules are intracellular aggregates composed of mRNAs, ribosomal subunits, and various proteins. Phosphorylation of eIF2α and consequent attenuation of protein translation increases the risk for the formation of stress granules [79]. Recent studies have shown that overall cellular stress can induce stress granule formation [80–82] and PERK-mediated phosphorylation of eIF2α initiates stress granule formation of TDP-43 [83, 84].
Accumulation of mutant SOD1 in the ER, which is implicated in some cases of ALS, induces ER stress through association with BiP. BiP is believed to chaperone the transport of SOD1 into the ER, with the protein-binding domain (residues 392–509) playing a critical role in the binding of SOD1 and BiP [68]. SOD1 is a protein that binds to copper or zinc ions to form cytoplasmic and mitochondrial isozymes that eliminate free superoxide radicals. In SOD1G93A transgenic mouse model of ALS, PERK is activated. The consequent phosphorylation of eIF2α leads to increased expression of ATF4. AtF4 activation as a result of the PERK pathway was evidenced in disease-affected areas such as the spinal cords. However, ATF4 was not activated in unaffected areas such as cerebellum, suggesting that PERK activation is selectively activated in discreet regions [68]. Mutant SOD1 also increases mitochondrial and oxidative stress, which in turn induces PERK-mediated phosphorylation of eIF2α [66].
Huntington’s Disease (HD)
Huntington’s disease is an autosomal dominant, neurodegenerative disease caused by a cytosine-adenine-guanine (CAG) repeat expansion within the huntingtin gene. This mutant gene leads to a pathogenic form of the huntingtin protein [85]. Mutant huntingtin aggregates and damages the striatum and cerebral cortex via mechanisms that are not yet clear. These pathological pathways lead to cognitive decline and motor impairment [86–88]. Generally, the life expectancy of HD is approximately 10–30 years after onset of symptoms but this prognosis can vary greatly depending on the number of repeats on the gene [5]. Along with HD, there is an array of Huntington’s disease-like (HDL) syndromes. HDL-1 is an autosomal dominant prion disease caused by additional octapeptide repeats in the prion protein gene. [89, 90]. Clinical symptoms such as ataxia are similar to those seen in HD; however, spongiosis is not prevalent [91, 92]. HDL-2 is autosomal dominant as well, but is caused by expansions of a CTG-CAG triplet repeat in the junctophilin-3 (JPH3) gene on chromosome 16q24.3 [93]. This form of HDL manifests very similarly to HD. HDL-3 an autosomal recessive disease that has only been seen in two Saudi Arabian families. It has an early age of onset (3–4 years) and very little is known about this disease [94]. HDL-4, also known as spinocerebellar ataxia type 17 (SCA17), is an autosomal disease caused by triplet repeat expansions in the TATA box-binding protein (TBP) gene located on chromosome 6q27. This is the most commonly inherited type of HDL with the most common clinically observed feature being cerebral ataxia [95]. Even though it is classified as an HDL syndrome, very few cases show a high level of similarity to symptoms seen in HD.
Aggregation of mutant huntingtin induces ER stress and activates the UPR. The UPR-PERK pathway has not been extensively studied in HD. However, levels of p-eIF2α are increased in cells expressing mutant huntingtin and addition of salubrinal showed decreased aggregates and increased cell viability [96]. This further suggests that sustained chronic eIF2α inhibition beyond that of the PERK pathway might show reversal of the PERK-mediated neurotoxic effects. In addition, p-eIF2α levels in wild type NIH 3T3 striatal cells are much lower than other cells types. However, p-eIF2α levels are increased upon transfection of mutant huntingtin into HEK 293T cells, suggesting that huntingtin over-expression induces the PERK pathway. Conversely, reduction of p-eIF2α levels by inhibition of the PERK pathway reverses toxicity of the mutant huntingtin protein [97]. In addition, functional assays in these studies showed that low levels of p-eIF2α were necessary for optimal striatal neuron function [98]. A later study by Li et al., showed that Grp78 is upregulated in cells expressing mutant huntingtin protein. The same study also showed that Grp78 reduces aggregation of mutant huntingtin protein [99]. Grp78 activation and dislodging form PERK suggests that the other UPR pathways are also activated, and in these cases, UPR activation is chronic. Still, very few results have been published about the PERK pathway specifically. More studies must be done to conclusively determine the role of PERK in HD and weigh the therapeutic potential of PERK.
Parkinson’s Disease (PD)
Parkinson’s disease is a neurodegenerative disease that is clinically diagnosed based on common symptoms: rest tremor, rigidity, bradykinesia, and postural instability. Confirmation of clinical diagnosis is done by post-mortem analysis. An estimated 1.5 million people in the US suffer from Parkinson’s. The classic hallmark of the disease is the presence of Lewy bodies, which typically include large accumulations of α-synuclein in neurons [100–103]. Norepinephrine deficiency is another contributing factor to the progression of the disease, which accounts for much of the impairment seen in the autonomic system of PD patients. Reduction in norepinephrine levels happens before reduction in dopamine levels [104]; however, it is unclear which neurotransmitter is the first or the major problem seen in the disease. The death of dopaminergic neurons in the substantia nigra is the most prominent and well-established sign of the disease and has provided a therapeutic target. Initially, dopamine deficiency leads to decline of motor skills that are accompanied by behavioral changes and dementia seen in late stages of the disease. The cause of death of dopaminergic neurons is largely unknown [56, 58]. The most common pharmacological treatment for PD is administration of Levadopa, which is converted to dopamine by L-dopa decarboxylase. While treatment with Levodopa has increased the standard of living for many patients with PD, there is substantial resistance to using the drug on the basis of dopamine dysregulation and its dire effects on reward stimulation and schizophrenic symptomatology. A compelling argument stems from the critical connectivity of the basal ganglia with other structures in the brain [105–109].
The most established location for a connection between the molecular basis of PD and chronic activation of the UPR was identified in dopaminergic neurons of Parkinson’s brain. Indeed, the levels of pPERK and p-eIF2α are significantly increased in dopaminergic neurons compared to age-matched control brains [56, 58]. However, there is no colocalization of α-synuclein and pPERK or of α-synuclein and p-eIF2α, suggesting that synuclein does not play a role in activating the UPR or its participation is indirect. A recent study by Le Masson et al. found that α-synuclein is localized in mitochondrial-associated ER membranes (MAM). Different mutations of α-synuclein determine the level of association seen with MAM and contribute to the overall level of mitochondrial dysfunction [110]. While not directly identifying the UPR or the PERK pathway, this discovery provides support to the idea α-synuclein association with the ER is detrimental in PD and confirms that the UPR is a potentially promising therapeutic target.
Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that is pathologically characterized by the appearance of amyloid plaques and tau tangles [111]. With current demographics showing 5.2 million Americans suffering from the disease, it is the most common cause of dementia in the US. Initial symptoms include difficulty with acquisition and recall of recent episodic memory. Progressive symptomatology expands to problems with overall cognitive function such as the inability to recognize places or family members. Late-stage AD patients show total loss of voluntary and involuntary muscle control [111].
The two major pathological hallmarks of Alzheimer’s disease are amyloid plaques and neurofibrillary tangles, which are composed of amyloid beta (Aβ) peptides and aberrantly folded microtubule-associated protein tau, respectively. Both Aβ and tau, and their diverse pathological conformations, interfere with cellular homeostasis. AD brains show signs of chronic UPR activation as evidenced by increased levels of Grp78, pPERK, pIRE1α, peIF2α, and ATF4 [57, 59, 112, 113]. As previously described, increased expression and availability of membrane-detached Grp78 suggests activation of the three branches of the UPR (Fig. 1). These studies corroborate activation of the IRE1 branch, but no other downstream effectors of this pathway are documented. Similarly, data suggesting activation of the ATF6 pathway is scant. Meanwhile, increased levels of pPERK, p-eIF2α, and ATF4 indicate that there is activation of the PERK pathway. Sustained ATF4 activity directly leads to activation of pro-apoptotic cascades [20]. Therefore, chronic activation of the PERK-ATF4 pathway could be a major mechanism mediating apoptotic cell death in AD. Nonetheless, the extent of UPR activation in AD has not been fully characterized. More information will help dissect the full regulation of the UPR and identify potential therapeutic targets to balance pro-survival vs. pro-apoptotic pathways.
The timing during which the UPR is initially activated in correlation with disease is a matter of debate, but at least in AD brains, pPERK levels accrue in hippocampal neurons that experience pre-tangle pathology [57]. Hoozemans et al., determined that pPERK directly co-localized with AT8-positive tau species and not with neurofibrillary tangles. In addition, pPERK levels positively correlate with the extent of tau pathology as marked by their Braak score [114]. Of importance is the identification of pPERK in hippocampal neurons where tau pathology begins in AD brains [114, 115]. These data argue for a temporal response to pathological tau aggregation where early pathological steps in the disease process activate the UPR. However, more recent data do not extricate early pathological tau species as indirect mediators of ER stress. Indeed, soluble tau is responsible for impairing ERAD, which in turn increases ER burden and activates the UPR [6].
Interestingly, UPR activation is associated with another pathological sign that is common in AD brains: granulo-vacuolar degenerating bodies (GVDs). These are deposits of autophagic vacuoles that seem to originate from smooth ER; their membranes contain lipid raft proteins like flotillin-1, and they encapsulate ubiquitinated proteins that are destined for autophagic destruction [116–118]. GVDs are not unique to AD pathology; they have been identified in brains suffering from Down’s syndrome, progressive supranuclear palsy (PSP), parkinsonism dementia complex of Guam, Pick’s disease, pallido-ponto-nigral degeneration, Parkinson’s disease, dementia with Lewy bodies, ALS, and elderly controls [119–121]. The common pathological hallmark associated with GVDs in these disorders is the colocalization with hyperphosphorylated tau. In turn, GVDs also show intense pPERK immunoreactivity in AD. Therefore, tau-mediated clogging of the autophagic pathway could result in another indirect mechanism by which tau induces ER stress in pretangle stages of disease. In addition, these findings support the idea that the UPR is active in other tauopathies. Depending on its conformation, tau clearance is directed in part by chaperone-co-chaperone interactions [122], which can direct tau for proteasomal or autophagic clearance [123].
The mechanism by which tau induces ER stress was recently established [6]. We previously showed increased levels of active PERK in the brains of rTg4510 mice, a tau transgenic mouse model that overexpresses FTD-related P301L mutant tau and show extensive neurofibrillary tangle pathology and cognitive deficits [124]. Hoozemans et al. shows co-localization of diffuse phosphorylated tau (AT8-positive) with phosphorylated PERK but neurofibrillary tangles show little to no PERK reactivity, suggesting that the mechanism of tau-mediated PERK activation is indirect. Indeed, we identified that pathological soluble tau species, some of the earliest and most neurotoxic tau species, are responsible for preventing egress of proteasome-bound ER substrates. As a result, ERAD is impaired, leading to an increased volume of nascent proteins in the ER lumen. In response, the UPR is activated.
More recent data suggest that there is a reverse link by which PERK increases the abundance of pathological tau species. In fact, three PERK-mediated pathways could lead to aberrant tau species (Fig. 2). First, pPERK activates GSK3β, a tau kinase that is implicated in tauopathy [125]. Second, PERK activates caspases that cleave tau [126]; the accumulation of caspase-cleaved tau (cTau) is an early indicator of pre-tangle pathology in AD and other tauopathies [122, 127, 128]. Mechanistically, cTau sensitizes neurons to ER stress-induced cell death, and it stimulates mitochondrial dysfunction [129, 130]. Moreover, preventing caspase cleavage of tau reduces tau filament formation [131]. Third, it was recently established that PERK itself could phosphorylate tau on residues that are associated with late stage pathology [132]. These data support the idea that PERK-mediated caspase activation could play a role in potentiating pathological tau species. This places PERK as a node in a bidirectional cycle of cell death whereby pathological tau species activate PERK and sustained PERK activity mediates cell death downstream while propagating the appearance of pathological tau species. In turn, increased pathological tau can cause neurotoxicity through many other pathways.
Fig. (2). PERK is a bimodal mediator of tau toxicity.
