

Abstract
Cells are a basic functional and structural unit of living organisms. Both unicellular communities and multicellular species produce an astonishing chemical diversity, enabling a wide range of divergent functions, yet each cell shares numerous aspects that are common to all living organisms. While there are many approaches for studying this chemical diversity, only a few are non-targeted and capable of analyzing hundreds of different chemicals at cellular resolution. Here, we review the non-targeted approaches used to perform comprehensive chemical analyses, provide chemical imaging information, or obtain high-throughput single cell profiling data. Single cell measurement capabilities are rapidly increasing in terms of throughput, limits of detection, and completeness of the chemical analyses; these improvements enable their application to understand ever more complex physiological phenomena such as learning, memory, and behavior.
Keywords: Analytical Methods, Bioanalysis, Imaging, Mass Spectrometry, Single Cell Analysis
Graphical Abstract:
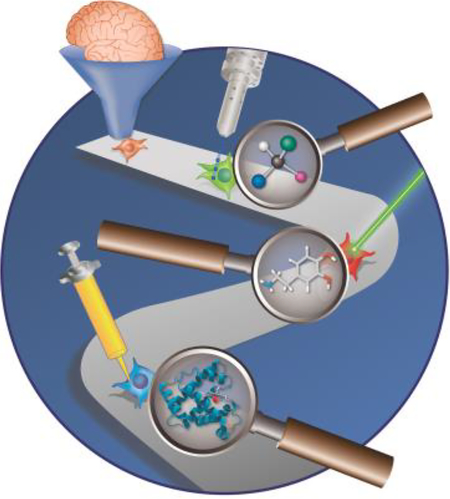
Cellular chemical heterogeneity enables divergent functions within unicellular communities and multicellular organisms. This review covers non-targeted techniques capable of identifying hundreds of chemicals at cellular resolution to begin understanding complex physiological phenomena, such as memory and cognition. We end the discussion with instrumentation improvements and multimodal approaches that enhance the chemical coverage of a single cell.
1. Introduction
Cell theory, first put forward in 1839 by Schleiden and Schwann,[1] originated from the concept that the cell is the basic structural and functional unit of all living organisms. Each cell is a marvel of detailed and complex architecture, with its history reflected in an inherited morphology. After observing the division of red blood cells in chicken embryos, Raspail and Remak[2] contributed an important tenet to cell theory with the knowledge that new cells are formed from pre-existing cells. The phrase “omnis cellula e cellula” − each cell stems from another cell – was popularized by Virchow,[3] and is a more precise version of Pasteur’s terse statement on biogenesis “omne vivum ex vivo” – all life is from life. A collection of individual cells may communicate, multiply, and differentiate to form a tissue. The large range of functions of individual cells, whether in complex ecological communities or within multicellular organisms, requires an astonishing chemical diversity. Chemical cues in the surrounding microenvironment govern cell division, morphogenesis, and aging, and have the ability to alter the phenotypes of even genetically identical cells. Although the concept that disease involves changes in normal cells was proposed by Virchow in the 1850s,[4] past technologies limited most research and clinical practices to bulk analyses of macroscopically homogeneous cell populations. Cell classification based on physical traits (e.g., size, shape, color, etc.) may be insufficient to unravel the complex nature of the chemical information obtained. Bulk measurement generates a population-averaged profile that hinders the investigation and identification of low-abundance cell subtypes that are implicated in disease etiology, progression, and exacerbation. In addition, extracting analytes from a homogenized sample dilutes analytes derived from rare cells while increasing the chemical complexity of the mixture. As such, the shift from bulk to single cell analysis is inevitable.
Given the importance of single cell chemical measurements, why are they rare? Efforts to measure the chemical contents of individual cells must overcome the challenges of sampling and characterizing the small quantity of chemically diverse analytes found within single cells. Due to their small volume, a technique with a femtomole limit of detection used to analyze a 1,000-μm3 cell (one picoliter; approximately the size of a typical mammalian cell) is only capable of detecting compounds at millimolar concentrations or above, which may be insufficient for most analytes. Other limitations include the ability to isolate single cells from tissues or cell cultures, the stability of cells, and difficult cell manipulations, which often lead to experimental artifacts.[5]
The nature of cellular heterogeneity creates additional challenges for single cell measurements. In contrast to the investigation of more widespread cells, targeting uncommon phenotypes requires the acquisition of larger samples to ensure a rare event with statistical confidence. Factors such as the ratio of cells to debris in the sample, the signal-to-noise ratio of detected signals to background, the frequency of rare cellular events, and the sensitivity of the instrument should all be considered. The number of total measurements depends on the desired standard error of the mean and the predicted frequency of the rare events occurring. Rare events can vary drastically from a few percent to one in several billion, as in the case of circulating tumor cells in the bloodstream of a metastatic cancer patient.[6] Researchers have devised clever approaches to overcome the challenge of finding rare cells, which include combining cell isolation/sorting schemes with analyses at single cell resolution. However, such schemes require a priori knowledge about the cells under investigation, and hence are limited to targeted analyses. Nevertheless, single cell analysis plays an essential role in modern cell biology, including measuring diversity within a population, identifying rare subpopulations, tracing cell lineages and phenotypes during normal development or disease stages, and discovering new cell types.[7]
Although a few earlier examples have been reported,[8] the current era of single cell chemical analysis began more than twenty years ago with several advances. For example, Jorgenson and Kennedy[9] used capillary separations to profile amino acids and neurotransmitters from three different neurons of the land snail Helix aspersa. Wightman and co-workers[10] monitored the secretion of catecholamines from single bovine chromaffin cells with a carbon-fiber microelectrode, and the Ewing group[11] estimated the free dopamine in the cytoplasm of the dopamine cell of Planorbis using voltammetry and capillary electrophoresis (CE). In 1992, Eberwine[12] performed more comprehensive single cell analysis and demonstrated that the molecular profile of a single, potentiated CA1 neuron depends on the abundance of multiple RNAs. These early examples highlighted the need for single cell studies by establishing previously unknown molecular variation within individual cells.
The links between genotype and phenotype are a consequence of the intricate pathways between gene transcription, mRNA translation, and protein-level regulation. Alone, the cellular genome does not fully explain the complexity and dynamics of the cellular peptidome and metabolome, which are important for intracellular function and intercellular communication. Therefore, while the genome and transcriptome continue to provide insights on cell function, single cell metabolomics and peptidomics (SCMP) measurement strategies are required to illuminate the identity, dynamics, and functions of key molecules and relate them to biological and physiological processes.
Although SCMP approaches have contributed to numerous advancements,[13] here we limit the scope of our discussion to non-targeted techniques, and several recent reviews complement this discussion with different focuses, such as neurometabolomics.[8, 14]As each cell is a small-volume, mass-limited sample, bioanalytical techniques have been downscaled and hyphenated to improve detection and characterization capabilities. The diverse repertoire of cells poses a tremendous challenge to obtaining distinct types of information associated with molecular processes that dictate cell-fate decisions. Matching biological questions with an appropriate SCMP technique requires balancing their sampling requirements and the incomplete datasets they generate (as no approach provides details on a majority of the chemicals present within each assayed cell). As illustrated in Figure 1, we categorize the approaches based on their ability to deeply catalog the contents of a few cells, assess the native anatomical context of cellular analytes via chemical imaging, and measure dissociated cells at high(er) throughput. In the first case, the complexity of biological samples warrants the leading role of separation techniques in a “divide and conquer” scheme to maximize the chemical information gained from each measurement. With slower analyses, fractionation coupled to mass spectrometry (MS) is practically limited to smaller numbers of cells. Because of this, the cells must be chosen carefully, further limiting the use of MS as a population-discovery technique. Anatomical information is maintained with chemical imaging approaches as they can map the two-dimensional (2D) or three-dimensional (3D) spatial distribution of biomolecules. However, without a separation, they tend to detect more abundant and easily ionizable compounds. Finally, cells that are isolated from tissue first may be analyzed faster and more completely, but often at the cost of the original cellular context. Profiling measurements performed at high throughput increase the odds of observing minor phenotypic differences and detecting rare cells. When cells are isolated from each other before measurements, there are relaxed constraints on the sampling probe, allowing greater flexibility in solvent extraction protocols and matrix-assisted ionization modalities.
Figure 1.

Analytical techniques for single cell analysis discussed in the review. (Top) Fractionation techniques include capillary electrophoresis combined with different detection methods. (Bottom) Cells are directly probed for their contents using a variety of ionization methods that offer a range of capabilities and performance specifications.
The outlook that native context, increased metabolic coverage, and assay speed are dependent and often mutually exclusive may not appear encouraging. However, researchers are overcoming limitations by improving instrumentation or coupling multiple analytical methods. We largely focus on recently developed MS and MS hyphenated approaches that have pushed the limits of both the breath and the depth of cellular analysis.
To an extent, improving chemical coverage, maintaining spatial integrity, and increasing throughput are active research areas. Moreover, single cell analysis results at times deviate from bulk analyses; as examples, glutamate and glutathione, which are abundant in bulk MS measures but are detected in lower abundance by single cell MS measurements. This may be because some compounds are released or degraded during cell isolation, or because compounds located extracellularly at high levels will not be detected from individual isolated cells.[14a, 15] After centuries of scientific investigation, we are linking the fundamental connections between individual cells through transport, breakdown, and formation of molecules to the emergent functions of memory, learning, and behavior. In what follows, we outline the sample preparation approaches followed by the lower throughput comprehensive approaches, chemical imaging, and finally, the high(er) throughput cell profiling techniques. As modern single cell analysis offers a glimpse at exploring biological systems at unprecedented resolution, we end our discussion on the outlook of non-targeted SCMP and its applications to biological research.
2. Sample Preparation for Single Cell Analysis
Successful single cell measurements require well-designed sample preparation protocols to ensure meaningful results. Cells from the same tissue region may require different isolation techniques. The optimal sample treatment must be selected for the analyte of interest and will determine both the methods available for analysis and which analytes will be preserved.[16] In general, single cell analyses with non-targeted SCMP techniques are performed either at the tissue level using technologies that provide subcellular spatial resolution, or on isolated cells using methods that improve molecular characterization, but at the expense of spatial information. For subcellular imaging, the integrity and spatial organization of tissues must be maintained during the collection, storage, and treatment procedures prior to sectioning. A recent review by Chughtai and Heeren[17] listed common biological sample handling protocols for MS analysis, with an emphasis on sample storage, that minimize tissue deformation and degradation. For dissociated single cell samples, protocols for whole tissue digestion should be optimized to maintain the viability of fragile cells while removing connective matrices that may hinder the study of individual cells. The suitability of treatment options is often evaluated on a per-case basis, with attention to maintaining endogenous distributions of analytes.
