

Summary
The structure-specific endonuclease ERCC1/XPF plays an important role in nucleotide excision repair and interstrand cross-link repair. In this study, we identified new functions of ERCC1/XPF in DNA double-strand break (DSB) repair. We found that the conserved function of ERCC1/XPF to remove non-homologous sequences at DSBs is a rate-limiting step for homologous recombination in mammalian cells, and more importantly, we uncovered an indispensable role of ERCC1/XPF in repair of DSBs containing DNA secondary structures, including the structure-prone AT-rich DNA sequences derived from common fragile sites and G-quadruplexes (G4s). We also demonstrated a synthetic lethal interaction of XPF with DNA translocase FANCM that is involved in removing DNA secondary structures. Furthermore, inactivation of XPF sensitizes FANCM-deficient cells to G4-interacting compounds. These results suggest an important function of ERCC1/XPF in protecting DNA secondary structures and provide a rationale for targeted treatment of FANCM-deficient tumors through inhibition of XPF.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
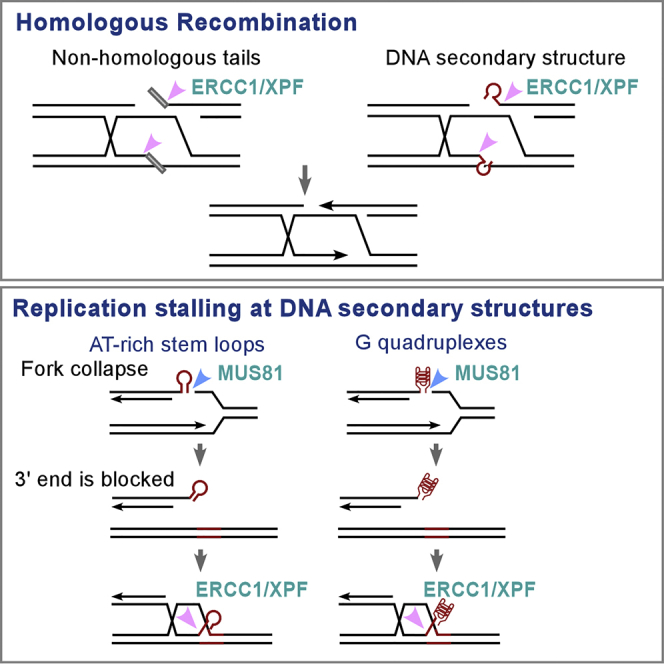
Highlights
-
•
ERCC1/XPF has a conserved role in removing non-homologous tails at DSBs during HR
-
•
ERCC1/XPF is required for repairing DSBs containing DNA secondary structures by HR
-
•
XPF is synthetic lethal with FANCM
-
•
XPF deficiency causes sensitivity to G4-interacting compounds
Biological Sciences; Molecular Biology; Cell Biology
Introduction
XPF forms a heterodimer with ERCC1, and is a highly conserved structure-specific endonuclease that plays an essential role in nucleotide excision repair (NER) (Manandhar et al., 2015). ERCC1/XPF incises double-stranded DNA 5′ specifically to the junction with the single-stranded DNA (ssDNA) (Sijbers et al., 1996) and is essential for removing bulky adducts and UV-induced pyrimidine dimers in NER (Staresincic et al., 2009). In addition to sensitivity to UV radiation, human patients with mutations in XPF often show neurological deterioration and, in some cases, accelerated aging, which are absent in other patients with xeroderma pigmentosum (XP) (Gregg et al., 2011). Mouse models also demonstrate that ERCC1 and XPF deficiencies lead to spontaneous development of symptoms characteristic of progressive neurodegeneration and signs of rapid aging (Gregg et al., 2011). These observations suggest that ERCC1/XPF possesses additional activities outside of NER. In support of this, ERCC1/XPF is required for removing DNA interstrand cross-links (ICLs), and deficiencies in XPF and ERCC1 cause extreme hypersensitivity to ICLs (Wood, 2010, Klein Douwel et al., 2014, Hodskinson et al., 2014).