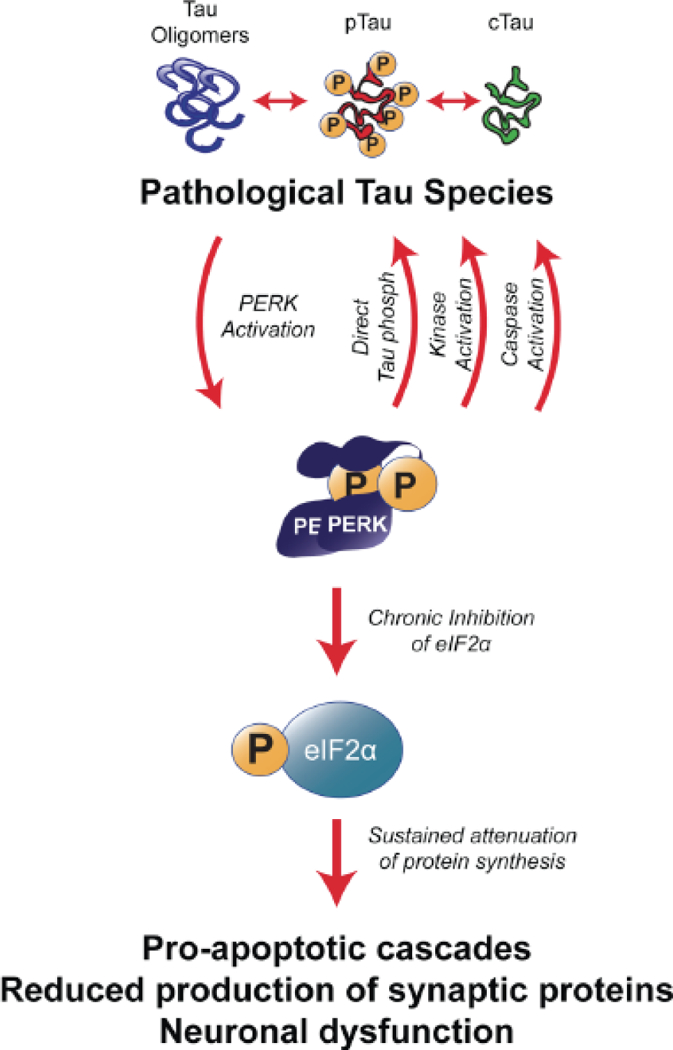
Pathological tau species chronically activate PERK leading to long-term reduction of protein synthesis via p-eIF2α. Chronic reduction of synaptic proteins causes neuronal dysfunction, which contributes to symptomatology in neurodegenerative disorders. Sustained levels of pPERK activate pro-apoptotic cascades downstream. Other downstream targets of the PERK pathway are identified to act on tau pathologically, such as GSK3β, caspase-3, and phosphorylate tau directly. Abbreviations: pTau: hyperphosphorylated tau; cTau: caspase-cleaved tau; PERK: protein kinase R-like ER kinase; eIF2α: eukaryotic initiation factor 2α; GSK3β: glycogen synthase kinase β.
UPR activation becomes evident in cells overexpressing APP, cells treated with Aβ oligomers, and iPSCs (induced-pluripotent stem cells) from familial and sporadic AD patients; albeit, the degree of UPR activation is discrepant in iPSCs [133–136]. The relationship between ER stress and Aβ appears to stem from retention and processing of APP in the ER. For instance, there is indication that APP-mediated activation of the UPR leads to enriched subcellular localization and abnormal processing of APP on the ER [137], which would cause strain on the ER membrane. Another potential mechanism could be mediated by APP processing into Aβ oligomers that would interfere with ER function; indeed, mechanisms of APP and Aβ-mediated ER dysfunction have been associated to impaired vesicle transport originating from the ER [138].
Another product of APP processing that is uniquely linked to the UPR is the APP-intracellular domain (AICD), a transcription factor that results from APP cleavage. AICD stimulates transcription of CHOP, which suggests a normal role of APP in mediating the UPR; an imbalance in this regulation (i.e. overproduction of APP) would induce further production of CHOP [136, 139, 140]. This could be one of the many mechanisms underlying neurotoxicity in AD, the consequences of which would be increased cell death.
While other ER stress markers are markedly changed by amyloid beta and APP, there is no current evidence linking amyloid beta or APP to PERK dysfunction. While human AD brains present increased levels of several UPR makers including pPERK, the Tg2576 mouse model of amyloidosis (a model overexpressing the Swedish mutation-containing APP gene), does not show pPERK reactivity [135]. Mean-while, rTg4510 mice show robust tau-mediated accumulation of pPERK and BiP [6]. Consistent with the finding that pPERK is abundant in tangle bearing neurons of FTD, PSP, and AD brains, it is likely that tau more directly links UPR activation than Aβ [60].
Progressive Supranuclear Palsy
Progressive supranuclear palsy is a neurodegenerative disease with a wide variety of clinical symptoms and seven different documented phenotypes [141]. The most common symptoms seen in early stages of all types of PSP are bradykinesia, increased falls, postural instability, tremors, cognitive changes, and supranuclear palsy of the vertical gaze [141, 142]. This disease is especially hard to diagnose, as early clinical symptoms overlap heavily with PD and other dementias [143, 144]. Approximately five per 100,000 people are affected by PSP with the average age of onset being 63 [145]. PSP is classified as a tauopathy due to the prevalence of tau aggregates in multiple brain regions including the basal ganglia, diencephalon, brainstem and cerebellum [146]. The hallmark characteristic that differentiates PSP from other tauopathies are the tufted astrocytes [147].
Activation of the PERK pathway has been seen in human tissue samples and amplified pPERK levels correlate with increasing levels of tau [60]. The most direct causal connection that links PERK with PSP onset is a genetic variation in EIF2AK3, the gene that codes for PERK. The largest genome-wide association study for PSP revealed a SNP in the EIF2AK3 that is associated with risk for the disease (rs7571971) [55]. Interestingly, this same SNP is in linkage disequilibrium with a series of other SNPs that change specific residues on the coding region of PERK [52–54]. The genetic impact of this risk for PSP seems to increase PERK activity, further suggesting that PERK inhibition could be an attractive therapeutic strategy. Furthermore, considering that inheritance of this SNP increases the risk for a wide spectrum of PSP phenotypes, AD, and lower bone mineral density, current efforts should strive to investigate the efficacy of PERK-inhibiting compounds. A noteworthy observation from these studies is that novel therapeutic targets for these disorders could exist downstream in the PERK pathway. For instance, inhibition of ATF4 could be an attractive therapeutic strategy that would have less of an overall effect on the UPR.
