

Abstract
Thyroid hormone (TH) is a key metabolic regulator that acts by coordinating short- and long-term energy needs. Accordingly, significant metabolic changes are observed depending on thyroid status. Although it is established that hyperthyroidism augments basal energy consumption, thus resulting in an enhanced metabolic state, the net effects on cellular respiration and generation of reactive oxygen species (ROS) remain unclear. To elucidate the effects of augmented TH signal in muscle cells, we generated a doxycycline-inducible cell line in which the expression of the TH-activating enzyme, type 2 deiodinase (D2), is reversibly turned on by the “Tet-ON” system. Interestingly, increased intracellular TH caused a net shift from oxidative phosphorylation to glycolysis and a consequent increase in the extracellular acidification rate. As a result, mitochondrial ROS production, and both the basal and doxorubicin-induced production of cellular ROS were reduced. Importantly, the expression of a set of antioxidant genes was up-regulated, and, among them, the mitochondrial scavenger Sod2 was specifically induced at transcriptional level by D2-mediated TH activation. Finally, we observed that attenuation of oxidative stress and increased levels of SOD2 are key elements of the differentiating cascade triggered by TH and D2, thereby establishing that D2 is essential in coordinating metabolic reprogramming of myocytes during myogenic differentiation. In conclusion, our findings indicate that TH plays a key role in oxidative stress dynamics by regulating ROS generation. Our novel finding that TH and its intracellular metabolism act as mitochondrial detoxifying agents sheds new light on metabolic processes relevant to muscle physiology.
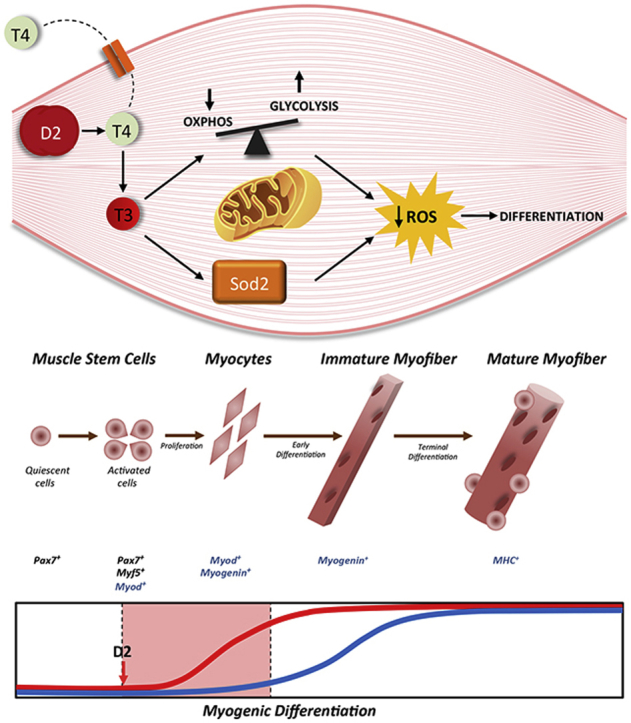
Graphical abstract
Highlights
-
•
Cell autonomous activation of TH by type 2 deiodinase induces a metabolic shift from oxidative phosphorylation to glycolysis.
-
•
D2 up-regulation reduces endogenous ROS production and mitochondrial ROS in muscle cells.
-
•
Induction of D2 increases TH-dependent transcription of the superoxide dismutase SOD2, thus contributing to reduced ROS balance. SOD2 silencing blocks the anti-oxidant effects of TH, thereby indicating that SOD2 mediates the detoxifying action of TH.
-
•
The detoxifying action of D2 contributes to myogenesis, linking the metabolic role of TH to its pro-differentiative effects.
1. Introduction
Reactive oxygen species (ROS) are bioproducts of various ubiquitous cellular processes, and were long considered deleterious moieties that generate oxidative stress, disease and aging [1]. However, it is now recognized that ROS is a molecular signal that regulates physiological cellular processes [2]. Skeletal muscle is one of the most active ROS-generating tissues, which is in line with its intense metabolic action. In physiologic conditions, mitochondrial ROS production is enhanced during muscle contraction, and superoxide generation can increase up to ∼100-fold during aerobic contractions [3]. However, excess ROS levels promote mitochondria fragmentation and dysfunction, thereby altering the physiological turnover of muscle fibers [3,4]. During muscle repair, ROS are physiologically active signaling molecules that trigger the cascade of events that enable correct muscle repair and activation of muscle stem cells (“satellite cells”) [5]. However, prolonged exposure to elevated ROS levels can exacerbate muscle injury by inducing oxidative toxic damage to regenerating myofibers [5]. Indeed, as in many other tissues, excess of ROS in skeletal muscle exacerbates muscular dystrophy [1], and other muscle diseases [4,6,7]. The relative prevalence of the aforementioned “positive” or “negative” effects of ROS depends on complex intracellular mechanisms that maintain the ROS threshold within physiological concentrations.
Thyroid hormone has a major impact on whole-body energy metabolism and on tissue-specific energy balance [8]. Thyroid hormone modulates all metabolic signaling pathways and consequently, altered TH concentrations in humans are associated with profound changes in energy status [8]. The extent of intramuscular TH action is determined both by the systemic levels of TH and by local regulation that modulates the nuclear availability of the hormone [9]. Indeed, although the hypothalamic–pituitary–thyroid axis efficiently regulates TH homeostasis, thus maintaining circulating TH levels in a constant steady-state, its intracellular concentration can rapidly be attenuated or increased independent of serum TH blood levels by the enzymatic control of the selenodeiodinases (D1, D2 and D3) that catalyze TH activation and catabolism [10]. The actions of the three deiodinases, together with the uptake of T3 and T4 into the cell by specific transporters, constitute a mechanism of pre-receptor control of TH action at cellular level regardless of the constant serum T3 levels [10]. The metabolic relevance of the deiodinases is exemplified by the effects of D2 on thermoregulation and the consequent energy expenditure in brown adipose tissue (BAT) [11,12]. Cold exposure causes an increase in the sympathetic activity of BAT, which, in turn, increases lipolysis, mitochondrial uncoupling and D2 activity [13]. In the absence of D2-generated T3, there is a decrease in Ucp1 gene expression and impaired adaptive thermogenesis [11]. In skeletal muscle, D2 expression is barely detectable under basal conditions, however its expression is markedly increased during muscle regeneration [14]. We previously demonstrated that D2 is functionally relevant in muscle stem cells and that loss of D2 severely impairs muscle repair after injury [14,15]. These findings raise the intriguing possibility that, similar to BAT, D2 is a “master regulator” of overall energy metabolism in skeletal muscle during the regeneration process.
To determine the metabolic relevance of intracellular D2-mediated TH changes in muscle cells, we generated a muscle cell line (pTRE-D2) in which D2 can be reversibly induced by doxycycline. We found that induction of D2 induces a shift toward glycolytic metabolism. Importantly, D2 induction decreased basal levels of ROS and induced a parallel up-regulation of the detoxifying gene Sod2. The D2-mediated TH activation led to detoxifying effects that were essential for differentiation. Taken together, these data reveal a novel metabolic role of TH in which this hormone regulates redox balance by inducing the transcription of Sod2 and by inducing a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis (Graphical Abstract).
