

Abstract
We conducted a multicenter pilot investigation of the safety and feasibility of bone marrow transplantation (BMT) in adults with severe sickle cell disease (SCD) (NCT 01565616) using a reduced toxicity preparative regimen of Busulfan (13.2 mg/kg), Fludarabine (175 mg/m2) and Thymoglobulin (6 mg/kg) and cyclosporine or tacrolimus and methotrexate for graft-versus-host disease (GVHD) prophylaxis. Twenty-two patients (median age 22 years; range 17–36) were enrolled at 8 centers. Seventeen patients received marrow from an HLA-identical sibling donor and 5 patients received marrow from an 8/8 HLA-allele matched unrelated donor. Before BMT, patients had stroke, acute chest syndrome, recurrent pain events, were receiving regular red blood cell transfusions, or had an elevated tricuspid regurgitant jet (TRJ) velocity, which fulfilled eligibility criteria. Four patients developed grade II-III acute GVHD (18%) and 6 developed chronic GVHD (27%) that was moderate in two and severe in one patient. One patient died of intracranial hemorrhage and one of GVHD. Nineteen patients had stable donor chimerism, 1-year post-transplant. One patient who developed secondary graft failure survives disease-free after a second BMT. The one-year overall survival and event-free survival (EFS) are 91% (95% CI 68–98%) and 86% (95% CI, 63–95%), respectively, and 3-year EFS is 82%. Statistically significant improvements in the pain interference and physical function domains of health-related quality of life were observed. The study satisfied the primary endpoint of 1-year EFS ≥70%. This regimen is being studied in a prospective clinical trial comparing HLA-matched donor BMT with standard of care in adults with severe SCD (NCT02766465).
Introduction
Sickle cell disease (SCD) is a hereditary anemia characterized by intermittent pain episodes and progressive damage to vital organs, which contribute to a diminished quality of life and premature mortality[1–3]. Newborn screening and comprehensive care, pneumococcal prophylaxis, hydroxyurea, transcranial Doppler screening, and chronic red blood cell (RBC) transfusions prevent severe infections, stroke, and other serious complications in childhood and have increased survival to adulthood. Bone marrow transplantation (BMT) from a human leukocyte antigen (HLA)--identical sibling donor is potentially curative, but has been applied quite sparingly and restricted largely to children[1–3].
Unlike children, young adults with SCD may experience rapid disease progression marked by renal insufficiency, abnormal pulmonary function, and ultimately, pulmonary hypertension, irreversible organ damage, and premature mortality. [4–13] Chronic pain impairs quality of life, and 40–60% of adults with SCD are unemployed[14]. Supportive care for adult patients ameliorates symptoms, but does not address the overwhelming and progressive nature of this disease. Refinements in conditioning regimens, improved post-BMT supportive care, and better donor selection have increased the safety of allogeneic BMT for SCD, but early transplant-related mortality remains a risk.
If safety and efficacy of BMT can be established, it may become a suitable, if not preferred, therapeutic option for adults with severe SCD. A less toxic regimen was sufficient for donor engraftment after HLA-ID sibling BMT in adults with severe SCD, but this regimen can be extended to alternate donor BMT [15, 16]. This is an important consideration because only 18% of persons with SCD in the USA will have an HLA-identical sibling donor and only 19% will have an HLA-identical unrelated donor (URD) [17–19]. A pre-BMT conditioning regimen consisting of busulfan (Bu), fludarabine(Flu) and anti-thymocyte globulin has been tested in patients with Thalassemia and with chronic granulomatous disease[20–23]. The elimination of Cyclophosphamide (Cy) reduces the risk of venocclusive disease and in comparison, of Bu-Cy regimens, Bu-Flu regimens has been demonstrated to be associated with associated with better overall, event free and non-relapse free survival. [20, 21]. The lack of studies comparing BMT to standard of care in SCD remains a major gap in evidence[4, 24]. As a part of planning for a multicenter clinical trial comparing HCT to standard of care we conducted a pilot investigation to determine the safety and feasibility of BMT with this conditioning regimen in adults with severe SCD (NCT 01565616). We now report on the safety and efficacy of this approach in young adults with severe SCD.
Methods
Patients
The clinical trial protocol was approved by the Institutional Review Board at each of the participating institutions. Written informed consent was obtained from parents or patients ≥18 years of age and assent (age 17 years or less) was obtained before enrollment. Eligibility for enrollment was confirmed by a rotating two-member eligibility review committee (ERC) representing members of the team of the study team with expertise in SCD and BMT. The study was monitored by an external data safety monitoring board (DSMB), which consisted of experts in SCD, BMT, and biomedical ethics.