Even though direct tissue analysis places a stringent requirement on imaging probes to provide subcellular spatial resolution, recent advances have focused on sampling dissociated single cells at the expense of native spatial information. Manual cell isolation remains effective when examining a limited number of cells. Single cells with known features, morphologies, or precise anatomical positions can be isolated and then placed manually or seeded randomly on the analysis surface. For example, the well-annotated central nervous system of Aplysia californica enables manual isolation with sharp tungsten needles after enzymatically degrading the connective tissues. After years of morphological, electrophysiological and biochemical research, neurons in many clusters have been characterized.[18] Unfortunately, many mammalian cell types are challenging to identify without the aid of staining techniques, such as immunohistochemistry, which are incompatible with most MS techniques.
On the other hand, enzymatic treatments are applicable to tissue types where some cell loss during isolation is not a major concern. For example, neurons can be dissociated and cultured in a few hours from dorsal root ganglia (DRG), and remain alive and growing for weeks. In general, enzymatic dissociation increases throughput and mitigates the expertise required to perform manual isolation for cell systems that can withstand more rigorous treatments. Each dissociation protocol involves the combination of proteases (e.g., collagenase, papain, trypsin, etc.), inert proteins (e.g., bovine serum albumin), balanced salt solutions, and other added ingredients to preserve or stabilize cell contents. Once dissociated, cells can be cultured, sorted with microfluidic devices, or simply deposited on a substrate for MS analysis. A recent review by Hosic et al.[19] is dedicated to microfluidic sample preparation for single cell analysis. In many cases, the use of cell-sorting devices is optional, as recent methodologies make it possible for thousands of dispersed cells and non-cellular targets to be assayed in one experiment, as discussed in the following sections.
3. Comprehensive Multimodal Chemical Analysis
Separation techniques have historically played a key role in both preparative and analytical cellular level studies as they ensure the necessary selectivity and chemical purity as well as reduce isobaric interferences. The retention time within a chromatographic column provides a metric for compound identification. Following fractionation, several detection modalities other than MS are available, including electrochemistry, ultraviolet–visible and laser-induced fluorescence (LIF) spectroscopies, among others. LIF generally requires derivatization to attach a fluorophore, unless the analyte of interest is intrinsically fluorescent. Fluorescence detection has the lowest limits of detection, often less than a hundred molecules.[20] Electrochemical detection provides attomole detection limits[21] without derivatization and can be miniaturized without sensitivity losses.[22] Nonetheless, de novo identification of chemical analytes via optical or electrochemical methods is difficult due to the lack of chemical resolution.[22]
Although fundamental chemical information from individual cells has been obtained by single cell liquid chromatography (LC),[23] most recent single cell separation experiments are performed with miniaturized CE systems that require only nL to pL quantities of solution and are suitable for analyses of whole cells, fractions of cells, or cellular extracts.[24] CE separates compounds based on differences in their electrophoretic mobility, which depends upon the number of charges, size, and shape of each compound.[25] A whole cell or portion of a cell can be manually injected onto the column using a microscope and micromanipulator with subsequent injection into the capillary using pressure or electroosmotic flow and lysis. Alternatively, analytes are extracted from the cell and injected directly into the capillary.[26]
CE analysis of cultured cells is especially challenging as cells can adhere to culture substrates, making them difficult to remove.[24] Mechanical or enzymatic removal of cells may lead to deformation and release of important compounds. The Allbritton laboratory[27] circumvented the challenges of mechanical isolation with a pulsed laser lysis system, allowing immediate capillary loading that reduces changes to cell physiology. An alternative, rapid lysis scheme utilizes an electrode to induce a 30 V potential difference across the cell membrane.[28] To improve the throughput of single cell CE, many laboratories have coupled microfluidic devices to the front end of a CE system.[29] For example, a microfluidic device was used for cell lysis and CE separation before flowing through an electrospray ionization (ESI) emitter into the mass spectrometer, increasing the analysis rate to 12 cells per minute.[29b]
MS remains an effective choice for non-targeted analysis because of its mass selectivity, robustness, sensitivity and capability to identify unknown structures. MS is further enhanced when combined with separations for multimodal analysis. Solvent-assisted ionization methods, such as ESI, help preserve the separation efficiency of capillary electrophoresis (CE) while achieving sensitive MS analysis.[30] Non-targeted CE-MS analysis has been successfully used to probe the metabolic contents in a variety of biological systems, such as neurons,[31] cancer cells,[32] plants,[33] and red blood cells.[34] CE-LIF and CE-ESI-MS of individual cells remain active areas of research, with samples ranging from developing embryos within the species Xenopus laevis[35] to human stomach cancer cells.[36] Recently, the Nemes group[37] quantified proteins using bottom-up CE-ESI-MS and detected over 438 non-redundant protein groups during different developmental stages of the X. laevis embryo. Further, they studied the metabolic contents within certain blastomeres that contribute to cell differentiation into neuronal, epidermal, and hindgut tissues within 16-cell X. laevis embryos. Overall, 80 metabolites were detected and used to develop predictive metabolic profiles for undifferentiated cells. This same group further investigated temporal changes by utilizing live-cell CE-MS to probe metabolic differences between the dorsal and ventral side of X. laevis embryos at eight- and sixteen-cell stages. Metabolic differentiation occurred as early as eight cells, which is earlier than originally predicted.[37]
Other metabolic investigations include quantitation of mono-, di-, and tri-nucleotides, along with other energy-related anionic molecules within single Aplysia R2 neurons. The energy balance within single cells from such measurements was found to be comparable to that obtained by bulk sampling.[38] CE is applicable to the separation and quantitation of isobaric metabolites, namely l- and d-amino acids. The Sweedler group incorporated sample stacking with single cell CE-LIF to allow chiral separations of the d- and l-forms of aspartate and glutamate within individual Aplysia neurons.[39] The separation power and large dynamic range of CE allowed statistically distinct levels of the d-amino acids to be quantitated within different cell types. CE-LIF has also been used to study proteins within individual cancer cells, discovering heterogeneity within the expression of cysteine cathepsins.[40]
While single cells are the smallest functional unit of life, subcellular organelles actively manage and participate in hierarchical organization, expression, and intercellular communication. Within a decade of the first single cell analysis, CE progressed to enable investigation of structures as small as single vesicles. As early as 1998, Lundqvist et al.[41] extracted single, secretory vesicles from the atrial gland of A. californica and utilized CE-LIF to detect abundant amounts of taurine, supporting the role of taurine as a neuromodulator or hormone. The Allbritton group[42] later applied their laser system to selectively disrupt a portion of a cell, allowing selected CE-LIF analysis of specific portions of cell membranes and processes for localized detection of chemicals. Another strategy for subcellular analysis utilized on-column treatment of cell membranes with digitonin to selectively remove nuclei for CE-LIF analysis.[43] The combination of trypsin and digitonin treatment allows on-column isolation of mitochondria for CE quantification.[44]
Single cell CE analyses have successfully targeted diverse biochemical compounds from an eclectic collection of organisms. CE will continue to benefit from improvements in sampling throughput, as well as the sensitivity of the various detection modalities. An exciting prospect for single cell CE is the repeated analysis of living single cells to monitor biological phenomena such as cell development or reactions to stimuli. In 2017, Nemes and colleagues[35a] used CE-ESI-MS to study the metabolic changes in the developing X. laevis embryo. They probed the V1R cell twice and obtained results comparable to a CE-MS analysis of the dissected cell (Figure 2A). Of note, they monitored metabolic changes during formation of a neural cell lineage (Figure 2B) and by continually analyzing the progenitor cell, detected 100 molecular features that displayed significant changes. For instance, daughter cells contained decreasing amounts of aspartate and increasing amounts of GABA. Coupling multiple techniques improves the overall chemical information obtained and is another exciting avenue for probing live cells. The Sweedler group[45] performed CE-MS analysis on thalamic neurons after patch clamp electrophysiology was used to characterize the electrical activity of selected cells. Figure 2C displays the cell recordings and metabolic profile from a ventral basal thalamocortical neuron as compared to the thalamic reticular nucleus shown in Figure 2D. Over 100 different metabolites were detected within the samples. Analysis of living single cells allows for dynamic chemical analysis, which will certainly provide a better understanding of many metabolic processes, particularly in disease states or developmental stages.
Figure 2.

Several examples of single cell CE-MS approaches highlighting the chemical information obtained. (A) Comparison between CE electropherograms of the V1R cell dissected and probed twice consecutively. Similar metabolites were detected using either approach. (B) Significant changes in GABA and aspartate were detected between the progenitor cell and daughter cells, while creatine was not determined to be significantly different. A and B are adapted with permission from Ref. [35a], Onjiko et al., Anal. Chem. 2017, 89, 7069–7076. Copyright 2017 American Chemical Society. (C and D) Patch clamp profiles of two different neurons. (C) Ventral basal thalamocortical and (D) thalamic reticular nucleus, as well as metabolic CE electropherograms of each cell. The authors detected ornithine, GABA, glycine, serine, tryptophan, glutamine, glutamate, tyrosine, and proline. C and D are adapted with permission from Ref. [45], Aerts et al., Anal Chem. 2014, 86, 3203–3208. Copyright 2014 American Chemical Society.
Given the importance of mapping individual cell phenotypes, separation of cell contents maximizes the depth of cellular profiling, albeit, at the expense of cell throughput. Continued improvements in scalability will enable proportional increases in the breadth of these experiments. CE-MS is especially advantageous as the orthogonal mass separation ameliorates degraded resolution during fractionation. Beyond accelerating separations, preparation and manipulation of cells or organelles prior to fractionation help stratify phenotypically distinct cells for comprehensive analysis, greatly improving the quality of data obtained in a limited time frame. From this aspect, antigen and microfluidic-based sorting are natural complements to the in-depth analysis provided by CE.
