

Abstract
Recent studies of multiple enzyme families collectively referred to as ene-reductases (ERs) have highlighted potential industrial application of these biocatalysts in the production of fine and speciality chemicals. Processes have been developed whereby ERs contribute to synthetic routes as isolated enzymes, components of multi-enzyme cascades, and more recently in metabolic engineering and synthetic biology programmes using microbial cell factories to support chemicals production. The discovery of ERs from previously untapped sources and the expansion of directed evolution screening programmes, coupled to deeper mechanistic understanding of ER reactions, have driven their use in natural product and chemicals synthesis. Here we review developments, challenges and opportunities for the use of ERs in fine and speciality chemicals manufacture. The ER research field is rapidly expanding and the focus of this review is on developments that have emerged predominantly over the last 4 years.
Keywords: Ene-reductases, asymmetric alkene reduction, biocatalysis, synthetic biology, Old Yellow Enzymes
1. Introduction
There is wide recognition of the valuable contribution that biocatalytic and chemo-enzymatic approaches play towards the production of fine chemicals, pharmaceuticals, agrochemicals and more recently biofuels production.1–6 This is particularly relevant in the synthesis of structurally complex natural and non-natural compounds, especially pharmaceutical compounds, where suitable chemosynthetic routes are technically challenging.2 Biocatalytic routes are complementary to traditional synthetic methods, where the relative strengths of each will lead to both an increased efficiency in both natural and non-natural product synthesis, and enable routes to industrially useful chemicals synthesis.
This explosion in the exploration of biocatalytic routes is led by knowledge of the wide range of organic reactions catalysed by enzymes, coupled with the often-remarkable chemo-, regio-, and stereo-selectivity achieved under relatively mild reaction conditions (temperature, pH and pressure).6–7 Increasingly, chemo-enzymatic approaches to fine chemicals synthesis are employed, where individual or multiple cascading reactions (i.e. ‘one-pot’ biotransformations)8–10 replace some traditional synthetic steps. Synthetic biology and metabolic engineering approaches have also become more prevalent, where valuable chemicals are produced in vivo from a variety of feedstocks by recombinant microorganisms containing de novo engineered metabolic pathways. Notably, this has been achieved in the semisynthesis of artemisinin, where the precursor artemisinic acid was generated by an Escherichia coli strain harboring genes encoding terpenoid biosynthesis genes.11 Overall, these approaches can lead to the use of non-petroleum-based synthons, environmentally friendly bio-sourced catalysts and decreases in waste and by-product generation, thereby satisfying the increasing legislative emphasis on the development of ‘green’ chemistry approaches to chemical production. This is particularly relevant to the food additive industry, where there is a legislative discrimination between chemically identical food additives from synthetic and ‘natural’ origin.
Suitable targets for biocatalytic approaches to chemical synthesis include the asymmetric reduction of activated C=C bonds, a widely employed synthetic step that has the potential to generate up to two stereogenic centers (Figure 1).6–7, 12 There has been extensive research into NAD(P)H-dependent biocatalytic alkene reduction catalysed by families of enzymes known collectively as ene-reductases (ERs), with many recent reviews describing their potential applications in industrial processes.e.g.2, 5–7, 13–15 This has identified enzymes capable of generating chiral mono- and di-carboxylic acids and esters, aldehydes, ketones, nitriles, nitro compounds protected acyloins and halogen-substituted acrylates.16
Figure 1.

Asymmetric activated alkene reduction catalysed by ene-reductases (ER). Enzyme classes: OYE = Old Yellow Enzyme; EnoR = oxygen-sensitive enoate reductases; SDR = short chain dehydrogenase/reductase salutaridine/menthone reductase-like subfamily; MDR = medium chain dehydrogenase/reductase leukotriene B4 dehydrogenase subfamily; QnoR = quinone reductase-like ene-reductase.
The most predominant family of ERs (Figure 1) are the FMN-containing Old Yellow Enzyme (OYE) family of oxidoreductases (EC 1.6.99.1).15 They catalyse the reduction of α,β-unsaturated compounds, with a high specificity for activating groups containing aldehydes, ketones or nitro groups. Another family of ERs are the oxygen-sensitive FAD and [4Fe-4S]-containing clostridial Enoate Reductases (EnoR; EC 1.3.1.31), specific for substrates containing carboxylic acids and esters.17 The leukotriene B4 dehydrogenase subfamily of medium chain dehydrogenases/reductases (MDR; EC 1.3.1)18–19 and the salutaridine/menthone reductase-like subfamily of short chain dehydrogenases/reductases (SDR; EC 1.1.1.207-8)20–21 are flavin-independent ERs, and are enzymes that have been investigated recently for their biocatalytic potential. Typical substrates include aromatic and monocyclic alkenes, containing aldehydes or ketones as activating groups (Figure 1).18, 20 In combination these enzyme families cover an exceptional range of substrate type, which emphasizes the catalytic versatility of the expanding ER toolbox.
This review summarises recent progress in developing ER-catalysed asymmetric alkene reductions, with a particular emphasis on the production of potentially useful synthons. It explores both the discovery of new biocatalysts and the application of existing ERs in engineering de novo (bio)synthetic routes to fine chemicals. The inclusion of ERs in synthetic biology and metabolic engineering programmes highlights also the potential use of ERs in whole-cell fermentations for the production of fine chemicals. Additional discussion on recent structural elucidation, mechanistic understanding and enzyme engineering of ERs is included to illustrate the impact of knowledge-based design and optimization of enzymes for industrial biotransformations.
2. Newly Discovered Ene Reductases
Prior extensive reviews of ERs exhibiting biocatalytic potential have shown that the majority are derived from either Eubacterial, fungal and plant sources, and function under typically mesophilic reaction conditions.e.g.22 However, the often modest turnover numbers with key synthons,23 poor substrate tolerance preventing high substrate loading, instability under industrial conditions and in some cases unsatisfactory enantioselectivity drives the search for new and improved ERs. In recent years, the range of ERs exploited for biocatalytic applications has been extended to other less well-studied classes of double bond reductases. This section discusses recent discoveries of new ERs belonging to different enzyme families, including the discovery of a new family of double bond reductases. These new biocatalysts augment the already extensive range of reactions possible using ER enzymes.22
Thermophilic enzymes are advantageous in industrial applications, in relation to enhanced temperature stability and solvent tolerance. A new ER activity has been discovered within the hyperthermophile Pyrococcus furiosus.24 As bona fide OYEs have not been discovered in extremophiles before, the class of enzyme(s) responsible for this activity is unknown. Studies into the whole cell reduction of conjugated carboxylic acids showed activity towards cyclohexen-2-one and trans-4-phenyl-3-buten-2-one. Further studies are now required to identify the ER(s) responsible for this activity but the discovery of additional thermophilic enzymes is a welcome addition to the ER biocatalytic toolkit.24
A recent structural bioinformatics method for predicting catalytic activities of database enzymes was described using a ER-like three-dimensional constellation of functional groups within active sites (so-called catalophores).26 The search parameters included a low sequence homology to OYEs, similar binding modes of phenolic ligands and substrates, C=C double bond reduction capability and a higher NADPH:NADH binding ratio. Two non-OYE thermophilic ERs were identified, which were previously annotated as a putative styrene monooxygenase reductase from Thermus thermophilus (TtENR) and a FMN-binding protein from Pyrococcus horikoshii (PhENR; Figure 2A). These enzymes exhibited double bond reduction towards α,β-unsaturated aldehydes and ketones including 2-methyl-2-pentenal, citral, 4-ketoisophorone and N-phenyl-2-methylmaleimide.26 Interestingly, the opposite enantiomeric products were obtained with the latter two substrates, compared to reactions with a classical OYE PETNR from Enterobacter cloacae PB2,27 suggesting an inverted stereo-preference.
Figure 2.

Comparison of ERs belonging to OYE and quinone reductase-like families. A) Overall crystal structures of PETNR (PDB: 1GVQ) and PhENR (PDB: 3ZOG). The structures are shown as rainbow cartoons, from blue to red for the N- to C-termini, respectively. The FMN and cyclohexen-2-one are shown as atom coloured sticks with yellow and magenta carbons, respectively, throughout the Figure. B) Comparative active site arrangement between cyclohexen-2-one-bound PETNR and PhENR (2 molecules bound). Residues are shown as atom coloured sticks with green carbons. The figure was generated using Pymol.25
Comparative studies between these newly discovered ERs and OYEs showed they exhibited a significantly altered substrate spectrum, more negative redox potential and a different kinetic mechanism.26 The three-dimensional crystal structures of each enzyme showed these ERs were homodimers, with a different overall protein fold and equivalent, yet mirror image substrate binding modes to OYEs (Figure 2B). Overall, PhENR and TtENR were described as NADPH-dependent quinone reductases with significant OYE-like side activities. This study illustrates an alternative and important approach in the discovery of novel ERs with biocatalytic potential.26
Genome mining studies with the acidophilic iron-oxidizing betaproteobacterium Ferrovum st. JA12 identified a thermophilic-like OYE (FOYE-1) with moderate thermostability (up to 50 °C).28 It displayed significant activity towards N-phenylmaleimide analogues (> 98% conversion), with high (R)-succinamide selectivity (Scheme 1A). Another thermophilic-like OYE was described from Rhodococcus opacus 1CP (OYERo2), exhibiting activity towards N-substituted maleimides (Scheme 1A) with near optical purity (>99% ee (R)).29 Unusually for a thermophilic-like OYE, it exhibited poor thermostability and solvent tolerance, although moderate improvements were gained by the incorporation of an intermolecular salt bridge characteristic of these enzymes by site-specific mutagenesis (t1/2 raised 3.1-fold at 32 °C).29
Scheme 1.

Biocatalytic α,β-unsaturated alkene reduction catalysed by novel A) thermophilic, B) fungal, C) plant ERs and D) clostridial EnoR.
Functional screening of microorganisms for ER activity towards useful compounds is an alternative approach to sequence mining for the discovery of new ER biocatalysts. For example, a whole-cell biotransformation screen of 28 fungi belonging to phyla Ascomycota, Basidiomycota, and Zygomycota was performed to detect ER activity towards three representative OYE substrates with different activating groups (ketone, nitro, and aldehyde) conjugated with the C=C double bond.30 In most cases, activity towards cyclohexen-2-one, α-methyl-β-nitrostyrene, and (E)-α-methylcinnamaldehyde resulted in the production of the respective saturated alcohol, presumably due to the action of constitutive alcohol dehydrogenases. While all the fungal strains exhibited some ER activity, the best performers were Gliomastix masseei, Mucor circinelloides, and Mucor plumbeus, in terms of high yields of all three substrates.30 Similarly, another study screened eight OYEs from non-conventional yeasts for biocatalytic potential by expressing them in an OYE-deletion strain of S. cerevisiae (BY4741ΔOye2).31 Target compounds were ketoisophorone, α-methyl-trans-cinnamaldehyde and trans-β-methyl-β-nitrostyrene, and studies were supported by crystal structure determination of the OYEs from Kluyveromyces lodderae and Candida castellii. Density functional theory (DFT) computational studies were also performed to investigate the mode of substrate discrimination.31
A further whole cell biotransformation study of M. circinelloides was performed with cyclohexen-2-one, α-methyl cinnamaldehyde and methyl cinnamate as model substrates.32 In silico analysis identified ten potential OYEs from M. circinelloides, and the expression of the individual genes were monitored parallel to product formation. This suggested the presence of at least eight ERs within M. circinelloides that showed high reduction of the industrially important α-methyl cinnamaldehyde (>99% conversion after 20 h; Scheme 1B).32
Several studies have investigated the properties of newly isolated fungal ERs, employing extensive genomic sequence mining in an attempt to identify and screen for novel robust ERs with increased turnover number/yields with commercially-useful synthons. For example, the yeast OYE from Meyerozyma guilliermondii (MgER)33 demonstrated its potential as an industrial biocatalyst with (R)-carvone by generating (2R,5R)-dihydrocarvone with high yields (99%) and optical purity (>99% de; Scheme 1B).33 Another fungal ER characterised recently is an OYE from Clavispora (Candida) lusitaniae (ClER).34 It displayed both high activity (>99% conversion) and stereospecificity (>99% ee or de) towards typical OYE substrates (R)- and (S)-carvone (Scheme 1B), ketoisophorone, N-phenylmethyl maleimide and cinnamaldehyde. It displayed high stereospecificity, generating the commercially useful (R)-levodione with near optical purity.34
Functional screening of ER activity has also been described with 13 plant tissue cultures, using cyclohexen-2-one as the model substrate.35 Plant cultures from Medicago sativa, Tessaria absinthioides calli and Capsicum annuum hairy roots displayed ER activity towards β-nitrostyrene and maleimides, rather than classical OYE-like substrates. They also displayed activity towards (E)-chalcone, generating dihydrochalcone (38% yield in 4 d) with high chemoselectivity, and small amounts of the equivalent alcohol side product due to the action of both ER and ADH activities, respectively (Scheme 1C).35
Recently discovered bacterial OYEs have been described in Achromobacter sp. JA81 (Achr-OYE4)36 and Bacillus subtilis str.168 (YqiG),37 the latter displaying C=C reduction towards substrates such as maleimides and acyclic ketones. Unlike most bacterial genera, Clostridia showed the presence of FAD and [4Fe-4S]-containing EnoR’s, which have not been exploited industrially due to their oxygen sensitivity.17 Recently the EnoR from Clostridium sporogenes DSM 795 (FldZ) was expressed in E. coli under anaerobic conditions, and it displayed C=C reduction towards a variety of aromatic enoates, such as α-cinnamic acid derivatives (Scheme 1D), but not short chain unsaturated aliphatic acids.38 This is in contrast to other ERs, as the single carboxyl functionality is thought to be too weak an activating group for OYEs.22 Therefore, these enzymes have great industrial potential to expand the biocatalytic toolbox available for enantioselective hydrogenation of carbon-carbon double bonds.