Patients 16 – 40 years of age (inclusive) with HbSS, SC or S/β thalassemia were eligible for the study if they had one or more of the following: a. Clinically significant neurologic event (stroke) or any neurological deficit lasting > 24 hours; b. History of two or more episodes of acute chest syndrome (ACS) in the 2-year period preceding enrollment despite supportive care measures, with ACS syndrome defined as a new pulmonary alveolar consolidation (or infiltrate) involving at least one complete lung segment associated with acute symptoms including one or more of the following: fever ≥ 38.5oC, chest pain, tachypnea per age adjusted normal, intercostal retractions/nasal flaring/use of accessory muscles of respiration, wheezing, rales or cough that is not attributed to asthma or bronchiolitis.; c. History of three or more severe painful vaso-occlusive episodes per year in the 2-year period preceding enrollment despite supportive care measures (i.e. a pain management plan and/or treatment with hydroxyurea). A painful vaso-occlusive episode was defined as a new onset of pain that lasted for at least 2 hours for which there was no other explanation and which was treated in a medical facility; d. Administration of regular RBC transfusions, which was defined as receiving 8 or more transfusions per year for 1 year to prevent vaso-occlusive clinical complications; e. Trans-thoracic echocardiographic evidence of tricuspid regurgitant jet (TRJ) velocity 2.7 m/sec or higher. If the patient had been receiving regular RBC transfusions for ≥1 year and had clinical or MRI evidence of iron overload or >20 lifetime transfusion exposures, a liver biopsy was performed to exclude cirrhosis, bridging fibrosis, or active hepatitis. The absence of bridging fibrosis was determined using the histological grading and staging scale as described by Ishak and colleagues. [25–27]
Eligible subjects had adequate physical function and organ function before BMT as defined by the following: a. Karnofsky performance score > or equal to 60. b. Cardiac function: Left ventricular ejection fraction (LVEF) > 40%; or LV shortening fraction > 26% by cardiac echocardiogram or by MUGA scan. c. Pulmonary function: Pulse oximetry with a baseline O2 saturation of 85% and DLCO > 40% (corrected for hemoglobin) d. Renal function: Serum creatinine ≤ 1.5 x upper limit of normal for age and GFR > 70 mL/min/1.73 m2. e. Hepatic function: Serum conjugated (direct) bilirubin < 2x upper limit of normal and ALT and AST < 5 times upper limit of normal as per local laboratory unless an increased serum bilirubin was the result of hyperhemolysis, or a significant drop in hemoglobin after RBC transfusion, which was acceptable. A related or unrelated bone marrow donor matched at 8 of 8 HLA loci (A, B, C, and DRB1) by allelic testing was required for participation in the study. Umbilical cord blood or peripheral blood stem cell donors were excluded. Patients were offered alternative treatment options such as hydroxyurea or automated RBC exchange transfusion. The hemoglobin S level was decreased to and maintained ≤ 30% by transfusion within 7 days of commencing transplant conditioning and chelation and Hydroxyurea were discontinued 48 hours before conditioning. Patients with cirrhosis of the liver, uncontrolled bacterial, viral or fungal infection, seropositive for HIV, had received a previous BMT, were currently receiving an investigational drug or device or off-label use of a drug or device, had demonstrated lack of compliance with prior medical care were unwilling to use approved contraception for at least 6 months after transplant, had a history of substance abuse that interfered with their care, or were pregnant or breast feeding were excluded from the study.
Treatment
Based on the experience that the substitution of fludarabine for cyclophosphamide in the myeloablative conditioning regimen improve safety and retain efficacy after HLA-matched sibling or URD BMT for leukemia[20] or thalassemia[28], we elected to test a reduced toxicity conditioning regimen of busulfan: 3.2 mg/kg administered as a single daily dose IV on Days –8 through −5 and dosing. Based upon busulfan (BU) population pharmacokinetics of once-daily IV busulfan described by Madden et al [29], we targeted busulfan pharmacokinetics (PK) to ensure a uniform dose exposure to achieve a steady state of 600–900ng/ml (target 750ng/ml) or a daily area under the curve of 3507–5261μM.min (target 4384 μM.min) on the subsequent day. The total regimen AUC was targeted to be in the range of 14,000 – 21,000 μM.min. Blood samples were collected and BU doses were adjusted according to Busulfan PK as per institutional standards. fludarabine: 35 mg/m2/day was administered IV on Days –7 through –3 (total fludarabine dose 175 mg/m2), and rabbit antithymocyte globulin (rATG) 0.5mg/kg was administered IV on day −6 1 mg/kg on day −5 and 1.5 mg/kg on days −4, −3 and −2 (total rATG dose of 6 mg/kg). There was a rest day on Day −1. Prophylaxis for graft-versus-host disease (GVHD) consisted of a calcineurin inhibitor (tacrolimus or cyclosporine) administered from Day –3 and extending through 180 days after BMT and methotrexate administered intravenously (7.5 mg/m2/day) on Days 1, 3 and 6 and 11 after BMT. The same methotrexate dosage schedule used in the BMT CTN 0601 study of URD BMT for SCD was adopted[30]. We targeted pre-BMT Hb S level to be ≤ 30% within 30 days prior to enrollment. This was achieved by exchange or simple transfusion according to local institutional practice. When doing an exchange transfusion, a hematocrit level of 30% was targeted, while not exceeding 36%. Donor-recipient ABO incompatibility was managed according to standard institutional practice. Individuals who were unable to safely receive PRBC transfusions because of severe allosensitization were not included in the study. Supportive care included weekly surveillance for cytomegalovirus (CMV) and Epstein-Barr virus (EBV) reactivation. Seizure prophylaxis commenced before conditioning and continued through 180 days after transplant, or until the calcineurin inhibitor was discontinued, whichever was later. In addition, supportive care measures included strict blood pressure control, RBC transfusions to keep hemoglobin between 9 and 11g/dL, and platelet transfusions to keep levels ≥50 × 109 /L to mitigate the risk of intracranial hemorrhage and posterior-reversible encephalopathy (PRES). Pre-emptive therapy for viral infections and bacterial prophylaxis during periods of neutropenia were also employed. Strategies to preserve fertility preservation were offered according to institutional practice.