Frontotemporal Dementia (FTD)
Frontotemporal dementia is a clinically and pathologically diverse neurodegenerative disease typically classified by tau aberrancy [148]. The term frontotemporal dementia is used to describe the clinical syndromes seen in the disease, while the term frontotemporal lobar degeneration (FTLD) is used to describe the neuropathology of the disease seen post mortem. In adults 45–65 years old, FTD is almost as prevalent as AD, occurring in about fifteen people per 100,000 [149]. The major syndromes associated with FTD include behavioral variant FTD (bvFTD), semantic dementia (SD), progressive non-fluent aphasia (PNFA), corticobasal syndrome (CBS), and progressive supranuclear palsy syndrome (PSPS). All of these diagnoses are based on behavioral changes including personality changes, change in language function, and motor changes. Post mortem diagnosis of FTLD based on pathological biomarkers of most FTD patients can be split into two categories: FTLD-TDP and FTLD-tau. The other cases that do not fall into either of those categories are then divided into the ubiquitin protea-some syndrome (FTLD-UPS), the fused in sarcoma protein (FTLD-FUS), ubiquitin only (FTLD-U), or lack inclusions completely (FTLD-ni) [148].
To date, there have been few studies exploring the link between PERK and FTLD. Correlation between increasing tau levels and increasing PERK levels is seen in FTLD-tau brains. Specifically, the region showing highest reactivity was the hippocampus. FTLD-TDP and FTLD-FUS brain tissue samples, however, show no increase in PERK activity [60]. Validation of the lack of PERK activation in FTLD-TDP was shown by Tong et al. Transgenic rats bred to express mutant human TDP-43 exclusively in the forebrain showed no activation of PERK (or any other UPR markers) [150]. The disparity in the results of these studies may be due to differences clinical symptoms of FTD and pathology seen in FTLD. This leaves a large niche to fill in regards to finding therapeutic treatments.
THERAPEUTICS
The data on PERK and its role in disease implicate its pathway as a therapeutic target for treatment of PERK-opathies. Further insight into the pathway itself and the molecular mechanisms through which it functions could yield significant insight into the understanding and treatment of many neuropathological and systemic diseases discussed in this review. Promising results in experimental phases of PERK inhibitors have fueled the development of PERK-targeting strategies [50, 151–153]. Despite the toxicity of on-and off-target effects, inhibitors such as GSK2606414 have shown exciting therapeutic (not just preventative) rescue of normal phenotypes.
Long-term PERK inhibition could have adverse consequences to ER homeostasis. To avoid this risk we propose to explore the therapeutic potential of inhibiting downstream effectors of the PERK pathway. For instance, inhibition of ATF4 could be an attractive therapeutic strategy that would have less of an overall effect on the UPR. Targeting down-stream effectors of the PERK pathway could mitigate potential side effects caused by prolonged PERK activation or inhibition throughout the body. However, development of these tools is still in its infancy, and more needs to be learned from the intricate regulation of the UPR. It remains to be seen whether inhibiting the PERK pathway exerts feedback regulation of other pathways such as the UPR and beyond; it is also not clear whether this phenomenon would be beneficial. It would be exciting to find intermittent strategies that would ultimately activate and inhibit PERK to offer optimal outcomes: activate PERK to alleviate the ER, but inhibit PERK to prevent cell death in a timely manner.
PERK targeting is an attractive strategy for cancer therapeutics due to antiproliferative properties of long-term inhibition. The effects of PERK knockdown continue to provide key insights into the physiology of different cancers, which could see advancements in treatment through therapeutic modification or modulation of the PERK pathway. Inhibiting PERK in cancer cells may limit their ability to thrive under hypoxic and nutrient deprived conditions, which in turn leads to apoptosis of cancerous cells or tumor growth inhibition [50]. This has been shown using PERK inhibitors in various cancer models where PERK inhibition precludes tumor cell growth [49]. Whether these data will lead to curative effects in humans still remains to be seen.
The general therapeutic strategy presented here is to inhibit PERK to de-repress protein synthesis. While chronic PERK activity shows neurotoxicity, inhibition of the eIF2α phosphatase GADD34 with salubrinal effectively increases phosphorylation of eIF2α and abrogates translation [154]. However, GADD34 inhibition improves motoneuron function and increases lifespan in SOD1G93A transgenic mice [154]. As discussed earlier in this review, interruption of protein synthesis for an extended period of time underlines neurotoxicity. Nonetheless, chronic PERK activity with increased eIF2α inhibition/phosphorylation in this model shows improved outcomes. It is possible that reducing overall translation might effectively reduce synthesis of mutant SOD1 thereby abrogating other SOD1-mediated neurotoxic insults. While this would be effective in models where protein over-expression is the culprit of the disease phenotype, it bears limited appeal to therapeutics in diseases where accumulation of the pathogenic protein is indifferent to protein synthesis. However, salubrinal might have important ramifications for individuals with trisomy 21, where expression of three copies of APP is ascribed to onset of Alzheimer’s-like pathology and cognitive decline [155].
CONCLUSIONS
The data reviewed suggest that PERK is a potent mediator of neuronal dysfunction that is linked to neurodegenerative disorders. Many of these disorders are tauopathies (AD, PSP, FTD, among others), which have no cure. Since tauopathies are the most devastating cognitive threat to the aging population, there is an urgent need to identify therapeutic targets like PERK. Notwithstanding, a vast amount of unresolved issues in the strategy, therapeutic window, and safety impede progress at this time.
Future studies aiming to identify the earliest time points in which the PERK pathway is active will help identify an early therapeutic window. Equally important is the identification of neuronal dysfunction and cell death after PERK activation. With this information, it is possible to carefully trace the timeline during which PERK stops being beneficial and initiates deleterious cascades.