Studies in yeast revealed an important function of Rad1/Rad10 (orthologs of ERCC1/XPF) in DNA double-strand break (DSB) repair. Rad1 and Rad10 are required for removing 3′ flaps in single-strand annealing (SSA) and for clipping off non-homologous tails in gene conversion (Fishman-Lobell and Haber, 1992, Bardwell et al., 1994, Ivanov and Haber, 1995). In mammalian cells, it has also been shown that ERCC1 deficiency results in a reduction in SSA efficiency (Al-Minawi et al., 2008, Bennardo et al., 2008), and ERCC1 is needed for removal of non-homologous tails in targeted gene replacement between alleles on plasmids and chromosomes (Adair et al., 2000). This suggests that removing non-homologous tails by ERCC1/XPF is a conserved mechanism. The role of ERCC1/XPF in DSB repair is consistent with observations that mice deficient in ERCC1/XPF and fibroblasts derived from XPF-deficient patients are sensitive to ionizing radiation (Ahmad et al., 2008). Genome instability and recombination-dependent deletion and rearrangement are elevated in ERCC1 knockout (KO) mice (Sargent et al., 1997, Melton et al., 1998).
DNA secondary structures often arise at certain genomic loci containing special DNA sequences and consequently, interfere with replication, transcription, and other cellular processes. These structure-forming DNA sequences are found at chromosomal rearrangement breakpoints in cancers and are associated with neurological diseases (Thys et al., 2015). Common fragile sites (CFSs) are large chromosomal regions prone to forming breaks and gaps at metaphase chromosomes upon replication stress (Durkin and Glover, 2007). CFSs often contain AT-rich sequences, which are predicted to form DNA secondary structures leading to replication stalling and fork collapse (Lukusa and Fryns, 2008, Wang et al., 2014, Zhang and Freudenreich, 2007). CFSs are a part of normal chromosome structure present in all human beings, but they are also hotspots for chromosomal rearrangements found in cancers (Durkin and Glover, 2007, Tsantoulis et al., 2008). Another group of DNA sequences that frequently adopt DNA secondary structures are guanine-rich sequences known as G-quadruplexes (G4s) and are often found at sites of genome instability (Wu and Brosh, 2010). In the human genome, more than 700,000 sequences are predicted to have the potential to form G4s, and they tend to locate in promoters, untranslated regions of mRNA, telomeres, and replication origins, where they play important regulatory roles (Rhodes and Lipps, 2015). It is critical to understand how genome stability is preserved at structure-prone DNA sequences.
In this study, we investigated the role of ERCC1/XPF in the repair of DNA DSBs. Surprisingly, we found that XPF is required for repairing not only DSBs containing non-homologous tails but also DSBs that are blocked by DNA secondary structures. In addition, we identified a synthetic lethal interaction of XPF with FANCM and showed that FANCM-deficient cells are sensitive to G4-stabilizing compounds.
Results
Homologous Recombination Frequency Is Reduced when DSB Ends Are Blocked by Non-homologous Sequences
To study the mechanism of tail removal during homologous recombination (HR) in mammalian cells, we established an EGFP-based reporter EGFP-HR-Luc, which carries a 455-bp luciferase-containing fragment (Luc) non-homologous to the donor template (Figure 1A). As revealed by guide RNA (gRNA)/Cas9 cleavage, when both DSB ends are blocked by non-homologous tails (232 bp/223 bp or 415 bp/40 bp), HR is less efficient than when one end is blocked (455 bp/0 bp) (Figure 1B). We further showed that HR frequency is comparable at DSBs with one long (415 bp/0 bp) or one short (35 bp/0 bp) non-homologous tail, but much lower than at the DSBs with no tail (0 bp/0 bp) (Figure 1C). Thus a non-homologous tail as small as 35 bp suppresses HR to a similar extent as a long tail (>400 bp). Upon DSB formation and end resection, 3′ single-stranded non-homologous tails can only be detected when they form flaps after adjacent homologous DNA sequences pair with the donor sequences (Figure S1, left). As end resection of 415 bp would take much longer than for 35-bp DNA, these results also suggest that for repairing DSB ends with non-homologous tails, cleavage of the non-homologous tails rather than end resection is the limiting step for HR.
Figure 1.

Non-homologous Tails at DSBs Suppress HR Frequency
(A) Schematic drawing of the EGFP-based HR-Luc reporter. A luciferase fragment (Luc, 455 bp) was inserted in the middle of EGFP open reading frame.
(B and C) U2OS cells containing EGFP-HR-Luc reporter were transfected with one gRNA/Cas9-containing plasmid (B) or two gRNA/Cas9-containing plasmids (C). The positions of gRNA cleavage sites are indicated by arrows on the reporter, and the length of the non-homologous tails on both sides of the gRNA cleavage sites is shown. HR efficiency was assayed by fluorescence-activated cell sorting analysis 5 days after transfection. Data are represented as mean ± SD. Statistical analysis was performed using two-tailed unpaired Student's t tests. **p < 0.01, ns, not significant.