Study Design
The primary endpoint was 1-year event-free survival (EFS). EFS was defined as survival with stable donor erythropoiesis with no new clinical evidence of SCD. Primary or late graft rejection, disease recurrence, and death were defined as events for this endpoint. Pre-specified secondary endpoints were overall survival, hematopoietic recovery as evidenced by cumulative incidence of neutrophil and platelet engraftment, lineage-specific chimerism following BMT, Grade II-IV and Grade III-IV acute GVHD, chronic GVHD, frequency of transplant-related complications such as infections, hepatic sinusoidal syndrome, interstitial pneumonitis, seizure, PRES, proportion of patients receiving immunosuppression 1 year after BMT, and health-related quality of life (HRQOL) as measured by the PROMIS-57 profile. The PROMIS 57 profile is a collection of short forms containing 8 items each from seven PROMIS domains (Depression, Anxiety, Physical Function, Pain interference, fatigue, Sleep Disturbance and Ability to Participate in Social Roles and activities) and a single item to assess pain intensity on a 0–10 numeric rating scale. Study evaluations at days 100 post-transplant, six months, and one-year post-transplant, consisted of HRQoL, Physical exam, CBC, differential, platelet count, and liver function tests. At one-year post-BMT serum ferritin, echocardiogram, GFR, pulmonary function testing, and a brain MRI were also obtained.
Lineage Specific Chimerism Analysis
To measure erythroid-lineage chimerism, pyrosequencing of the sickle cell mutation was used to quantify the relative levels of normal and sickle beta-globin mRNA in patient blood samples as previously described [31–33]. Peripheral blood samples were sorted on post-transplant days 28, 100, and 180 days by multidimensional FACS into CD3+CD56- (T-lymphocytes) and CD45+side scatter mid-hi (granulocytes). Genomic DNA extracted from peripheral blood and peripheral blood CD3+CD56- (T-lymphocytes) were analyzed for a variable number of tandem repeats (VNTR) to detect donor engraftment in mononuclear cells and CD3+ lymphocytes. [33] Percent donor chimerism was analyzed by PCR-based VNTR analysis and quantified by routine phosphorimager analysis.
Statistical analysis
The primary goal of this multicenter Phase II study was to determine the feasibility of achieving > 70% rate of survival with donor erythropoiesis at 1-year post transplant using a conditioning regimen with Bu/Flu/ATG in adult patients with severe SCD. The original target sample size of this pilot study was 15 patients with the goal of demonstrating that 11 or more patients would survive one year with donor hematopoiesis( EFS 73%). Because of high demand for enrollment on the study, this was revised, with the permission of the DSMB, to 20 patients. The DSMB subsequently gave permission to further expand enrollment to 22 patients in order to gain further experience with unrelated donors.
EFS and overall survival (OS) were calculated using the method of Kaplan and Meier. The cumulative incidence of acute graft versus host disease (GVHD) and chronic GVHD was calculated from the time of transplant to the time of each event with death as a competing risk using the cmprsk package in R version 3.5.1. Changes in continuous measures between two time points were assessed using a paired t-test. HRQOL measurement was based on the PROMIS-57. Raw scores for each domain were transformed to a standardized T-score using the PROMIS assessment manual (version 1.0). The depression, anxiety, physical function, pain interference, and fatigue domains were normed to a score of 50, standard deviation 10, based on the average for the US populations.[34] Ability to participate in social roles and activities and sleep disturbance were normed to a population enriched with patients with chronic disease. A higher PROMIS T-score represents more of the concept being measured. Pain intensity was collected as a raw score on a 0 to 10 scale. PROMIS 57 domains were analyzed using a two-sided paired t-test; p-values were adjusted for multiplicity of testing by a modified Holm approach in which the primary endpoint of changes between baseline and one-year post-transplant was tested at the 0.05 level. Changes between baseline and 6 months, and between 6 months and one year, were each tested at the nominal 0.0125 level to provide significance at the 0.05 level after adjustment.
Results
Subject characteristics
The trial enrolled 22 patients between July 2012 and June 2015. The median age was 22.5 (range 17–36) years; 36% of patients were male. Patient characteristics are summarized in Table 1. Subjects were enrolled in the study only if eligibility was confirmed by ERC review. Indications endorsed by the ERC for study enrollment were stroke (2), recurrent ACS (2), recurrent pain events (15), chronic transfusions (4) or elevated TRJ velocity (4). Seventeen patients were enrolled based on having a single criterion; 5 patients were enrolled with having 2 criteria. However, the participating sites reported that 11 patients had more than one eligibility criterion. Six patients who were receiving regular RBC transfusions had a liver biopsy before BMT, but none had bridging fibrosis or liver cirrhosis. Seventeen patients received bone marrow from an HLA-identical sibling donor and five from an HLA-matched URD. The median total nucleated cell dose was 3.07 × 108 /kg (range 0.3 – 29.7). The median CD34 dose was 4× 106 /kg (range 1.29 –19.73).
Table 1.