3. Individual and Cascading Biocatalytic Reactions
As more studies investigate the potential of a wide range of biocatalysts, there is an increasing trend towards one-pot multi-enzyme applications, chemoenzymatic approaches and whole cell catalysed biotransformations. As more fine chemical syntheses are beginning to exploit the stereo- and enantio-specific abilities of enzymes, chemoenzymatic approaches in particular are increasing in popularity. This section will discuss recent studies in the application of recombinant ER biocatalysts either individually, or in multiple enzyme cascades in the biosynthesis of industrially-useful synthons.
Many studies employing enzymatic biotransformations often use native or recombinant cell extracts or whole cells as the source of the enzyme(s).39 This approach was exploited in the production of the chiral building block (2R,5R)-dihydrocarvone from (R)-carvone, using recombinant E. coli expressing the ER from Nostoc sp. PCC 7120 (NoER).40 In vivo cofactor regeneration was improved by the co-expression of variant formate dehydrogenase from Mycobacterium vaccae. To minimise substrate and/or product toxicity, both in situ substrate feeding and product removal techniques were employed, the latter using either ionic liquids or hydrophobic adsorbent resins. Under optimised conditions, (2R,5R)-dihydrocarvone was obtained (97% conversion) with high stereoselectivity (97% de).40 Ionic liquids also improved the whole cell bioreduction of ethyl (E/Z)-2-acetyl-3-phenylpropenoate and derivatives to the corresponding α-benzyl-β-ketoesters using Baker’s yeast (Scheme 2A).41 Comparative reactions using aqueous and biphasic water/[(bmim)PF6] media showed the presence of the ionic liquid prevented the formation of the respective ethyl 2-benzyl-3-hydroxybutanoate due to the absence of constitutive ADH activity.41
Scheme 2.

Whole cell catalysed α,β-unsaturated alkene reduction for the production of A) α-benzyl-β-ketoesters and B) chiral γ-oxo esters.
Chiral γ-oxo esters are industrially important synthons as they are employed as precursors of bicyclic (spiro)lactams and γ-butyrolactone found in therapeutic drugs and natural products.42 Recently, a three-step biocatalytic approach to chiral cyclic 3-oxoesters from methyl and ethyl cyclopentene- and cyclohexene-carboxylates was described using alginate entrapped Rhizopus oryzae resting cells. 43 This two-stage process incorporated two biooxidation and one alkene reduction step (Scheme 2B), with the intial allylic hydroxylation step performed by R. oryzae. The latter two steps are performed sequentially using a laccase/TEMPO system for carbonyl group formation, and hydrogenation of the alkene bond by a panel of OYEs.43 Another study described a screen of the bioreduction of 18 unsaturated γ-oxo esters using 9 isolated OYEs. The optimal substrate:ER pair was determined for each γ-oxo ester to maximise yields and optical purity. In some cases, both enantiomeric products were achieved via enzyme- or substrate-based stereocontrol.42 This approach was applied to the two-step chemoenzymatic bioreduction-deprotection of benzyl 2-(6-oxocyclohex-1-en-1-yl)acetate to generate (R)-2-(2-oxocyclohexyl)acetic acid, using the OYE from Zymomonas mobilis (NCR) followed by deprotection using the H2/Pd/C catalyst. (Scheme 2B). The enzymatic bioreduction step was optimised to generate 81% conversion with high optical purity (>97% ee).42
The prostaglandin analogue Latanoprost is a valuable pharmaceutical used in controlling the progression of glaucoma.44 A chemoenzymatic approach to its synthesis has been described (Scheme 3A), utilising Pichia anomala in one-pot whole cell biocatalysis to produce the Lactondiol L precursor. This process involved three highly stereoselective steps, namely ER, esterase and ketoreductase activities, to generate high yields of Lactondiol L.44 Fungal biotransformation approaches have also been applied to the chemoselective biohydrogenation of α,β- and α,β,γ,δ-unsaturated ketones and bis-α,β-unsaturated ketones.45–46 For example, biphasic reactions with whole hyphae of Penicillium citrinum CBMAI 1186 successfully reduced the C=C bond of α,β-, di- α,β-, and mono-α,β,γ,δ-unsaturated ketones (Scheme 3B) using phosphate buffer and n-hexane (9:1).46 In addition, monophasic biotransformations of a variety of bis-α,β-unsaturated ketones were converted to the respective dihydro products, by P. citrinum and other fungal mycelia from Trichoderma sp. CBMAI 932 and Aspergillus sp. FPZSP 146 and 152.45 Up to 84% conversions were obtained, and led to the production of analogs of the natural product curcumin, known for its anti-inflammatory and antioxidant properties.45
Scheme 3.

Whole cell fungal biotransformations for the generation of A) Latanoprost precursor and B) α,β- and α, β, γ, δ-unsaturated ketones and derivatives.
Screening approaches of isolated ERs towards target compounds enables the identification of biocatalysts with higher stereo- and enantio-selectivity and increased product yields, while eliminating competing reactions commonly encountered when using whole cell biocatalysts. Often the contaminating activity in ER-containing whole cell biotransformations is ketoreduction,7 leading to formation of the respective saturated alcohol product. However, a recent example describes the production of cinnamyl alcohol from L-phenylalanine, incorporating the enzymes phenylalanine ammonia lyase, carboxylic acid reductase (CAR) and alcohol dehydrogenase.47 A significant proportion of 3-phenylpropanol was also generated, due to the constitutive E. coli ER activity on the CAR-generated cinnamaldehyde.47
The isolated enzyme screening approach was applied to the bioreduction of the 2-arylpropenoic acids and their esters in the chemoenzymatic synthesis of the pharmacologically-relevant profen class.48 Classically, the reduction of α,β-unsaturated carboxylic acids and esters by ERs is challenging, requiring the presence of a second conjugated electron withdrawing group. However, a screen of eight OYEs and one MDR showed both XenA (Pseudomonas putida) and GYE (Gluconobacter oxydans) catalysed the direct bioreduction of α,β-unsaturated carboxylic acids to generate Naproxen and analogues 2-phenylpropionic acid and 2-(4-propylphenyl)propanoic acid (Table 1 entry 1).48 Also, additional OYEs catalysed the bioreduction of 2-arylpropenoic acids methyl esters, enabling the generation of the equivalent saturated acids via base hydrolysis. Unfortunately, the non-pharmacologic (R)-enantiomeric products were obtained, but several studies have clearly established that enantiocomplementaty forms of robust variant OYEs can be generated (vide infra section on Engineered Enzymes).49–55 Therefore there is potential for OYEs to be used for (S)-profen production.
Table 1. Biocatalytic and chemo-enzymatic routes to industrially-useful compounds.
| Entry | Final product (s) | Uses | ER(s) | Ref. |
|---|---|---|---|---|
| 1 |
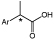 Ar = Ph, propylphenyl, 6-methoxynapthyl |
Non-steroidal anti-inflammatory drugs | XenA GYE | 48 |
| 2 |
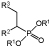 R1 = Me, Et; R2 = Me, Bn; R3 = C=O, CN, CO2R |
Potential cancer drug, antibiotics and chemical applications | OYE3 | 56 |
| 3 |
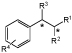 R1 = CO2H, NO2; R2 = H, CO2Et; R3 = H, Me, C=C; R4 = H, Br |
Industrially-useful synthons | 23 ER screen | 57 |
Like 2-arylpropenoic acids, vinylphosphonate derivatives were considered to be poor substrates for OYEs due to the insufficient activation by the neighbouring phosphonate group. However, the addition of an additional electron-withdrawing group (EWG) at the β-position enabled these compounds to be substrates for OYEs in the production of α- and β-functionalised phosphonate antibiotic analogues.56 In this study, a large panel of purified ERs was screened against tri-substituted α,β-unsaturated phosphonates (Table 1 entry 2). Successful bioreduction by multiple OYEs was seen with substrates containing either a carbonyl, nitrile or ester secondary β-EWG, with near optical purity but variable yields. Compounds unreactive with the OYEs were ones containing diethyl phosphonate groups, aromatic groups in position R3 (Table 1 entry 2) or a cyano group attached to the same carbon as the phosphonate group. It is thought steric hinderance was responsible for the lack of product formation in these cases. Interestingly, a deuterium labelling study of OYE3 with the phosphonate substrate containing a cyano group at R3 showed the EWG was exclusively the cyano group, supporting the assumption that the phosphoryl moiety is too weak to serve as an effective EWG for OYEs. Overall, the study found the most successful biocatalyst was OYE3 from S. cerevisiae, generating β-keto-, cyano- and ester phosphonates from the equivalent (E)-isomers of α,β-unsaturated phosphonates (≤ 72% isolated yield) with high enantiopurity (> 99% ee).56
To investigate the potential application of biocatalytic hydrogenations, a library of 23 ERs were screened for C=C reduction against 21 activated alkenes from different compound classes (Table 1 entry 3).57 The substrate scope included α,- or β-methyl, β-substituted arylnitroalkenes, α,β-unsaturated carboxylic acids or esters with or without β-substituents, cyclic α,β-unsaturated ketones and an α,β-unsaturated boronic acid. To further assess industrial potential of biohydrogenations, selected reactions were screened against 41 reaction parameters, such as the presence of chaotrophic salts, polyols, amino acids and organic solvents. Multiple preparative reactions between OYEs and selected substrates were performed under optimised conditions, demonstrating high conversions and high enantioselectivity, where applicable. For example, Gox-ER from Gluconobacter oxydans catalysed the reduction of 2-phenylacrylic acid to (R)-2-phenylpropanoic acid with an 85% yield and >99% enantiopurity in the presence of 10% PEG, 20% cyclohexane and a cofactor recycling system.
Enzymatic routes employing ERs have been shown for the sustainable production of polymer precursors. For example, a chemo-enzymatic approach to the biodegradable poly((±)-β-methyl-δ-valerolactone polymer production was described, incorporating an ER-catalysed reduction of anhydromevalonolactone.58 The generation of optically enriched (+)- and (-)-β-methyl-δ-valerolactones was achieved using the ERs YqjM variant C26D/I69T from Bacillus subtilis and OYE2 from S. cerevisiae, respectively (Scheme 4A). These products were used to generate atactic and isotactic amorphous poly((±)-β-methyl-δ-valerolactone polymers with a low softening temperature.58 Interestingly, an alternative fermentation approach to valerolactone, levulinic acid, and derivatives of both was recently patented, utilising organisms such as engineered S. cerevisiae and Pichia stipitis.59 This included a step whereby an ER was employed for the reduction of 4-hydroxy-2-oxo-pentanoic acid into levulinic acid.58
Scheme 4.

Routes to polymer precursors A) (+)-β-methyl-δ-valerolactones, B) lactones, C) adipic acid and D) 4-aminohydrocinnamic acid via chemoenzymatic and multienzyme cascade reactions.