(D) PCR strategy for detection of DNA synthesis initiation from DSBs containing non-homologous tails with PCR primers indicated by black arrows (left). Homology between the donor and recipient cassettes is shown in green, and non-homologous tails are marked in blue. A nucleotide change in the homology of the donor versus recipient is shown in red, and this nucleotide difference creates specificity of the left primer to the donor but not to the recipient cassette in the PCR reaction. PCR was performed using extracted genomic DNA at indicated days after transfection of EGFP-HR-Luc cells with specific gRNA/Cas9 plasmids cutting at different positions on Luc (right). PCR products were resolved by agarose gel and quantified by ImageJ with the density of the sample (223 bp/1.5 day) set as 1 as a reference.
See also Figure S1.
To study whether the tail length influences the repair kinetics, we examined the start of DNA synthesis after strand invasion, which can only occur after non-homologous tails are clipped off (Figure S1, left). This was achieved by PCR analysis using one primer on the recipient EGFP::Luc cassette (cleaved) containing no (0 bp), 40-, or 223-bp non-homologous tails and one primer on the donor template cassette (Figure 1D, left). The start of DNA synthesis on the strand without non-homologous tails (0 bp) occurs earlier than on the strand with tails (40 bp or 223 bp), and tail length does not have an obvious effect on the timing of DNA synthesis initiation (Figure 1D, right). This is consistent with the model suggesting that removal of the non-homologous tails but not end resection is the rate-limiting step for HR when the ends are blocked with non-homologous tails.
ERCC1/XPF Is Required for HR when DSB Ends Are Blocked by Non-homologous Sequences
To study the role of ERCC1/XPF in tail removal, we depleted XPF or ERCC1 by short hairpin RNAs (shRNAs) in cells carrying different HR reporters. We showed that when XPF or ERCC1 is depleted, I-SceI-induced HR is significantly reduced in the EGFP-HR-Luc reporter where I-SceI cleavage generates two non-homologous tails, 391 and 68 bp in length, whereas HR levels remain mostly unchanged in the EGFP-HR reporter where 12- and 13-bp non-homologous tails are generated (Figures 2A and S2A). The nuclease activity of XPF and the interaction of XPF with SLX4 are important for this HR-mediated repair of DSBs with non-homologous tails, as expression of XPF-WT but not the mutants, XPF-D687A, impaired in nuclease activity (corresponding to D676A in the previously used XPF clone with 11 amino acid deletion at the N terminus, Enzlin and Scharer, 2002), and XPF-L230R, deficient in associating with SLX4 (Klein Douwel et al., 2017), suppresses HR defect in XPF KO cells as assayed by the EGFP-HR-Luc reporter after I-SceI cleavage (Figures 2B and S10A). To further define the length of non-homologous tails that require XPF activity for HR, we generated EGFP-HR-Luc reporters containing 20-, 40-, and 390-bp non-homologous sequences on both sides of I-SceI (20/20, 40/40, and 390/390) to the donor. All other reporter cell lines contain a single and stably integrated reporter cassette, whereas for the analysis of EGFP-HR-Luc reporters (20/20, 40/40 and 390/390), we used pooled cell populations after transfection of the reporter to avoid the effect of genomic locus on repair efficiency. We showed that the EGFP-HR-20/20 cell line does not require XPF for HR, but the 40/40 and 390/390 cell lines exhibit significant dependence on XPF (Figure 2C). These data suggest that XPF is required for removing not only long non-homologous tails, but also tails as short as 40 bp. When non-homologous tails are 20 bp or shorter, tail removal requires other mechanisms that are independent of XPF.
Figure 2.

XPF Is Required for HR when DSBs Contain Non-homologous Sequences
(A) HR was assayed in U2OS (EGFP-HR) and U2OS (EGFP-HR-Luc) cell lines expressing XPF shRNA, ERCC1 shRNA, or vector (Ctrl) after I-SceI lentiviral infection. The I-SceI cleavage site on the EGFP-HR and EGFP-HR-Luc reporters is shown on top. Depletion of XPF and ERCC1 by shRNAs is shown by western blot.