Characteristics of Donors and Recipients
| Donor Characteristics | N |
|---|---|
| Age, median (range), years | 32 (10, 48) |
| Race/ethnicity | |
| Caucasian | 1 |
| African American | 20 |
| Multirace | 0 |
| Hispanic/Latino | 0 |
| Not reported | 1 |
| Sex | |
| Male | 12 |
| Female | 10 |
| Recipient Characteristics | |
| Age (mean, median, range) years | 22, 24,17–36 |
| Sex | |
| Male | 8 |
| Female | 14 |
| Race/ethnicity | |
| African American | 22 |
| Hispanic | 0 |
| Transplant Characteristics | |
| CMV seronegative donor and recipient | 7 |
| CMV seropositive donor and recipient | 7 |
| CMV seronegative donor and CMV seropositive recipient | 7 |
| CMV seropositive donor and CMV seronegative recipient | 1 |
Event-free Survival, Overall Survival, and Engraftment
For the entire cohort, the 1-year EFS was 86% (95% CI, 63–95%) and OS was 91% (95% CI, 68–98%), which satisfied the pre-specified primary endpoint threshold of EFS ≥70% (Figure 1A; Table 2). All 22 patients had primary engraftment of donor cells. The median time to neutrophil and platelet recovery was 17 days (range 12–26) and 21 days (range 13–26), respectively. One of 22 patients (4.5%) experienced a secondary graft failure with marrow aplasia after URD BMT. This patient, who had an ABO major mismatched donor, developed severe hemolysis after BMT and acute renal damage and did not receive methotrexate prophylaxis for this reason. The patient had prolonged cytopenia with 100% donor chimerism in CD33 and 100% recipient chimerism in the CD3 compartment and was diagnosed with secondary graft rejection on day+252. The patient subsequently received a second BMT on day +316 from a different 10/10 HLA allele-matched URD for secondary graft failure and had stable full donor chimerism after conditioning with 4mg/m2 pentostatin and 2 daily fractions of 200 cGy total body irradiation. Two of 22 patients (9%) died after BMT. One patient developed PRES 80 days after HLA-identical sibling BMT and had a sudden, catastrophic increase in blood pressure and a fatal intracranial hemorrhage while recovering from sedation for a bone marrow biopsy to evaluate unexplained drop in blood levels of platelets, WBC and Hb. This patient was receiving cyclosporine and corticosteroids for upper GI GVHD. The patient had a remote history of seizures, and had been seizure free and off anti-epileptic medications for 12 years. Cerebral vasculopathy was suspected based on narrowing of cerebral blood vessels on an MRI/MRA 7 years prior to BMT. Subsequent TCDs and pre-transplant MRI/MRA were normal. The patient was never started on chronic transfusion, and was instead prescribed aspirin for the suspected cerebral vasculopathy. The patient admitted to being off aspirin for at least several months prior to BMT.
Figure 1.
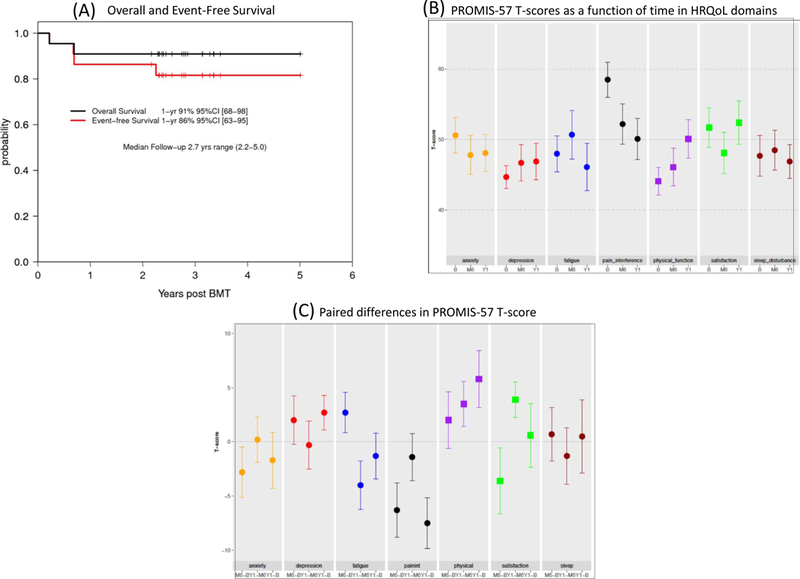
A. Overall and event-free survival. B. T-scores as a function of time in quality of life domains. C. Paired Differences in PROMIS-57 T-score.
Table 2.
Outcome following BMT
| All Patients N (%) |
MRD N (%) |
MURD N (%) |
|
|---|---|---|---|
| N | 22 | 17 | 5 |
| Death | 2 (9) | 1 | 1 |
| Cause of Death | PRES, GVHD | PRES | GVHD |
| Secondary Graft Failure | 1 (4.5) | 1 (5.9) | 0 (0) |
| 1-yr Overall Survival [95% CI] | 91 [68–98] | 94 [65–99] | 80 [20–97] |
| 1-yr Event-free Survival [95% CI] | 86 [63–95] | 94 [65–99] | 60 [13–88] |
|
3-yr Event-free Survival [95% CI] * estimate at 2.3 years |
82 [58–93] | 88 [61–97] | 60 [13–88]* |
| grade II-IV acute GVHD | 4 (18) | 3 (18) | 1 (20) |
| Day 180 Cumulative Incidence gr II-IV acute GVHD [95% CI] | 18 [5–37] | 18 [4–39] | 20 [1–62] |
| grade III acute GVHD | 1 (4.5) | 0 (0) | 1 (20) |
| chronic GVHD | 6 (27) | 5 (29) | 1 (20) |
| mild | 3 | 3 | 0 |
| moderate | 2 | 2 | 0 |
| severe | 1 | 0 | 1 |
| 1-yr Cumulative Incidence of chronic GVHD [95% CI] | 27 [11–47] | 29 [10–52] | 20 [1–62] |
| Patients receiving immunosuppression at 1 year after BMT | 8 (36) | 6 (35) | 2 (40) |
| Patients receiving immunosuppression at last follow-up | 2 (d+843, d+1005 post BMT) |
1 | 1 |
| Bacteremia | 8 (36) | 6 (29) | 2 (40) |
| Viral reactivation | 13 (59) | 10 (59) | 3 (60) |
| CMV | 10 | 7 | 3 |
| EBV | 3 | 3 | 0 |
| Secondary bone marrow aplasia | 2(9) | 1 (6) | 1(20) |
A second patient died of severe chronic GVHD after an HLA-matched URD BMT. The clinical course also was accompanied by pulmonary failure secondary to bronchiolitis obliterans. This patient died of progressive respiratory failure 252 days post-transplant. Autopsy revealed bilateral diffuse alveolar damage and bronchiolitis obliterans. There was also an incidental finding of post-transplant lymphoproliferative foci in the lung associated with EBV reactivation.