2. Results
2.1. D2 up-regulation induces a metabolic shift toward glycolysis and reduces oxidative phosphorylation
To determine the metabolic effects of D2 in muscle cells and its contribution to the rate of cellular respiration and ROS production, we generated an inducible C2C12 cell line (pTRE-D2) in which the D2 gene can be turned on by tetracycline administration (tetracycline regulation system, TET-ON, Figure S1). Doxycycline treatment (2 μg/ml) (Fig. 1A) induced up-regulation of D2 mRNA (Fig. 1B), protein (Fig. 1B and C) and enzymatic activity (Fig. 1D). The D2 surge determined increased TH action as indicated by activation of the TRE-LUC-response plasmid (TRE3TK-Luc) transiently transfected in the pTRE-D2 cells (Fig. 1E). This effect was strongly reduced when cells were cultured in the absence of TH (charcoal stripped medium, CH, Fig. 1F), thus confirming that the increased promoter activity is effectively due to the D2-mediated T4-to-T3 conversion. Accordingly, the TH-target genes MyoD, Myogenin and FoxO3a were up regulated in C2C12 cells grown with normal TH-containing FBS after D2 induction, but not in C2C12 cells cultured in the absence of TH (Fig. 1G).
Fig. 1.

D2 is upregulated in C2C12 cells at mRNA and protein level by the tetracycline regulation system, TET-ON.(A) Schematic representation of the doxycycline treatment of proliferating pTRE-D2 C2C12 cells. (B) D2 mRNA and D2-Flag protein expression was evaluated by real time PCR and Western blot analysis in C2C12 pTRE-D2 cells ± 2 μg/ml doxycycline for 24 h. (C) Immunofluorescence analysis of D2-Flag protein (red) in C2C12 pTRE-D2 cells treated as in A revealed the correct expression and localization of D2 by confocal microscopy. (D) D2 enzymatic activity was measured in pTRE-D2 cells ± 2 μg/ml doxycycline for 24 h and 48 h as described in Material and Methods. (E) T3 transactivation activity was measured indirectly by using the T3-reporter plasmid T3TRE-Tk-Luc. C2C12 pTRE-D2 ± 2 μg/ml doxycycline for 24 h were transfected with the T3TRE-TkLuc and CMV-Renilla as internal control. The results are shown as means ± SD of the luciferase/renilla (LUC/Renilla) ratios from at least 3 separate experiments, performed in duplicate. (F) pTRE-D2 cultured in normal serum (NS) or in TH-depleted medium (Charcoal stripped, CH) ±2 and 4 μg/ml doxycycline for 24 h were transfected with the T3TRE-TkLuc and CMV-Renilla as internal control. (G) mRNA expression of the indicated genes was measured by real time PCR in C2C12 pTRE-D2 cells grown in normal serum or in TH-depleted serum (CH). All the experiments were performed in proliferating cells. The results are shown as means ± SD from at least 3 separate experiments. *P < 0.05, **P < 0.01. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Consistent with the finding that TH favors a shift from type I (oxidative) fibers to type II (glycolytic) fibers [10], induction of D2 promoted glycolytic metabolism as demonstrated by a reduced oxygen consumption rate (OCR), and an increased acidification rate (ECAR) (Fig. 2A and S2A and B). Coupling efficiency and spare capacity were reduced, which indicates that an increase in D2, and the consequent production of active TH, down-modulated the overall contribution of OXPHOS to the cellular energy demand (Figure S2C and D). Maximal respiration was accordingly reduced, thus confirming the reduced ability to address the energy demands in conditions of increased D2-mediated TH up-regulation (Fig. 2B). Furthermore, the mitochondrial uncoupling protein UCP3 was strongly upregulated, thus supporting the reduced respiration rate in D2-overexpressing muscle cells (Fig. 2C). D2 did not affect total ATP production as demonstrated by both Seahorse assay and quantitation of ATP levels (Fig. 2D). However, the differential contribution to the ATP production rate of OXPHOS, glycolysis and other metabolic pathways, differed between control cells and D2-overexpressing cells. Treatment with 2-deoxyglucose, an inhibitor of ATP production by glycolysis had a more potent effect on ATP production in D2-expressing cells, while conversely, treatment with oligomycin which inhibits ATP production by OXPHOS did not decrease ATP production in D2-expressing cells (Fig. 2D). Overall these data indicate that D2 induction reduced the contribution of OXPHOS to ATP production and increased the contribution of glycolysis (Fig. 2E).
Fig. 2.

Thyroid hormone activation via D2 induces a shift from OxPhos to glycolysis.(A, B) pTRE-D2 cells were treated with 2 μg/ml doxycycline for 24 h and 48 h and the rate of OXPHOS and glycolysis was measured with the Seahorse analyzer. OCR was measured continuously throughout the experiment at baseline and in the presence of the indicated drugs. (C) UCP3 mRNA was measured by real time PCR analysis in pTRE-D2 cells treated with doxycycline for 48 h. Cyclophilin A served as internal control. Normalized copies of UCP3 in untreated cells (CTR) were set as 1. (D, E) ATP production was measured in pTRE-D2 cells treated with doxycycline for 48 h by using the ATPlite kit. (F) Expression levels of the myosin heavy chain isoforms (MHC) were measured by real time PCR analysis in pTRE-D2 cells treated with 2 μg/ml doxycycline for 24 h. All the experiments were performed in proliferating cells; the results are shown as means ± SD from at least 3 separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Analysis of myosin heavy chain expression showed that D2 induction drastically reduces the expression of the slow, oxidative fibers (type I fibers, MHC I) and increases the expression of the fast, glycolytic fibers (type II, MHC IIa and IIb) (Fig. 2F). These results confirmed that the metabolic shift induced by D2 up-regulation is associated with changes in MHC expression towards faster, more glycolytic isoforms.
Taken together, the data reported above demonstrate that D2 reduces the rate of OXPHOS in muscle C2C12 cells.
2.2. Basal ROS production is reduced by D2 induction
We evaluated if the observed metabolic shift from OXPHOS to glycolysis induced by D2 affects ROS homeostasis. We found that the intracellular basal ROS level was significantly reduced by D2 induction (Fig. 3A). Moreover, the rate of ROS production induced by doxorubicin was similarly reduced by D2 (Fig. 3B). These findings suggest that D2 and TH activation protects against basal and doxorubicin-induced oxidative stress.
Fig. 3.

D2 reduces endogenous ROS production.(A) Basal ROS were measured by FACS analysis in pTRE-D2 cells treated with doxycycline (DOX) and in C2C12 control cells. Bottom panel shows the relative mean fluorescence compared to CTR cells arbitrarily set as 1. (B) ROS production was evaluated as in A in pTRE-D2 cells treated with doxycycline and/or with 0.5, 1 or 2 μM doxorubicin (DB) as indicated. Bottom panel represents relative mean fluorescence compared to CTR cells arbitrarily set as 1. (C) Mitochondrial ROS were measured by fluoresecence-activated cell sorting using MitoSOX dye in pTRE-D2 cells treated with doxycycline for the indicated time points, and in non treated pTRE-D2 cells (controls). Right panel shows the relative mean fluorescence versus CTR cells arbitrarily set at 1. (D, E) mRNA expression of a panel of antioxidant genes (D) and OXPHOS genes (E) was measured in pTRE-D2 cells treated with doxycycline for 24 h and in control cells. Cyclophilin A was used as internal control. Normalized copies of the indicated gene in untreated cells (CTR) were set as 1. All the experiments were performed in proliferating cells; the results are shown as means ± SD from at least 3 separate experiments. *P < 0.05, **P < 0.01.