Another strength of CE is its flexibility in performing several types of separations, which can target specific biochemical classes or separate enantiomers and other kinds of similar structures. Clever additives and covalent modifications will allow separation of currently unresolved peaks. Specific to single cell analysis, automated sorting and repeated analyses will likely find more applications and be adapted to commercial systems. Continued work with multimodal analyses will help streamline fractionation and optimize workflows, leading to more complete descriptions of single cells from smaller samples than ever before.
4. Imaging Methods for Chemical Analysis of Single Cells
Individual cells often have distinct roles that depend on their position within a tissue. Chemical imaging allows for recognition and identification of cells within their biological context. A prominent, non-targeted chemical imaging modality is MS imaging (MSI), which we highlight here. MSI methods raster a desorption probe across a tissue sample at cellular spatial resolutions to acquire mass spectra from cells while recording their positions within the context of their native environment. The resulting images ideally represent the spatio-chemical distribution of ionizable compounds in the tissue. Realistically, the limit of detection determines observable analytes and is affected by instrumentation and sample complexity, among other factors.
4.1. Secondary Ionization Mass Spectrometry (SIMS) Imaging
One of the oldest chemical imaging methods used to obtain cellular resolution is SIMS, which uses a primary ion beam that can achieve submicron footprints to sputter a sample surface. The collisions of primary ions with the sample surface cause ejection and ionization of analytes that are subsequently detected by a mass analyzer. Traditionally, SIMS analyses are categorized into two operating modes: dynamic and static.[46] The commercial CAMECA SIMS instruments were developed for dynamic SIMS analysis, with primary ion sources focused down to 50 – 200 nm probe diameters while maintaining high energy fluence. The term “NanoSIMS” was coined for many dynamic SIMS instruments, including those commercialized by CAMECA. Static SIMS is usually described as being less destructive due to its lower primary ion dose (below 1012 ions/cm2). Originally used for analyzing organic molecules on the surface monolayer, static SIMS is becoming widely used in biological MSI.[47]
SIMS imaging has been used to probe halogens, enriched metals, and isotopically labeled additives;[48] measure the cellular uptake and distribution of drugs in different co-cultured cell types;[49] and visualize different stages of cell cycles.[50] NanoSIMS has found wide application in biological geochemistry, cell biology, and microbiology to study the uptake, assimilation, storage, and translocation of trace elements.[51] A recent review by Musat et al.[51] highlighted the use of NanoSIMS-based methodologies to identify, quantify, and visualize the incorporation of labeled substrates, many of which were performed at single cell resolution. In one pioneering work, Lechene et al.[52] used NanoSIMS imaging to quantify N2 fixation by individual bacterium inhabiting the gills of the ship-worm Lyrodus pedicellatus (i.e., single bacterium within an animal cell). Popa et al.[53] used NanoSIMS to characterize cellular development and metabolite exchange in and between individual vegetative and heterocyst cells of Anabaena oscillarioides, a filamentous freshwater cyanobacterium. The two cell types of A. oscillarioides coexist in the same filament, but only heterocyst cells specialize in nitrogen fixation. Other vegetative cells participate in oxygenic photosynthesis and CO2 fixation. By adding tracer-levels of 13C and 15N, tracking stable isotopes from inorganic pools to their cellular fate was achieved with 100-nm spatial resolution using NanoSIMS. In a similar study, Kuypers and colleagues[54] measured the assimilation of H13CO3- and 15NH4+ by individual cells of the anaerobic, phototropic bacterial species Chromatium okenii, Lamprocystis purpurea, and Chlorobium clathratiforme. The study revealed a large range of uptake rates of ammonium and inorganic carbon for single cells of the same species, which might result from genomic diversity in phylogenetically identical but physiologically distinct populations.
SIMS can be combined with other single cell approaches. In environmental microbiology, NanoSIMS has been combined with fluorescence in situ hybridization (FISH),[55] Raman spectroscopy,[56] and high resolution optical microscopy.[57] McGlynn et al.[55a] coupled FISH and NanoSIMS to investigate the metabolic activities of single archaeal and bacterial cells within a consortium (see Figure 3A). Saka et al.[57a] correlated stimulated emission depletion microscopy with NanoSIMS imaging to visualize and quantify the turnover of isotopically labeled proteins in different organelles from cultured hippocampal neurons. These are a few of many examples where the combined methods allow simultaneous activity measurements and identification of single microbial cells.
Figure 3.

A series of cellular chemical images demonstrating the information that can be gained from SIMS. (A) Anaerobic oxidation of methane of consortia of anaerobic methanotrophic archaea (green) paired with Deltaproteobacteria (pink) identified by fluorescence in situ hybridization (FISH) (left panels) and NanoSIMS imaging (middle panels). Single cell activities (right panels) are measured as 15N atom percentages for regions of interest representing the FISH-identified archaea and bacteria in each consortium. Lighter shaded cells are more enriched in 15N, which corresponds with higher levels of anabolic activity and 15NH4+ assimilation. Scale bars, 3 μm. Adapted with permission from Ref. [55a], McGlynn et al., Nature 2015, 526, 531–535. Copyright 2015 Nature Publishing Group. (B) Multidimensional, composite image obtained from multiplexed ion beam imaging (MIBI) illustrating quantitatively protein expression and colocalization of E-cadherin (green), actin (red) and vimentin (blue), qualitatively categorizing cell nuclei into two subpopulations: ERα+PR+Ki-67+ (yellow) and ERα+PR+(aqua). Scale bars, 25 μm. Adapted with permission from Ref. [59], Angelo et al., Nat. Med. 2014, 20, 436–442. Copyright 2014 Nature Publishing Group.
NanoSIMS has recently been applied in the field of biomedical imaging. Nolan and co-workers[58] employed mass cytometry labels (e.g., affinity probes with metal isotope tags) in an imaging application referred to as multiplex ion beam imaging (MIBI). MIBI has several advantages over conventional immunohistochemistry techniques, including the absence of background auto-fluorescence signals, an order of magnitude larger dynamic range, and a higher plexity of up to 60 targets. In a recent report, Angelo et al.[59] used MIBI to image human breast tumors and revealed immunophenotypes of cell subpopulations that could be related back to the original clinical pathology of the tissue (Figure 3B). Another recent, exciting development is the use of atomic recombination to measure distances that are smaller than instrumental imaging resolution. Atomic recombination occurs when atoms from different molecules combine to form diatomic ions and is sensitive to the pairwise distance between molecules. Moss and Boxer[60] exploited the atomic recombination of 13C and 15N to form 13C15N- to assess the lateral distribution heterogeneity of lipids at a spatial resolution of 100 nm. The recombination phenomenon may eventually allow probing the proximity of a lipid to a protein of interest at nanometer-length scales. NanoSIMS has also been used to quantitate synaptic proteins within individual rodent neurons.[61]
While biological information has been and will continue to be gained from NanoSIMS, time-of-flight (TOF)-SIMS instruments are suitable for non-targeted analysis because of improved detection of intact molecular ions over a larger mass range. Rather than mapping internal, atomic species using dynamic mode, early TOF-SIMS experiments were performed in static mode and focused on the identification and localization of cell surface compounds. Pioneering work by the Winograd and Ewing groups[62] developed the unique strength of SIMS in biological imaging of single mammalian cells and intact tissues. The authors used a 15-keV In+ liquid metal ion beam to examine the lipid distribution along the conjugation junction of mating Tetrahymena, a model for studying membrane fusion. The images revealed a decrease in abundance of phosphatidylcholine and an increase in 2-aminoethylphosphonolipid at highly curved fusion pores, suggesting that Tetrahymena direct lipids to adjust membrane structure during conjugation. Shortly after, Monroe et al.[63] studied the subcellular localization of vitamin E in the soma-neurite junction of single, isolated neurons from A. californica using a TRIFT III instrument equipped with a 22-keV Au+ liquid metal ion source. Using the same instrument, Tucker et al.[64] imaged the cultured neurons under different treatment conditions and mapped the distributions of cholesterol, vitamin E, and phosphocholine head groups in cell soma, neurites, and growth cones.
The development of polyatomic ion beams, such as the C60+ and Ar1000–4000+, marked a new era for TOF-SIMS imaging of biological samples. The C60+ ion beam developed by the Vickerman group and Ionoptika[65] provides greater high mass ion yields, molecular depth profiling and reduced subsurface chemical damage compared to early Ga+ and SF5+ ion sources. Advances in instrumentation have improved the compromise between high mass resolution and high spatial resolution, while further reducing analysis time.[66] The authors modified the Ionoptika J105 3D Chemical Imager by equipping the instrument with a 40-keV C60+ ion gun with a minimum spot size of 200 nm. The capabilities of the instrument were demonstrated by performing 3D imaging of single benign prostatic hyperplasia (BPH) cells and 2D imaging of single HeLa, human cheek cells, and sectioned Xenopus blastomers (0.8 – 1.3 mm in diameter). The images of single cells revealed the distributions of adenine and lipids. The tandem MS capability of the J105 allowed the identification of several intact lipids.[66] Passarelli et al.[67] and Lanni et al.[68] further demonstrated the capability of TOF-SIMS with tandem MS in mapping various intact lipids across the cell surface of single neurons of A. californica.
The use of cluster ion sources has demonstrated great potential for molecular 3D imaging of single cells (for a recent review on 3D imaging with SIMS, see[69]). A dual ion source system was shown to be useful in reports by Breitenstein et al.[70] and Nygren et al.,[71] who performed 3D TOF-SIMS imaging of normal rat kidney and anaplastic thyroid carcinoma cells, respectively, at a spatial resolution of 300–350 nm using a 25-keV Bi3+ liquid metal ion beam for imaging and a 10-keV C60+ for etching and removing the damaged layers caused by the Bi3+ beam. A variety of endogenous amino acids, cholesterol, and intact phospholipids were detected (see Figure 4). Castner and colleagues[72] used the same ion beam combination but at higher energies (25 keV-Bi3+ and 20 keV C602+) to image NIH/3T3 fibroblasts in 2D and 3D. Brison et al.[73] applied a similar dual beam mode with the sputter depth calibrated using atomic force microscopy to perform 3D imaging of native and non-native chemical species in HeLa cells.
Figure 4.

SIMS imaging can be used to determine the localization of a variety of different chemical classes within single cells. (A) Pooled signals of amino acid fragment ions are represented in red, (B) phospholipids in green, and (C) substrate-derived secondary ions in blue. (D–F) Red–green–blue color overlay for correlation analysis of a confluent monolayer of normal rat kidney cells. Both horizontal x,y and vertical x,z sections are shown. Adapted from with permission from Ref. [70], Breitenstein et al., Angew. Chem. Int. Ed. 2007, 46, 5332–5335. Copyright 2007 Wiley InterScience.