(B) HR was assayed after I-SceI lentiviral infection in U2OS (EGFP-HR-Luc) WT cells or XPF knockout (KO cells reconstituted with XPF-WT, XPF-D678A, or XPF-L230R alleles or vector. The expression of XPF is shown by western blot.
(C) U2OS cells carrying different EGFP-HR reporters (EGFP-HR-Luc [20/20], [40,40], and [(390/390]), with indicated length of non-homologous tails (20, 40, and 390 bp on both sides of the I-SceI site) expressing XPF shRNAs or vector (Ctrl), were infected with I-SceI lentiviruses, and HR was assayed. Depletion of XPF by shRNAs is shown by Western blot.
(D and E) The U2OS (EGFP-HR-Luc) cell line expressing XPF shRNAs or vector (Ctrl) was transfected with one (D) or two indicated gRNA/Cas9 plasmids (E), and HR was assayed. Depletion of XPF by shRNAs is shown by western blot.
(F) qPCR with the primers shown in Figure 1D was performed to quantify DNA synthesis from indicated tails in U2OS (EGFP-HR-Luc) cells expressing XPF shRNAs or vector (Ctrl) 1.5 days after transfection of corresponding gRNA/Cas9 plasmids as shown in Figure 1B. The hygromycin-resistance gene present in the reporter was used as an internal reference for normalization. XPF expression is shown by western blot analysis using KU70 as a loading control.
Data are represented as mean ± SD. Statistical analysis was performed using two-tailed unpaired Student's t tests. *p < 0.05, **p < 0.01, ns, not significant.
See also Figure S2.
To test whether XPF is also needed when only one non-homologous tail is present, we inactivated XPF in U2OS (EGFP-HR-Luc) cells and used gRNA/Cas9 to create one or two non-homologous tails (455/0 bp, 232/223 bp, and 415/40 bp). We found that XPF is required for repairing DSBs not only with two non-homologous tails but also with one non-homologous tail (Figures 2D and S2B). We further showed that XPF is required when a single non-homologous tail is 415 bp (415/0 bp) or 35 bp in length (35/0 bp), but not when DSBs do not have tails (0/0 bp) (Figures 2E and S2C). Thus XPF is important for the removal of single non-homologous tails as small as 35 bp.
We also showed that the presence of non-homologous tail delays DNA synthesis during HR (Figure 1D). We silenced XPF by shRNAs and showed by quantitative PCR (qPCR) that inactivation of XPF further delays initiation of DNA synthesis from the strand containing a non-homologous tail (40 or 223 bp), but has no effect when the invading strand does not have a non-homologous tail (0 bp) (Figure 2F). These results demonstrate that XPF-dependent removal of non-homologous tails is critical for the initiation of DNA synthesis during HR.
XPF Is Required for HR-Mediated DSB Repair at DNA Sequences Containing Secondary Structures
Besides non-homologous tails, DSB ends may also be blocked by DNA sequences with secondary structures, which need to be removed before DNA synthesis can occur for HR (Figure S1, right). Flex1, an AT-rich sequence derived from FRA16D, is prone to forming DNA secondary structures and causes replication stalling and DSB formation (Zhang and Freudenreich, 2007, Wang et al., 2014, Wang et al., 2018). We showed that Flex1 induces HR-mediated mitotic recombination, as revealed by an EGFP-HR-Flex reporter (Wang et al., 2014). Interestingly, Flex1-induced mitotic recombination is significantly decreased when XPF is depleted by shRNAs (Figure 3A). Hydroxyurea (HU)-induced mitotic recombination at Flex1 is also compromised when XPF or ERCC1 is depleted (Figure 3B). Reduction of mitotic recombination can be due either to reduced DSB formation or impaired DSB repair after DSB formation (Figure S3).
Figure 3.

DSBs Accumulate at Flex1 upon Replication Stress when XPF is Depleted
(A) Schematic drawing of EGFP-HR-Flex reporter (top). Spontaneous recombination was examined in U2OS (EGFP-HR-Flex) cells expressing XPF shRNA#1 or #2 or control vector (Ctrl) (bottom) at indicated days.
(B) U2OS (EGFP-HR-Flex) cells were infected with lentiviruses expressing XPF shRNA#1 or #2, ERCC1 shRNAs#1 or #2, and a control vector (Ctrl) and assayed for mitotic recombination after HU (2 mM, 24 h) treatment. Depletion of XPF and ERCC1 by shRNAs is shown by western blot.