The compound GSK2606414 is a potent and selective PERK inhibitor [50]. It was recently used in a study to show alleviation of motor deficits in a rodent model of prion disease [151]. The compound successfully reduced the prion phenotype both in preventative and therapeutic interventions. However, treatment for more than three months caused the mice to lose 20% of their body weight and moderately increase their blood glucose levels. These data suggest that the compound has on-target effects on a pancreatic homolog of PERK, PKR. These interactions preclude current PERK inhibitors to be funneled through to clinical testing. These data also frame the proof-of-concept that inhibition of the PERK pathway is an attractive and novel therapeutic strategy, and they substantiate efforts to develop next generation PERK inhibitors that are more specific.
PERK’s fundamental role in the cell suggests that inhibition of its pathway could lead to unexpected deleterious consequences. As described in the first part of this article, the UPR is a complex pathway with three branches that self regulate. Two potential outcomes of long-term PERK suppression on overall UPR function are that the UPR system could reset itself after chronic inhibition of PERK or that the UPR will not correct itself upon PERK inhibition and pro-apoptotic cascades will ensue. It is possible that cross talk between all UPR players could elicit a compensatory state where future ER stress activates a moderate UPR (without activation of the PERK pathway) without initiating pro-apoptotic cascades.
Another caveat to PERK-mediated treatments for neurodegenerative disorders is the varied etiology of these diseases. For instance, not all tauopathic brains show a similar amount of UPR activation status [156]. This is likely because of different stages of disease; however, it is also possible that based on the diverse molecular mechanisms through which the UPR can be induced, different identities and conformations of pathogenic proteins could activate/inhibit the UPR in different ways [157]. In the case of PERK, this is important because it could help identify diverse activity levels that favor one branch over another. In addition, modulating the UPR could help prevent pro-apoptotic cascades thereby promoting cell survival in neurodegenerative disorders.
Despite these concerns, it is clear that PERK activity is common to many systemic and neurodegenerative disorders. Recent genetic and biochemical data suggest that PERK plays a pathogenic role in some cases, while in others it potentiates disease progression. It is not clear yet what would be the long-term cost of inhibiting the UPR. Nonetheless, important and exciting studies to characterize and challenge the PERK pathway of the UPR are currently underway, and they could unlock not only clues into the mechanisms of disease onset and progression, but also effective therapeutic paradigms.
ACKNOWLEDGEMENTS
We acknowledge funding support from the Alzheimer’s Association NIRG-14–322441. MCB and SEM contributed equally to the work.
LIST OF ABBREVIATIONS
- AD
Alzheimer’s disease
- ASK1
apoptosis signal-regulated kinase 1
- ATF4
cyclic AMP-dependent transcription factor-4
- ATF6
cyclic AMP-dependent transcription factor-6
- BiP
immunoglobulin heavy chain-binding protein
- CHOP
C-EBP-Homologous Protein
- cTau
caspase-cleaved tau
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- FTD
fronto-temporal dementia
- GADD153
growth arrest DNA damage-inducible transcript 3
- GADD34
growth arrest dna damage-inducible GADD34
- Grp
glucose-regulated protein
- GSK3β
glycogen synthase kinase β
- HD
Huntington’s disease
- IRE1
inositol-requiring enzyme 1
- JNK
c-Jun N-terminal kinase
- Nrf2
nuclear factor erythroid 2-related factor 2
- PD
Parkinson’s disease
- PERK
protein kinase R-like ER kinase
- PSP
progressive supranuclear palsy
- pTau
hyperphosphorylated tau
- uORF
upstream open reading frames
- UPR
unfolded protein response
- XBP1
X box-binding protein 1
Biography

Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- [1].Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica 2012: 857516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Matus S, Valenzuela V, Medinas DB, Hetz C. ER Dysfunction and Protein Folding Stress in ALS. Intern J Cell Biol 2013: 674751 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15(5): 481–90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest 110(10): 1389–98 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shineman DW, Basi GS, Bizon JL, Colton CA, Greenberg BD, Hollister BA, et al. Accelerating drug discovery for Alzheimer’s disease: best practices for preclinical animal studies. Alzheimers Res Ther 3(5): 28 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Abisambra JF, Jinwal UK, Blair LJ, O’Leary JC 3rd, Li Q, Brady S, et al. Tau Accumulation Activates the Unfolded Protein Response by Impairing Endoplasmic Reticulum-Associated Degradation. J Neurosci 33(22): 9498–507 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313(5785): 324–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Develop 22(23): 3308–19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yoshida H ER stress and diseases. FEBS J 274(3): 630–58 (2007). [DOI] [PubMed] [Google Scholar]
- [10].Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta 1843(10): 2143–9 (2014). [DOI] [PubMed] [Google Scholar]
- [11].Spindler SR, Crew MD, Mote PL, Grizzle JM, Walford RL. Dietary energy restriction in mice reduces hepatic expression of glucose-regulated protein 78 (BiP) and 94 mRNA. J Nutr 120(11): 1412–7 (1990). [DOI] [PubMed] [Google Scholar]
- [12].Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell 132(1): 24–6 (2008). [DOI] [PubMed] [Google Scholar]
- [13].Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21(1): 81–93 (2004). [DOI] [PubMed] [Google Scholar]
- [14].Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Develop 16(11): 1345–55 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453): 664–6 (2000). [DOI] [PubMed] [Google Scholar]
- [16].Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol 625(1–3): 234–46 (2009). [DOI] [PubMed] [Google Scholar]
- [17].Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Develop Cell 13(3): 365–76 (2007). [DOI] [PubMed] [Google Scholar]
- [18].Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33(1): 75–89 (2008). [DOI] [PubMed] [Google Scholar]
- [19].Parmar VM, Schroder M. Sensing endoplasmic reticulum stress. Adv Exp Med Biol 738: 153–68 (2012). [DOI] [PubMed] [Google Scholar]
- [20].Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, et al. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res 72(20): 5396–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6(5): 1099–108 (2000). [DOI] [PubMed] [Google Scholar]