Fletcher et al.[74] performed 3D TOF-SIMS analysis of individual oocyte cells mounted on copper tape using a single 40-kV C60+ beam. Aside from common ions such as cholesterol and oleic acid, they observed groups of mass spectral peaks occurring at m/z 540–700 and m/z 800–1000 that might originate from glycosphingolipids of the membrane constituents, although unambiguous identifications were not possible. Nonetheless, the signals at low mass, such as phosphocholine and adenine, produced 3D visualizations of the membrane and nucleus, as demonstrated by Vickerman and co-workers[75] for HeLa-M cells. The localization of small molecules and metabolites in single bacterial cells was recently reported by Tian et al.,[76] who showed direct localization of unlabeled tetracycline and ampicillin in single, antibiotic-dosed E. coli cells (about 2 μm long and 0.25–1.0 μm in diameter) using TOF-SIMS 3D imaging at 300 nm spatial resolution. Similarly, 3D orbitrap-SIMS has been recently developed for subcellular imaging of neurotransmitters and lipids for enhanced mass resolution as compared to TOF-SIMS.[77]
4.2. MALDI MSI
SIMS imaging offers superior spatial resolution, but achieves a limited mass range because the ion beams can cause fragmentation. A complementary imaging approach, MALDI MSI, is a soft ionization method that can ionize large proteins, although the use of a matrix can cause other issues. MALDI MS directly probes analytes from a solid sample, such as dispersed cells or tissue sections,[78] and is a highly sensitive analytical technique capable of attomole detection limits.[79] MALDI MS is uniquely suited for single cell analysis because it can probe cells directly with minimal perturbations and can analyze low amounts of many chemical compounds. Most of the detected analytes are singly-charged positive or negative molecular ions.[80] The sample is prepared by “dissolving” analyte compounds in the matrix, typically a small, organic compound that absorbs UV light. Irradiating the sample with a focused UV laser causes the matrix to erupt and generate gas-phase matrix and analyte ions representative of the ablated area. In addition, MALDI is more tolerant of high concentrations of salt often found in cells as compared to other ionization techniques, such as ESI.[81] The robustness of MALDI MS is particularly appreciated in the analysis of complex biological specimens, where sample treatments frequently fail to remove all analyte interference.
Matrix application is critical for MALDI MS as the nature of matrix compounds and the methods used to apply the matrix determine the chemical coverage, analyte migration, and the overall quality of measurements. Several strategies for applying the matrix compound to the sample have been established, including sublimation,[82] nebulization,[83] inkjet printing,[84] and electrospray.[85] Each MALDI MS measurement strikes a balance between extraction efficiency and analyte diffusion, which is especially problematic for tissue imaging.[83] The choices for MALDI matrices and their application continue to expand as novel compounds are found to be suitable matrices for specific analyses or classes of analytes. MALDI MS provides broad chemical coverage of analytes, including proteins,[86] peptides,[87] lipids,[88] and small metabolites.[89] A frequently cited limitation is isobaric interference from the MALDI matrix, which complicates metabolite detection without high-resolution mass analyzers. While MALDI MS is a destructive technique, it often consumes only a fraction of the sample,[90] allowing repeated or follow-up analyses to enhance the amount of information acquired from the same target.[91] This is especially useful for prescreening cells to determine rare cells or to classify cells into subpopulations based on representative markers for subsequent analysis.
Cellular and subcellular imaging (<10 μm) with MALDI MS is still not common using most commercial instruments. Laboratories have achieved single cell and subcellular analysis of tissue samples by using custom instrumentation capable of significantly smaller spatial resolution (several microns). High spatial resolution is dependent not only on the laser spot size, but also matrix crystal size and the sensitivity of the mass spectrometer. In 2010, 5-μm spatial resolution mouse bladder tissue imaging was accomplished by the Spengler group.[92] In 2012, Schober et al.[78b] developed an atmospheric MALDI source with 7-μm spatial resolution, capable of subcellular lipid and metabolite localization within HeLa cell cultures. Only months later, the Caprioli laboratory[93] demonstrated a 1-μm beam spot size achieved with transmission geometry illumination. The source arrangement was used to image individual HEK-293 cells and detected insulin differences between nuclei and cytoplasm within human pancreatic islets. More recently, in 2017 the Spengler group[94] developed an atmospheric pressure MALDI imaging source capable of 1.4-μm lateral resolution. The source capabilities were demonstrated by probing the subcellular lipid, metabolite, and peptide chemical differences between different sections of the Paramecium caudatum cellular membrane with a 3-μm pixel size (Figure 5A–F).
Figure 5.

High spatial resolution MALDI-MSI can be also be used to determine subcellular localization of chemical classes that are often different than those obtained with SIMS imaging. (A, C and E) Optical images of P. caudatum before MALDI-MSI analysis. (B, D and F) MSI images (RGB mode) of different lipids (i.e. [DG(31:0) + NH4]+, [PS(32:1) + H - H2O]+, [DG(38:1) + NH4]+) or small peptides (e.g., [(Lys3Ala) + H]+) present within the cell. MS images were acquired at 3-μm spatial resolution, showing discrete subcellular locations of the different lipids or small peptides. Some of the metabolites correlated well with structures such as cilia. Scale bar: 100 μm. Adapted with permission from Ref. [94], Kompauer et al., Nat. Methods 2017, 14, 90–96. Copyright 2017 Nature Publishing Group.
4.3. Alternative Imaging Approaches
SIMS and MALDI MSI are the most common MSI techniques capable of cellular imaging, but there are alternatives being developed. The Zare group[95] created laser desorption/ionization droplet delivery mass spectrometry, which is capable of 3-μm spatial resolution, to image tissue samples as well as measure both single cell apoptosis and live cell exocytosis. Laskin’s group[96] has been pushing the limit of NanoDESI, recently achieving a spatial resolution of 11 μm. In another example, Yang and colleagues[97] developed single-probe MS, which is effectively a miniaturized probe constructed from a fused silica capillary with an ESI emitter placed inside a dual-bore needle. The dual-bore needle can puncture cell membranes, gaining direct access to cell cytoplasm. Single-probe MS was first used to interrogate individual HeLa cells and detect many small molecules and lipids within individual cells. Single-probe MS has since been used within an imaging context, achieving an 8.5-μm spatial resolution,[98] allowing image acquisition of lipids within single cells. Single-probe MS offers the highest spatial resolution among solvent-based, ambient approaches.
When choosing a chemical imaging modality, it is important to understand that the selection of an ionization technique requires balancing a number of factors: the required spatial resolution and mass range of interest for the specific analyte, as well as instrument availability and cost. For example, SIMS provides superior spatial resolution compared to other imaging modalities, but most systems are too cost prohibitive to be owned by a single lab. Moreover, fragmentation prevents analysis of peptides, proteins, and larger nucleic acids. Despite the limitations, the “out-of-box” capabilities of SIMS imaging are impressive, and are becoming even more so. MALDI MS systems are more readily available, even if many of the above examples require modifications, and can more easily analyze higher mass compounds without fragmentation. Achieving the spatial resolution required to analyze single cells in the context of tissues is currently being performed by a handful of laboratories using either MALDI MS or ambient ionization.
5. Single Cell Profiling
Performing subcellular resolution MSI introduces a host of challenges for sample preparation and instrument development, and is generally slow. An alternative method for probing the contents of single cells is to isolate them from their microenvironment and individually interrogate each cell. Even though the spatial information is lost, profiling single cells is often faster and has a better limit of detection than conventional MSI at single cell resolution. Commercial instruments can be used without alteration, providing access to single cell measurements to more researchers. Moreover, the latest developments allow the interrogation of thousands of cells for population studies and analysis of cell heterogeneity.
5.1. ESI
ESI is a solvent-based, soft ionization technique that pairs well with many mass analyzers. In probe electrospray ionization (PESI), an ESI emitter is directly inserted into or touches the sample, effectively performing a surface extraction. Following extraction, the emitter (often a stainless steel needle) is repositioned directly in front of a mass spectrometer inlet for ESI analysis without further addition of liquid.[99] PESI allows live cell sampling as well as enrichment of metabolites by probing the sample multiple times before the MS analysis. The Zhang laboratory[100] used the approach to analyze the outer and inner epidermal cells from Allium cepa. Several chemical differences were noted between the cell types, including a higher diversity of fructans localized within the inner epidermal cells and distinct lipid profiles.
In 2018, the Vertes group[101] combined fluorescence microscopy with capillary microsampling to perform ESI analysis of cells at different mitotic stages. The Huang lab[102] used a patch clamp capillary as an ESI emitter to study metabolic changes within single mammalian neurons after electrophysiological analysis. They determined that cells that had unusual patching profiles also had unusual chemical profiles, demonstrating the need to measure both physiology and chemistry on the same, living neuron, perhaps multiple times.
A related method, live-cell MS, was developed by the Masujima group;[103] it utilizes a sharp nano-ESI emitter to puncture a cell, extracting contents (even from an organelle) into the emitter. The emitter is then transferred to the inlet for MS analysis. Due to the size of the probe, extraction does not appreciably perturb a living cell, allowing repeated sampling of the same cell. In a typical experiment, cells are observed under a microscope and when an event of interest occurs, such as a physical response to stimuli, the nano-ESI emitter is placed into the cell for extraction. This technique allows for high selectivity of the analyzed region of the cell, but does require the ability to execute fine manipulations. Masujima and colleagues[103] have detected over 700 analytes within an individual cell with live-cell MS and applied it to study metabolites within plants.[104] Similarly, the Huang lab[105] demonstrated the analysis of proteins from live cells without destroying the cell. The Laskin group[106] used localized electro-osmotic extraction to drive picoliter volumes out of live cells into a nanopipette compatible with nano-ESI analysis. They detected more than 50 metabolites, including sugars and flavonoids, some of which could be quantified using sequential extraction of a known volume of an aqueous solution containing deuterated standards. These approaches hold great promise for measuring the changes in chemical content of the same cell as a function of disease or growth. Nevertheless, it is critical to carry out parallel measurements to monitor cell conditions, and to avoid mechanically induced changes in cell states.