All survivors, at 1-year post-BMT, were alive at the last date of contact at a median of 2.7 years post -BMT (range, 2.2–5.0 yrs). All patients had stable donor chimerism at year 1 (Table 3). One patient developed pancytopenia 823 days post-transplant, but had donor chimerism of 100% and 80%, in the myeloid and lymphoid compartments, respectively. This patient had EBV reactivation following BMT and was treated with Rituximab. When the patient presented with pancytopenia on day+823 EBV early (D) Ag IgG, EBV VCA Ag IgM, and EBV PCR were negative, while EBV nuclear Ag IgG, and EBV VCA IgG were positive. EBV (D) early antigen IgG became positive a month later. At the discretion of the site investigator, the patient received a T-lymphocyte depleted peripheral blood stem cell infusion from the same sibling donor. The patient received no conditioning before receiving the donor cells and no GVHD prophylaxis. He had recovery with full hematopoiesis engraftment, stable donor chimerism, and no evidence of GVHD 6 months post-infusion. Since the patient received a second stem cell infusion we adjudicated this as a secondary graft failure event.
Table 3.
Post-transplant Donor Chimerism (%) by Hematopoietic Lineage
| Type | Day Post- BMT |
N | Mean | SD | Min | p25th | Median | p75th | Max |
|---|---|---|---|---|---|---|---|---|---|
| Myeloid | 28 | 14 | 100 | 0 | 100 | 100 | 100 | 100 | 100 |
| 100 | 13 | 100 | 0 | 100 | 100 | 100 | 100 | 100 | |
| 180 | 13 | 99.92 | 0.28 | 99 | 100 | 100 | 100 | 100 | |
| 365 | 12 | 99.83 | 0.58 | 98 | 100 | 100 | 100 | 100 | |
| RBC | 28 | 20 | 97.55 | 3.55 | 89 | 95.75 | 100 | 100 | 100 |
| 100 | 20 | 92.05 | 17.75 | 24 | 91 | 100 | 100 | 100 | |
| 180 | 17 | 96.47 | 6.60 | 77 | 96 | 100 | 100 | 100 | |
| 365 | 16 | 81.75 | 17.13 | 48 | 70.25 | 83.5 | 100 | 100 | |
| T-cell | 28 | 21 | 58.05 | 24.70 | 12 | 43 | 55 | 78 | 100 |
| 100 | 22 | 69.59 | 27.83 | 0 | 57.5 | 75.5 | 87.5 | 100 | |
| 180 | 18 | 70.72 | 30.51 | 0 | 59 | 77 | 97.75 | 100 | |
| 365 | 19 | 82.26 | 23.19 | 12 | 76 | 87 | 100 | 100 | |
| Whole Blood | 28 | 8 | 97.75 | 1.58 | 95 | 97 | 97.5 | 99 | 100 |
| 100 | 8 | 97.38 | 1.92 | 95 | 96 | 97 | 98.5 | 100 | |
| 180 | 8 | 95.88 | 4.91 | 86 | 94.75 | 98 | 98.5 | 100 | |
| 365 | 8 | 96.50 | 6.21 | 82 | 97 | 99 | 100 | 100 |
SD=standard deviation; p=percentile
For patients who underwent BMT from matched siblings the one-year OS was 94% (95% CI, 65–99%) one-year EFS 94% (95% CI, 65–99%) and 3-year EFS 88% (95% CI, 61–97%). Among 5 patients who had an 8/8 HLA allele-matched URD transplant, one patient developed grade III acute GVHD and one patient died due to severe, chronic GVHD, one patient had secondary graft failure followed by a successful second URD BMT, and 3 patients survive event-free. Four of 5 URD recipients survive disease-free. One-year OS 80% (95% CI, 20–97%) EFS 60% (95% CI, 13–88%)
Graft-Versus-Host Disease
Four of 22 patients developed acute GVHD and the day 180 cumulative incidence was 18% (95% CI, 5–37) (Table 2). This included three, grade II (upper and lower GI, skin) and one, grade III (liver). The four patients that developed acute GVHD ranged in age from 23–26 years. Six of 21 evaluable patients developed chronic GVHD and the 1-year cumulative incidence was 27% (95% CI, 11–47). This included three mild (mouth, musculoskeletal, liver), two moderate (skin, GI, mouth, liver), one severe that was fatal (pulmonary) as noted above). For the three mild, chronic GVHD cases, the ages ranged from 21–24, moderate (age 20, 21), and one severe was aged 17. among the 5 patients who underwent BMT from an unrelated donor, 1 developed grade III, acute GVHD (20%) and 1 developed chronic GVHD (20%). Among patients who underwent BMT from a matched, sibling donor, three (18%) developed grade II acute. Among patients who underwent BMT from a matched, unrelated donor, one (20%) developed severe, chronic GVHD. Among patients who underwent BMT from a matched, sibling donor, five (29%) developed chronic GVHD, which was moderate in 2 (12%); while, none had severe, chronic GVHD. Eight of 19 patients (42%) were receiving immunosuppression at the 1-year time point and 6 of these 8 patients subsequently stopped immunosuppressive therapy uneventfully. At last follow-up, 2 patients were receiving immunosuppressive therapy at 886 and 444 days after BMT.
Health Related Quality of Life
HRQOL studies comparing one-year post-BMT to baseline in the 17 patients with paired measurements showed significant improvement in physical function (p=0.044) and significant reduction in pain interference (p=0.006) (Figures 1B and 1C and Supplementary Tables 1 and 2). Both domains were significantly compromised at baseline. No other changes were statistically significant. Pain intensity was highly variable over time within patients; no significant changes were observed in this measure.