- [22].Wethmar K, Smink JJ, Leutz A. Upstream open reading frames: molecular switches in (patho)physiology. BioEssays 32(10): 885–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51(3): S455–61 (2002). [DOI] [PubMed] [Google Scholar]
- [24].Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J 23(1): 169–79 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PloS one 4(1): e4170 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059): 1081–6 (2011). [DOI] [PubMed] [Google Scholar]
- [27].Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J 24(6): 1243–55 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yamaguchi Y, Larkin D, Lara-Lemus R, Ramos-Castaneda J, Liu M, Arvan P. Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J Biol Chem 283(25): 17020–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Biol Chem 153(5): 1011–22 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K, et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J 17(11): 1573–5 (2003). [DOI] [PubMed] [Google Scholar]
- [31].De Angelis PM, Svendsrud DH, Kravik KL, Stokke T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Can 5: 20 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23(20): 7198–209 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Nat Acad Sci USA 97(23): 12475–80 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 22(22): 4390–405 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Intern Med 42(1): 7–14 (2003). [DOI] [PubMed] [Google Scholar]
- [36].Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell 7(6): 1153–63 (2001). [DOI] [PubMed] [Google Scholar]
- [37].Maly DJ, Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol 10(11): 892–901 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Feng N, Ma X, Wei X, Zhang J, Dong A, Jin M, et al. Common variants in PERK, JNK, BIP and XBP1 genes are associated with the risk of prediabetes or diabetes-related phenotypes in a Chinese population. Chin Med J 127(13): 2438–44 (2014). [PubMed] [Google Scholar]
- [39].Ozbek MN, Senee V, Aydemir S, Kotan LD, Mungan NO, Yuksel B, et al. Wolcott-Rallison syndrome due to the same mutation (W522X) in EIF2AK3 in two unrelated families and review of the literature. Pediatr Diabetes 11(4): 279–85 (2010). [DOI] [PubMed] [Google Scholar]
- [40].Rubio-Cabezas O, Patch AM, Minton JA, Flanagan SE, Edghill EL, Hussain K, et al. Wolcott-Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metabol 94(11): 4162–70 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Julier C, Nicolino M. Wolcott-Rallison syndrome. Orphanet J Rare Dis 5: 29 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wolcott CD, Rallison ML. Infancy-onset diabetes mellitus and multiple epiphyseal dysplasia. J Pediatr 80(2): 292–7 (1972). [DOI] [PubMed] [Google Scholar]
- [43].Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metabol 10(1): 13–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716): 271–4 (1999). [DOI] [PubMed] [Google Scholar]
- [45].Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306(5695): 457–61 (2004). [DOI] [PubMed] [Google Scholar]
- [46].Zito E, Chin KT, Blais J, Harding HP, Ron D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J Cell Biol 188(6): 821–32 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kosakowska-Cholody T, Lin J, Srideshikan SM, Scheffer L, Tarasova NI, Acharya JK. HKH40A downregulates GRP78/BiP expression in cancer cells. Cell Death Dis 5: e1240\(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29(27): 3881–95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC, et al. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Can Res 15(1): R2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl] acetyl}−2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55(16): 7193–207 (2012). [DOI] [PubMed] [Google Scholar]
- [51].Mujcic H, Rzymski T, Rouschop KM, Koritzinsky M, Milani M, Harris AL, et al. Hypoxic activation of the unfolded protein response (UPR) induces expression of the metastasis-associated gene LAMP3. Radiotherapy Oncol 92(3): 450–9 (2009). [DOI] [PubMed] [Google Scholar]
- [52].Ferrari R, Ryten M, Simone R, Trabzuni D, Nicolaou N, Hondhamuni G, et al. Assessment of common variability and expression quantitative trait loci for genome-wide associations for progressive supranuclear palsy. Neurobiol Aging 35(6): 1514 e1–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Group PSPGS, et al. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun 1(1): 31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu J, Hoppman N, O’Connell JR, Wang H, Streeten EA, McLenithan JC, et al. A functional haplotype in EIF2AK3, an ER stress sensor, is associated with lower bone mineral density. JOF Bone Min Res 27(2): 331–41.(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43(7): 699–705 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun 354(3): 707–11 (2007). [DOI] [PubMed] [Google Scholar]
- [57].Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174(4): 1241–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neuro-degenerative Dis 10(1–4): 212–5 (2012). [DOI] [PubMed] [Google Scholar]
- [59].Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, et al. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110(2): 165–72 (2005). [DOI] [PubMed] [Google Scholar]
- [60].Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 226(5): 693–702 (2012). [DOI] [PubMed] [Google Scholar]
- [61].Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485(7399): 507–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Duvarci S, Nader K, LeDoux JE. De novo mRNA synthesis is required for both consolidation and reconsolidation of fear memories in the amygdala. Learning Mem 15(10): 747–55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the “common” neurologic disorders? Neurology 68(5): 326–37 (2007). [DOI] [PubMed] [Google Scholar]
- [64].Mehta P, Antao V, Kaye W, Sanchez M, Williamson D, Bryan L, et al. Prevalence of amyotrophic lateral sclerosis -United States, 2010–2011. Morb Mortal Weekly Rep Surveill Summ 63(7): 1–14 (2014). [PubMed] [Google Scholar]
- [65].Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PloS One 8(11): e81170 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130(Pt 12): 3111–23 (2007). [DOI] [PubMed] [Google Scholar]
- [67].Grosskreutz J, Van Den Bosch L, Keller BU. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Cal 47(2): 165–74 (2010). [DOI] [PubMed] [Google Scholar]
- [68].Kikuchi H, Almer G, Yamashita S, Guegan C, Nagai M, Xu Z, et al. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Nat Acad Sci USA 103(15): 6025–30 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Amato AA, Russell JA. Neuromuscular disorders. New York: McGraw-Hill Medical (2008). [Google Scholar]
- [70].Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. New Eng J Med 344(22): 1688–700 (2001). [DOI] [PubMed] [Google Scholar]
- [71].Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10(11): 661–70 (2014). [DOI] [PubMed] [Google Scholar]