Using the mitochondrial indicator, mitoSOX, we also evaluated the effects of D2 on mitochondrial ROS. Unsurprisingly, mitochondrial ROS levels were reduced by D2 induction (Fig. 3C). To determine whether the effects exerted by D2 on endogenous ROS production were due to enhanced antioxidant potential (scavenger pathway) or to a reduced ROS production rate (oxidative stress pathway), we measured the expression of a panel of antioxidant genes and OXPHOS genes. While OXPHOS gene expression was unchanged by D2 induction, the expression of three antioxidant genes (Sod2, Gpx1 and Gclc) was up-regulated by D2 (Fig. 3D and E).
2.3. D2-mediated TH activation potentiates antioxidant fluxes by up-regulating SOD2
The analysis of ChIP-Seq data in T3-treated C2C12 cells versus untreated C2C12 cells demonstrated that, among the three up-regulated genes in Fig. 3D, superoxide-dismutase SOD2 is a putative direct target of T3 (Fig. 4A). Chromatin immunoprecipitation assay confirmed that the thyroid receptor TRα physically binds to the Sod2 promoter region (Fig. 4B). Moreover, TH treatment and D2 induction caused up-regulation of the SOD2 protein in muscle cells (Fig. 4C–E). Accordingly, in vivo SOD2 levels were higher in hyperthyroid gastrocnemius (GC) muscles and lower in hypothyroid GC muscles than in control muscles, (Fig. 4F and G). SOD2 expression was also higher and mitochondrial ROS expression lower in muscle fibers isolated from hyperthyroid muscles (Fig. 4H, I and S3). These data reveal that the scavenger protein SOD2 is a novel TH-target gene, which, in turn, suggests that TH plays a specific role in the regulation of oxidative stress.
Fig. 4.

Thyroid hormone is a positive regulator of SOD2 transcription.(A) ChIP seq analysis of T3-target genes performed in C2C12 cells showed that the SOD2 gene is a direct target of T3. The mammalian conservation, the gene structure and position from Genome Browser and the ChIP Seq results (black bars) are indicated for the SOD2 gene. (B) Chromatin immunoprecipitation assay was performed in C2C12 cells. Immunoprecipitation of the chromatin using the anti-TRα antibody revealed that the SOD2 gene is a direct target of T3. (C, D) Protein levels of SOD2 were measured by Western blot analysis in pTRE-D2 cells treated or not with doxycycline and in cells treated with TH for 10 h and 24 h. (E) Immunofluorescence analysis of SOD2 expression in pTRE-D2 cells treated or not with doxycycline. (F, G) Real time PCR analysis and Western blot analysis of SOD2 expression in gastrocnemius (GC) muscles of hyper- and hypothyroid mice (see Methods). (H) SOD2 expression was analyzed in muscle fibers isolated from control (euthyroid) and hyperthyroid mice. (I) mitoSOX analysis in the same muscle fibers as in H was assessed by immunofluorescence. The results are shown as means ± SD from at least 3 separate experiments. *P < 0.05, **P < 0.01.
Given that FoxO proteins play a role in inducing muscle differentiation [18] thereby repressing oxidative stress and regulating SOD2 expression [19], and also given the reciprocal cross talk between TH-FoxO3 and D2 [14], we asked whether FoxO3 is involved in the mechanism by which TH regulates ROS production in muscle cells. To this aim, we transfected C2C12 cells with FoxO3 dominant negative (FoxO DN) or empty vector (CMV-Flag) and treated them with TH. We also transfected C2C12 cells with a FoxO3 shRNA or control shRNA (as described in [14]) and treated them with TH. Although, as expected, basal levels of SOD2 and GCLC were reduced by FoxO3 inhibition, TH treatment induced the expression of these antioxidant genes to a similar extent regardless of FoxO3 expression, which suggests that the expression of TH-induced detoxifying genes is independent of FoxO3 (Figure S4A, B). Accordingly, intracellular ROS levels were similarly down-regulated by TH in the presence and absence of FoxO3 (Figure S4C). These data indicate that TH and FoxO3 exert cooperative, but not synergistic effects, in protecting muscle cells from oxidative stress (Figure S4D).
2.4. The antioxidant action of TH augments myogenic differentiation
The cell-signaling role of ROS includes the ability to inhibit myogenic differentiation [[20], [21], [22]], whereas antioxidant genes are positive regulators of myogenic differentiation [23]. Since TH and its activation by D2 are critical mediators of myogenic differentiation, we evaluated whether reduction of ROS levels is part of the TH-dependent cascade that triggers myogenic differentiation. To this aim, we first analyzed the effects of D2 induction on muscle cell proliferation and differentiation. As expected, D2 induction led to reduced proliferation and enhanced differentiation. Indeed, cyclin D1 and EdU positive cells were down regulated (Fig. 5A–C), while the differentiation markers Myogenin, desmin and MHC were up regulated by D2 (Fig. 5D and E). Notably, the differentiation observed in doxycycline-treated cells reflected the levels of D2 induction in a dose- and time-dependent manner. Indeed, the expression of Myogenin was up regulated by doxycycline-induced D2 up to 8 μg, 24 h after doxycycline treatment (Fig. 5F–H). ROS levels paralleled D2 expression, being down regulated by D2 induction in a dose-dependent manner (Fig. S5). Moreover, time-course induction of D2 showed that the SOD2 and ROS levels were inversely regulated by D2 in a time-dependent manner in myoblasts exposed to doxycycline for 48 h, whereas prolonged exposure of myoblasts to high D2 levels reversed these effects which suggests that prolonged activation of D2 might have deleterious effects on oxidative stress (Figure S6).
Fig. 5.

D2 upregulation induces myogenic differentiation.(A) Schematic representation of the doxycycline treatment of proliferating and differentiating pTRE-D2 C2C12 cells. (B) Cyclin D1 mRNA and protein levels were measured in proliferating pTRE-D2 cells treated with 2 μg/ml doxycycline for 24 h. (C) EdU incorporation was assessed by immunofluorescence analysis of the same cells as in A. (D) Myogenin and MHC mRNA levels were measured in pTRE-D2 cells grown in proliferative or differentiative conditions as indicated in A and treated or not with 2 μg/ml doxycycline for 24 h. (E) Immunofluorescence analysis of Myogenin, Desmin and MHC expression was performed in differentiated pTRE-D2 cells grown as shown in A. Scale bars represent 200 μm and 50 μm, respectively. (F) mRNA D2 expression levels were measured in proliferating pTRE-D2 cells treated for 24 h with doxycycline at the indicated concentrations and in pTRE-D2 non treated cells (control cells). (G) D2, Myogenin and SOD2 protein levels were measured by Western blot in the same cells as in F. (H) Quantification of the single protein levels versus tubulin levels is represented by histograms. The results are shown as means ± SD from at least 3 separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, to assess if the antioxidant effects of D2 and TH are mediated by the superoxide dismutase SOD2, we down-regulated SOD2 in C2C12 cells using the siRNA method. Two siRNAs (shSOD.1 and shSOD.2) were selected and validated for effective silencing of SOD2 by greater than 50% (Fig. S7). Importantly, SOD2 silencing reduced the ability of D2 to trigger cellular differentiation, as indicated by reduced levels of myogenic differentiation markers (Fig. 6A and B). Furthermore, SOD2 inhibition potently reduced the antioxidant effects of D2 on ROS and mitochondrial ROS (Fig. 6C), which confirms that SOD2 is the effector of TH-dependent ROS scavenger activity. These results indicate that the intracellular activation of TH by D2 is a metabolic modulator of muscle cells, and that activation of D2 in proliferating muscle cells induces cell differentiation by regulating the ROS balance, thereby linking the pro-differentiative action of TH to its metabolic action (Fig. 6D).