Donor Chimerism after BMT
Because mixed chimerism is frequently observed after BMT for SCD, the percentage of donor cells was measured at 28, 100, 180, and 365 days after BMT (Table 2). Full donor myeloid chimerism was observed at all time points. Initially, mixed donor chimerism in the lymphoid compartment was observed in 21 of 22 patients. The T-cell donor chimerism increased over time with a mean of 82% at day +365. The delay in full donor T-cell engraftment was not associated with graft rejection or GVHD.
Other SCD-related measurements:
Of 17 patients who had a brain MRI performed at baseline and at one year after BMT, all had a stable MRI. A small saccular aneurysm by MRA was observed in one patient 1 year after BMT, but there was no baseline MRA for comparison. Pulmonary function tests were obtained both at baseline and 1 year in 18 patients, although all components were not repeated one year after BMT. There were no statistically significant changes in the mean FEV1 (n=18, p=0.92), FEV1/FVC (n=16, p=0.40), and TLC (n=11, p=0.69). TRJ velocity was measured at baseline in 18 patients, but TRJ was recorded at one year in only 11 patients. Of the 18 patients with pre-BMT assessments, TRJ was normal in 13 and ≥ 2.7 m/sec in 5 patients. Three patients with TRJ > 2.7m/sec did not have TRJ recorded in follow-up studies while in the other two with TRJ= 2.7 m/sec pre-BMT, decreased <2.7m/sec. In one patient, TRJ of 2.4 m/sec at baseline increased to 2.7m/sec at 1-year post BMT.
Other BMT toxicities:
There were eight episodes of bacteremia (Table 3). There were no instances of invasive fungal infection. CMV reactivation, without active CMV disease, occurred in 10 patients. EBV viremia was detected in three patients, and EBV-related post-transplant lymphoproliferative disease was observed in one patient who died of GVHD. No patient developed sinusoidal obstruction syndrome. One patient with PRES died of an intracranial hemorrhage and the patient with pulmonary failure and bronchiolitis obliterans also had severe interstitial fibrosis noted on autopsy.
Discussion
We report the results of the first NIH-supported, multicenter BMT trial for and young adult patients with SCD. The reduced toxicity myeloablative regimen of Busulfan, Fludarabine, and ATG was sufficient for primary engraftment in all but one case and did not cause significant toxicity in this group of young adults. We exceeded a target of achieving a one-year endpoint of ≥ 70% EFS that we had reasoned would be necessary before pursuing a comparative clinical trial of the regimen. These results are comparable to the 81% OS and EFS in patients >16 years of age after HLA identical sibling BMT for SCD as reported in a recent international survey [35] and 87% EFS after HLA-identical sibling in adult SCD recipients BMT reported in a single center by Hsieh et al[16]. The EFS probability we observed also is similar to results in children with SCD treated by HLA-identical sibling BMT[35].These outcomes, if reproduced in a larger trial, may be considered acceptable to support broader adoption of allo-BMT for adult patients with severe SCD compared with the risk of premature mortality in adults with SCD.
Despite these promising results, there is a prevailing view that a reduced intensity conditioning regimen in lieu of myeloablation would be more suitable in this patient population[36]. This is supported in part by a perception that efficacy and safety are optimized after reduced intensity conditioning, particularly in patients with significant vaso-occlusive end-organ co-morbidities. However, this view is challenged by the favorable toxicity profile we observed and the low incidence of graft rejection. An important barrier to a successful transplant outcome in SCD is graft rejection, which can be mitigated by the intensity of the regimen. Several reports of reduced intensity or minimal toxicity regimens suggest these are not sufficiently intensive for sustained donor engraftment, particularly after HLA-mismatched or URD HCT[30, 37–39], or engraftment is extended only by the administration of long-term immunosuppressive therapy in many patients[16, 40]. In addition, in an analysis of 95 patients with SCD who received related or URD HCT between 2002 and 2013, all 13 graft rejection events occurred in patients who received reduced intensity conditioning. There were no rejections in patients who received myeloablative conditioning[41]. Thus, we conclude that this pilot investigation warrants further investigation in adults with severe SCD. While it would also be useful to compare the reduced intensity and myeloablative regimens in SCD directly, sample size requirements might constrain the feasibility of a comparison trial.
This study completed enrollment in 28 months compared to 6–9 years for full accrual in previous multicenter studies of BMT for SCD[1, 30]. This, in part, reflects the increasing interest in BMT as a curative therapy for adults with SCD in the sickle cell community, which is based upon excellent reports described in pediatric and adult patients[1, 16, 40, 42].
The pattern of early mixed lymphoid chimerism, which improved over time in this study, is similar to our previous observations.[31, 43, 44] Most patients had mixed donor chimerism in the lymphoid compartment that probably reflects the effect of rabbit ATG on donor T-cell engraftment. The donor lymphoid chimerism increased over time. Mixed lymphoid chimerism did not appear to predict graft rejection or protect from GVHD. Notably, there was full donor erythroid chimerism at nearly all post-transplant time points, indicating a survival advantage of donor erythroid precursors.
We observed a fatal episode of PRES in a single patient who was receiving a combination of a calcineurin inhibitor and corticosteroid, which underscores the well-known risk of PRES in patients with SCD[30, 45]. The sudden onset of PRES, with no preceding hypertension or prodromal manifestations, also suggests that PRES may not be avoidable even when preventive measures are employed. The rate of PRES observed in this study was substantially lower than observed in the recent pediatric URD BMT trial in SCD[30].