(C) Phosphorylation of H2AX (S139), CHK1 (S317), and RPA2 (S4/S8) was examined by western blot analysis in WT or MUS81 KO cells with or without depletion of XPF by shRNA#1 after HU (2 mM, 24 h) treatment. KU70 was used as a loading control.
(D) U2OS (EGFP-HR-Flex) cells were infected with lentiviruses expressing XPF shRNA#1 or a control vector (Ctrl), followed by treatment with or without 2 mM HU for 9 h. γH2AX ChIP at Flex1 on the EGFP-HR-Flex reporter was performed and analyzed by qPCR. ChIP value without HU treatment is set as 1 for normalization. Depletion of XPF was shown by western blot analysis using KU70 as a loading control.
Data are represented as mean ± SD. Statistical analysis was performed using two-tailed unpaired Student's t tests. **p < 0.01.
See also Figure S3.
MUS81 has been reported to be involved in cleaving stalled replication forks, leading to DSB formation upon replication stress (Hanada et al., 2007). Inactivation of MUS81 significantly reduces γH2AX levels after HU treatment, whereas XPF depletion does not reduce but rather increases γH2AX levels (Figure 3C). However, such increase of γH2AX level caused by depletion of XPF is not observed in MUS81 KO cells (Figure S10B), suggesting that XPF functions downstream of MUS81 to prevent DSB accumulation. Loss of MUS81 does not prevent S-phase checkpoint activation as revealed by phosphorylation of RPA2 and CHK1, which is consistent with the previous observation that ATR pathway is activated in MUS81-deficient cells upon camptothecin treatment (Regairaz et al., 2011). More specifically, by chromatin immunoprecipitation (ChIP) analysis, we further showed that γH2AX is significantly increased at Flex1 site after HU treatment when XPF is depleted by shRNAs (Figure 3D). These data suggest that XPF does not play an essential role in cleaving stalled replication forks upon replication stress. Reduced mitotic recombination observed at Flex1 in XPF-silencing cells is not due to a decrease in DSB formation at Flex1, but rather caused by a defect in HR as XPF has a role in repairing DSBs generated by MUS81-mediated cleavage of stalled replication forks.
To examine the role of XPF in the repair of DSBs generated at structure-prone Flex1 more directly, we created a DSB by I-SceI cleavage at the side of Flex1 in the EGFP-HR-Flex reporter, which contains an insert of Flex1 (0.34 kb) compared with the donor, and in the EGFP-HR reporter, which does not have an insert (Figure 4A, left). Although XPF is not required for HR-mediated DSB repair in the EGFP-HR reporter, XPF depletion by shRNAs significantly reduces HR in the EGFP-HR-Flex reporter after DSBs are generated by I-SceI (Figure 4A, right). This suggests that XPF is required for HR after DSB formation when the AT-rich structure-prone Flex1 is present at the end of a DSB.
Figure 4.

XPF Is Required for HR Repair of DSBs Containing DNA Secondary Structures
(A) Schematic drawing of EGFP-HR and EGFP-HR-Flex reporters is shown (left). I-SceI-induced HR was assayed in U2OS (EGFP-HR) and U2OS (EGFP-HR-Flex) reporter cell lines with expression of XPF shRNA#1 or a control vector (Ctrl, right).
(B) Schematic drawing of HR reporters HR-Flex/D-Flex and HR-Luc/D-Luc described previously (Wang et al., 2018) (top). See also Figure S4. The BamHI and EcoRI sites in the donor are at the corresponding position of the I-SceI site in the recipient cassette. U2OS (HR-Flex/D-Flex) or U2OS (HR-Luc/D-Luc) cells expressing XPF shRNAs or a control vector (Ctrl) were infected with I-SceI lentiviruses. After 6 days, genomic DNA was extracted and digested with I-SceI, followed by PCR using indicated primers (shown as arrows). PCR products with or without BamHI and EcoRI digestion were resolved on agarose gel, and the percentage of BamHI- and EcoRI-digestible PCR products among the total DNA was calculated (bottom).
Data are represented as mean ± SD. Statistical analysis was performed using two-tailed unpaired Student's t tests. *p < 0.05, **p < 0.01, ns, not significant.