An in vitro cascading enzymatic pathway to lactone monomers has been developed, using an alcohol dehydrogenase (Lk-ADH; Lactobacillus kefir), ER (e.g. XenB from Pseudomonas sp.) and CHMOAcineto (Acinetobacter sp.; Scheme 4B).60–61 This route leads to the formation of biorenewable polyesters, such as carvolactone, a precursor to thermoplastic polyesters.61 Later studies investigated the effect of cascade reactions containing an Lk-ADH and XenB-CHMOAcineto fusion protein on dihydrocarvone lactone formation, which found a 40% increase in product yield over reactions with the three individual enzymes.62 A similar multi-enzyme cascade to lactone formation was described employing three OYE homologues from Pseudomonas putida NCIMB 10007 (XenA, XenB and NemA) with CHMOAcineto.63
Other recent studies have applied the use of EnoRs (ER-Ca and ca2ENR, both sourced from C. acetobutylicum) in the production of adipic acid64 and 4-aminohydrocinnamic acid (4AHCA),65 which are monomers of Nylon 6 and poly(4AHCA), respectively (Scheme 4C-D). In the case of ER-Ca, other di-acid substrates were reduced, such as 2-hexenedioic acid isomers and cis, cis- muconic acid.64
The use of pheromones for European elm bark beetle (Scolytus multistriatus) control is a more sustainable and less environmentally damaging approach than the application of toxic pesticides. A one-pot cascading biosynthesis of the four stereoisomers of the pheromone 4-methylheptan-3-ol (Table 2 entry 1) from (E)-4-methylhept-4-en-3-ol has been described, utilising initially an ER followed by an ADH.66 In each step, one of two different enzyme variants was employed, which generated products of the opposite enantiomer. This enabled all 4 stereoisomers to be generated, which was advantageous given that each was active against a different species of beetle.66
Table 2. Multi-step biocatalysis and chemo-enzymatic routes to industrially-useful compounds.
| Entry | Final product (s) | Uses | ER(s) | Other enz. | Ref. |
|---|---|---|---|---|---|
| 1 |
 4-methylheptan-3-ol1 |
Insect pest management | OYE2.6 OYE1 W116V | ADH440 ADH270 | 66 |
| 2 |
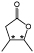 3-Methyl-4-pentanolide1 |
Fragrance industry | OYE2 | KRED READH | 67 |
| 3 |
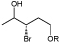 R=Ac, Bz, Bn |
Precursors of drugs, flavours, and agrochemicals | OYE3 | EVO030 EVO270 | 68 |
| 4 |
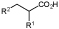 R1=Me, Ph, NHCHO; R2=Et, Ph, 4-Cl-Ph |
Cosmetics, food additives, and pharmaceuticals | OYE2 | EcAldDH | 12 |
| 5 |
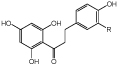 R=H, OH, OMe |
Antioxidants and flavor enhancers | sERED | CHI | 69 |
| 6 |
 Butanol |
Biofuel | YqjM | PDC, ADH | 70 |
| 7 |
 (R) and (S) forms |
Industrially-useful synthons | YqjM C26D/I69T variant | ATA | 71 |
Four stereoisomers produced.
Highlighted chiral centres. ER sources: OYE2.6 = P. stipitis; OYE1 = Saccharomyces pastorianus. Other enz = other biocatalysts utilised within the cascading reactions, excluding cofactor regenerating enzymes.
Along a similar line, the production of the four naturally occurring stereoisomers of 4,5-dimethyl substituted γ-lactones from Nicotiana tabacum has been investigated via stereoselective chemoenzymatic synthesis utilising both an ER and ADH (Table 2 entry 2).67 These fragrant molecules differ in their odour profiles dependent on their absolute stereochemistry. The 2-step reduction of the α,β-unsaturated ketoester generated 4 equivalent hydroxyesters, which was followed by trifluoroacetic acid treatment to generate the lactones. The enantiodivergent nature of the initial ER-catalysed step by OYE2 was achieved via a substrate-engineering strategy supported by computational studies to reliably generate the required enantiomeric product. In contrast, the enantiopreference of the ADH-catalysed step was determined by which enzyme homologue was used.67
The stereoselective biosynthesis of 2-methyl-3-substituted tetrahydrofurans is commercially useful due to their applications as drugs, flavours and agrochemicals.68 One synthetic route incorporates a two-enzyme cascade reduction of α-bromo-α,β-unsaturated ketones, where C=C and C=O reductions are catalysed by OYE3 and pro (R)- or pro (S)-ADHs, respectively (Table 2 entry 3). This generated the equivalent bromohydrins, which were latterly converted into tetrahydrofuran synthons using either enzymatic (lipase) or chemical isomerisation (camphor sulfonic acid) routes. This approach was applied to the chemoenzymatic synthesis of (2S,3R)-2-methyl-3-thioacetate tetrahydrofuran, an aroma chemical found in roasted meat.68 This double biocatalyst strategy (ER followed by ADH activity) overcame the poor specificity of the ADHs for unsaturated ketones, enabling stereoselective control of each step.
Similarly, another study sought to overcome the lack of OYE C=C reduction activity towards α,β-unsaturated carboxylic acids containing only one activating group by utilising a two enzyme concurrent reductive-oxidative biocatalytic cascade.12 The starting substrate was the respective α,β-unsaturated aldehyde, a class of compounds OYEs are generally highly specific towards. The final carboxylic acid product is obtained by the action of aldehyde dehydrogenases (Table 2 entry 4). This approach had the advantage of a self-sufficient hydrogen-borrowing cascade, where no external hydride source from a sacrificial substrate is needed. This process has applicability towards the production of chiral substituted hydrocinnamic acids, aliphatic acids and heterocycles, potentially with elevated yield, chemo- and stereo-selectivity.12
Flavonoids are polyphenolic compounds that have important commercial applications due to their antioxidant and flavour enhancer roles.69 The biosynthetic flavonoid pathway in the gut bacterium Eubacterium ramulus includes a chalcone isomerase (CHI) and an oxygen-sensitive EnoR. Recombinant forms of these enzymes were used in a 2-step cascade, where flavanones were isomerised to chalcones (CHI), followed by C=C reduction to the respective dihydrochalcones (EnoR; Table 2 entry 5). This process generated naringenin, eriodictyol, and homoeriodictyol with medium to high conversions within 17 h under anaerobic conditions (93, 72, 63%, respectively).69
Conventional routes to bio-butanol, a next generation biofuel and platform chemical, rely on anaerobic ABE fermentations, typically by solventogenic Clostridia strains.70 The natural biosynthetic pathway from glucose incorporates 16 enzyme activities, 5 CoA-dependent intermediates, 3 cofactors, and several ATP-dependent conversion steps. A recent study established a cell-free chemoenzymatic n-butanol producing process, involving only three enzyme activities and one amino acid catalyzed enamine–aldol condensation, eliminating the need for acetyl-CoA and employing NADH as the sole redox shuttle. One enzymatic step is catalysed by YqjM, namely the reduction of (E)-but-2-enal to butyraldehyde (Table 2 entry 6).70 This demonstrates a new commercial application of ERs in the in vitro production of bio-based fuel additives.
YqjM variant Cys26Asp/Ile69Thr was recently utilised in a two-enzyme cascade with amine transaminase (ATA-VibFlu) variant Leu56Ile from Vibrio fluvialis for the production of diastereoisomers (1R,3R)- and (1S,3R)-1-amino-3-methylcyclohexane (Table 2 entry 7).71 Multiple variants of both enzymes were screened, as all known wildtype EREDs show (S)-selectivity towards 3-methylcyclohex-2-enone, and wild-type ATA-VibFlu shows only modest enantioselectivity. The transaminase reaction was increased by the addition of the lactate dehydrogenase/glucose dehydrogenase recycling system, by the removal of pyruvate generated by alanine deamination (amine donor).71 This process led to the production of (1R,3R)-1-amino-3-methylcyclohexane with high optical purity (97% de).
4. Engineered Enzymes
Research into determining successful stereo- and enantio-specific ER-substrate pairs is not limited to biocatalytic screening of existing enzymes or the discovery of novel enzymes. Biocatalyst engineering can open up a wide range of potentially useful enzymes with increased substrate specificity, improved reaction rate, increased product stereo- and/or enantio-specificity and increased tolerance to reaction conditions, such as solvent and temperature. Recent advances in the development of engineered ERs will be discussed here, highlighting their potential commercial application.
One approach is to build on the properties of existing enzymes, by modifying them to increase product yield and/or enantio-specificity, increase the substrate scope to work with novel compounds, and modulate enzyme mechanism(s) to introduce new functionality.27 The generation of enzyme variants has become routine, and its application in determining improved functionality is often limited by the ability to perform high-throughput product analysis to detect changes in yield and/or product enantiospecificity. A recent study tackled this issue by designing a rapid, high-throughput colorimetric assay for ERs, taking advantage of the presence of the GDH/NADP+/glucose cofactor recycling system commonly incorporated in bioreductions.72 In this process, activated olefin reduction by ERs generates NAD(P)+, which requires reoxidation GDH concomitant with the consumption of glucose. A latter colorimetric FRED (fast and reliable ene-reductases detection) assay detects the levels of glucose remaining after the bioreduction stage via glucose oxidase/peroxidase reactions, leading to the formation of the quinoneimine dye, with an εmax at 500 nm. This coupled assay has great potential in the high-throughput screening of variant ER libraries and enzyme ‘fingerprinting’.72
A key aspect in the design of enzyme variants is the identification of ‘hot spots’; that is enzyme regions where catalytic functioning is likely to be altered upon residue substitution. A recent fast protein engineering strategy was applied to ER enzymes to access stereo-complementary pairs of products, where improved biocatalysts were obtained from relatively small numbers of variants.50 In this strategy, it is assumed that the impact of residue changes detected in some ER variants will have the same impact when transferred to other family members. The scaffolds chosen were representative members of the classical and thermophilic-like OYEs, and three groups of variants (C26D/I69T, C26G and W116I) were identified (‘hotspots’ 1-3 C26X, I69X and W116X, respectively) and screened with three substrates (Scheme 5A). These positions were targeted based on prior studies with OYE1, YqjM and OYE2.6, where changes in product stereochemistry was observed.49, 51–55 In general, classical OYEs tend to have bulky groups in hotspots 2-3, leading to a facial (S)-product selectivity with 3-methylcyclohex-2-en-1-one, while the thermophilic-like OYE YqjM generated the opposite enantiomer. This control of facial selectivity of YqjM was transferrable to NCR, RmER, DrER and TsER with variants C26G and C26D/I69T. This approach was successful in generating stereocomplementary pairs of products of (S)-3-methylcyclohexan-1-one, 2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-one and methyl 3-hydroxy-2-methylpropanoate. In many cases, the opposite enantiomeric products were obtained between wild-type and double variant enzymes of the same scaffold. For example, the unusual monomeric thermophilic-like RmER from Ralstonia metallidurans73 generated (2R,5S)- and (2S,5S)-2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-one from wild-type and C25G/I66T variant, respectively. Overall, the study found that hotspot positions 1 and 2 containing Asp/Thr and Gly/(Thr or wild-type), respectively, generated stereo- complementary pairs of thermophilic-like OYE variants. Additionally, incorporation of Ile into hotspot position III affected NCR than any thermophilic-like OYE. This study highlighted that systematic prediction of absolute enantio-selectivity of OYE variants remains challenging, but a scaffold sampling approach could increase the likelihood of designing variants with the desired properties from minimal library sizes.50
Scheme 5.

Generation of stereo-complementary pairs of products by engineering of ERs using A) scaffold sampling and B) site-saturated mutagenesis approaches. OYE sources: NCR = Zymomonas mobilis ; DrER = Deinococcus radiodurans ; TsER = Thermus scotoductus SA-01; RmER = Ralstonia metallidurans; OYE2.6 = P. stipitis.