As described in previous reports, this study also demonstrated stable organ function as measured by stable, brain MRI and PFTs following BMT. A significant limitation in interpreting the impact of BMT on pulmonary hypertension is the inconsistent measurement and documentation of TRJ on the post-transplant echocardiogram exam. Another limitation of this pilot study is that we did not capture biomarkers of hemolysis or details of healthcare utilization related to sickle cell disease post-BMT. We observed a significant improvement in physical functioning and a significant decrease in pain interference domains, at one-year post-BMT, while changes in fatigue, anxiety, depression, sleep disturbance, satisfaction with participation in social roles, and pain intensity domains did not change significantly. We note that, at study entry, physical functioning and pain interference were the domains of HRQoL in which the subjects were significantly more compromised than the reference population.
Pain intensity in this study was ascertained using a single question on PROMIS-57 measure. This question is limited to pain intensity in the previous seven days, and is subject to recall bias. A limitation of this study is that it did not capture detailed patient reported outcomes of residual pain burden among survivors of BMT for SCD. The use of an electronic pain diary has the potential to capture patient-reported dimensions of pain in real-time and is not subject to recall bias. Such studies are more able to determine the trajectory of chronic pain as well as the course of its resolution following BMT. Insights gained from the detailed study of the resolution of pain following BMT will inform patients about the likely impact of BMT on their experience of pain and help design suitable interventions for the management of chronic pain following BMT.
Recent reports of the impact of BMT of SCD on HRQOL[30, 46–49] suggest that the change in different domains of physical and psychosocial functioning is variable. For e.g., in the study reported by Saraf et al, while there were greater than minimally significant improvement in the mean scores on SF36 from baseline to day 28, 100 and 365, statistically significant changes were limited to bodily pain, general health and vitality domains. Similarly, in the current study in the first year post-BMT, multiple domains of HRQOL remain unchanged; though, the significance of this observation is unclear because these values did not differ substantially from population norms at baseline. The interpretation of studies of HRQOL following BMT for SCD is, however, limited by small sample size and the lack of long-term follow-up. They are also difficult to compare across clinical trials because of the heterogeneity of patient populations, conditioning regimen, donor types, and HRQOL instruments used. The use of PROMIS measures of HRQOL, as was done in this study, supplemented by selected domains of the ASCQ-Me sickle cell-specific measures[50, 51] in adult SCD patients in large prospective studies of BMT with long-term follow-up, have the potential to determine the impact of BMT on patient-reported outcomes across clinical trials.
The results of this pilot trial support our conclusion that a multicenter clinical trial testing a reduced-toxicity conditioning regimen of Bu/Flu/ATG is feasible and effective in adults with SCD. This study also generated BMT pilot experience of HLA-matched URD BMT for SCD. Taken together these observations provided the rationale for testing this regimen in a larger multicenter study BMT CTN 1503 (NCT02766465), which compares BMT to standard of care in individuals without a suitably matched related or URD.
Supplementary Material
Acknowledgements:
Supported by a grant from the National Heart, Lung, and Blood Institute (R34-HL108761).
Footnotes
Conflict of Interest: The authors have no relevant disclosures.
References
- 1.Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2010;16:263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King AA, Kamani N, Bunin N, et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am J Hematol 2015;90:1093–1098. [DOI] [PubMed] [Google Scholar]
- 3.Walters MC, De Castro LM, Sullivan KM, et al. Indications and Results of HLA-Identical Sibling Hematopoietic Cell Transplantation for Sickle Cell Disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2016;22:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Decision Memo for Stem Cell Transplantation (Multiple Myeloma, Myelofibrosis, and Sickle Cell Disease) (CAG-00444R). Centers for Medicare and Medicaid Services, Baltimore, MD: 2016. [Google Scholar]
- 5.Darbari DS, Kple-Faget P, Kwagyan J, et al. Circumstances of death in adult sickle cell disease patients. Am J Hematol 2006;81:858–863. [DOI] [PubMed] [Google Scholar]
- 6.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. The New England journal of medicine 2004;350:886–895. [DOI] [PubMed] [Google Scholar]
- 7.Guasch A, Navarrete J, Nass K, et al. Glomerular involvement in adults with sickle cell hemoglobinopathies: Prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol 2006;17:2228–2235. [DOI] [PubMed] [Google Scholar]
- 8.Koumbourlis AC, Lee DJ, Lee A. Longitudinal changes in lung function and somatic growth in children with sickle cell disease. Pediatric pulmonology 2007;42:483–488. [DOI] [PubMed] [Google Scholar]
- 9.Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Rep 2013;128:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McClish DK, Penberthy LT, Bovbjerg VE, et al. Health related quality of life in sickle cell patients: the PiSCES project. Health and quality of life outcomes 2005;3:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panepinto JA. Health-related quality of life in patients with hemoglobinopathies. Hematology Am Soc Hematol Educ Program 2012;2012:284–289. [DOI] [PubMed] [Google Scholar]
- 12.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. The New England journal of medicine 1994;330:1639–1644. [DOI] [PubMed] [Google Scholar]
- 13.Powars DR, Chan LS, Hiti A, et al. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine 2005;84:363–376. [DOI] [PubMed] [Google Scholar]
- 14.Bediako SM. Predictors of employment status among African Americans with sickle cell disease. J Health Care Poor Underserved 2010;21:1124–1137. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. The New England journal of medicine 2009;361:2309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. Jama 2014;312:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walters MC, Patience M, Leisenring W, et al. Barriers to bone marrow transplantation for sickle cell anemia. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 1996;2:100–104. [PubMed] [Google Scholar]
- 18.Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. The New England journal of medicine 2014;371:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mentzer WC, Heller S, Pearle PR, et al. Availability of related donors for bone marrow transplantation in sickle cell anemia. Am J Pediatr Hematol Oncol 1994;16:27–29. [PubMed] [Google Scholar]
- 20.Andersson BS, de Lima M, Thall PF, et al. Once daily i.v. busulfan and fludarabine (i.v. Bu-Flu) compares favorably with i.v. busulfan and cyclophosphamide (i.v. BuCy2) as pretransplant conditioning therapy in AML/MDS. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2008;14:672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.La Nasa G, Argiolu F, Giardini C, et al. Unrelated bone marrow transplantation for beta-thalassemia patients: The experience of the Italian Bone Marrow Transplant Group. Ann N Y Acad Sci 2005;1054:186–195. [DOI] [PubMed] [Google Scholar]
- 22.Gungor T, Teira P, Slatter M, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet 2014;383:436–448. [DOI] [PubMed] [Google Scholar]
- 23.Key Features of the Affordable Care Act By Year. DHHS; 2014.