Flex1 forms DNA secondary structures at DSB ends. However, because it is not present in the donor template in the EGFP-HR-Flex reporter, Flex1 would also become a non-homologous tail after I-SceI cleavage (Figure 4A, left). To more directly test whether XPF is needed for repairing DSBs containing DNA secondary structures at the ends, we used DSB repair substrates (HR-Flex/D-Flex and HR-Luc/D-Luc) with Flex1 or Luc inserted in both the I-SceI cleavage recipient cassette and the donor cassette (Figure 4B, top) (Wang et al., 2018), so that the recipient and donor cassettes contain perfect homologies. DSBs generated by I-SceI cleavage in the HR-Flex/D-Flex and HR-Luc/D-Luc reporters can be repaired by HR or non-homologous end joining (NHEJ). The donor sequences (D-Flex and D-Luc) are marked with BamHI and EcoRI cleavage sites. If HR is used, BamHI and EcoRI sites would be transferred to the recipient cassette to replace the I-SceI site in the repair products, whereas NHEJ products would not contain BamH1 and EcoRI sites. After I-SceI expression in vivo, genomic DNA was purified and digested with I-SceI in vitro to remove the parental EGFP::Flex1 or EGFP::Luc recipient cassettes (uncut by I-SceI or perfectly relegated after I-SceI cleavage in cells). The ratio of BamHI and EcoRI cleavable and non-cleavable recipient cassettes (Figure S4) would indicate the ratio of repair products by HR and by imperfect end joining. Depletion of XPF substantially reduces the percentage of using HR in U2OS (HR-Flex/D-Flex) cells, but not in U2OS (HR-Luc/D-Luc) cells (Figure 4B, bottom), suggesting that XPF is required for the repair of DSBs when the ends are blocked by DNA secondary structures, even when DSB ends contain perfect homology to the donor sequences.
XPF and FANCM Are Synthetic Lethal
DNA translocase FANCM plays a significant role in preventing DSB formation at Flex1 by promoting fork reversal to remove DNA secondary structures at Flex1 (Wang et al., 2018). Loss of FANCM activity significantly increases Flex1-induced mitotic recombination. We further showed that increased mitotic recombination caused by FANCM deficiency depends on XPF (Figure 5A). This suggests that DSBs that have accumulated in FANCM-deficient cells cannot be repaired sufficiently in the absence of XPF. We thus asked whether inactivation of both genes would cause cell death. Indeed, depletion of XPF by shRNAs in FANCM KO cells drastically suppresses cell proliferation (Figure 5B), suggesting a synthetic lethal interaction between XPF and FANCM. Thus inactivation of XPF is potentially a good strategy for targeted treatment to kill FANCM-deficient tumors.
Figure 5.

XPF and FANCM Are Synthetically Lethal
(A) Spontaneous recombination was examined at indicated days in the U2OS (EGFP-HR-Flex) reporter cell line expressing XPF shRNA#1 and FANCM shRNA alone or simultaneously, compared with a control vector. Data are represented as mean ± SD. Statistical analysis was performed using two-tailed unpaired Student's t tests. **p < 0.01.
(B) Growth curve of HCT116 WT or FANCM KO cells was plotted after expressing XPF shRNA#1 or a control vector (Ctrl). XPF expression is shown by western blot analysis with KU70 as a loading control.
Deficiency in XPF and FANCM Causes Cell Sensitivity to G4-Interacting Compounds
We reasoned that the roles of FANCM in protecting structure-prone DNA sequences and XPF in processing DSB ends with secondary structures may not be limited to CFS-derived AT-rich sequences. G4-forming DNA sequences are abundantly present in the human genome (Hansel-Hertsch et al., 2017). To test whether XPF and FANCM are also important for protecting G4s, we treated XPF KO and FANCM KO cells with G4-interacting compound pyridostatin (PDS) (McLuckie et al., 2013). Both XPF KO and FANCM KO are more sensitive to PDS than wild-type (WT) cells (Figure 6A). Upon PDS treatment, γH2AX is significantly increased in FANCM KO cells compared with WT cells (Figure 6B). We performed comet assay to more directly monitor DSB formation and found that DSB formation is also more significant in XPF KO cells after PDS treatment (Figures 6C and S5). These data suggest that FANCM and XPF are important for preventing DSB accumulation at G4s and support the model that FANCM has an important role in protecting G4s from DSB formation while XPF is involved in repairing DSBs generated at G4s.
Figure 6.

XPF- and FANCM-Deficient Cells Are Sensitive to PDS
(A) U2OS WT and XPF KO cells (left) or HCT116 WT and FANCM KO cells (right) were treated with the indicated concentration of PDS, and cell viability assays were performed after 5 days. Data are represented as mean ± SD.