5.2. MALDI MS
MALDI MS is the primary analytical technique capable of obtaining direct chemical information from cells and has been used more than the other approaches. Here we divide MALDI MS approaches into lower throughput manual isolation and higher throughput acquisitions. Until the last decade, single cell MALDI MS required a combination of manual cell isolation, placement, and MS acquisition. Two methods for high-throughput acquisition of single cells were recently reported: high-density microarrays for mass spectrometry (MAMS) with defined locations (Figure 6A)[88a] and image-guided analysis of cells randomly seeded on a transparent substrate (Figure 6B).[107] Each method offers unique performance advantages and enables the analysis of hundreds to thousands of cells within a single experiment.
Figure 6.

Two approaches are highlighted that provide high-throughput single cell measurements via direct profiling MALDI MS. (A) Schematic of the MAMS process, where cells are seeded within evenly spaced wells for MS analysis, and of (B) optically guided MS analyses, where cells are randomly seeded on a glass slide. (C) MAMS has facilitated multimodal analysis of algal cells by both MALDI MS and Raman spectroscopy. Adapted with permission from Ref. [88a], Urban et al., Lab Chip 2010, 10, 3206–3209. Copyright 2010 The Royal Society of Chemistry. (D) Similarly, microMS has facilitated the analysis of the same pancreatic islet cell by both MALDI MS and CE-MS. This specific method enabled the use of MALDI MS to prescreen single islet cells for CE-MS analysis of ß-cells. Adapted with permission from Ref. [91], Comi et al., Anal. Chem. 2017, 89, 7765–7772. Copyright 2017 American Chemical Society.
5.2.1. Direct MALDI MS Profiling
The Van der Greef group[108] reported the earliest examples of single cell MALDI MS in 1993, where they profiled peptides from individual Lymnaea stagnalis neurons. Improvements to mass spectrometer sensitivity allowed detection of more analytes from single cells and even tandem MS for structural identification without a priori knowledge. Important neuroscience findings have direct roots in the single cell MALDI MS experiments pioneered by Van der Greef, Geraerts, Sweedler, Burke, and others.[18b, 87b, 109] Despite the limitations of this early research, hundreds of biologically important neuropeptides, neuromodulators, and neurotransmitters (among many other molecules) were discovered and correlated with physiological function. Discoveries were performed using samples ranging from large invertebrate neurons to small peptidergic mammalian cells.[87a, 110] One of the most recent studies involving low throughput MALDI MS was performed in 2018 by the Setou lab,[111] where they measured phosphocholine lipids in single neurons as they extended neurites.
5.2.2. MAMS
The first single cell profiling using MAMS was reported in 2010 by the Zenobi group[88a] where they detected metabolites, such as adenosine triphosphate and uridine diphosphate glucose, from single Saccharomyces cerevisiae cells and lipids within single Chlamydomonas reinhardtii cells. MAMS chips are fabricated from a conductive slide covered with a hydrophobic coating. Wells, approximately 100 μm in diameter, are patterned by laser ablation of the coating. Each well contains a small volume that restricts diffusion during matrix application. When cells are added to the surface, they settle only within the wells. Since the wells are located at known coordinates, sampling proceeds at a rate of about two samples per second. Additionally, restricting each cell to a known address simplifies correlation between different analyses, such as native fluorescence and Raman spectroscopy (Figure 6C). In a follow-up study, the Zenobi group[79] characterized metabolites within yeast cells, facilitating investigation of metabolic heterogeneity at the single cell level. MAMS has since been applied to a variety of cell systems, such as human blood peripheral mononuclear cells[112] and Haematococcus pluvialis.[113]
5.2.3. Optically Guided Profiling
Ong et al.[107] reported high-throughput, optical microscopy-guided MALDI-TOF MS of randomly seeded cells in 2015. They analyzed several thousand pituitary cells, frequently detecting pro-opiomelanocortin peptides. The protocol begins by incubating cells with Hoechst 33342, a fluorescent DNA intercalator, and acquiring a fluorescence image to locate the cells over the area of a microscope slide. Using the low background inherent in fluorescence images, the location of each cell is determined automatically. Registering fiducial marks that are visible in both the microscopy image and instrument camera enables the conversion of pixel positions into stage coordinates to automate the MALDI-TOF MS analysis. Knowing the location of each cell allows for mass spectral acquisition at defined points at high throughput.[107] After the initial demonstration of this approach, Jansson and coauthors[114] performed additional studies of single cells isolated from pancreatic islets of Langerhans and detected peptide hormones unique to cell types within the islet.
To expand optically guided single cell profiling beyond MALDI-TOF instrumentation, Comi et al.[115] further developed the open source software, microMS, for analyzing microscopy images and correlating cell locations to instrument positions. The software provides automatic cell finding and filtering by attributes such as size, fluorescence intensity, and distance between cells, and is simple to tailor to a variety of instruments, expanding the availability of single cell profiling. The ease of switching between output coordinate systems also facilitates analysis of the same cell on multiple instruments; an example being MALDI-TOF MS analysis with follow-up CE-MS analysis (Figure 6D).[115] Do et al.[116] recently used microMS to set up sequential MALDI MS analyses of rat DRG cells. Lipids, peptides, and small proteins were detected in the same cells, many of which were not previously detected in tissue homogenates or releasates. The DRG populations were stratified with multivariate statistical analysis using peptides and proteins as potential markers. A similar method utilizing flexImaging software from Bruker Corp. was recently published by the Caprioli lab[117] to determine heterogeneity within cultured macrophage cells using a Bruker solariX XR FT-ICR mass spectrometer. The authors imaged single cells by rastering across the slide and comparing the ion images with optical images acquired beforehand. They observed changes in lipid expression of macrophages upon chemical stimulation.
The optically guided single cell profiling approach was recently demonstrated for SIMS using microMS software.[115] In the study, Do et al.[118] utilized ionic liquid matrix-assisted 20-keV C60+ TOF-SIMS to profile metabolites and intact lipids in three cell types: large A. californica pedal neurons, medium-sized cell bodies of rat DRG, and small rat cerebellar cells at the rate of 600 cells/h. The single cell mass spectral datasets were subjected to multivariate statistical analyses, such as principal component analysis and t-distributed stochastic neighboring embedding, which helped distinguish cell types with similar lipid profiles, including DRG and cerebellum. In addition, these analyses suggested that lipid ratios could be endogenous markers to define brain regions. Overall, advanced statistical analysis strategies are necessary to fully harness the utility of high-throughput data.
The performance metrics of SIMS and MALDI MS imaging carry over to single cell analysis, as the limitations and advantages are inherent to the ionization technique. Moreover, the MALDI MS and SIMS examples discussed here largely sacrifice cellular connections, which can be consequential when the spatial organization of the system determines the function of a cell, such as the brain. To circumvent this loss of information, cells and organelles can be manually isolated from selected cells; this allows for cellular and even subcellular chemical analysis, but it is low throughput. ESI-based methods do not always require that structures of interest be removed from tissues, but these methods are also often laborious and low throughput. Furthermore, while ESI benefits from multiply charged ions, improving detection of proteins of higher mass, it is less salt tolerant than MALDI or SIMS. If fundamental single cell profiling of cellular heterogeneous systems, such as the brain, plant tissues, or disease states, is to be widely adopted, each approach—whether it be low throughput with known spatial location or high throughput without spatial information—will need to be developed further.
6. Summary and Outlook
Among the many available methods that can provide information on individual cells, we have emphasized those capable of non-targeted chemical analysis. Single cell chemical analysis has evolved rapidly over recent decades to provide ‘omics-scale molecular details at the individual cell level. Front-end techniques, such as CE and LC, have been downscaled to enhance separation on minute samples, and in some cases, to perform repeated sampling of the same cell to obtain real-time metabolomic information.[26, 35a, 119] MAMS and optically guided MS profiling now facilitate the assay of several thousands of cells within a couple of hours,[79, 114, 118] and cellular and subcellular spatial resolution chemical imaging can be achieved by selected MS probes such as NanoSIMS,[51–55, 59] SIMS[46c, 66, 69–70, 74–76, 120] and MALDI.[94] Future work will be aimed at achieving faster analyses with higher mass and spatial resolutions and developing more sensitive instrumentation. Several groups as discussed above have achieved detection of 10 zeptomoles of material with modern MS instruments, equivalent to about 6000 molecules. Fluorescence detectors can detect single selected molecules. These reported detection limits for MS should be sufficient for detecting most compounds within a single cell or most subcellular organelles. Why then is the scientific community unable to realize the limits of detection in ideal systems when applied to single cell chemical analyses? We attribute the lowered performance largely to limitations caused by the inherent chemical complexity of cells and less than ideal sampling protocols. Obtaining isolated single cells from a tissue requires dissection, dissociation, plating, rinsing, and finally, analysis. During every single step, different compounds are potentially released or removed from cells, reducing the number of molecules remaining for detection, as well as introducing other non-native contaminants. Sampling issues are compounded by ion suppression effects, transmission efficiency through the spectrometer or column, and the native background. As a final consideration, complex systems, such as single cells, require a high dynamic range to detect molecules at abundances over many orders of magnitude. The exciting aspect is that newer instruments meet the necessary performance requirements. In addition, at least for some analytes, experiments are being performed that sample living cells within their native environment, ameliorating several of the issues related to sampling. Techniques such as CE and live-probe MS enable live cell analysis and could be applied to native chemical analysis. Repeated sampling of individual cells can allow monitoring of the progression of disease, such as cancer and diabetes, facilitating a better understanding of disease development on the single cell scale. In situ monitoring is not a panacea however; many cell environments would require labeling of cells to track their position and allow reliable, repeated sampling, particularly for systems containing millions to billions of cells.