An earlier site-saturated mutagenesis study of OYE 2.6 was performed to develop variants with reversed stereo-selectivities of the wild-type enzyme with three Baylis-Hillman adducts.55 Multiple rounds of mutagenesis generated double and triple mutants that possessed inverted stereoselectivities for two of the three substrates, with conversions >99% and high enantiomeric purity (91% (R) ee) compared to wild-type enzyme (>90% (S)). Crystal structures of two of the variants (Y78W and Y78W/I113C) combined with computer modeling suggested a flipped substrate binding conformation in the variants compared to the wild-type enzyme, which could explain the reversal in the product enantiomeric identity.55 These variants were later screened against a panel of 16 structurally-diverse electron-deficient alkenes, to look for changes in product yields and a reversal in product enantiomeric identity (Scheme 5B).74 For example, β,β-di-substituted 2-cyclohexenones are typically poor substrates for wild-type OYE2.6, due to steric clashes between the bulky β-substituent and the protein. Variants Y78W + changes at residue 113 led to near complete conversion of 3-methylcyclohex-2-en-1-one after 24 hours, compared to wild-type (55%). Additional variants showed a switch to generating the (R)-enantiomeric product, compared to the (S)-selective wild-type enzyme. Therefore these combined studies have generated variants of OYE2.6 with an increased substrate scope and the ability to generate stereocomplementary pairs of previously inaccessible compounds.74
The stereoselective, two-step multi-order bioreduction of ketoisophorone to (4R,6R)-actinol by ERs and (6R)-levodione reductase (LVR; Corynebacterium aquaticum M-13) generates only 67% product, due to the accumulation of (4S)-phorenol, a poor substrate for wild-type ERs (Scheme 6A).75 The OYE from Candida macedoniensis (CmOYE) was crystallised, and underwent mutagenesis to introduce point mutations into the substrate-recognition loop. The variant CmOYE P295G showed 2- and 12-fold increased catalytic activity towards both ketoisophorone and (4S)-phorenol, respectively. This enabled an efficient bioconversion to (4R,6R)-actinol in either order of the reactions, leading to an overall 90% yield of product. The study highlighted the substrate-recognition loop in OYEs as a potential hot-spot for further mutagenesis studies for increasing substrate binding.75
Scheme 6.

Multi-step enzymatic bioconversions of A) ketoisophorone to (4R,6R)-actinol and B) cinnamyl alcohol to 3-phenylpropan-1-ol.
The bioreduction of the C=C bond of cinnamyl alcohol has recently been performed in a multi-enzyme cascade composed of alcohol dehydrogenase (alcohol to aldehyde), ER (C=C reduction) and a second ADH step (aldehyde to alcohol).76 The process contained completely internal cofactor recycling, provided the ER homologue was NADH-dependent (Scheme 6B). Both OYE1 and morphinone reductase (MR) from Pseudomonas putida M10 catalysed the reduction of cinnamaldehyde, but NCR showed no activity with this substrate. To overcome this, four rationally designed β/α-loop variants based on OYE1 and MR were incoporated into NCR, resulting in successful ADH-containing cascade reactions of cinnamyl alcohol, geraniol and (S)-(-)-perillyl alcohol to their respective saturated alcohols in the presence of ADH.76 This shows another example of successful loop grafting in OYEs, resulting in the introduction of new substrate acceptance.
Another target for enzyme mutagenesis studies is the improvement of protein stability to enable their application within industrial processes. Further loop modulation variants were designed for NCR to increase its temperature and solvent stability, based on sequence and structural comparisons between mesophilic and thermophilic ERs.77 The targeted regions were two exposed flexible loop regions, which were shortened by 7 and 4 amino acids, respectively. Variants showed increased thermostability, but a loss in the conversion rate with some substrates (e.g. ketoisophorone, cinnamaldehyde and geranial). Further studies revealed one loop segment close to the active site was tolerant of extensive modifications, generating a biocatalyst with increased thermostability and solvent tolerance.77
Circular permutation is a more extensive mutagenesis strategy whereby the amino acid composition remains unchanged, but the primary sequence is reorganised by the covalent linkage of the existing termini and the introduction of new N- and C-termini through backbone cleavage elsewhere in the protein.78 This approach was taken with OYE1 where 228 circular permutants were generated, targeting several loop regions, helix α1 that partially covers the bound FMN and the central part of the (βα)-8-barrel protein.79 Interestingly, 70 variants showed equal or higher bioreduction of ketoisophorone to (R)-levodione (up to over 10-fold), with no compromise in product enantiopurity. Further biophysical and X-ray crystal structural investigations on selected variants showed termini relocation into the loop β6 region (active site lid) enhanced local flexibility and increased the environmental exposure of the active site.80 These changes did not perturb the conformation of other key active site residues with the exception of Y375. However, the additive effect of permutants with diastereoselectivity inversion mutations W116X highlighted the contribution of loop β6 towards OYE stereoselectivity, which may drive further engineering efforts.80
The FMN-containing OYE family is known for its ability to act as both an ene-reductase and to a lesser extent as a nitroreductase.22 Recently KYE1 from Kluyveromyces lactis underwent rationally designed site-directed and iterative mutagenesis to increase the native nitroreductase activity by increasing the binding site cavity volume.81 Variant F296A/Y375A showed a 100-fold increase in nitroreductase over ER activity, with a catalytic rate of 1.2 s-1 with 4-nitrobenzenesulfonamide. An additional mutation of the catalytic H191 to alanine abolished ER activity, but also significantly reduced NADPH binding. This newly improved nitroreductase has potential commercial applications in therapeutic treatments such as protein self-assembly with leucine zippers.81
5. Cofactor Regeneration
An important consideration in the application of ERs for laboratory and industrial-scale biocatalysis is the choice of a cost-effective hydride source. This is critical for industrial application of ER bioreductions as the costs of supplying high levels of NAD(P)H can be prohibitive. This section will review the range of options currently available for hydride supply, spanning effective nicotinamide coenzyme recycling, and more direct coenzyme-independent FMN reduction techniques for OYEs.
The simplest solution to provide a near inexhaustible hydride supply is to incorporate a cofactor recycling system, where NAD(P)H is continually replenished by the addition of a second NAD(P)+-dependent enzyme/substrate pair.13, 82 Commonly used cofactor recycling systems include GDH/glucose, formate dehydrogenase/formate and phosphite dehydrogenase/phosphite.82–83 A recent study investigated co-immobilisation techniques of ER with its cofactor recycling enzyme, to improve its stability and reusability in biocatalytic reactions.84 An unspecified ER and GDH were co-immobilised within cross-linked enzyme aggregates (CLEAs) and biomimetic silica (biosilicification), generating reusable biocatalysts with higher thermal and pH stability. Activity towards 4-(4-methoxyphenyl)-3-buten-2-one was increased compared to free enzyme reactions, making this approach an inexpensive solution to generating stable and reusable self-sufficient biocatalysts.84
A novel system for cofactor regeneration has been described, where three equivalents of NADH are generated using a cascade of ADH, formaldehyde dismutase (FDM) and FDH to oxidise methanol to carbon dioxide (Scheme 7A).85 This system was applied to the reduction of ketoisophorone to (R)-levodione by TsER from Thermus scotoductus, which showed near complete conversion with high enantiopurity (92% ee). However, this system is currently limited by the relatively low catalytic efficiency of the initial ADH oxidation step, leading to the requirement of relatively high methanol concentrations.85
Scheme 7.

Alternative ER-FMN reduction strategies using A) co-substrates + enzymes, B) sacrificial substrates and C) nicotinamide biomimetics.
In multi-enzyme cascade reactions, it is advantageous to choose biocatalysts that rely on the same oxidised/reduced nicotinamide cofactor. This generates a closed loop system, where the catalytic activity of one enzyme generates the coenzyme of the second biocatalyst, and vice versa. This was demonstrated in the production of the semisynthetic analgesic opiate hydromorphone from morphine, using E. coli cells expressing the NAD(H)-dependent morphine dehydrogenase and MR from Pseudomonas putida (Scheme 7A).83, 86 The lack of activity of MR towards morphine led to 90% hydromorphone yields, with no dihydromorphine by-product.86 A similar closed-loop coenzyme regeneration scheme was demonstrated with the reduction of allylic alcohols to the corresponding saturated ketone by an ADH from Thermus sp. and TsER.87
An alternative nicotinamide-independent, coupled substrate C=C bioreduction system has been developed, whereby a second sacrificial substrate is dehydrogenated, which acts as the hydrogen donor for the direct recycling of the flavin cofactor in OYEs (Scheme 7B).88 However, dehydrogenation of cyclohex-2-enone substrates generates dienones that quickly tautomerise to form the corresponding phenol, which in turn inhibits ER bioreduction activity. To overcome this, a recent study tested potential co-substrates that would form (quasi)aromatic, but non-inhibiting dehydrogenation co-products with a variety of OYEs.89 The co-substrates were grouped as substituted cyclohexen-2-ones, cyclohexanediones, ketoheterocycles and other H-donors (non-phenolic forming). Six low cost and highly potent co-substrates were identified, such as menthone which enabled CrS from Thermus scotoductus SA-01 to reduce ketoisophorone to 80% conversion.89
Nicotinamide biomimetics (mNADHs) are synthetic nicotinamide analogues of natural cofactors used by oxidoreductases as redox equivalents.90 Recently, a study was performed to compare the effect of mNADHs versus natural cofactors on the bioreduction of ketoisophorone by 12 OYEs (Scheme 7C).91 It showed that overall the ‘better than nature’ biomimetics were successful in replacing NAD(P)H in biotransformations, without having a significant influence on the overall product enantiomeric excess. An efficient mNADH recycling system was established (1 mM mNADH) using the rhodium complex [Cp*Rh(bpy)(H2O)]2+ and formate as the hydride source. Further improvements in cofactor recycling schemes will allow mNADHs to be used in catalytic amounts, increasing the cost efficiency of the reaction.91
Other studies with mNADHs have focussed on the design of alternative cofactor recycling systems, to decrease the costs associated with OYE-dependent bioreductions. For example, the NADH-oxidase from Lactobacillus pentosus (LpNox) catalyses the reduction of mNADHs with water as a by-product.92 Interestingly, free FAD also reduced mNADHs, however the by-product was the reactive oxygen species hydrogen peroxide.92 In another case, the TsER-catalysed reduction of 2-methylbut-2-enal to 2-methylbutanal with mNADH was successful by the incorporation of the recycling enzyme glucose dehydrogenase from Sulfolobus solfataricus.93 A related study investigated the reduction of 4 mNADHs by free FAD, iron-porphyrin and the water-forming NADH-oxidase from Lactobacillus pentosus, thereby increasing the range of available biomimetic recycling strategies.94
Electroenzymatic synthesis is a less well-studied biotransformation where a cofactor or co-substrate is regenerated by an electrochemical reaction in the presence of chemical mediators.95 The OYE-catalysed bioreduction of (-)-carvone to (+)-dihydrocarvone was investigated, where NADPH regeneration was performed electrochemically in the presence of the mediators [Cp*Rh(bpy)(H2O)]2+, cobalt sepulchrate or safranin T.96 High productivities and high current efficiencies (~90%) were achieved, but only under anaerobic conditions as the required cathode potentials mediator reduction are more negative than the O2 reduction potential.96
Photosynthetic organisms utilise solar energy to catalyse the reduction of carbon dioxide to carbohydrates, concomittant with the reduction of nicotinamide cofactors. Therefore, there is the potential of harnessing photosynthetic capture of ‘free’ energy to power ER-catalysed bioreductions.97 An interesting photobiocatalytic bioreduction approach was described where engineered Synechocystis sp. PCC 6803 expressing YqjM was used in the bioreduction of unsaturated cyclic ketones, such as 2-methylmaleimide.98 Enzyme expression was under the control of a light-induced promoter, and reduced nicotinamide cofactor formation was driven by the natural photosynthesis pathway. The latter was inferred by the significant decrease in 2-methylsuccinimide production in the presence of the photosynthesis inhibitor 3-(3,4-dichlorophenyl)-1,1-dimethylurea. Overall, under optimised conditions, high yields of enantiopure 2-methylsuccinimide were generated (80% yield).98 A related study described a similar approach to the YqjM-catalysed bioreduction of unsaturated ketones and lactones within Synechocystis sp. PCC 6803 whole cells, with NADPH recycling coupled to photosynthesis.99
A nicotinamide-independent route to OYE-catalysed chiral alkanes has been described, employing a photoelectrochemical (PEC) cell for enzyme-bound FMN reduction. The PEC cell consisted of a protonated graphitic carbon nitride (p-g-C3N4) and carbon nanotube hybrid (CNT/p-g-C3N4) lm cathode, with a FeOOH-deposited bismuth vanadate (FeOOH/BiVO4) photoanode.100 In this process, photoexcited electrons from FeOOH/BiVO4 are transferred to the cathode, which transfers electrons to FMNox to facilitate the enantioselective conversion of ketoisophorone to (R)-levodione by TsOYE (Scheme 8A). This suggests that biocatalytic PEC could potentially be used to generate industrially useful synthons, using water as an electron donor.