(B) HCT116 WT and FANCM KO cells were treated by PDS (50 μM, 2 days) and lysed for western blot analysis of γH2AX using KU70 as a loading control.
(C) U2OS WT and XPF KO cells were treated with PDS (50 μM) for 4 days, and comet assays were performed and quantified. Data are presented in box-and-whisker plots of tail moment. The comet images of cell population are shown in Figure S5.
(D) HCT116 WT or FANCM KO cells infected by a low titer of lentiviruses expressing XPF shRNA#1 or a control vector (Ctrl) were treated with the indicated concentration of PDS for 5 days, and cell viability assays were performed. XPF expression is shown by western blot using KU70 as a loading control. Data are represented as mean ± SD.
As a deficiency in either XPF or FANCM results in sensitivity to PDS, we were interested in whether inhibition of XPF would further sensitize FANCM-deficient cells to PDS. We used a relatively low titer of lentiviruses encoding XPF shRNAs, which reduces endogenous XPF to about one-third of the original level (Figure 6D, left). This level of XPF inhibition did not cause significant sensitivity of WT HCT116 cells to PDS, but synergistically killed FANCM KO-deficient cells with PDS (Figure 6D, right). These results suggest that a potential treatment strategy could use low level of XPF inhibition in combination with PDS to achieve specific killing of FANCM-deficient tumors with low toxicity to normal cells.
PDS Induces Acute Replication Stress in XPF-Deficient Cells
It has been suggested that PDS causes replication-associated DNA damage (Rodriguez et al., 2012), which is consistent with the notion that G4s are formed during DNA replication when ssDNA is present. We hypothesized that loss of XPF would impair DSB repair at G4s that are formed during DNA replication, causing an obstruction of replication fork progression and replication stress. Indeed, PDS induces γH2AX accumulation and ATR activation as revealed by phosphorylation of CHK1 and RPA2, which are further enhanced in XPF KO cells (Figure 7A, left). This suggests that replication stress induced by chemical stabilization of G4s is potentiated in XPF-deficient cells. S-phase checkpoint activation is increased when MUS81 is deficient on PDS treatment. However, different from in WT cells, depleting XPF does not further enhance γH2AX accumulation and S-phase checkpoint in MUS81 KO cells (Figure 7A, right), further supporting the notion that XPF functions downstream of MUS81 to repair DSBs generated by MUS81 at stalled replication forks.
Figure 7.

PDS Induces Acute Replication Stress in XPF-Deficient Cells
(A) U2OS cells with or without XPF KO (left) or MUS81 KO (right) combined with or without XPF depletion by shRNA#1 as indicated were treated with PDS (20 μM, 2days), and phosphorylation of H2AX (S139), CHK1 (S317), and RPA2 (S4/S8) was examined by western blot analysis using KU70 as a loading control.
(B) U2OS WT or XPF KO cells labeled with CldU (30 min) were incubated in IdU for 60 min with or without PDS (20 μM) and processed for DNA fiber analysis. Scale bar, 10 μm. The replication velocity in kilobases per minute of treated cells was quantified by analyzing the replication tract length of 110–130 fibers for each condition. The middle line represents median, and p values were calculated using a Mann-Whitney test. ****p < 0.0001, ns, not significant. See also Figure S6.
(C) Proposed model for the role of ERCC1/XPF in removing DNA secondary structures at DSB ends for HR repair and replication restart from collapsed forks. FANCM deficiency or PDS treatment leads to an accumulation of DNA secondary structures on replication forks and causes formation of DSBs that require XPF to repair. The concerted roles of FANCM in protecting DNA secondary structures and XPF in repairing DSBs with blocked DSB ends underlie the synthetic lethality interactions between these two proteins.
To address further how replication is disturbed by PDS in XPF-deficient cells, we performed single molecular DNA fiber analysis. We pulse labeled cells with chlorodeoxyuridine (CldU), followed by second pulse labeling with iododeoxyuridine (IdU) in the presence of PDS. Relative track length is significantly reduced in XPF KO cells after PDS treatment compared with the cells without treatment (Figure 7B). Track length is also reduced in MUS81 KO cells, but inactivation of XPF in MUS81 KO cells does not lead to significant further reduction of fork length (Figure S6). These results support the model that the major function of XPF is downstream of MUS81 to repair DSBs that are generated by MUS81, thereby antagonizing the effect of PDS in stabilizing G4s and preventing DSB accumulation (Figure 7C).