An alternative route for increasing the chemical information detected within a single cell is to combine two or more techniques for multimodal analysis of individual cells. Multiple orthogonal approaches can increase the chemical information gained from one sample by merging the assets of each method. While simple in concept, multimodal analysis requires attention to detail; each analytical technique has specific sample preparation requirements, substrates, and conditions that may be incompatible with the other sampling protocols, and of course, the approaches must leave sufficient sample behind for the next measurement. In 2011, the Zenobi group[121] developed a method for coupling laser desorption/ionization MS with Raman and fluorescence imaging to analyze carotene and phospholipids within individual algal cells randomly seeded on a metal target. Furthermore, they coupled Raman and fluorescence microscopy with MALDI MS to study adenosine triphosphate, adenosine diphosphate, and beta-carotene within individual algal cells.[113] NanoSIMS and electron microscopy can be correlated and allow for quantitation of small molecules[122] in addition to internalization and drug localization at the subcellular level.[123] These studies demonstrate that not only is the combination of MS with optical approaches possible, the union can enhance the chemical coverage of a single cell or organelle. Our group has hyphenated various analytical techniques to obtain more complete measurements on small samples. In 2013, we used immunocytochemistry to label neurons within the insect, Periplaneta americana, with follow-up peptide analysis using MALDI MS.[124] Combining different toolsets, patch clamp electrophysiology was coupled to CE-MS in 2014 to study the relation between the physiological activity of gamma-aminobutyric acid-containing neurons within the rat thalamus to the neurochemical condition.[45] More recently, we combined several MS approaches together, such as MALDI MS and SIMS, on the same cells.[115] In 2017 an optically guided liquid microjunction extraction probe was utilized to hyphenate MALDI MS with CE-MS for analysis of single α and β pancreatic islet cells.[91] In that work, MALDI MS was used first to prescreen cells for peptides indicative of a specific cell type, and CE-MS was used to analyze metabolite profiles of cells representative of a cell type. This combination can be further employed to study both the small metabolite and peptide heterogeneity of different cell types. We expect multimodal measurements to remain a rapidly developing approach for increasing chemical coverage.[125] By coupling single cell MS techniques with complementary targeted and non-targeted techniques, such as single cell transcriptomics, spectroscopy, and staining, even more information can be garnered.
Looking back to the days of Schleiden and Schawnn[1] when the theory of the cell was first described, the scientific community has learned much about this fundamental unit of life. Numerous technological advances have greatly expanded the information content that can be obtained from single cell analyses. Systems that have been explored include algae, plants, sea slugs and mammals, suggesting that single cell analysis has been effectively transforming many areas of biological science. Future advancements may allow scientists to probe more complex systems and glean information on some of the least understood emergent properties of organisms, such as memory within the brain. Despite the challenges, probing the chemical profiles and activity of individual cells is most certainly a field worth pursuing.
Acknowledgements
The authors gratefully acknowledge support from the National Institutes of Health, Award Number P30 DA018310 from the National Institute on Drug Abuse, and from the National Institute of Mental Health, Award Number 1U01 MH109062. E.K.N. and T.J.C. acknowledge support from the National Science Foundation Graduate Research Fellowship Program and the Springborn Fellowship. T.J.C. received additional support through the Training Program at Chemistry-Interface with Biology (T32 GM070421).
References
- [1].Schwann T, Microscopic Investigations on the Accordance in theStructure and Growth of Plants and Animals, English ed., The Sydenham Society: London, Berlin, 1839. [Google Scholar]
- [2].Schmiedebach HP, Z. Arztl Fortbild (Jena) 1990, 84, 889–894. [PubMed] [Google Scholar]
- [3].Virchow RLK, Cellular Pathology, JohnChurchill, London, 1859. [Google Scholar]
- [4].Schultz M, Emerg. Infect. Dis 2008, 14, 1480–1481. [Google Scholar]
- [5].a) Crow M, Paul A, Ballouz S, Huang ZJ, Gillis J, Genome Biol. 2016, 17, 101; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hodne K, Weltzien FA, Int. J. Mol. Sci 2015, 16, 26832–26849; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shapiro E, Biezuner T, Linnarsson S, Nat. Rev. Genetics 2013, 14, 618–630. [DOI] [PubMed] [Google Scholar]
- [6].Haber DA, Velculescu VE, Cancer Discov. 2014, 4, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen X, Love JC, Navin NE, Pachter L, Stubbington MJT, Svensson V, Sweedler JV, Teichmann SA, Nat. Biotechnol 2016, 34, 1111–1118. [DOI] [PubMed] [Google Scholar]
- [8].Comi TJ, Do TD, Rubakhin SS, Sweedler JV, J. Am. Chem. Soc 2017, 139, 3920–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kennedy RT, Jorgenson JW, Anal. Chem 1989, 61, 436–441. [DOI] [PubMed] [Google Scholar]
- [10].Wightman RM, Jankowski JA, Kennedy RT, Kawagoe KT, Schroeder TJ, Leszczyszyn DJ, Near JA, Diliberto EJ Jr., Viveros OH, Proc. Natl. Acad. Sci. U. S. A 1991, 88, 10754–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chien JB, Wallingford RA, Ewing AG, J. Neurochem 1990, 54, 633–638. [DOI] [PubMed] [Google Scholar]
- [12].Mackler SA, Brooks BP, Eberwine JH, Neuron 1992, 9, 539–548. [DOI] [PubMed] [Google Scholar]
- [13].Zenobi R, Science 2013, 342, 1243259. [DOI] [PubMed] [Google Scholar]
- [14].aZhang L, Vertes A, Angew. Chem. Int. Ed 2018, 57, 4466–4477; [DOI] [PubMed] [Google Scholar]; b) Pozebon D, Scheffler GL, Dressler VL, J. Anal. At. Spectrom 2017, 32, 890–919; [Google Scholar]; c) Qi M, Philip MC, Yang N, Sweedler JV, ACS Chem. Neurosci 2018, 9, 40–50; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Murphy TW, Zhang Q, Naler LB, Ma S, Lu C, Analyst 2018, 143, 60–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goodwin RJA, J. Proteomics 2012, 75, 4893–4911. [DOI] [PubMed] [Google Scholar]
- [16].Cecala C, Sweedler JV, Analyst 2012, 137, 2922–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chughtai K, Heeren RMA, Chem. Rev 2010, 110, 3237–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Ye X, Xie F, Romanova EV, Rubakhin SS, Sweedler JV, ACS Chem. Neurosci 2010, 1, 182–193; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Floyd PD, Li LJ, Rubakhin SS, Sweedler JV, Horn CC, Kupfermann I, Alexeeva VY, Ellis TA, Dembrow NC, Weiss KR, Vilim FS, J. Neurosci 1999, 19, 7732–7741; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miao H, Rubakhin SS, Scanlan CR, Wang L, Sweedler JV, J. Neurochem 2006, 97, 595–606; [DOI] [PubMed] [Google Scholar]; d) Moroz LL, Edwards JR, Puthanveettil SV, Kohn AB, Ha T, Heyland A, Knudsen B, Sahni A, Yu F, Liu L, Jezzini S, Lovell P, Iannucculli W, Chen M, Nguyen T, Sheng H, Shaw R, Kalachikov S, Panchin YV, Farmerie W, Russo JJ, Ju J, Kandel ER, Cell 2006, 127, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hosic S, Murthy SK, Koppes AN, Anal. Chem 2016, 88, 354–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Johnson ME, Landers JP, Electrophoresis 2004, 25, 3513–3527. [DOI] [PubMed] [Google Scholar]
- [21].Wallingford RA, Ewing AG, Anal. Chem 1988, 60, 1972–1975. [DOI] [PubMed] [Google Scholar]
- [22].Ewing AG, Mesaros JM, Gavin PF, Anal. Chem 1994, 66, A527–A537. [DOI] [PubMed] [Google Scholar]
- [23].Sun L, Dubiak KM, Peuchen EH, Zhang Z, Zhu G, Huber PW, Dovichi NJ, Anal. Chem 2016, 88, 6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Woods LA, Roddy TP, Ewing AG, Electrophoresis 2004, 25, 1181–1187. [DOI] [PubMed] [Google Scholar]
- [25].Altria KD, Methods Mol. Biol 1996, 52, 3–13. [DOI] [PubMed] [Google Scholar]
- [26].Nemes P, Knolhoff AM, Rubakhin SS, Sweedler JV, Anal. Chem 2011, 83, 6810–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sims CE, Meredith GD, Krasieva TB, Berns MW, Tromberg BJ, Allbritton NL, Anal. Chem 1998, 70, 4570–4577. [DOI] [PubMed] [Google Scholar]
- [28].Han FT, Wang Y, Sims CE, Bachman M, Chang RS, Li GP, Allbritton NL, Anal. Chem 2003, 75, 3688–3696. [DOI] [PubMed] [Google Scholar]