- 24.Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2016:CD007001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anthony PP, Ishak KG, Nayak NC, et al. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J Clin Pathol 1978;31:395–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishak KG. Chronic hepatitis: morphology and nomenclature. Mod Pathol 1994;7:690–713. [PubMed] [Google Scholar]
- 27.Ishak KG. Pathologic features of chronic hepatitis. A review and update. Am J Clin Pathol 2000;113:40–55. [DOI] [PubMed] [Google Scholar]
- 28.La Nasa G, Littera R, Locatelli F, et al. The human leucocyte antigen-G 14-basepair polymorphism correlates with graft-versus-host disease in unrelated bone marrow transplantation for thalassaemia. British journal of haematology 2007;139:284–288. [DOI] [PubMed] [Google Scholar]
- 29.Madden T, de Lima M, Thapar N, et al. Pharmacokinetics of once-daily IV busulfan as part of pretransplantation preparative regimens: a comparison with an every 6-hour dosing schedule. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2007;13:56–64. [DOI] [PubMed] [Google Scholar]
- 30.Shenoy S, Eapen M, Panepinto JA, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood 2016;128:2561–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu CJ, Gladwin M, Tisdale J, et al. Mixed haematopoietic chimerism for sickle cell disease prevents intravascular haemolysis. British journal of haematology 2007;139:504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu CJ, Krishnamurti L, Kutok JL, et al. Evidence for ineffective erythropoiesis in severe sickle cell disease. Blood 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu CJ, Hochberg EP, Rogers SA, et al. Molecular assessment of erythroid lineage chimerism following nonmyeloablative allogeneic stem cell transplantation. Experimental hematology 2003;31:924–933. [DOI] [PubMed] [Google Scholar]
- 34.Gershon RC, Rothrock N, Hanrahan R, et al. The use of PROMIS and assessment center to deliver patient-reported outcome measures in clinical research. J Appl Meas 2010;11:304–314. [PMC free article] [PubMed] [Google Scholar]
- 35.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsieh MM. A Standard Nonmyeloablative Transplantation Regimen for Adults with Sickle Cell Disease: Are We There Yet? Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2016;22:397–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamani NR, Walters MC, Carter S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2012;18:1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv 2017;1:652–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saraf SL, Oh AL, Patel PR, et al. Haploidentical Peripheral Blood Stem Cell Transplantation Demonstrates Stable Engraftment in Adults with Sickle Cell Disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saraf SL, Oh AL, Patel PR, et al. Nonmyeloablative Stem Cell Transplantation with Alemtuzumab/Low-Dose Irradiation to Cure and Improve the Quality of Life of Adults with Sickle Cell Disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2016;22:441–448. [DOI] [PubMed] [Google Scholar]
- 41.Abraham A, Hsieh M, Eapen M, et al. Relationship between Mixed Donor-Recipient Chimerism and Disease Recurrence after Hematopoietic Cell Transplantation for Sickle Cell Disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2017;23:2178–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Non-Myeloablative Allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT) for Patients with Severe Sickle Cell Disease (SCD). Blood 2011;118:10–10. [Google Scholar]
- 43.Krishnamurti L, Kharbanda S, Biernacki MA, et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2008;14:1270–1278. [DOI] [PubMed] [Google Scholar]
- 44.Wu CJ, Krishnamurti L, Kutok JL, et al. Evidence for ineffective erythropoiesis in severe sickle cell disease. Blood 2005;106:3639–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walters MC, Sullivan KM, Bernaudin F, et al. Neurologic complications after allogeneic marrow transplantation for sickle cell anemia. Blood 1995;85:879–884. [PubMed] [Google Scholar]
- 46.McHorney CA, Ware JE Jr., Lu JF, et al. The MOS 36-item Short-Form Health Survey (SF-36): III. Tests of data quality, scaling assumptions, and reliability across diverse patient groups. Med Care 1994;32:40–66. [DOI] [PubMed] [Google Scholar]
- 47.McHorney CA, Ware JE Jr., Raczek AE The MOS 36-Item Short-Form Health Survey (SF-36): II. Psychometric and clinical tests of validity in measuring physical and mental health constructs. Med Care 1993;31:247–263. [DOI] [PubMed] [Google Scholar]
- 48.Ware JE Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care 1992;30:473–483. [PubMed] [Google Scholar]
- 49.Bhatia M, Kolva E, Cimini L, et al. Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2015;21:666–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Treadwell MJ, Hassell K, Levine R, et al. Adult sickle cell quality-of-life measurement information system (ASCQ-Me): conceptual model based on review of the literature and formative research. The Clinical journal of pain 2014;30:902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evensen CT, Treadwell MJ, Keller S, et al. Quality of care in sickle cell disease: Cross-sectional study and development of a measure for adults reporting on ambulatory and emergency department care. Medicine 2016;95:e4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.