- [72].Chio A, Calvo A, Moglia C, Mazzini L, Mora G, group Ps. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatr 82(7): 740–6 (2011). [DOI] [PubMed] [Google Scholar]
- [73].Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry 78(6): 629–31 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20(7): 1774–84 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283(19): 13302–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Nat Acad Sci USA 99(21): 13583–8 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jinwal UK, Abisambra JF, Zhang J, Dharia S, O’Leary JC, Patel T, et al. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J Biol Chem 287(29): 24814–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Winton MJ, Van Deerlin VM, Kwong LK, Yuan W, Wood EM, Yu CE, et al. A90V TDP-43 variant results in the aberrant localization of TDP-43 in vitro. FEBB Lett 582(15): 2252–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res 1462: 16–25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PloS one 5(10): e13250 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, et al. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20(7): 1400–10 (2011). [DOI] [PubMed] [Google Scholar]
- [82].Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P 3rd, Good SK, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol 31(5): 1098–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol 147(7): 1431–42 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kedersha N, Anderson P. Regulation of translation by stress granules and processing bodies. Prog Mol Biol Transl Sci 90: 155–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Group THsDCR. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72(6): 971–83 (1993). [DOI] [PubMed] [Google Scholar]
- [86].Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 85(15): 5733–7 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324(5932): 1327–30 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Roze E, Cahill E, Martin E, Bonnet C, Vanhoutte P, Betuing S, et al. Huntington’s Disease and Striatal Signaling. Front Neuroanat 5: 55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edstrom L, et al. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet 69(6): 1385–8 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Schneider SA, Bhatia KP. Huntington’s disease look-alikes. Handbook Clin Neurol 100: 101–12 (2011). [DOI] [PubMed] [Google Scholar]
- [91].Xiang F, Almqvist EW, Huq M, Lundin A, Hayden MR, Edstrom L, et al. A Huntington disease-like neurodegenerative disorder maps to chromosome 20p. Am J Hum Genet 63(5): 1431–8 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Laplanche JL, Hachimi KH, Durieux I, Thuillet P, Defebvre L, Delasnerie-Laupretre N, et al. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain 122 (Pt 12): 2375–86 (1999). [DOI] [PubMed] [Google Scholar]
- [93].Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29(4): 377–8 (2001). [DOI] [PubMed] [Google Scholar]
- [94].Kambouris M, Bohlega S, Al-Tahan A, Meyer BF. Localization of the gene for a novel autosomal recessive neurodegenerative Huntington-like disorder to 4p15.3. Am J Hum Genet 66(2): 445–52.(2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Santos C, Wanderley H, Vedolin L, Pena SD, Jardim L, Sequeiros J. Huntington disease-like 2: the first patient with apparent European ancestry. Clin Gen 73(5): 480–5 (2008). [DOI] [PubMed] [Google Scholar]
- [96].Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res 314(5): 950–60 (2008). [DOI] [PubMed] [Google Scholar]
- [97].Wang H, Blais J, Ron D, Cardozo T. Structural determinants of PERK inhibitor potency and selectivity. Chem Biol Drug Des 76(6): 480–95.(2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Leitman J, Barak B, Benyair R, Shenkman M, Ashery U, Hartl FU, et al. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PloS one 9(3): e90803(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jiang Y, Lv H, Liao M, Xu X, Huang S, Tan H, et al. GRP78 counteracts cell death and protein aggregation caused by mutant huntingtin proteins. Neurosci Lett 516(2): 182–7 (2012). [DOI] [PubMed] [Google Scholar]
- [100].Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 388(6645): 839–40 (1997). [DOI] [PubMed] [Google Scholar]
- [101].Abdullah R, Basak I, Patil KS, Alves G, Larsen JP, Moller SG. Parkinson’s disease and age: The obvious but largely unexplored link. Exp Gerontol (2014). [DOI] [PubMed] [Google Scholar]
- [102].Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646): 841 (2003). [DOI] [PubMed] [Google Scholar]
- [103].Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364(9440): 1167–9 (2004). [DOI] [PubMed] [Google Scholar]
- [104].Rommelfanger KS, Weinshenker D. Norepinephrine: The redheaded stepchild of Parkinson’s disease. Biochem Pharmacol 74(2): 177–90 (2007). [DOI] [PubMed] [Google Scholar]
- [105].Denny-Brown D Diseases of the basal ganglia. Their relation to disorders of movement. Lancet 2(7161): 1155–62 (1960). [DOI] [PubMed] [Google Scholar]
- [106].Mettler FA. Substantia nigra and Parkinsonism. Arch Neurol 11: 529–42 (1964). [DOI] [PubMed] [Google Scholar]
- [107].Seiden LS, Carlsson A. Brain and Heart Catecholamine Levels after L-Dopa administration in reserpine treated mice: correlations with a conditioned avoidance response. Psychopharmacologia 5: 178–81 (1964). [DOI] [PubMed] [Google Scholar]
- [108].Bertler A, Rosengren E. Possible role of brain dopamine. Pharmacol Rev 18(1): 769–73 (1966). [PubMed] [Google Scholar]
- [109].Vogt M Functional aspects of the role of catecholamines in the central nervous system. Br Med Bull 29(2): 168–72.(1973). [DOI] [PubMed] [Google Scholar]
- [110].Le Masson G, Przedborski S, Abbott LF. A computational model of motor neuron degeneration. Neuron 83(4): 975–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Thies W, Bleiler L, Alzheimers A. 2013 Alzheimer’s disease facts and figures. Alzheimer’s Dement 9(2): 208–45 (2013). [DOI] [PubMed] [Google Scholar]
- [112].Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA. Expression of heat shock proteins in Alzheimer’s disease. Neurology 41(3): 345–50 (1991). [DOI] [PubMed] [Google Scholar]
- [113].Kohler C, Dinekov M, Gotz J. Granulovacuolar degeneration and unfolded protein response in mouse models of tauopathy and Abeta amyloidosis. Neurobiol Dis 71: 169–79.(2014). [DOI] [PubMed] [Google Scholar]
- [114].Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4): 239–59.(1991). [DOI] [PubMed] [Google Scholar]
- [115].Braak H, Braak E. Alzheimer’s disease affects limbic nuclei of the thalamus. Acta Neuropathol 81(3): 261–8.(1991). [DOI] [PubMed] [Google Scholar]
- [116].Okamoto K, Hirai S, Iizuka T, Yanagisawa T, Watanabe M. Reexamination of granulovacuolar degeneration. Acta Neuropathol 82(5): 340–5 (1991). [DOI] [PubMed] [Google Scholar]