Scheme 8.

Strategies of cofactor-independent photoactivation of FMN using photosensitisers A) Rose bengal and B) ruthenium complexes.
Alternative cofactor- and PEC-free OYE-catalysed bioreductions have been reported, incorporating direct photoactivation of the FMN in the presence of photosensitisers. For example, the reduction of cinnamaldehyde to 3-phenylpropanal by TOYE from Thermoanaerobacter pseudethanolicus E39 was facilitated by direct hydride transfer to enzyme-bound FMNox after light activation of Rose bengal in the presence of the electron donor triethanolamine (TEA).101 A variety of xanthine dyes were tested, and the reactivity was significantly affected by halogen atom substitutions. Additional reactions with 2-methylcyclohexen-2-one generated high conversions (80-90%) of enantiopure (R)-2-methylcyclohexanone.101 An earlier light-driven biocatalytic study of PETNR with α,β-unsaturated aldehydes, ketones and nitroalkenes was performed using transition metal complexes as photosensitisers.102 Nicotinamide-independent flavin reduction was achieved via light activated Ru(II) or Ir(III) complexes in the presence of the electron donor (TEA) and mediator methyl viologen (Scheme 8B). In many cases, product yields and enantiopurities were at least comparable to reactions using NADPH as the hydride donor.102
6. Synthetic Biology Applications
Traditional methodologies for fine chemical biosynthesis are centered on existing technologies of one-pot single and multi-enzyme in vitro biotransformations, or fermentation of recombinant microorganisms expressing the biocatalyst(s). The application of synthetic biology techniques is rapidly becoming an attractive alternative approach towards sustainable (bio)chemical production, as recombinant organisms are engineered with all the necessary genes to generate high value products during fermentations on simple and cost-effective carbon sources.103 This section will examine recent fine chemical syntheses utilising a synthetic biology approach, where ERs catalyse one of the biocatalytic steps.
The aromatic acids 3-phenylpropionic acid (3PPA) and 3-(4-hydroxyphenyl) propionic acid (HPPA) are important commodities used in the food, pharmaceutical and chemical industries. A biosynthetic route to these compounds was devised by combining the E. coli phenylalanine pathway with the non-native enzymes tyrosine ammonia lyase (TAL) and clostridial EnoR (Scheme 9A).104 The full pathway was assembled in E. coli, which led to the efficient production of cinnamyl alcohol and HPPA, as well as the cytotoxic intermediates cinnamic acid and p-coumaric acid, respectively. Optimisation of individual enzyme expression levels led to titres of 3PPA and HPPA of 367 and 225 mg/L, respectively. Interestingly, the oxygen sensitive EnoRs were catalytically active under the microaerophilic fermentation conditions.104
Scheme 9.

Introduced pathways into host microorganisms for the production of the industrially-useful compounds A) hydrocinnamic acids from amino acids, B) 2-methyl succinic acid from pyruvate and C) (2R,5S)-carvolactone from (R)-limonene. pheA = chorismate mutase/prephenate dehydratase; tyrA = chorismate mutase/prephenate dehydrogenase; TAL = tyrosine ammonia lyase; CimA = citramalate synthase; LeuCD = 3-isopropylmalate dehydratase; CHMO = cyclohexanone monooxygenase.
Polymers based on 2-methylsuccinic acid (2-MSA) have applications as coatings, cosmetic solvents and bioplastics. A de novo pathway was designed in E. coli by combining native pyruvate and acetyl-CoA biosynthesis with the methanogenic citramalate synthase (CimA), isopropylmalate isomerase (LeuCD) and ERs YqjM or KpnER from Klebsiella pneumoniae (Scheme 9B).105 Successful production of 2-MSA in E. coli was achieved with a titre of 0.96 g/L when using KpnER. Subsequent optimisation of the cofactor regeneration, host-strain engineering and a switch to microaerophilic conditions increased the titre nearly 4-fold.47
The biosynthetic production of chiral carvolactone from limonene would be a promising step towards cost-effective bio-derived thermoplastic polyester production.61 To achieve this, a one-pot mixed culture approach was taken, with two recombinant organisms containing the genes required for the bioconversion. The first organism was recombinant P. putida S12 containing cumene dioxygenase (CumDO) from Rhodococcus opacus PWD4, which catalysed the first stage of the hydroxylation of (R)-limonene to (S)-carvone (Scheme 9C). The remaining stages were catalysed by an ADH, XenB and CHMOAcineto expressed in E. coli.60 This mixed culture approach enabled the near complete conversion of limonene from valorised waste orange peel to the monomer carvolactone, highlighting the power of cascade biocatalysis.61
Existing biosynthetic routes to the commercially important dihydrochalcone phlorizin is via precursor feeding in microbial strains engineered for flavonoid production.106 A key step in its production is the bioreduction of p-coumaroyl-CoA to p-dihydrocoumaroyl-CoA catalysed by the very long chain (VLC) enoyl-CoA reductase ScTsc13 from S. cerevisiae. This enzyme catalyses the double bond reduction in each cycle of the VLC fatty acid elongation pathway. Therefore, a biosynthetic pathway to phlorizin was constructed in S. cerevisiae, which was later extended to include the production of nothofagin (antioxidant), phlorizin (antidiabetic molecule) and naringin dihydrochalcone (sweetener) by the inclusion of additional enzymes.106 Interestingly the clostridial EnoR, described earlier in p-coumaric acid bioreduction (Scheme 9A), was also tested in place of ScTsc13 in the S. cerevisiae pathway to phlorizin. However, it was not successful, as the naringen levels generated were the same as the control construct (no ScTsc13).106
7. Mechanistic Insights
Comprehensive studies into the crystal structures and catalytic mechanism of ERs provides valuable insight into substrate discrimination of related homologues, and likely reasons behind the observed stereo- and enantio-specificity of the products. It drives rationally designed mutagenesis studies to improve catalytic functioning and even enable alternative mechanisms to be catalysed. This section will focus on recent structural and mechanistic insights, and the discovery of novel catalytic abilities, primarily within the OYE family.
The mechanism of activated C=C reduction by OYEs has been the subject of exhaustive study over the last few decades. However, additional insights into the redox-dependent substrate and cofactor interactions of XenA have been obtained by investigating the reactions using Raman spectroscopy and X-ray crystallography.107 The application of Raman spectroscopy enables the impact of reactive cofactor binding on the Michaelis-complex, rather than traditional studies using non-reactive oxidised cofactor mimics. This study revealed that substrates bind to oxidised and reduced flavin in different orientations, but only the authentic Michaelis complexes showed a rich vibrational band pattern attributed to a strong donor–acceptor complex between reduced flavin and the substrate. It is postulated that this complex likely activates the ground state of the reduced flavin, thereby accelerating the overall reaction.107
The increasing application of synthetic mNADHs for ER-catalysed bioreductions necessitates an investigation into the mechanisms of reduced biomimetic hydride transfer to the ER flavin. A recent study investigated the origins of the enhanced performance of selected mNADHs with PETNR, XenA, and TOYE by studying the isotope dependence of reaction rate as a function of temperature.108 The mNADH typically have lower activation enthalpies compared to NAD(P)H, however the higher activation entropies suggests that enzyme−mNADH complexes are more disordered than the natural complexes. A strong correlation was found between the rate constants and the temperature dependence of the kinetic isotope effect (KIE) for H/D transfer, suggesting the rate acceleration by mNADHs may be associated with enhanced donor-acceptor distance sampling, and hydride transfer is likely to proceed via quantum mechanical tunneling. Therefore, this emphasises the important contribution of donor-acceptor distance sampling in OYE-catalysed bioreductions.108
Detailed mechanistic studies with PETNR have revealed that quantum mechanical tunneling contributes to the enzymatic hydride transfer step from NAD(P)H to the FMN cofactor, and fast protein dynamics likely play a key role in catalysis.109–110 However the mechanism of preferential binding of NADPH over NADH is not clearly understood, and is likely related to fast protein dynamics. Therefore, near-complete 1H, 15N and 13C backbone resonance assignments (97%) of oxidised PETNR-FMNox were determined using heteronuclear multidimensional NMR spectroscopy to investigate the role of fast protein dynamics in catalysis.111 The NMR structural prediction was in very good agreement with the crystallographic data, providing high confidence in the assignments of the PETNR:FMNox complex. This is the first NMR structural study of an OYE apoenzyme, which will provide a basis for further structural and functional studies by NMR spectroscopy to increase understanding of the role of protein dynamics in catalysis.111
Flavins are ubiquitous natural cofactors, capable of both 1- and 2-electron chemistry, however enzyme bound forms tend to react via 2-electron mechanisms.7 However OYE1 was found to possess a single electron mechanism in the reduction of menadione to the corresponding radical anion.112 Therefore, the OYE family was targeted to determine if they could catalyse the radical-based enantioselective dehalogenation of α-bromoesters, as opposed to the traditional 2-electron biohydrogenation reaction (Scheme 10A).113 A screen of 9 OYEs with ethyl 2-bromo-2-phenylpropanoate found that all homologues possessed dehalogenation activity to varying degrees (19-89% yield). Further studies with the GluER variant Y177F (G. oxydans) showed a dramatic improvement in product enantiopurity. Mechanistic studies suggested that dehalogenation likely proceeds via electron transfer from the flavin hydroquinone (FMNHhq) to the substrate, followed by mesolytic cleavage to form an α-acyl radical with the release of the halogen moiety (Scheme 10A). The radical species abstracts a hydrogen atom from FMNsq to generate the reduced product.113 This new catalytic functionality for OYEs opens up a new potential applicability in the detoxification of halogenated organic compounds.
Scheme 10.

Insights into the catalytic mechanisms of different ER families. A) Proposed mechanism of ER-catalysed radical dehalogenation. B) Hydride-independent C=C migration vs ene-reduction catalysed by OYE2.
OYEs are known to display unusual catalytic mechanisms towards cyclic compounds, such as α,β-unsaturated cycloalkenones or lactones. For example, nicotinamide-independent dismutation reactions of sacrificial substrates are exploited to provide a hydride source for a second alkene substrate bioreduction (Scheme 7B). Additionally, the redox-neutral C=C isomerisation of α-Angelica lactone to thermodynamically more stable 3-methylfuran-2(5H)-one by OYE2 required only catalytic amounts of NADH to activate the flavin, triggering intermolecular hydride transfer from substrate endo-Cβ onto exo-Cβ through FMN (Scheme 10B).114 This latter reaction is in effect converting non-activated compounds (C=C not conjugated to the activating group) into OYE substrates. Therefore, a two-step, one enzyme method was applied for the production of γ-valerolactone from α-Angelica lactone, via the initial generation of the OYE substrate 3-methylfuran-2(5H)-one.115 This approach opens up new possibilities of OYE-catalysed bioreductions on non-activated compounds, as the same biocatalyst catalyses both the activation and bioreduction steps.
Mechanistic information about bioreductions catalysed by the flavin-independent salutaridine/menthone reductase subfamily of SDRs is limited, as there very few reports of C=C bioreductions by these ERs. A recent study investigated the origin(s) of the mechanistic discrimination between two ketoreductases and one ER from peppermint (Mentha piperita) exhibiting high sequence homology. The ER isopiperitenone reductase (IPR) catalyses the C=C reduction of isopiperitenone to cis-isopulegone, while ketoreductases (—)-menthone:(—)-menthol reductase (MMR) and (—)-menthone:(+)-neomenthol reductase (MNMR) reduce menthone and isomenthone to menthol isomers (Scheme 11).20 The identification of a catalytic residue substitution (IPR E238 to MNMR Y244) followed by residue-swapping mutagenesis and X-ray structure determination suggested this change was a likely cause of the switch from ene-reduction to ketoreduction, respectively.
Scheme 11.

Comparison of the proposed ene-reduction and ketoreduction mechanisms catalysed by the highly conserved SDR enzymes IPR and MMR, respectively.