Fig. 6.

SOD2 silencing attenuates TH-mediated myogenic differentiation.(A) mRNA expression of Desmin, Myogenin and MHC were measured in differentiated pTRE-D2 cells treated or not with doxycycline and transfected with SOD2.1 shRNA or CTR shRNA as indicated. (B) Immunofluorescence analysis of Desmin and Myogenin expression of the same cells as in A. Scale bars represent 200 μm and 50 μm, respectively. (C) Levels of total ROS and mitochondrial ROS were measured by FACS analysis of the same cells as in A. (D) D2 up-regulation increases myogenic differentiation and reduces ROS production. SOD2 silencing attenuates the pro-differentiative ability of D2. **P < 0.01, ***P < 0.001.
3. Discussion
Despite the large body of data on the multiple roles of TH signaling in muscle metabolism, several fundamental questions remain unanswered: (i) Since TH increases the cellular metabolic rate, how do TH-dependent metabolic changes affect oxidative stress? (ii) What is the tissue-specific role played by TH modulators and deiodinases in cellular respiration and oxidative stress? (iii) What are the cross-implications between the metabolic and pro-differentiative roles of TH in muscle?
In this study we demonstrate that TH and its activating enzyme, D2, attenuate intracellular ROS production in C2C12 muscle cells. Our study shows that TH alters the intracellular metabolism of muscle cells by reducing the extent of OCR and inducing ECAR. This is in line with the ability of TH to induce a shift from slow-to-fast muscle fibers [10]. Significantly, the same shift was associated with increased uncoupling power and UCP3 expression, and a reduced rate of OXPHOS, thus suggesting that the intracellular activation of TH might reduce oxidative stress. This concept is further validated by the finding that the endogenous production of ROS was markedly reduced in muscle cells overexpressing D2. This role was unexpected given the pro-apoptotic action of TH in muscle stem cells [24]. Similarly, since TH is commonly regarded as a hyper-metabolic agent, one might expect that it should be associated with enhanced cellular respiration and increased oxidative damage, which was not confirmed by our study.
Our finding that D2-mediated TH activation reduces oxidative stress in muscle cells is in line with reports that TH and its activation by D2 is critical for the maintenance of cellular homeostasis during stress responses as occurs in idiopathic pulmonary fibrosis [25]. In contrast, a study performed with hepatic HepG2 cells showed that an increased T3 signal leads to increased mitochondrial activity, OXPHOS rate and fission [26]. These discrepancies highlight the complicate interconnections between TH and cellular metabolism, and can be partially explained by speculating that TH exerts dose-dependent control over ROS dynamics, thereby reducing ROS production under physiological circumstances, while increasing ROS production and apoptosis in conditions of thyrotoxicosis. Compatible with this speculation, we found that, while D2-TetON cells displayed ROS reduction, increased levels of proteins involved in ROS-detoxification, and greater susceptibility to the myogenic process, prolonged exposure to D2 or excessive levels of D2 induction produced the opposite effects.
Mitochondria are the predominant site of ROS production in skeletal muscle [27]. Contextually, TH is a central regulator of mitochondrial biogenesis (via the peroxisome proliferator-activated receptor gamma, coactivator 1α) [28] and of dynamics (via DRP-1) [26]. Moreover, the existence of a mitochondrial-specific TH receptor (p43) emphasizes the important role of TH as a mitochondrial regulator [29]. These examples illustrate that TH affects cellular respiration in mitochondria through multiple genomic and non-genomic pathways [30].
Our finding that mitochondrial ROS production was reduced by TH confirms the intimate relationship between TH and mitochondria, and is in accordance with the concept that TH enhances muscle cell differentiation. We also found that SOD2, the primary mitochondrial oxidative scavenger, is a novel TH-target gene. Our data show that the genomic action of TH via TRα increased SOD2 transcription and protein synthesis in vitro and in vivo and importantly, SOD2 silencing interfered with TH-dependent decrease in ROS, which indicates that SOD2 is a TH-downstream mediator of ROS dynamics.
To explore the connection between TH and ROS in protein regulation, we evaluated whether the detoxifying action of TH requires FoxO3, which is an upstream regulator of D2 expression [14], and, contextually, a TH-target gene [24]. Surprisingly, we found that, although both FoxO3 and D2 reduced intracellular ROS and that both induced SOD2 expression, the effects of D2 on ROS generation remained unchanged in the absence of FoxO3. These data indicate that the TH signal and FoxO3 play convergent roles in oxidative stress regulation, but do not synergistically reduce endogenous ROS.
Radical oxygen species are no longer viewed solely as damaging agents. Indeed, in many physiological processes, ROS are intracellular signaling molecules [2]. In muscle cells, increased ROS levels inhibit myotube formation while favoring cell proliferation [2]. These effects raise the possibility that TH-dependent regulation of myogenic differentiation is linked to its detoxifying action. Our data demonstrate that D2-mediated TH activation triggers muscle differentiation by reducing oxidative stress. Indeed, SOD2 inhibition partially reduces the ability of D2 to induce muscular differentiation.
In conclusion, under physiological conditions, intracellular activation of TH via D2 attenuates cellular reliance on aerobic glycolysis, thus reducing mitochondrial respiration and ROS production, which in turn minimizes oxidative stress and augments myogenic differentiation. In addition, TH induces the increased expression of the ROS scavenger SOD2. Here we describe a model whereby D2-mediated TH activation regulates redox balance by inducing SOD2 transcription and by inducing a metabolic shift from OXPHOS to glycolysis (Graphical Abstract). This suggests that D2 is a key player in the induction of fast-twitch fibers in muscle tissue and that it is involved in activation of alternative, uncoupling processes.