- [29].a) Munce NR, Li JZ, Herman PR, Lilge L, Anal. Chem 2004, 76, 4983–4989; [DOI] [PubMed] [Google Scholar]; b) Mellors JS, Jorabchi K, Smith LM, Ramsey JM, Anal. Chem 2010, 82, 967–973; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hargis AD, Alarie JP, Ramsey JM, Electrophoresis 2011, 32, 3172–3179; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dickinson AJ, Armistead PM, Allbritton NL, Anal. Chem 2013, 85, 4797–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brügger B, Annu. Rev. Biochem 2014, 83, 79–98. [DOI] [PubMed] [Google Scholar]
- [31].Moroz LL, Dahlgren RL, Boudko D, Sweedler JV, Lovell P, J. Inorg. Biochem 2005, 99, 929–939. [DOI] [PubMed] [Google Scholar]
- [32].Hu S, Le Z, Krylov S, Dovichi NJ, Anal. Chem 2003, 75, 3495–3501. [DOI] [PubMed] [Google Scholar]
- [33].Lochmann H, Bazzanella A, Kropsch S, Bachmann K, J. Chromatogr. A 2001, 917, 311–317. [DOI] [PubMed] [Google Scholar]
- [34].Zhi Q, Xie C, Huang X, Ren J, Anal. Chim. Acta 2007, 583, 217–222. [DOI] [PubMed] [Google Scholar]
- [35].a) Onjiko RM, Portero EP, Moody SA, Nemes P, Anal. Chem 2017, 89, 7069–7076; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Onjiko RM, Plotnick DO, Moody SA, Nemes P, Anal. Methods 2017, 9, 4964–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang X, Ma Y, Zhao M, Zhou M, Xiao Y, Sun Z, Tong L, J. Chromatogr. A 2016, 1469, 128–134. [DOI] [PubMed] [Google Scholar]
- [37].Lombard-Banek C, Reddy S, Moody SA, Nemes P, Mol. Cell. Proteomics 2016, 15, 2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu J-X, Aerts JT, Rubakhin SS, Zhang X-X, Sweedler JV, Analyst 2014, 139, 5835–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Patel AV, Kawai T, Wang L, Rubakhin SS, Sweedler JV, Anal. Chem 2017, 89, 12375–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen D, Fan F, Zhao X, Xu F, Chen P, Wang J, Ban L, Liu Z, Feng X, Zhang Y, Liu B-F, Anal. Chem 2016, 88, 2466–2471. [DOI] [PubMed] [Google Scholar]
- [41].Chiu DT, Lillard SJ, Scheller RH, Zare RN, Rodriguez-Cruz SE, Williams ER, Orwar O, Sandberg M, Lundqvist JA, Science 1998, 279, 1190–1193. [DOI] [PubMed] [Google Scholar]
- [42].Li HN, Sims CE, Wu HY, Allbritton NL, Anal. Chem 2001, 73, 4625–4631. [DOI] [PubMed] [Google Scholar]
- [43].Gunasekera N, Olson KJ, Musier-Forsyth K, Arriaga EA, Anal. Chem 2004, 76, 655–662. [DOI] [PubMed] [Google Scholar]
- [44].Johnson RD, Navratil M, Poe BG, Xiong GH, Olson KJ, Ahmadzadeh H, Andreyev D, Duffy CF, Arriaga EA, Anal. Bioanal. Chem 2007, 387, 107–118. [DOI] [PubMed] [Google Scholar]
- [45].Aerts JT, Louis KR, Crandall SR, Govindaiah G, Cox CL, Sweedler JV, Anal. Chem 2014, 86, 3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].a) Benninghoven A, Angew. Chem. Int. Ed 1994, 33, 1023–1043; [Google Scholar]; b) Guerquin-Kern JL, Wu TD, Quintana C, Croisy A, Biochim. Biophys. Acta 2005, 1724, 228–238; [DOI] [PubMed] [Google Scholar]; c) Fletcher JS, Analyst 2009, 134, 2204–2215. [DOI] [PubMed] [Google Scholar]
- [47].a) Boxer SG, Kraft ML, Weber PK, Annu. Rev. Biophys 2009, 38, 53–74; [DOI] [PubMed] [Google Scholar]; b) Gamble LJ, Anderton CR, Micros. Today 2016, 24, 24–31; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pacholski ML, Winograd N, Chem. Rev 1999, 99, 2977–3006; [DOI] [PubMed] [Google Scholar]; d) Winograd N, Anal. Chem 2005, 77, 142 A–149 A; [Google Scholar]; e) Fletcher JS, Vickerman JC, Winograd N, Curr. Opin. Chem. Biol 2011, 15, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Levi-Setti R, Chabala JM, Gavrilov K, Espinosa R 3rd, Le Beau MM, Microsc. Res. Tech 1997, 36, 301–312; [DOI] [PubMed] [Google Scholar]; b) Levi-Setti R, Gavrilov KL, Strissel PL, Strick R, Appl. Surf. Sci 2004, 231-232, 479–484; [Google Scholar]; c) Romer W, Wu T-D, Duchambon P, Amessou M, Carrez D, Johannes L, Guerquin-Kern J-L, Appl. Surf. Sci 2006, 252, 6925–6930. [Google Scholar]
- [49].a) Zha X, Ausserer WA, Morrison GH, Cancer Res. 1992, 52, 5219–5222; [PubMed] [Google Scholar]; b) Lorey DR, Morrison GH, Chandra S, Anal. Chem 2001, 73, 3947–3953. [DOI] [PubMed] [Google Scholar]
- [50].Chandra S, Lorey II DR, Int. J. Mass Spectrom 2007, 260, 90–101. [Google Scholar]
- [51].Musat N, Foster R, Vagner T, Adam B, Kuypers MM, FEMS Microbiol. Rev 2012, 36, 486–511. [DOI] [PubMed] [Google Scholar]
- [52].Lechene CP, Luyten Y, McMahon G, Distel DL, Science 2007, 318, 570–570. [DOI] [PubMed] [Google Scholar]
- [53].Popa R, Weber PK, Pett-Ridge J, Finzi JA, Fallon SJ, Hutcheon ID, Nealson KH, Capone DG, ISME J 2007, 1, 354–360. [DOI] [PubMed] [Google Scholar]
- [54].Musat N, Halm H, Winterholler B, Hoppe P, Peduzzi S, Hillion F, Horreard F, Amann R, Jorgensen BB, Kuypers MM, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 17861–17866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].a) McGlynn SE, Chadwick GL, Kempes CP, Orphan VJ, Nature 2015, 526, 531–535; [DOI] [PubMed] [Google Scholar]; b) Orphan VJ, House CH, Hinrichs KU, McKeegan KD, DeLong EF, Science 2001, 293, 484–487; [DOI] [PubMed] [Google Scholar]; c) Dekas AE, Poretsky RS, Orphan VJ, Science 2009, 326, 422–426; [DOI] [PubMed] [Google Scholar]; d) Fike DA, Gammon CL, Ziebis W, Orphan VJ, ISME J 2008, 2, 749–759. [DOI] [PubMed] [Google Scholar]
- [56].Eichorst SA, Strasser F, Woyke T, Schintlmeister A, Wagner M, Woebken D, FEMS Microbiol. Ecol. 2015, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].a) Saka SK, Vogts A, Krohnert K, Hillion F, Rizzoli SO, Wessels JT, Nat. Commun 2014, 5, 3664; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Phan NTN, Li X, Ewing AG, Nat. Rev. Chem 2017, 1; [Google Scholar]; c) Legin AA, Schintlmeister A, Jakupec MA, Galanski M, Lichtscheidl I, Wagner M, Keppler BK, Chem. Sci 2014, 5, 3135–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Spitzer MH, Nolan GP, Cell 2016, 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, Natkunam Y, Nolan GP, Nat. Med 2014, 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Moss FR 3rd, Boxer SG, J. Am. Chem. Soc 2016, 138, 16737–16744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claßen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO, Science 2014, 344, 1023–1028. [DOI] [PubMed] [Google Scholar]
- [62].Ostrowski SG, Van Bell CT, Winograd N, Ewing AG, Science 2004, 305, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Monroe EB, Jurchen JC, Lee J, Rubakhin SS, Sweedler JV, J. Am. Chem. Soc 2005, 127, 12152–12153. [DOI] [PubMed] [Google Scholar]
- [64].Tucker KR, Li Z, Rubakhin SS, Sweedler JV, J. Am. Soc. Mass Spectrom 2012, 23, 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].a) Wong SCC, Hill R, Blenkinsopp P, Lockyer NP, Weibel DE, Vickerman JC, Appl. Surf. Sci 2003, 219, 203–204; [Google Scholar]; b) Weibel D, Wong S, Lockyer N, Blenkinsopp P, Hill R, Vickerman JC, Anal. Chem 2003, 75, 1754–1764. [DOI] [PubMed] [Google Scholar]
- [66].Fletcher JS, Rabbani S, Henderson A, Blenkinsopp P, Thompson SP, Lockyer NP, Vickerman JC, Anal. Chem 2008, 80, 9058–9064. [DOI] [PubMed] [Google Scholar]
- [67].Passarelli MK, Ewing AG, Winograd N, Anal. Chem 2013, 85, 2231–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lanni EJ, Dunham SJ, Nemes P, Rubakhin SS, Sweedler JV, J. Am. Soc. Mass Spectrom 2014, 25, 1897–1907. [DOI] [PubMed] [Google Scholar]
- [69].Fletcher JS, Vickerman JC, Anal. Chem 2013, 85, 610–639. [DOI] [PubMed] [Google Scholar]
- [70].Breitenstein D, Rommel CE, Mollers R, Wegener J, Hagenhoff B, Angew. Chem. Int. Ed 2007, 46, 5332–5335. [DOI] [PubMed] [Google Scholar]
- [71].Nygren H, Hagenhoff B, Malmberg P, Nilsson M, Richter K, Micros. Res. Tech 2007, 70, 969–974. [DOI] [PubMed] [Google Scholar]
- [72].Robinson MA, Graham DJ, Castner DG, Anal. Chem 2012, 84, 4880–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Brison J, Robinson MA, Benoit DSW, Muramoto S, Stayton PS, Castner DG, Anal. Chem 2013, 85, 10869–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fletcher JS, Lockyer NP, Vaidyanathan S, Vickerman JC, Anal. Chem 2007, 79, 2199–2206. [DOI] [PubMed] [Google Scholar]
- [75].Fletcher JS, Rabbani S, Henderson A, Lockyer NP, Vickerman JC, Rapid Commun. Mass Spectrom 2011, 25, 925–932. [DOI] [PubMed] [Google Scholar]
- [76].Tian H, Six DA, Krucker T, Leeds JA, Winograd N, Anal. Chem 2017, 89, 5050–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Passarelli MK, Pirkl A, Moellers R, Grinfeld D, Kollmer F, Havelund R, Newman CF, Marshall PS, Arlinghaus H, Alexander MR, West A, Horning S, Niehuis E, Makarov A, Dollery CT, Gilmore IS, Nat. Methods 2017, 14, 1175. [DOI] [PubMed] [Google Scholar]