The proposed SDR-like ene-reduction likely proceeds via an ordered sequential ternary compex medium, where NAD(P)H binds first and leaves last. The co-crystal structure of isopiperitenone-IPR shows that residue E238 positions the substrate to allow hydride attack on the C=C bond rather than the carbonyl group. Hydride transfer from NAD(P)H is likely to generate the respective enolate ion, followed by proton donation to produce the more stable enol form. Proton abstraction from the substrate then initiates carbonyl-group reformation and the formation of cis-isopulegone. This study highlighted that there is potential to develop a new group of ERs by transforming the extensive family of SDR ketoreductases into ERs by generating homologous Y244E variants, in addition to other active site modulations to improve substrate binding in a position allowing C=C reduction.115
8. Future Perspectives
The increasing acceptance of biocatalytic, chemoenzymatic and more recently synthetic biology approaches towards fine chemical synthesis has been reflected in a shift in research funding from primarily enzyme characterisation/mechanistic studies to non-natural substrate screening with commercially useful synthons. This coincides with the commercial and legislative-led drive towards more cost-effective, yet environmentally ethical routes to high value products. As the focus switches towards more sustainable manufacturing techniques, synthetic biology approaches are likely to become more prevalent as they enable the production of high value products from non-petroleum, sustainable waste feed stocks.
Asymmetric bioreduction of activated C=C bonds by ERs has been researched for many decades as the generation of up to two stereogenic centres within a product is of high value. Biocatalytic approaches towards both chiral natural and non-natural product synthesis are particularly useful as traditional asymmetric synthetic routes can be lengthy, low yielding and often requiring expensive and/or toxic catalysts. Recent developments have led to the expansion of the toolbox of available ERs, both from the exploration of their existence in alternative host organisms and the generation of libraries of variants of existing enzymes. This has broadened the available substrate scope for asymmetric bioreductions, and in some cases identified new catalytic activities with potential for exploitation. The development of alternative, non-natural approaches to reducing equivalent (‘hydride’) supply has revolutionised the application of ERs within chemoenzymatic routes to fine chemicals. It is likely that this trend towards the acceptance and industrialisation of biocatalytic approaches can only increase, and future developments will continue to find novel synthetic niches and overcome current constraints in their commercial application.
Acknowledgment
N.S.S. was an Engineering and Physical Sciences Research Council (EPSRC; EP/J020192/1) Established Career Fellow and work in the author’s laboratory is funded by the UK Engineering and Physical Sciences Research Council and the Biotechnology and Biological Sciences Research Council.
Funding Sources
Funding was provided by the Biotechnology and Biological Sciences Research Council (BBSRC) under grant BB/M017702/1 (SYNBIOCHEM).
Work in the authors laboratory is funded by the Biotechnology and Biological Sciences Research Council (BB/M017702/1; BB/K00199X/1) and the Engineering and Physical Sciences Research Council (EP/M013219/1; EP/J020192/1)
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
ORCID
Helen S. Toogood: 0000-0003-4797-0293; Nigel S. Scrutton: 0000-0002-4182-3500.
References
- (1).Blamey JM, Fischer F, Meyer H-P, Sarmiento F, Zinn M. In: Biotechnology of Microbial Enzymes. Brahmachari G, Demain A, Adrio JL, editors. Academic Press; Cambridge, MA USA: 2017. pp. 347–403. [Google Scholar]
- (2).Classen T, Pietruszka J. Complex molecules, clever solutions – Enzymatic approaches towards natural product and active agent syntheses. Bioorg Med Chem. 2017 doi: 10.1016/j.bmc.2017.06.045. In press. [DOI] [PubMed] [Google Scholar]
- (3).Honda K. In: Biotechnology of Microbial Enzymes. Brahmachari G, Demain A, Adrio JL, editors. Vol. 16. Academic Press; Cambridge, MA USA: 2017. pp. 433–450. [Google Scholar]
- (4).Kataoka M, Miyakawa T, Shimizu S, Tanokura M. Enzymes useful for chiral compound synthesis: structural biology, directed evolution, and protein engineering for industrial use. Appl Microbiol Biotechnol. 2016;100:5747–5757. doi: 10.1007/s00253-016-7603-8. [DOI] [PubMed] [Google Scholar]
- (5).Vidal LS, Kelly CL, Mordaka PM, Heap JT. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim Biophys Acta, Proteins Proteomics. 2018;1866:327–347. doi: 10.1016/j.bbapap.2017.11.005. [DOI] [PubMed] [Google Scholar]
- (6).Winkler CK, Faber K, Hall M. Biocatalytic reduction of activated CC-bonds and beyond: emerging trends. Curr Opin Chem Biol. 2017;43:97–105. doi: 10.1016/j.cbpa.2017.12.003. [DOI] [PubMed] [Google Scholar]
- (7).Toogood HS, Scrutton NS. New developments in 'ene'-reductase catalysed biological hydrogenations. Curr Opin Chem Biol. 2014;19:107–115. doi: 10.1016/j.cbpa.2014.01.019. [DOI] [PubMed] [Google Scholar]
- (8).Faber K. Biotransformations in Organic Chemistry A Textbook. Springer; Graz, Austria: 2018. pp. 31–313. [Google Scholar]
- (9).Köhler V, Turner NJ. Artificial concurrent catalytic processes involving enzymes. Chem Commun. 2014;51:450–464. doi: 10.1039/c4cc07277d. [DOI] [PubMed] [Google Scholar]
- (10).Quin MB, Wallin KK, Zhang G, Schmidt-Dannert C. Spatial organization of multienzyme biocatalytic cascades. Org Biomol Chem. 2017;15:4260–4271. doi: 10.1039/c7ob00391a. [DOI] [PubMed] [Google Scholar]
- (11).Chang MCY, Eachus RA, Trieu W, Ro D-K, Keasling JD. Engineering Escherichia coli for production of functionalized terpenoids using plant P450s. Nat Chem Biol. 2007;3:274–277. doi: 10.1038/nchembio875. [DOI] [PubMed] [Google Scholar]
- (12).Knaus T, Mutti FG, Humphreys LD, Turner NJ, Scrutton NS. Systematic methodology for the development of biocatalytic hydrogen-borrowing cascades: Application to the synthesis of chiral α-substituted carboxylic acids from α-substituted α,β-unsaturated aldehydes. Org Biomol Chem. 2015;13:223–233. doi: 10.1039/c4ob02282c. [DOI] [PubMed] [Google Scholar]
- (13).Knaus T, Toogood HS, Scrutton NS. In: Green Biocatalysis. Patel RN, editor. John Wiley & Sons, Inc; New Jersey, USA: 2016. pp. 473–488. [Google Scholar]
- (14).Muschiol J, Peters C, Oberleitner N, Mihovilovic MD, Bornscheuer UT, Rudroff F. Cascade catalysis – strategies and challenges en route to preparative synthetic biology. Chem Commun. 2015;51:5798–5811. doi: 10.1039/c4cc08752f. [DOI] [PubMed] [Google Scholar]
- (15).Scholtissek A, Tischler D, Westphal A, van Berkel W, Paul C. Old Yellow Enzyme-catalysed asymmetric hydrogenation: Linking family roots with improved catalysis. Catalysts. 2017;7:130. [Google Scholar]
- (16).Pesic M, Fernández Fueyo E, Hollmann F. Characterization of the Old Yellow Enzyme homolog from Bacillus subtilis (YqjM) ChemistrySelect. 2017;2:3866–3871. [Google Scholar]
- (17).Rohdich F, Wiese A, Feicht R, Simon H, Bacher A. Enoate reductases of Clostridia. J Biol Chem. 2001;276:5779–5787. doi: 10.1074/jbc.M008656200. [DOI] [PubMed] [Google Scholar]
- (18).Mansell DJ, Toogood HS, Waller J, Hughes JMX, Levy CW, Gardiner JM, Scrutton NS. Biocatalytic asymmetric alkene reduction: Crystal structure and characterization of a double bond reductase from Nicotiana tabacum. ACS Catalysis. 2013;3:370–379. doi: 10.1021/cs300709m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nordling E, Jörnvall H, Persson B. Medium-chain dehydrogenases/reductases (MDR) Eur J Biochem. 2002;269:4267–4276. doi: 10.1046/j.1432-1033.2002.03114.x. [DOI] [PubMed] [Google Scholar]
- (20).Lygidakis A, Karuppiah V, Hoeven R, Ní Cheallaigh A, Leys D, Gardiner JM, Toogood HS, Scrutton NS. Pinpointing a mechanistic switch between ketoreduction and “Ene” reduction in Short-Chain Dehydrogenases/Reductases. Angew Chem, Int Ed Engl. 2016;55:9596–9600. doi: 10.1002/anie.201603785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Moummou H, Kallberg Y, Tonfack LB, Persson B, van der Rest BT. The plant Short-Chain Dehydrogenase (SDR) superfamily: genome-wide inventory and diversification patterns. BMC Plant Biol. 2012;12:1–1. doi: 10.1186/1471-2229-12-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Toogood HS, Mansell D, Gardiner JM, Scrutton NS. Reduction: Enantioselective bioreduction of carbon-carbon double bonds. In: Carreira EM, Yamamoto H, editors. Comprehensive Chirality. 1 ed. Vol. 7. Elsevier Science; Oxford: 2011. pp. 216–260. [Google Scholar]
- (23).Durchschein K, Hall M, Faber K. Unusual reactions mediated by FMN-dependent ene- and nitro-reductases. Green Chemistry. 2013;15:1764–1772. [Google Scholar]
- (24).Ni Y, Hagedoorn P-L, Xu J-H, Arends IWCE, Hollmann F. Pyrococcus furiosus-mediated reduction of conjugated carboxylic acids: Towards using syngas as reductant. J Mol Catal B: Enzym. 2014;103:52–55. [Google Scholar]
- (25).DeLano WL. The PyMOL Molecular Graphics System,Version 1.2r3pre. Schrödinger, LLC; 2002. [Google Scholar]
- (26).Steinkellner G, Gruber CC, Pavkov-Keller T, Binter A, Steiner K, Winkler C, Lyskowski A, Schwamberger O, Oberer M, Schwab H, Faber K, et al. Identification of promiscuous ene-reductase activity by mining structural databases using active site constellations. Nat Commun. 2014;5:4150. doi: 10.1038/ncomms5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Amato ED, Stewart JD. Applications of protein engineering to members of the Old Yellow Enzyme family. Biotechnol Adv. 2015;33:624–631. doi: 10.1016/j.biotechadv.2015.04.011. [DOI] [PubMed] [Google Scholar]
- (28).Scholtissek A, Ullrich SR, Mühling M, Schlömann M, Paul CE, Tischler D. A thermophilic-like ene-reductase originating from an acidophilic iron oxidizer. Appl Microbiol Biotechnol. 2017;101:609–619. doi: 10.1007/s00253-016-7782-3. [DOI] [PubMed] [Google Scholar]
- (29).Riedel A, Mehnert M, Paul CE, Westphal AH, Van Berkel WJH, Tischler D. Functional characterization and stability improvement of a thermophilic-like ene-reductase from Rhodococcus opacus 1CP. Front Microbiol. 2015;6:1073. doi: 10.3389/fmicb.2015.01073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Romagnolo A, Spina F, Brenna E, Crotti M, Parmeggiani F, Varese GC. Identification of fungal ene-reductase activity by means of a functional screening. Fungal Biol. 2015;119:487–493. doi: 10.1016/j.funbio.2015.01.006. [DOI] [PubMed] [Google Scholar]