4. Materials and methods
4.1. Cell cultures
C2C12 cells were obtained from ATCC and cultured in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% FBS (Microgem), 2 mM glutamine, 50 i.u. penicillin, and 50 μg/ml streptomycin (proliferating medium). C2C12 TET-ON D2 cells were cultured in DMEM supplemented with 10% tetracycline-free Fetal Bovine Serum (FBS) (Clontech, Mountain View, CA, USA), 100 μg/ml geneticin (Biowest, Nuaillé, France) and 0.8 μg/ml puromycin (Invitrogen, Carlsbad, CA, USA). For studies in proliferative conditions (PROL), cells were grown at 40–50% confluence and treated with the indicated amounts of doxycycline for 16, 24 or 48 h as indicated. For experiments in differentiation conditions (DIFF), cells were grown at 60%–70% confluence, treated with doxycycline and then induced to differentiate in Differentiation Medium (DMEM with 2% horse serum, insulin 10 μg/ml and transferrin 5 μg/ml). D2 expression was turned on by 2 μg/ml doxycycline (Clontech, Mountain View, CA, USA). In some experiments, TH (T3 and T4, Sigma-Aldrich S. Louis, Missouri, USA) were added in culture medium at a 30 nM final concentration or removed from the FBS by charcoal absorption (Larsen, 1972).
4.2. Constructs and transfections
The set of vectors for tetracycline-inducible transgene expression, constituted by the response pTRE3G, the regulator pCMV-Tet3G and the pTRE3G-Luc control plasmids, was purchased from Clontech Laboratories. The full-length coding sequence of Dio2 gene, the 3′-UTR containing the SECIS and three repetitions of the Flag tag at the N-terminal, was inserted into the EcoRV site of the pTRE3G vector (Fig. S1). A puromycin resistance gene was also cloned in the XhoI site of the same vector for the screening of positive clones. Direct sequencing was carried out to verify the correct sequence (Nucleic acid sequencing, CEINGE-Biotecnologie Avanzate s.c. a r.l.).
4.3. Conditional Dio2 expression in C2C12 cells
Transfection experiments were performed using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. Briefly, C2C12 cells were stably transfected with the pCMV-Tet3G plasmid that encodes the reverse transactivator and the neomycin phosphotransferase gene. Transfected cells were selected in media with 100 μg/ml G418 and single clones were isolated and expanded. Clones with a low basal activity and high transactivator inducibility in the presence of doxycycline were identified by transient transfections with the pTRE3G-Luc control vector. Two clones displaying approximately 2500-fold (clone 6) and 3800-fold (clone 8) luciferase induction were chosen for the second round of transfection with the pTRE3G-D2 plasmid. Cells were selected in media with 100 μg/ml G418 and 0.8 μg/ml puromycin, yielding a total of 90 clones. Dio2 expression was induced in all selected clones by supplementing the medium with 2 μg/mL doxycycline. Cells were recovered after 48 h of induction and Western blot using a monoclonal anti-Flag M2 antibody (Sigma-Aldrich) was performed to detect the D2-Flag in the cell lysates. The clone with the highest fold induction of the D2 protein (clone 7) was selected for propagation and further testing. Conditional Dio2 expression was also confirmed by mRNA analysis and Luciferase (Luc) expression assays.
4.4. Luciferase (Luc) expression assays
The luciferase reporter plasmids TRE3-TkLuc or the pTRE3G-Luc and CMV-Renilla were co-transfected into cells. Luc activities were measured 48 h after transfection with the Dual Luciferase Reporter Assay System (Promega, Madison, Wisconsin, USA), and differences in transfection efficiency were corrected relative to the level of Renilla Luciferase. Each construct was studied in triplicate in at least three separate transfection experiments.
4.5. D2 deiodination assays
Cells were homogenized on ice in phosphate buffer containing 0.25 M sucrose, 1 mm EDTA, and 10 mM DTT and complete protease inhibitor cocktail from Roche. Reactions were incubated at 37 °C for 4 h in 0.1 ml PE buffer with 1 nM or 500 nM T4 (blank), 20 mM DTT, 1 mM propylthiouracil, with the addition of approximately 2 × 105 cpm [3ʹ-5ʹ-125I]T4 [14]. Blank values were subtracted before calculation of deiodinase activities.
4.6. In vivo experiments
12-week-old C57BL/6 male mice (The Jackson Laboratory, Bar Harbor, Maine, USA) (WT) were made hyperthyroid by intraperitoneally injection of TH (5 μg/mouse T3 + 20 μg/mouse T4) 3 days before harvesting the muscles. Hypothyroidism was obtained by adding 0.1% MMI and 1% KClO4 to the drinking water for 6 weeks. Animal experimental protocols were approved by the Animal Research Committee of the University of Naples Federico II.
4.7. Western blot analysis
Total protein extracts from cells and muscles were run on a 10% SDS-PAGE gel and transferred onto an Immobilon-P transfer membrane (Millipore, Burlington, MA, USA). The membrane was then blocked with 5% non-fat dry milk in PBS, probed with anti-SOD2 (sc-133134) and anti-Cyclin D1 (sc-20044) antibodies overnight at 4 °C, washed, and incubated with horseradish peroxidase–conjugated anti-mouse immunoglobulin G secondary antibody (1:3000), and detected by chemiluminescence (Millipore, cat. WBKLS0500). After extensive washing, the membrane was incubated with anti-tubulin (sc-8035) antibody as loading control. All Western blots were run in triplicate, and bands were quantified with ImageJ software.
4.8. Chromatin immunoprecipitation (ChIP) assay
Approximately 2 × 106 C2C12 cells were fixed for 10 min at 37 °C by adding 1% formaldehyde to the growth medium. Fixed cells were harvested and the pellet was resuspended in 1 ml of lysis buffer containing protease inhibitors (200 mM phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin). The lysates were sonicated to obtain DNA fragments of 200–1000 bp. Sonicated samples were centrifuged and the soluble chromatin was diluted 10-fold in dilution buffer and used directly for ChIP assays. An aliquot (1/10) of sheared chromatin was further treated with proteinase K, extracted with phenol/chloroform and precipitated to determine DNA concentration and shearing efficiency (“input DNA”). Briefly, the sheared chromatin was pre-cleared for 2 h with 1 μg of non-immune IgG (Calbiochem, Burlington, MA, USA) and 30 μL of Protein G Plus/Protein A Agarose suspension (Calbiochem) saturated with salmon sperm (1 mg/mL). Precleared chromatin was divided in aliquots and incubated at 4 °C for 16 h with 1 μg of anti-THR antibody (C3) (ab2743, Abcam). After five rounds of washing, bound DNA-protein complexes were eluted by incubation with 1% sodium dodecyl sulfate-0.1 M NaHCO3 elution buffer. Formaldehyde cross-links were reversed by incubation in 200 mM NaCl at 65 °C. Samples were extracted twice with phenol-chloroform and precipitated with ethanol. DNA fragments were recovered by centrifugation, resuspended in 50 μl H2O, and used for real-time PCRs.
4.9. Real-time PCR
Messenger RNAs were extracted with Trizol reagent (Life Technologies). Complementary DNAs were prepared with Vilo reverse transcriptase (Life Technologies) as indicated by the manufacturer. The cDNAs were amplified by PCR in an iQ5 Multicolor Real Time Detector System (BioRad) with the fluorescent double-stranded DNA-binding dye SYBR Green (BioRad). Specific primers for each gene were designed to work under the same cycling conditions (95 °C for 10 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min), thereby generating products of comparable sizes (about 200 bp for each amplification). Primer combinations were positioned whenever possible to span an exon–exon junction and the RNA digested with DNAse to avoid genomic DNA interference. Primer sequences are reported in the Supplementary material (Table 1). For each reaction, standard curves for reference genes were constructed based on six four-fold serial dilutions of cDNA. All samples were run in triplicate. The template concentration was calculated from the cycle number when the amount of PCR product passed a threshold established in the exponential phase of the PCR. The relative amounts of gene expression were calculated with cyclophilin A expression as an internal standard (calibrator). The results, expressed as N-fold differences in target gene expression, were determined as follows: N *target = 2(ΔCt sample−ΔCt calibrator).