- [117].Funk KE, Mrak RE, Kuret J. Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol App Neurobiol 37(3): 295–306.(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Nishikawa T, Takahashi T, Nakamori M, Yamazaki Y, Kurashige T, Nagano Y, et al. Phosphatidylinositol-4,5-bisphosphate is enriched in granulovacuolar degeneration bodies and neurofibrillary tangles. Neuropathol App Neurobiol 40(4): 489–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Lagalwar S, Berry RW, Binder LI. Relation of hippocampal phospho-SAPK/JNK granules in Alzheimer’s disease and tauopathies to granulovacuolar degeneration bodies. Acta Neuropathol 113(1): 63–73.(2007). [DOI] [PubMed] [Google Scholar]
- [120].Schwab C, DeMaggio AJ, Ghoshal N, Binder LI, Kuret J, McGeer PL. Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol Aging 21(4): 503–10 (2000). [DOI] [PubMed] [Google Scholar]
- [121].Yamazaki Y, Matsubara T, Takahashi T, Kurashige T, Dohi E, Hiji M, et al. Granulovacuolar degenerations appear in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders. PloSOne 6(11): e26996 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Abisambra JF, Jinwal UK, Suntharalingam A, Arulselvam K, Brady S, Cockman M, et al. DnaJA1 antagonizes constitutive Hsp70-mediated stabilization of tau. J Mol Biol 421(4–5): 653–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem 285(29): 21978–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733): 476–81 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 147(1): 58–62 (1992). [DOI] [PubMed] [Google Scholar]
- [126].Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 280(14): 14189–202 (2005). [DOI] [PubMed] [Google Scholar]
- [127].Newman J, Rissman RA, Sarsoza F, Kim RC, Dick M, Bennett DA, et al. Caspase-cleaved tau accumulation in neurodegenerative diseases associated with tau and alpha-synuclein pathology. Acta Neuropathol 110(2): 135–44 (2005). [DOI] [PubMed] [Google Scholar]
- [128].Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol J 11(2): 341–54.(2002). [DOI] [PubMed] [Google Scholar]
- [129].Matthews-Roberson TA, Quintanilla RA, Ding H, Johnson GV. Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res 1234: 206–12 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem 284(28): 18754–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, et al. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry 42(27): 8325–31 (2003). [DOI] [PubMed] [Google Scholar]
- [132].Cavallini A, Brewerton S, Bell A, Sargent S, Glover S, Hardy C, et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem 288(32): 23331–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12(4): 487–96 (2013). [DOI] [PubMed] [Google Scholar]
- [134].Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, et al. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 75(5): 824–37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park UJ, et al. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med 42(5): 386–94 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Copanaki E, Schurmann T, Eckert A, Leuner K, Muller WE, Prehn JH, et al. The amyloid precursor protein potentiates CHOP induction and cell death in response to ER Ca2+ depletion. Biochim Biophys Acta 1773(2): 157–65 (2007). [DOI] [PubMed] [Google Scholar]
- [137].Tomimoto H, Akiguchi I, Wakita H, Nakamura S, Kimura J. Ultrastructural localization of amyloid protein precursor in the normal and postischemic gerbil brain. Brain Res 672(1–2): 187–95 (1995). [DOI] [PubMed] [Google Scholar]
- [138].Abisambra JF, Fiorelli T, Padmanabhan J, Neame P, Wefes I, Potter H. LDLR expression and localization are altered in mouse and human cell culture models of Alzheimer’s disease. PloS one 5(1): e8556 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Takahashi K, Niidome T, Akaike A, Kihara T, Sugimoto H. Amyloid precursor protein promotes endoplasmic reticulum stress-induced cell death via C/EBP homologous protein-mediated pathway. J Neurochem 109(5): 1324–37 (2009). [DOI] [PubMed] [Google Scholar]
- [140].Kogel D, Concannon CG, Muller T, Konig H, Bonner C, Poeschel S, et al. The APP intracellular domain (AICD) potentiates ER stress-induced apoptosis. Neurobiol Aging 33(9): 2200–9 (2012). [DOI] [PubMed] [Google Scholar]
- [141].Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, et al. The phenotypic spectrum of progressive supranuclear palsy: A retrospective multicenter study of 100 definite cases. Movement Disord 29(14): 1758–66 (2014). [DOI] [PubMed] [Google Scholar]
- [142].Respondek G, Roeber S, Kretzschmar H, Troakes C, Al-Sarraj S, Gelpi E, et al. Accuracy of the National Institute for Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Movement Disord 28(4): 504–9 (2013). [DOI] [PubMed] [Google Scholar]
- [143].Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 54(5): S15–9 (2003). [DOI] [PubMed] [Google Scholar]
- [144].Liscic RM, Srulijes K, Groger A, Maetzler W, Berg D. Differentiation of progressive supranuclear palsy: clinical, imaging and laboratory tools. Acta Neurolog Scand 127(5): 362–70 (2013). [DOI] [PubMed] [Google Scholar]
- [145].Litvan I, Mangone CA, McKee A, Verny M, Parsa A, Jellinger K, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 60(6): 615–20 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23(4): 394–400 (2010). [DOI] [PubMed] [Google Scholar]
- [147].Nishimura M, Namba Y, Ikeda K, Oda M. Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett 143(1–2): 35–8 (1992). [DOI] [PubMed] [Google Scholar]
- [148].D’Alton S, Lewis J. Therapeutic and diagnostic challenges for frontotemporal dementia. Front Aging Neurosci 6: 204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Riedl L, Mackenzie IR, Forstl H, Kurz A, Diehl-Schmid J. Frontotemporal lobar degeneration: current perspectives. Neuropsychiatr Dis Treat 10: 297–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Tong J, Huang C, Bi F, Wu Q, Huang B, Zhou H. XBP1 depletion precedes ubiquitin aggregation and Golgi fragmentation in TDP-43 transgenic rats. J Neurochem 123(3): 406–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Trans Med 5(206): 206ra138 (2013). [DOI] [PubMed] [Google Scholar]
- [152].Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73(6): 1993–2002 (2013). [DOI] [PubMed] [Google Scholar]
- [153].Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46(2): 152–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12(5): 627–36 (2009). [DOI] [PubMed] [Google Scholar]
- [155].Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature 331(6156): 528–30 (1988). [DOI] [PubMed] [Google Scholar]
- [156].Unterberger U, Hoftberger R, Gelpi E, Flicker H, Budka H, Voigtlander T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol 65(4): 348–57 (2006). [DOI] [PubMed] [Google Scholar]
- [157].Castillo-Carranza DL, Zhang Y, Guerrero-Munoz MJ, Kayed R, Rincon-Limas DE, Fernandez-Funez P. Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem Res 37(8): 1707–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]