- (31).Romano D, Contente ML, Molinari F, Eberini I, Ruvutuso E, Sensi C, Amaretti A, Rossi M, Raimondi S, Recombinant S. cerevisiae expressing Old Yellow Enzymes from non-conventional yeasts: an easy system for selective reduction of activated alkenes. Microb Cell Fact. 2014;13:60. doi: 10.1186/1475-2859-13-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Romagnolo A, Spina F, Poli A, Risso S, Serito B, Crotti M, Monti D, Brenna E, Lanfranco L, Varese GC. Old Yellow Enzyme homologues in Mucor circinelloides: expression profile and biotransformation. Sci Rep. 2017;7:12093. doi: 10.1038/s41598-017-12545-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang B, Zheng L, Lin J, Wei D. Characterization of an ene-reductase from Meyerozyma guilliermondii for asymmetric bioreduction of α,β-unsaturated compounds. Biotechnol Lett. 2016;38:1527–1534. doi: 10.1007/s10529-016-2124-1. [DOI] [PubMed] [Google Scholar]
- (34).Ni Y, Yu H-L, Lin G-Q, Xu J-H. An ene reductase from Clavispora lusitaniae for asymmetric reduction of activated alkenes. Enzyme Microb Technol. 2014;56:40–45. doi: 10.1016/j.enzmictec.2013.12.016. [DOI] [PubMed] [Google Scholar]
- (35).Magallanes-Noguera C, Cecati FM, Mascotti ML, Reta GF, Agostini E, Orden AA, Kurina-Sanz M. Plant tissue cultures as sources of new ene- and ketoreductase activities. J Biotechnol. 2017;251:14–20. doi: 10.1016/j.jbiotec.2017.03.023. [DOI] [PubMed] [Google Scholar]
- (36).Wang H-B, Pei X-Q, Wu Z-L. An enoate reductase Achr-OYE4 from Achromobacter sp. JA81: characterization and application in asymmetric bioreduction of C=C bonds. Appl Microbiol Biotechnol. 2013;98:705–715. doi: 10.1007/s00253-013-4899-5. [DOI] [PubMed] [Google Scholar]
- (37).Sheng X, Yan M, Wei M. Identification and characterization of a novel Old Yellow Enzyme from Bacillus subtilis str.168. J Mol Catal B: Enzym. 2016;130:18–24. [Google Scholar]
- (38).Mordaka PM, Hall SJ, Minton N, Stephens G. Recombinant expression and characterisation of the oxygen-sensitive 2-enoate reductase from Clostridium sporogenes. Microbiology. 2017;164:122–132. doi: 10.1099/mic.0.000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Noda S, Kondo A. Recent advances in microbial production of aromatic chemicals and derivatives. Trends Biotechnol. 2017;35:785–796. doi: 10.1016/j.tibtech.2017.05.006. [DOI] [PubMed] [Google Scholar]
- (40).Castiglione K, Fu Y, Polte I, Leupold S, Meo A, Weuster-Botz D. Asymmetric whole-cell bioreduction of (R)-carvone by recombinant Escherichia coli with in situ substrate supply and product removal. Biochem Eng J. 2017;117:102–111. [Google Scholar]
- (41).de Paula BRS, Zampieri DS, Nasário FD, Rodrigues JAR, Moran PJS. Regioselectivity control of enone reduction mediated by aqueous Baker’s Yeast with addition of ionic liquid [bmim(PF6)] Biocatal Agric Biotechnol. 2017;12:166–171. [Google Scholar]
- (42).Turrini NG, Cioc RC, van der Niet DJH, Ruijter E, Orru RVA, Hall M, Faber K. Biocatalytic access to nonracemic γ-oxo esters via stereoselective reduction using ene-reductases. Green Chem. 2017;19:511–518. [Google Scholar]
- (43).Brenna E, Crotti M, Gatti FG, Monti D, Parmeggiani F, Pugliese A, Tentori F. Biocatalytic synthesis of chiral cyclic γ-oxoesters by sequential C–H hydroxylation, alcohol oxidation and alkene reduction. Green Chem. 2017;19:5122–5130. [Google Scholar]
- (44).Contente ML, Zambelli P, Galafassi S, Tamborini L, Pinto A, Conti P, Molinari F, Romano D. A new chemoenzymatic approach to the synthesis of Latanoprost and Bimatoprost. J Mol Catal B: Enzym. 2015;114:7–12. [Google Scholar]
- (45).Ferreira IM, de Vasconcellos SP, da Cruz JB, Comasseto JV, Porto ALM, Rocha LC. Hydrogenation of bis-α,β-unsaturated enones mediated by filamentous fungi. Biocatal Agric Biotechnol. 2015;4:144–149. [Google Scholar]
- (46).Ferreira IM, Meira EB, Rosset IG, Porto ALM. Chemoselective biohydrogenation of α,β- and α,β,γ,δ-unsaturated ketones by the marine-derived fungus Penicillium citrinum CBMAI 1186 in a biphasic system. J Mol Catal B: Enzym. 2015;115:59–65. [Google Scholar]
- (47).Klumbys E, Zebec Z, Weise NJ, Turner NJ, Scrutton NS. Bio-derived production of cinnamyl alcohol via a three step biocatalytic cascade and metabolic engineering. Green Chem. 2018;20:658–663. doi: 10.1039/C7GC03325G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Waller J, Toogood HS, Karuppiah V, Rattray NJW, Mansell DJ, Leys D, Gardiner JM, Fryszkowska A, Ahmed ST, Bandichhor R, Reddy GP, et al. Structural insights into the ene-reductase synthesis of profens. Org Biomol Chem. 2017;15:4440–4448. doi: 10.1039/c7ob00163k. [DOI] [PubMed] [Google Scholar]
- (49).Bougioukou DJ, Kille S, Taglieber A, Reetz MT. Directed evolution of an enantioselective enoate-reductase: Testing the utility of iterative saturation mutagenesis. Adv Synth Catal. 2009;351:3287–3305. [Google Scholar]
- (50).Nett N, Duewel S, Richter AA, Hoebenreich S. Revealing additional stereocomplementary pairs of Old Yellow Enzymes by rational transfer of engineered residues. ChemBioChem. 2017;18:685–691. doi: 10.1002/cbic.201600688. [DOI] [PubMed] [Google Scholar]
- (51).Padhi SK, Bougioukou DJ, Stewart JD. Site-saturation mutagenesis of tryptophan 116 of Saccharomyces pastorianus old yellow enzyme uncovers stereocomplementary variants. J Am Chem Soc. 2009;131:3271–3280. doi: 10.1021/ja8081389. [DOI] [PubMed] [Google Scholar]
- (52).Ruthlein E, Classen T, Dobnikar L, Scholzel M, Pietruszka J. Finding the selectivity switch – A rational approach towards stereocomplementary variants of the ene reductase YqjM. Adv Synth Catal. 2015;357:1775–1786. [Google Scholar]
- (53).Stueckler C, Winkler C, Bonnekessel M, Faber K. Asymmetric synthesis of (R)-3-hydroxy-2-methylpropanoate (‘Roche Ester’) and derivatives via biocatalytic C=C-bond reduction. Adv Synth Catal. 2010;352 [Google Scholar]
- (54).Walton AZ, Conerly WC, Pompeu Y, Sullivan B, Stewart JD. Biocatalytic reductions of Baylis-Hillman adducts. ACS Catal. 2011;1:989–993. [Google Scholar]
- (55).Walton AZ, Sullivan B, Patterson-Orazem AC, Stewart JD. Residues controlling facial selectivity in an alkene reductase and semirational alterations to create stereocomplementary variants. ACS Catal. 2014;4:2307–2318. doi: 10.1021/cs500429k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Janicki I, Kiełbasiński P, Turrini NG, Faber K, Hall M. Asymmetric bioreduction of β-activated vinylphosphonate derivatives using Ene-Reductases. Adv Synth Catal. 2017;359:4190–4196. [Google Scholar]
- (57).Reß T, Hummel W, Hanlon SP, Iding H, Gröger H. The organic–synthetic potential of recombinant Ene Reductases: Substrate-scope evaluation and process optimization. ChemCatChem. 2015;7:1302–1311. [Google Scholar]
- (58).Zhang C, Schneiderman DK, Cai T, Tai Y-S, Fox K, Zhang K. Optically active β-methyl-δ-valerolactone: Biosynthesis and polymerization. ACS Catal. 2016;4:4396–4402. [Google Scholar]
- (59).Zanghellini AL, CORP A Fermentation route for the production of levulinic acid, levulinate esters and valerolactone and derivatives thereof. 2017 Sep 28; WO2012030860A1; https://patents.google.com/patent/US20170275657A1/en. [Google Scholar]
- (60).Oberleitner N, Peters C, Rudroff F, Bornscheuer UT, Mihovilovic MD. In vitro characterization of an enzymatic redox cascade composed of an alcohol dehydrogenase, an enoate reductases and a Baeyer–Villiger monooxygenase. J Biotechnol. 2014;192:393–399. doi: 10.1016/j.jbiotec.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Oberleitner N, Ressmann AK, Bica K, Gärtner P, Fraaije MW, Bornscheuer UT, Rudroff F, Mihovilovic MD. From waste to value – direct utilization of limonene from orange peel in a biocatalytic cascade reaction towards chiral carvolactone. Green Chem. 2017;19:367–371. [Google Scholar]
- (62).Peters C, Rudroff F, Mihovilovic MD, Bornscheuer UT. Fusion proteins of an enoate reductase and a Baeyer-Villiger monooxygenase facilitate the synthesis of chiral lactones. Biol Chem. 2017;398:31–37. doi: 10.1515/hsz-2016-0150. [DOI] [PubMed] [Google Scholar]
- (63).Peters C, Kölzsch R, Kadow M, Skalden L, Rudroff F, Mihovilovic MD, Bornscheuer UT. Identification, characterization, and application of three enoate reductases from Pseudomonas putida in in vitro enzyme cascade reactions. ChemCatChem. 2014;6:1021–1027. [Google Scholar]
- (64).Joo JC, Khusnutdinova AN, Flick R, Kim T, Bornscheuer UT, Yakunin AF, Mahadevan R. Alkene hydrogenation activity of enoate reductases for an environmentally benign biosynthesis of adipic acid. Chem Sci. 2017;8:1406–1413. doi: 10.1039/c6sc02842j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kawasaki Y, Aniruddha N, Minakawa H, Masuo S, Kaneko T, Takaya N. Novel polycondensed biopolyamide generated from biomass-derived 4-aminohydrocinnamic acid. Appl Microbiol Biotechnol. 2017;102:631–639. doi: 10.1007/s00253-017-8617-6. [DOI] [PubMed] [Google Scholar]
- (66).Brenna E, Crotti M, Gatti F, Monti D, Parmeggiani F, Pugliese A. One-pot multienzymatic synthesis of the four stereoisomers of 4-methylheptan-3-ol. Molecules. 2017;22:1591. doi: 10.3390/molecules22101591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Brenna E, Gatti FG, Monti D, Parmeggiani F, Sacchetti A, Valoti J. Substrate-engineering approach to the stereoselective chemo-multienzymatic cascade synthesis of Nicotiana tabacum lactone. J Mol Catal B: Enzym. 2015;114:77–85. [Google Scholar]
- (68).Brenna E, Crotti M, Gatti FG, Marinoni L, Monti D, Quaiato S. Exploitation of a multienzymatic stereoselective cascade process in the synthesis of 2-methyl-3-substituted tetrahydrofuran precursors. ACS Catal. 2017;82:2114–2122. doi: 10.1021/acs.joc.6b02927. [DOI] [PubMed] [Google Scholar]
- (69).Gall M, Thomsen M, Peters C, Pavlidis IV, Jonczyk P, Grünert PP, Beutel S, Scheper T, Gross E, Backes M, Geißler T, et al. Enzymatic conversion of flavonoids using bacterial chalcone isomerase and enoate reductase. Angew Chem, Int Ed Engl. 2014;53:1439–1442. doi: 10.1002/anie.201306952. [DOI] [PubMed] [Google Scholar]
- (70).Reiße S, Haack M, Garbe D, Sommer B, Steffler F, Carsten J, Bohnen F, Sieber V, Brück T. In vitro bioconversion of pyruvate to n-butanol with minimized cofactor utilization. Front Bioeng Biotechnol. 2016;4:74. doi: 10.3389/fbioe.2016.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Skalen L, Peters C, Ratz L, Bornscheuer UT. Synthesis of (1R,3R)-1-amino-3-methylcyclohexane by an enzyme cascade reaction. Tetrahedron. 2016;72:7207–7211. [Google Scholar]
- (72).Forchin MC, Crotti M, Gatti FG, Parmeggiani F, Brenna E, Monti D. A rapid and high-throughput assay for the estimation of conversions of Ene-Reductase-catalysed reactions. ChemBioChem. 2015;16:1571–1573. doi: 10.1002/cbic.201500219. [DOI] [PubMed] [Google Scholar]
- (73).Opperman DJ. Structural investigation into the C-terminal extension of the ene-reductase from Ralstonia (Cupriavidus) metallidurans. Proteins, Struct, Funct, Bioinf. 2017;85:2252–2257. doi: 10.1002/prot.25372. [DOI] [PubMed] [Google Scholar]