4.10. Cellular bioenergetics
Cellular bioenergetics was analyzed on a Seahorse extracellular flux bioanalyzer (XF96) according to the manufacturer’s instructions. Briefly, C2C12 TET-ON D2 cells were seeded at an appropriate density in a Seahorse XF96 plate. Cells were cultured for 24 h and 48 h in growth media supplemented with doxycycline. Immediately prior to the assay, all cells were equilibrated in minimal assay media (Seahorse Biosciences, 37 °C, pH 7.40) supplemented with 25 mM glucose and 1 mM sodium pyruvate for 45–60 min in a non-CO2 incubator. The basal rates of oxygen consumption (OCR) and extracellular-acidification (ECAR) were measured in triplicate for 2 min (for a total of 15mins) from the rate of decline in O2 partial pressure (OCR), and the rate of change in assay pH (ECAR). Mitochondrial respiration and glycolytic activity were monitored at basal level and after sequential injection of the mitochondrial modulators oligomycin (3 μM) for the oxygen consumed for ATP production, FCCP (6 μM) for maintenance of the proton gradient and antimycin (2.5 μM) that induce mitochondrial stress (spare respiratory capacity and non-mitochondrial respiration). ECAR is expressed in milli pH (mpH) units representing the change in pH per minute and is measured simultaneously with OCR in the Seahorse assay. ATP levels were determined using the ATPlite 1 step kit (PerkinElmer) following the manufacturer’s recommendations.
4.11. Short hairpin RNA-mediated knockdown of SOD2
C2C12 TET-ON D2 cells were grown until they reached 60% confluence and then they were transfected with two SOD2 shRNAs (SOD.1 ID 152022; SOD.2 ID 152023, Thermofisher Scientific) and control shRNA (AM4635, Thermofisher Scientific). Forty-eight hours after transfection, total protein lysate was collected and analyzed by Western blot, total RNA was extracted and analyzed by real-time PCR and intracellular ROS were measured by Flow Cytometry.
4.12. Immunofluorescence
For immunofluorescence staining, cells were fixed with 4% formaldehyde and permeabilized in 0.1% Triton X-100, then blocked with 0.2% BSA/PBS and washed in PBS. Cells were then incubated with primary antibody overnight at 4 °C. Secondary antibody incubation was carried out at room temperature for 1 h, followed by washing in 0.2% Tween/PBS. Images were acquired with an IX51 Olympus microscope and the Cell*F Olympus Imaging Software.
4.13. Measurement of cellular ROS
Total and mitochondrial ROS levels were measured using the 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate CM-H2DCFDA probe and mitoSOX dye, respectively (Molecular Probes, USA), according to the manufacturer’s instructions. Briefly, after trypsinization with 0.25% (w/v) Trypsin–EDTA (Life Technologies), cells were collected and rinsed with PBS. Cells were then resuspended and incubated in pre-warmed PBS containing 5 μM CM-H2DCFDA or 5 μM mitoSOX in the dark for 20 min at 37 °C. As positive control, total ROS levels were evaluated after treating with 200 μM H2O2. Intracellular fluorescence was then quantified using a FACS Canto2 (Becton Dickinson, USA). To evaluate the contribution of D2 on induced ROS production, cells were treated for 16 h with 0.5 μM, 1 μM and 2.5 μM of doxorubicin (Sigma Aldrich).
4.14. Confocal microscopy
C2C12 TET-OND2 cells were grown on coverslips and treated with doxycycline for 48 h. Cells were labeled with mitoSOX Red reagent, which fluoresces when oxidized by superoxide, and nuclei were stained with the blue fluorescent dye Hoechst 33342 (Sigma Aldrich). Images were acquired on a Zeiss 510 confocal microscope (Carl Zeiss, Oberkochen, Germany) using an x60 1.4 NA oil immersion objective using the same instrument settings for each image.
4.15. Isolation and loading of single skeletal muscle fibers with CM-H2DCFDA and mitoSOX
Male C57BL/6 mice, 3 months old, were euthanized and the extensor digitorum longus (EDL) muscles were removed and placed into 0.1% type 1 collagenase (C0130, Sigma–Aldrich Co. St. Louis, MO) solution in DMEM. Both EDL muscles from each mouse were incubated in collagenase solution at 37 °C for 1 h. Fiber bundles that had not been released during the incubation were separated using a wide-bore glass pipette. The fibers were washed four times in fresh culture medium. Cleaned fibers were plated onto 60-mm dishes in medium for satellite cells (50% DMEM, 50% MCDB, 20% FBS, 1% Ultroser G, 2 mM glutamine, 50 i.u. penicillin, and 50 μg/ml streptomycin). Fibers were incubated for 30 min at 37 °C. The medium was then replaced by PBS containing 5 μM mitoSOX and incubated for 20 min at 37 °C. The fibers were then washed and analyzed by fluorescence microscopy. Animal experimental protocols were approved by the Animal Research Committee of the University of Naples Federico II.
4.16. Statistics
The results are shown as means ± SD throughout. Differences between samples were assessed by the Student’s two-tailed t-test for independent samples. Relative mRNA levels (in which the control sample was arbitrarily set as 1) are reported as results of real-time PCR, in which the expression of cyclophilin A served as housekeeping gene. In all experiments, differences were considered significant when p was less than 0.05. Asterisks indicate significance at *P < 0.05, **P < 0.01, and ***P < 0.001 throughout.
Conflicts of interest
The authors have declared no conflict of interest.
Acknowledgments
This work was supported by grants from AIRC to M.D. (IG 13065) and by the ERCStG2014 grant from European Research Council under the European Union's Horizon2020 Programme (STARS – 639548) to M.D; by the grant from the European Research Council under the European Union's Horizon2020 Programme – EU FP7 contract Thyrage (grant number 666869) to D.S. and M.D; R01 DK044128 to A. M. Z. from NIH; A.N.O.W. is supported by T32 DK007529 from NIH. The authors have declared that no conflict of interest exists. We thank Jean Ann Gilder (Scientific Communication srl., Naples, Italy) for writing assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101228.