- [78].a) Kruse RA, Rubakhin SS, Romanova EV, Bohn PW, Sweedler JV, J. Mass Spectrom 2001, 36, 1317–1322; [DOI] [PubMed] [Google Scholar]; b) Schober Y, Guenther S, Spengler B, Rompp A, Anal. Chem 2012, 84, 6293–6297. [DOI] [PubMed] [Google Scholar]
- [79].Ibanez AJ, Fagerer SR, Schmidt AM, Urban PL, Jefimovs K, Geiger P, Dechant R, Heinemann M, Zenobi R, Proc. Natl. Acad. Sci. U.S.A 2013, 110, 8790–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].El-Aneed A, Cohen A, Banoub J, App. Spec. Rev 2009, 44, 210–230. [Google Scholar]
- [81].Garden RW, Moroz LL, Moroz TP, Shippy SA, Sweedler JV, J. Mass Spectrom 1996, 31, 1126–1130. [DOI] [PubMed] [Google Scholar]
- [82].Yang JH, Caprioli RM, Anal. Chem 2011, 83, 5728–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Caldwell RL, Caprioli RM, Mol. Cell. Proteomics 2005, 4, 394–401. [DOI] [PubMed] [Google Scholar]
- [84].Baluya DL, Garrett TJ, Yost RA, Anal. Chem 2007, 79, 6862–6867. [DOI] [PubMed] [Google Scholar]
- [85].Caprioli RM, Farmer TB, Gile J, Anal. Chem 1997, 69, 4751–4760. [DOI] [PubMed] [Google Scholar]
- [86].Labas V, Teixeira-Gomes AP, Bouguereau L, Gargaros A, Spina L, Marestaing A, Uzbekova S, J. Proteomics 2017, 175, 56–74. [DOI] [PubMed] [Google Scholar]
- [87].a) Rubakhin SS, Churchill JD, Greenough WT, Sweedler JV, Anal. Chem 2006, 78, 7267–7272; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jimenez CR, Vanveelen PA, Li KW, Wildering WC, Geraerts WPM, Tjaden UR, Vandergreef J, J. Neurochem 1994, 62, 404–407. [DOI] [PubMed] [Google Scholar]
- [88].a) Urban PL, Jefimovs K, Amantonico A, Fagerer SR, Schmid T, Madler S, Puigmarti-Luis J, Goedecke N, Zenobi R, Lab Chip 2010, 10, 3206–3209; [DOI] [PubMed] [Google Scholar]; b) Hossen MA, Nagata Y, Waki M, Ide Y, Takei S, Fukano H, Romero-Perez GA, Tajima S, Yao I, Ohnishi K, Setou M, Anal. Bioanal. Chem 2015, 407, 5273–5280. [DOI] [PubMed] [Google Scholar]
- [89].a) Amantonico A, Oh JY, Sobek J, Heinemann M, Zenobi R, Angew. Chem. Int. Ed 2008, 47, 5382–5385; [DOI] [PubMed] [Google Scholar]; b) Amantonico A, Urban PL, Fagerer SR, Balabin RM, Zenobi R, Anal. Chem 2010, 82, 7394–7400. [DOI] [PubMed] [Google Scholar]
- [90].Page JS, Sweedler JV, Anal. Chem 2002, 74, 6200–6204. [DOI] [PubMed] [Google Scholar]
- [91].Comi TJ, Makurath MA, Philip MC, Rubakhin SS, Sweedler JV, Anal. Chem 2017, 89, 7765–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rompp A, Guenther S, Schober Y, Schulz O, Takats Z, Kummer W, Spengler B, Angew. Chem. Int. Ed 2010, 49, 3834–3838. [DOI] [PubMed] [Google Scholar]
- [93].Zavalin A, Todd EM, Rawhouser PD, Yang J, Norris JL, Caprioli RM, J. Mass Spectrom 2012, 47, 1473–1481. [DOI] [PubMed] [Google Scholar]
- [94].Kompauer M, Heiles S, Spengler B, Nat. Methods 2017, 14, 90–96. [DOI] [PubMed] [Google Scholar]
- [95].Lee JK, Jansson ET, Nam HG, Zare RN, Anal. Chem 2016, 88, 5453–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yin R, Kyle J, Burnum-Johnson K, Bloodsworth KJ, Sussel L, Ansong C, Laskin J, Anal. Chem 2018, 90, 6548–6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Pan N, Rao W, Kothapalli NR, Liu RM, Burgett AWG, Yang ZB, Anal. Chem 2014, 86, 9376–9380. [DOI] [PubMed] [Google Scholar]
- [98].Rao W, Pan N, Yang ZB, J. Am. Soc. Mass Spectrom 2015, 26, 986–993. [DOI] [PubMed] [Google Scholar]
- [99].Chen LC, Nishidate K, Saito Y, Mori K, Asakawa D, Takeda S, Kubota T, Terada N, Hashimoto Y, Hori H, Hiraoka K, Rapid Commun. Mass Spectrom 2008, 22, 2366–2374. [DOI] [PubMed] [Google Scholar]
- [100].Gong XY, Zhao YY, Cai SQ, Fu SJ, Yang CD, Zhang SC, Zhang XR, Anal. Chem 2014, 86, 3809–3816. [DOI] [PubMed] [Google Scholar]
- [101].Zhang L, Sevinsky CJ, Davis BM, Vertes A, Anal. Chem 2018, 90, 4626–4634. [DOI] [PubMed] [Google Scholar]
- [102].Zhu H, Zou G, Wang N, Zhuang M, Xiong W, Huang G, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 2586–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Mizuno H, Tsuyama N, Harada T, Masujima T, J. Mass Spectrom 2008, 43, 1692–1700. [DOI] [PubMed] [Google Scholar]
- [104].a) Fujii T, Matsuda S, Tejedor ML, Esaki T, Sakane I, Mizuno H, Tsuyama N, Masujima T, Nat. Protoc 2015, 10, 1445–1456; [DOI] [PubMed] [Google Scholar]; b) Tejedor ML, Mizuno H, Tsuyama N, Harada T, Masujima T, Anal. Chem 2012, 84, 5221–5228. [DOI] [PubMed] [Google Scholar]
- [105].Li G, Yuan S, Zheng S, Liu Y, Huang G, Anal. Chem 2018, 90, 3409–3415. [DOI] [PubMed] [Google Scholar]
- [106].Yin R, Prabhakaran V, Laskin J, Anal. Chem 2018, 90, 7937–7945. [DOI] [PubMed] [Google Scholar]
- [107].Ong TH, Kissick DJ, Jansson ET, Comi TJ, Romanova EV, Rubakhin SS, Sweedler JV, Anal. Chem 2015, 87, 7036–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].van Veelen PA, Jimenez CR, Li KW, Wildering WC, Geraerts WPM, Tjaden UR, van der Greef J, Org. Mass Spectrom 1993, 28, 1542–1546. [Google Scholar]
- [109].a) Jimenez CR, Li KW, Dreisewerd K, Spijker S, Kingston R, Bateman RH, Burlingame AL, Smit AB, van Minnen J, Geraerts WPM, Biochemistry 1998, 37, 2070–2076; [DOI] [PubMed] [Google Scholar]; b) Kellett E, Perry SJ, Santama N, Worster BM, Benjamin PR, Burke JF, J. Neurosci 1996, 16, 4949–4957; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li KW, Kingston R, Dreisewerd K, Jimenez CR, vanderSchors RC, Bateman RH, Geraerts WPM, Anal. Chem 1997, 69, 563–565; [Google Scholar]; d) Perry SJ, Dobbins AC, Schofield MG, Piper MR, Benjamin PR, Eur. J. Neurosci 1999, 11, 655–662; [DOI] [PubMed] [Google Scholar]; e) Garden RW, Moroz TP, Gleeson JM, Floyd PD, Li LJ, Rubakhin SS, Sweedler JV, J. Neurochem 1999, 72, 676–681; [DOI] [PubMed] [Google Scholar]; f) van Strien FJ, Jespersen S, van der Greef J, Jenks BG, Roubos EW, FEBS Lett. 1996, 379, 165–170. [DOI] [PubMed] [Google Scholar]
- [110].a) Li L, Garden RW, Romanova EV, Sweedler JV, Anal. Chem 1999, 71, 5451–5458; [DOI] [PubMed] [Google Scholar]; b) Li L, Garden RW, Sweedler JV, Trends Biotechnol. 2000, 18, 151–160; [DOI] [PubMed] [Google Scholar]; c) Boggio KJ, Obasuyi E, Sugino K, Nelson SB, Agar NY, Agar JN, Expert Rev. Proteomics 2011, 8, 591–604; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li L, Romanova EV, Rubakhin SS, Alexeeva V, Weiss KR, Vilim FS, Sweedler JV, Anal. Chem 2000, 72, 3867–3874; [DOI] [PubMed] [Google Scholar]; e) Page JS, Rubakhin SS, Sweedler JV, Anal. Chem 2002, 74, 497–503. [DOI] [PubMed] [Google Scholar]
- [111].Sugiyama E, Yao I, Setou M, Biochem. Biophys. Res. Commun 2017, 495, 1048–1054. [DOI] [PubMed] [Google Scholar]
- [112].Pabst M, Fagerer SR, Kohling R, Eyer K, Krismer J, Jefimovs K, Ibanez AJ, Zenobi R, J. Am. Soc. Mass Spectrom 2014, 25, 1083–1086. [DOI] [PubMed] [Google Scholar]
- [113].Fagerer SR, Schmid T, Ibanez AJ, Pabst M, Steinhoff R, Jefimovs K, Urban PL, Zenobi R, Analyst 2013, 138, 6732–6736. [DOI] [PubMed] [Google Scholar]
- [114].Jansson ET, Comi TJ, Rubakhin SS, Sweedler JV, ACS Chem. Biol 2016, 11, 2588–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Comi TJ, Neumann EK, Do TD, Sweedler JV, J. Am. Soc. Mass Spectrom 2017, 28, 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Do TD, Ellis JF, Neumann EK, Comi TJ, Tillmaand EG, Lenhart AE, Rubakhin SS, Sweedler JV, Chemphyschem 2018, 19, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yang B, Patterson NH, Tsui T, Caprioli RM, Norris JL, J. Am. Soc. Mass Spectrom 2018, 29, 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Do TD, Comi TJ, Dunham SJ, Rubakhin SS, Sweedler JV, Anal. Chem 2017, 89, 3078–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Nemes P, Rubakhin SS, Aerts JT, Sweedler JV, Nat. Protoc 2013, 8, 783–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Colliver TL, Brummel CL, Pacholski ML, Swanek FD, Ewing AG, Winograd N, Anal. Chem 1997, 69, 2225–2231. [DOI] [PubMed] [Google Scholar]
- [121].Urban PL, Schmid T, Amantonico A, Zenobi R, Anal. Chem 2011, 83, 1843–1849. [DOI] [PubMed] [Google Scholar]
- [122].Lovrić J, Dunevall J, Larsson A, Ren L, Andersson S, Meibom A, Malmberg P, Kurczy ME, Ewing AG, ACS Nano 2017, 11, 3446–3455. [DOI] [PubMed] [Google Scholar]
- [123].Jiang H, Passarelli MK, Munro PMG, Kilburn MR, West A, Dollery CT, Gilmore IS, Rakowska PD, Chem. Commun 2017, 53, 1506–1509. [DOI] [PubMed] [Google Scholar]
- [124].Neupert S, Rubakhin SS, Sweedler JV, Chem. Biol 2012, 19, 1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Neumann EK, Comi TJ, Spegazzini N, Mitchell JW, Rubakhin SS, Gillette MU, Bhargava R, Sweedler JV, Anal. Chem 2018, 90, 11572–11580. [DOI] [PMC free article] [PubMed] [Google Scholar]