- (74).Patterson-Orazem A, Sullivan B, Stewart JD. Pichia stipitis OYE 2.6 variants with improved catalytic efficiencies from site-saturation mutagenesis libraries. Bioorg Med Chem. 2014;22:5628–5632. doi: 10.1016/j.bmc.2014.07.001. [DOI] [PubMed] [Google Scholar]
- (75).Horita S, Kataoka M, Kitamura N, Nakagawa T, Miyakawa T, Ohtsuka J, Nagata K, Shimizu S, Tanokura M. An engineered old yellow enzyme that enables efficient synthesis of (4R,6R)-Actinol in a one-pot reduction system. ChemBioChem. 2015;16:440–445. doi: 10.1002/cbic.201402555. [DOI] [PubMed] [Google Scholar]
- (76).Reich S, Nestl BM, Hauer B. Loop-grafted Old Yellow Enzymes in the bienzymatic cascade reduction of allylic alcohols. ChemBioChem. 2016;17:561–565. doi: 10.1002/cbic.201500604. [DOI] [PubMed] [Google Scholar]
- (77).Reich S, Kress N, Nestl BM, Hauer B. Variations in the stability of NCR ene reductase by rational enzyme loop modulation. J Struct Biol. 2014;185:228–233. doi: 10.1016/j.jsb.2013.04.004. [DOI] [PubMed] [Google Scholar]
- (78).Yu Y, Lutz S. Circular permutation: A different way to engineer enzyme structure and function. Trends Biotechnol. 2011;29:18–25. doi: 10.1016/j.tibtech.2010.10.004. [DOI] [PubMed] [Google Scholar]
- (79).Daugherty AB, Govindarajan S, Lutz SJ. Improved Biocatalysts from a Synthetic Circular Permutation Library of the Flavin-Dependent Oxidoreductase Old Yellow Enzyme. J Am Chem Soc. 2013;135:14425–14432. doi: 10.1021/ja4074886. [DOI] [PubMed] [Google Scholar]
- (80).Daugherty AB, Horton JR, Cheng X, Lutz S. Structural and functional consequences of circular permutation on the active site of Old Yellow Enzyme. ACS Catal. 2015;5:892–899. doi: 10.1021/cs501702k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Park JT, Gómez Ramos LM, Bommarius AS. Engineering towards nitroreductase functionality in ene-reductase scaffolds. ChemBioChem. 2015;16:811–818. doi: 10.1002/cbic.201402667. [DOI] [PubMed] [Google Scholar]
- (82).Toogood HS, Knaus T, Scrutton NS. Alternative hydride sources for Ene-Reductases: Current trends. ChemCatChem. 2014;6:951–954. [Google Scholar]
- (83).Hummel W, Groger H. Strategies for regeneration of nicotinamide coenzymes emphasizing self-sufficient closed-loop recycling systems. J Biotechnol. 2014;191:22–31. doi: 10.1016/j.jbiotec.2014.07.449. [DOI] [PubMed] [Google Scholar]
- (84).Li H, Xiao W, Xie P, Zheng L. Co-immobilization of enoate reductase with a cofactor-recycling partner enzyme. Enzyme Microb Technol. 2018;109:66–73. doi: 10.1016/j.enzmictec.2017.09.013. [DOI] [PubMed] [Google Scholar]
- (85).Kara S, Schrittwieser JH, Gargiulo S, Ni Y, Yanase H, Opperman DJ, Van Berkel WJH, Hollmann F. Complete enzymatic oxidation of methanol to carbon dioxide: Towards more eco-efficient regeneration systems for reduced nicotinamide cofactors. Adv Synth Catal. 2015;357:1687–1691. [Google Scholar]
- (86).Boonstra B, Rathbone DA, Bruce NC. Engineering novel biocatalytic routes for production of semisynthetic opiate drugs. Biomol Eng. 2001;18:41–47. doi: 10.1016/s1389-0344(01)00084-3. [DOI] [PubMed] [Google Scholar]
- (87).Gargiulo S, Opperman DJ, Hanefeld U, Arends IWCE, Hollmann F. A biocatalytic redox isomerisation. Chem Commun. 2012;48:6630–6632. doi: 10.1039/c2cc31947k. [DOI] [PubMed] [Google Scholar]
- (88).Stueckler C, Reiter TC, Baudendistel N, Faber K. Nicotinamide-independent asymmetric bioreduction of CC-bonds via disproportination of enones catalyzed by enoate reductases. Tetrahedron. 2010;66:663–667. doi: 10.1016/j.tet.2009.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Winkler CK, Clay D, Entner M, Plank M, Faber K. NAD(P)H-Independent asymmetric CC bond reduction catalyzed by Ene Reductases by using artificial co-substrates as the hydrogen donor. Chem - Eur J. 2014;20:1403–1409. doi: 10.1002/chem.201303897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Paul CE, Hollmann F. A survey of synthetic nicotinamide cofactors in enzymatic processes. Appl Microbiol Biotechnol. 2016;100:4773–4778. doi: 10.1007/s00253-016-7500-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Knaus T, Paul CE, Levy CW, de Vries S, Mutti FG, Hollmann F, Scrutton NS. Better than Nature: Nicotinamide biomimetics that outperform natural coenzymes. J Am Chem Soc. 2016;138:1033–1039. doi: 10.1021/jacs.5b12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Nowak C, Beer B, Pick A, Roth T, Lommes P, Sieber V. A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front Microbiol. 2015;6:957. doi: 10.3389/fmicb.2015.00957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Nowak C, Pick A, Lommes P, Sieber V. Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: Providing a regeneration system for artificial cofactors. ACS Catal. 2017;7:5202–5208. [Google Scholar]
- (94).Nowak C, Pick A, Csepei LI, Sieber V. Characterization of biomimetic cofactors according to stability, redox potentials, and enzymatic conversion by NADH oxidase from Lactobacillus pentosus . ChemBioChem. 2017;18:1944–1949. doi: 10.1002/cbic.201700258. [DOI] [PubMed] [Google Scholar]
- (95).Schmitz LM, Rosenthal K, Lütz S. In: Adv Biochem Eng/Biotechnol. Scheper Th, Belkin S, Bley Th, Bohlmann J, Gu MB, Hu W-S, Mattiasson B, Nielsen J, Seitz H, Ulber R, Zeng A-P, et al., editors. Springer; Berlin, Germany: 2017. pp. 1–48. [Google Scholar]
- (96).Tosstorff A, Kroner C, Opperman DJ, Hollmann F, Holtmann D. Towards electroenzymatic processes involving old yellow enzymes and mediated cofactor regeneration. Eng Life Sci. 2017;17:71–76. doi: 10.1002/elsc.201600158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Lee SH, Choi DS, Kuk SK, Park CB. Photobiocatalysis: Activating redox enzymes by direct or indirect transfer of photoinduced electrons. Angew Chem Int Ed Engl. 2017 doi: 10.1002/anie.201710070. In Press. [DOI] [PubMed] [Google Scholar]
- (98).Köninger K, Gómez Baraibar Á, Mügge C, Paul CE, Hollmann F, Nowaczyk MM, Kourist R. Recombinant Cyanobacteria for the asymmetric reduction of C=C bonds fueled by the biocatalytic oxidation of water. Angew Chem Int Ed Engl. 2016;55:5582–5585. doi: 10.1002/anie.201601200. [DOI] [PubMed] [Google Scholar]
- (99).List B, Gatzenmeier T. Photobiocatalytic reduction of α,β-unsaturated enones and amides. Synfacts. 2016;12:0745–0745. [Google Scholar]
- (100).Son EJ, Lee SH, Kuk SK, Pesic M, Choi DS, Ko JW, Kim K, Hollmann F, Park CB. Carbon nanotube-graphitic carbon nitride hybrid films for flavoenzyme-catalyzed photoelectrochemical cells. Adv Funct Mater. 2017;1705232:1–9. [Google Scholar]
- (101).Lee SH, Choi DS, Pesic M, Lee YW, Paul CE, Hollmann F, Park CB. Cofactor-Free, Direct photoactivation of enoate reductases for the asymmetric reduction of C=C bonds. Angew Chem Int Ed Engl. 2017;56:8681–8685. doi: 10.1002/anie.201702461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Peers MK, Toogood HS, Heyes DJ, Mansell D, Coe BJ, Scrutton NS. Light-driven biocatalytic reduction of α,β-unsaturated compounds by ene reductases employing transition metal complexes as photosensitizers. Catal Sci Technol. 2016;6:169–177. doi: 10.1039/c5cy01642h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Chang MCY, Keasling JD. Production of isoprenoid pharmaceuticals by engineered microbes. Nat Chem Biol. 2006;2:674–681. doi: 10.1038/nchembio836. [DOI] [PubMed] [Google Scholar]
- (104).Sun J, Lin Y, Shen X, Jain R, Sun X, Yuan Q, Yan Y. Aerobic biosynthesis of hydrocinnamic acids in Escherichia coli with a strictly oxygen-sensitive enoate reductase. Metab Eng. 2016;35:75–82. doi: 10.1016/j.ymben.2016.02.002. [DOI] [PubMed] [Google Scholar]
- (105).Wang J, Yang Y, Zhang R, Shen X, Chen Z, Wang J, Yuan Q, Yan Y. Microbial production of branched-chain dicarboxylate 2-methylsuccinic acid via enoate reductase-mediated bioreduction. Metab Eng. 2017;45:1–10. doi: 10.1016/j.ymben.2017.11.007. [DOI] [PubMed] [Google Scholar]
- (106).Eichenberger M, Lehka BJ, Folly C, Fischer D, Martens S, Simon E, Naesby M. Metabolic engineering of Saccharomyces cerevisiae for de novo production of dihydrochalcones with known antioxidant, antidiabetic, and sweet tasting properties. Metab Eng. 2017;39:80–89. doi: 10.1016/j.ymben.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Werther T, Wahlefeld S, Salewski J, Kuhlmann U, Zebger I, Hildebrandt P, Dobbek H. Redox-dependent substrate-cofactor interactions in the Michaelis-complex of a flavin-dependent oxidoreductase. Nat Commun. 2017;8 16084. [Google Scholar]
- (108).Geddes A, Paul CE, Hay S, Hollmann F, Scrutton NS. Donor-acceptor distance sampling enhances the performance of “Better than Nature” nicotinamide coenzyme biomimetics. ACS Catal. 2016;138:11089–11092. doi: 10.1021/jacs.6b05625. [DOI] [PubMed] [Google Scholar]
- (109).Pudney CR, Guerriero A, Baxter NJ, Johannissen LO, Waltho JP, Hay S. Scrutton, Fast protein motions are coupled to enzyme H-transfer reactions. J Am Chem Soc. 2013;135:2512–2517. doi: 10.1021/ja311277k. [DOI] [PubMed] [Google Scholar]
- (110).Longbotham JE, Hardman SJO, Görlich S, Scrutton NS, Hay S. Untangling heavy protein and cofactor isotope e ects on enzyme-catalyzed hydride transfer. J Am Chem Soc. 2016;138:13693–13699. doi: 10.1021/jacs.6b07852. [DOI] [PubMed] [Google Scholar]
- (111).Iorgu AI, Baxter NJ, Cliff MJ, Waltho JP, Hay S, Scrutton NS. 1H, 15N and 13C backbone resonance assignments of pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2. Biomol NMR Assignments. 2018 doi: 10.1007/s12104-017-9791-2. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Yamano T, Kuroda K, Fujii S, Miura R. Characterization of the electron acceptors of Old Yellow Enzyme: Mechanistic approach to the mode of one electron transfer from the enzyme to menadione or dyestuffs. J Biochem. 1993;114:879–884. doi: 10.1093/oxfordjournals.jbchem.a124271. [DOI] [PubMed] [Google Scholar]
- (113).Sandoval BA, Meichan AJ, Hyster TK. Enantioselective hydrogen atom transfer: Discovery of catalytic promiscuity in flavin-dependent ‘Ene’-Reductases. ACS Catal. 2017;139:11313–11316. doi: 10.1021/jacs.7b05468. [DOI] [PubMed] [Google Scholar]
- (114).Durchschein K, Wallner S, Macheroux P, Zangger K, Fabian WMF, Faber K. Unusual C=C bond isomerization of an α,β-unsaturated γ-butyrolactone catalysed by flavoproteins from the Old Yellow Enzyme family. ChemBioChem. 2012;13:2346–2351. doi: 10.1002/cbic.201200475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Turrini NG, Eger E, Reiter TC, Faber K, Hall M. Sequential enzymatic conversion of α-angelica lactone to γ-valerolactone through hydride-independent C=C bond isomerization. ChemSusChem. 2016;9:3393–3396. doi: 10.1002/cssc.201601363. [DOI] [PMC free article] [PubMed] [Google Scholar]