Author contributions
S.S., A.G.C., R.A., C.M., D.D.G., A.N., G.M., M.A.D.S. and C.L. performed the in vitro and in vivo experiments and prepared the figures; E.D.C. performed histochemistry and immunofluorescence; M.R. performed the FACS analysis studies; A.N.O.W. and A.M.Z. performed enzymatic activity assays, S.P. performed confocal microscopy experiments, A.M.Z and S.P. provided observations and scientific interpretations; D.S. and M.D. designed the overall study, supervised the experiments, analyzed the results, and wrote the paper. All authors discussed the results and provided input on the manuscript.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Murphy M.E., Kehrer J.P. Activities of antioxidant enzymes in muscle, liver and lung of chickens with inherited muscular dystrophy. Biochem. Biophys. Res. Commun. Jan 29 1986;134:550–556. doi: 10.1016/s0006-291x(86)80455-7. [DOI] [PubMed] [Google Scholar]; M. E. Murphy and J. P. Kehrer, "Activities of antioxidant enzymes in muscle, liver and lung of chickens with inherited muscular dystrophy," Biochem Biophys Res Commun, vol. 134, pp. 550-556, Jan 29 1986. [DOI] [PubMed]
- 2.Barbieri E., Sestili P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012:982794. doi: 10.1155/2012/982794. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]; E. Barbieri and P. Sestili, "Reactive oxygen species in skeletal muscle signaling," J Signal Transduct, vol. 2012, p. 982794, 2012. [DOI] [PMC free article] [PubMed]
- 3.Kanter M. Free radicals, exercise and antioxidant supplementation. Proc. Nutr. Soc. Feb 1998;57:9–13. doi: 10.1079/pns19980004. [DOI] [PubMed] [Google Scholar]; M. Kanter, "Free radicals, exercise and antioxidant supplementation," Proc Nutr Soc, vol. 57, pp. 9-13, Feb 1998. [DOI] [PubMed]
- 4.Rochard P., Rodier A., Casas F., Cassar-Malek I., Marchal-Victorion S., Daury L. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. J. Biol. Chem. Jan 28 2000;275:2733–2744. doi: 10.1074/jbc.275.4.2733. [DOI] [PubMed] [Google Scholar]; P. Rochard, A. Rodier, F. Casas, I. Cassar-Malek, S. Marchal-Victorion, L. Daury, et al., "Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors," J Biol Chem, vol. 275, pp. 2733-2744, Jan 28 2000. [DOI] [PubMed]
- 5.Tidball J.G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. Feb 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]; J. G. Tidball, "Inflammatory processes in muscle injury and repair," Am J Physiol Regul Integr Comp Physiol, vol. 288, pp. R345-R353, Feb 2005. [DOI] [PubMed]
- 6.Adhihetty P.J., Irrcher I., Joseph A.M., Ljubicic V., Hood D.A. Plasticity of skeletal muscle mitochondria in response to contractile activity. Exp. Physiol. Jan 2003;88:99–107. doi: 10.1113/eph8802505. [DOI] [PubMed] [Google Scholar]; P. J. Adhihetty, I. Irrcher, A. M. Joseph, V. Ljubicic, and D. A. Hood, "Plasticity of skeletal muscle mitochondria in response to contractile activity," Exp Physiol, vol. 88, pp. 99-107, Jan 2003. [DOI] [PubMed]
- 7.Sestili P., Barbieri E., Martinelli C., Battistelli M., Guescini M., Vallorani L. Creatine supplementation prevents the inhibition of myogenic differentiation in oxidatively injured C2C12 murine myoblasts. Mol. Nutr. Food Res. Sep 2009;53:1187–1204. doi: 10.1002/mnfr.200800504. [DOI] [PubMed] [Google Scholar]; P. Sestili, E. Barbieri, C. Martinelli, M. Battistelli, M. Guescini, L. Vallorani, et al., "Creatine supplementation prevents the inhibition of myogenic differentiation in oxidatively injured C2C12 murine myoblasts," Mol Nutr Food Res, vol. 53, pp. 1187-1204, Sep 2009. [DOI] [PubMed]
- 8.Cicatiello A.G., Di Girolamo D., Dentice M. Metabolic effects of the intracellular regulation of thyroid hormone: old players, new concepts. Front. Endocrinol. 2018;9:474. doi: 10.3389/fendo.2018.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]; A. G. Cicatiello, D. Di Girolamo, and M. Dentice, "Metabolic Effects of the Intracellular Regulation of Thyroid Hormone: Old Players, New Concepts," Front Endocrinol (Lausanne), vol. 9, p. 474, 2018. [DOI] [PMC free article] [PubMed]
- 9.Gereben B., Zavacki A.M., Ribich S., Kim B.W., Huang S.A., Simonides W.S. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. Dec 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]; B. Gereben, A. M. Zavacki, S. Ribich, B. W. Kim, S. A. Huang, W. S. Simonides, et al., "Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling," Endocr Rev, vol. 29, pp. 898-938, Dec 2008. [DOI] [PMC free article] [PubMed]
- 10.Salvatore D., Simonides W.S., Dentice M., Zavacki A.M., Larsen P.R. Thyroid hormones and skeletal muscle--new insights and potential implications. Nat. Rev. Endocrinol. Apr 2014;10:206–214. doi: 10.1038/nrendo.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]; D. Salvatore, W. S. Simonides, M. Dentice, A. M. Zavacki, and P. R. Larsen, "Thyroid hormones and skeletal muscle--new insights and potential implications," Nat Rev Endocrinol, vol. 10, pp. 206-214, Apr 2014. [DOI] [PMC free article] [PubMed]
- 11.Christoffolete M.A., Linardi C.C., de Jesus L., Ebina K.N., Carvalho S.D., Ribeiro M.O. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. Mar 2004;53:577–584. doi: 10.2337/diabetes.53.3.577. [DOI] [PubMed] [Google Scholar]; M. A. Christoffolete, C. C. Linardi, L. de Jesus, K. N. Ebina, S. D. Carvalho, M. O. Ribeiro, et al., "Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis," Diabetes, vol. 53, pp. 577-584, Mar 2004. [DOI] [PubMed]
- 12.Bianco A.C., Maia A.L., da Silva W.S., Christoffolete M.A. Adaptive activation of thyroid hormone and energy expenditure. Biosci. Rep. Jun-Aug 2005;25:191–208. doi: 10.1007/s10540-005-2885-6. [DOI] [PubMed] [Google Scholar]; A. C. Bianco, A. L. Maia, W. S. da Silva, and M. A. Christoffolete, "Adaptive activation of thyroid hormone and energy expenditure," Biosci Rep, vol. 25, pp. 191-208, Jun-Aug 2005. [DOI] [PubMed]
- 13.Heaton G.M., Wagenvoord R.J., Kemp A., Jr., Nicholls D.G. Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur. J. Biochem. Jan 16 1978;82:515–521. doi: 10.1111/j.1432-1033.1978.tb12045.x. [DOI] [PubMed] [Google Scholar]; G. M. Heaton, R. J. Wagenvoord, A. Kemp, Jr., and D. G. Nicholls, "Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation," Eur J Biochem, vol. 82, pp. 515-521, Jan 16 1978. [DOI] [PubMed]
- 14.Dentice M., Marsili A., Ambrosio R., Guardiola O., Sibilio A., Paik J.H. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J. Clin. Investig. Nov 2010;120:4021–4030. doi: 10.1172/JCI43670. [DOI] [PMC free article] [PubMed] [Google Scholar]; M. Dentice, A. Marsili, R. Ambrosio, O. Guardiola, A. Sibilio, J. H. Paik, et al., "The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration," J Clin Invest, vol. 120, pp. 4021-4030, Nov 2010. [DOI] [PMC free article] [PubMed]
- 15.Marsili A., Tang D., Harney J.W., Singh P., Zavacki A.M., Dentice M. Type II iodothyronine deiodinase provides intracellular 3,5,3'-triiodothyronine to normal and regenerating mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. Nov 2011;301:E818–E824. doi: 10.1152/ajpendo.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; A. Marsili, D. Tang, J. W. Harney, P. Singh, A. M. Zavacki, M. Dentice, et al., "Type II iodothyronine deiodinase provides intracellular 3,5,3'-triiodothyronine to normal and regenerating mouse skeletal muscle," Am J Physiol Endocrinol Metab, vol. 301, pp. E818-E824, Nov 2011. [DOI] [PMC free article] [PubMed]
- 18.Peng X.L., So K.K., He L., Zhao Y., Zhou J., Li Y. MyoD- and FoxO3-mediated hotspot interaction orchestrates super-enhancer activity during myogenic differentiation. Nucleic Acids Res. Sep 6 2017;45:8785–8805. doi: 10.1093/nar/gkx488. [DOI] [PMC free article] [PubMed] [Google Scholar]; X. L. Peng, K. K. So, L. He, Y. Zhao, J. Zhou, Y. Li, et al., "MyoD- and FoxO3-mediated hotspot interaction orchestrates super-enhancer activity during myogenic differentiation," Nucleic Acids Res, vol. 45, pp. 8785-8805, Sep 6 2017. [DOI] [PMC free article] [PubMed]
- 19.Ferber E.C., Peck B., Delpuech O., Bell G.P., East P., Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. Jun 2012;19:968–979. doi: 10.1038/cdd.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]; E. C. Ferber, B. Peck, O. Delpuech, G. P. Bell, P. East, and A. Schulze, "FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression," Cell Death Differ, vol. 19, pp. 968-979, Jun 2012. [DOI] [PMC free article] [PubMed]
- 20.Ardite E., Barbera J.A., Roca J., Fernandez-Checa J.C. Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-kappaB activation. Am. J. Pathol. Sep 2004;165:719–728. doi: 10.1016/s0002-9440(10)63335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; E. Ardite, J. A. Barbera, J. Roca, and J. C. Fernandez-Checa, "Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-kappaB activation," Am J Pathol, vol. 165, pp. 719-728, Sep 2004. [DOI] [PMC free article] [PubMed]
- 21.Fulle S., Protasi F., Di Tano G., Pietrangelo T., Beltramin A., Boncompagni S. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp. Gerontol. Jan 2004;39:17–24. doi: 10.1016/j.exger.2003.09.012. [DOI] [PubMed] [Google Scholar]; S. Fulle, F. Protasi, G. Di Tano, T. Pietrangelo, A. Beltramin, S. Boncompagni, et al., "The contribution of reactive oxygen species to sarcopenia and muscle ageing," Exp Gerontol, vol. 39, pp. 17-24, Jan 2004. [DOI] [PubMed]
- 22.Hansen J.M., Klass M., Harris C., Csete M. A reducing redox environment promotes C2C12 myogenesis: implications for regeneration in aged muscle. Cell Biol. Int. Jun 2007;31:546–553. doi: 10.1016/j.cellbi.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]; J. M. Hansen, M. Klass, C. Harris, and M. Csete, "A reducing redox environment promotes C2C12 myogenesis: implications for regeneration in aged muscle," Cell Biol Int, vol. 31, pp. 546-553, Jun 2007. [DOI] [PMC free article] [PubMed]
- 23.Hidalgo M., Marchant D., Quidu P., Youcef-Ali K., Richalet J.P., Beaudry M. Oxygen modulates the glutathione peroxidase activity during the L6 myoblast early differentiation process. Cell. Physiol. Biochem. 2014;33:67–77. doi: 10.1159/000356650. [DOI] [PubMed] [Google Scholar]; M. Hidalgo, D. Marchant, P. Quidu, K. Youcef-Ali, J. P. Richalet, M. Beaudry, et al., "Oxygen modulates the glutathione peroxidase activity during the L6 myoblast early differentiation process," Cell Physiol Biochem, vol. 33, pp. 67-77, 2014. [DOI] [PubMed]
- 24.Dentice M., Ambrosio R., Damiano V., Sibilio A., Luongo C., Guardiola O. Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression. Cell Metabol. Dec 2 2014;20:1038–1048. doi: 10.1016/j.cmet.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; M. Dentice, R. Ambrosio, V. Damiano, A. Sibilio, C. Luongo, O. Guardiola, et al., "Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression," Cell Metab, vol. 20, pp. 1038-1048, Dec 2 2014. [DOI] [PMC free article] [PubMed]
- 25.Yu G., Tzouvelekis A., Wang R., Herazo-Maya J.D., Ibarra G.H., Srivastava A. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. Jan 2018;24:39–49. doi: 10.1038/nm.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]; G. Yu, A. Tzouvelekis, R. Wang, J. D. Herazo-Maya, G. H. Ibarra, A. Srivastava, et al., "Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function," Nat Med, vol. 24, pp. 39-49, Jan 2018. [DOI] [PMC free article] [PubMed]
- 26.Sinha R.A., Singh B.K., Zhou J., Wu Y., Farah B.L., Ohba K. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy. 2015;11:1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]; R. A. Sinha, B. K. Singh, J. Zhou, Y. Wu, B. L. Farah, K. Ohba, et al., "Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling," Autophagy, vol. 11, pp. 1341-1357, 2015. [DOI] [PMC free article] [PubMed]
- 27.Koren A., Sauber C., Sentjurc M., Schara M. Free radicals in tetanic activity of isolated skeletal muscle. Comp. Biochem. Physiol. B. 1983;74:633–635. doi: 10.1016/0305-0491(83)90241-9. [DOI] [PubMed] [Google Scholar]; A. Koren, C. Sauber, M. Sentjurc, and M. Schara, "Free radicals in tetanic activity of isolated skeletal muscle," Comp Biochem Physiol B, vol. 74, pp. 633-635, 1983. [DOI] [PubMed]
- 28.Weitzel J.M., Iwen K.A. Coordination of mitochondrial biogenesis by thyroid hormone. Mol. Cell. Endocrinol. Aug 6 2011;342:1–7. doi: 10.1016/j.mce.2011.05.009. [DOI] [PubMed] [Google Scholar]; J. M. Weitzel and K. A. Iwen, "Coordination of mitochondrial biogenesis by thyroid hormone," Mol Cell Endocrinol, vol. 342, pp. 1-7, Aug 6 2011. [DOI] [PubMed]
- 29.Wrutniak-Cabello C., Casas F., Cabello G. Thyroid hormone action: the p43 mitochondrial pathway. Methods Mol. Biol. 2018;1801:163–181. doi: 10.1007/978-1-4939-7902-8_14. [DOI] [PubMed] [Google Scholar]; C. Wrutniak-Cabello, F. Casas, and G. Cabello, "Thyroid Hormone Action: The p43 Mitochondrial Pathway," Methods Mol Biol, vol. 1801, pp. 163-181, 2018. [DOI] [PubMed]
- 30.Davis P.J., Goglia F., Leonard J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016;12:111–121. doi: 10.1038/nrendo.2015.205. [DOI] [PubMed] [Google Scholar]; P. J. Davis, F. Goglia, and J. L. Leonard, “Nongenomic actions of thyroid hormone”, Nat Rev Endocrinol, vol. 12, pp. 111-121, 2016. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.