Discussion
In NER, ERCC1/XPF is responsible for making an incision 5′ to the lesion (Mu et al., 1995, Sijbers et al., 1996), and in ICL repair, ERCC1/XPF is required for unhooking incision of ICLs (Hodskinson et al., 2014, Klein Douwel et al., 2017, De Silva et al., 2000, Niedernhofer et al., 2004). In this study, we analyzed the role of XPF in processing DSBs with blocked ends. We found that XPF is required for removing not only non-homologous tails but also DNA secondary structures present at DSB ends for HR. Identifying this important role of XPF in repairing DSBs associated with DNA secondary structures has implications for understanding clinical features of XPF deficiency and for establishing targeted cancer treatments.
The Role of ERCC1/XPF in Processing DSB Ends with Non-homologous Tails
Study in yeast has revealed that Rad1 and Rad10 are required for the removal of 3′ non-homologous tails during SSA-mediated recombination of direct repeats (Ivanov and Haber, 1995, Fishman-Lobell and Haber, 1992). Similarly, Chinese hamster ovary (CHO) ERCC1- cells are deficient in SSA (Sargent et al., 1997, Sargent et al., 2000). It has also been shown that ERCC1 is required for removing non-homologous tails in the “ends-in” gene targeting in CHO cells (Adair et al., 2000), but in a separate report, it was suggested that in mouse embryonic stem cells, ERCC1/XPF is involved in targeted gene replacement with “ends-out” constructs irrespective of whether ends contain homologous or non-homologous sequences to the genomic locus (Niedernhofer et al., 2001). We thus used our EGFP-HR reporter to systematically analyze the role of XPF in gene conversion at the DSBs with or without non-homologous tails in mammalian cells.
In the repair of DSBs by HR, one or both 3′ ends of a DSB invade the homologous template (Mehta and Haber, 2014). The invaded 3′ DNA strand is then used as a primer for new DNA synthesis, but the initiation of new DNA synthesis requires removal of any non-homologous DNA sequences present at the 3′ end (Figure S1). We demonstrated that the presence of short (∼35–40 bp) or long (∼200–400 bp) non-homologous tails has a similar effect in blocking DSB ends and in delaying the initiation of DNA synthesis for HR. As the tail length does not affect HR repair frequency, we anticipate that the removal of non-homologous tails rather than end resection is the limiting step for HR at DSBs containing non-homologous sequences (Figure S1). We also showed that XPF is required for removing terminal non-homology of more than 20 bp in length for HR-mediated repair. This is almost identical to observations in yeast that efficient clipping of non-homologous sequences longer than 20 bp depends on Rad1/Rad10 endonuclease, but the Rad1/Rad10-independent pathway is used for processing shorter terminal non-homology (Ivanov and Haber, 1995). Thus the role of ERCC1/XPF in removal of terminal non-homology at DSBs is a well-conserved process that shares a common mechanism in yeast and human beings, and is essential for gene conversion in mammalian cells. Currently, it is not clear what nucleases are involved in removing small non-homologous tails (<20 bp).
In yeast, removing Ya or Yα sequences non-homologous to the donor is essential for mating type switching (Lyndaker et al., 2008). Nearly half of the human genome is composed of repetitive DNA sequences such as Alu elements (Li et al., 2001, Lander et al., 2001), and non-allelic recombination between these repeated sequences would involve processing of non-homologous tails if DSBs are generated outside of repeats, or at sites with small stretches of non-homologous sequences within repeats (Figure S7). Furthermore, development of CRISPR/Cas9-directed gene replacement or targeted gene correction for disease therapy would also benefit from the comprehensive understanding of processing of non-homologous ends present at target and donor sequences (Figure S8).
The Role of XPF in Processing DSB Ends with Secondary Structures
We demonstrated that XPF is not only involved in removing non-homologous tails but also in processing DNA secondary structures at DSB ends during gene conversion even if terminal sequences are completely homologous to the donor template. These studies suggest an important role of XPF in protection of genomic loci, which contain DNA secondary structures.
Upon DSB formation, 5′ ends undergo end resection to generate long 3′ ssDNA tails, allowing AT-rich or G4 sequences to form secondary structures at DSB ends (Figure S1, right). Such secondary structures block DNA polymerases from accessing the 3′ ends and need to be removed before initiation of DNA synthesis. Like non-homologous tails, DNA secondary structures present at DSBs also form flap-like structures after internal homologous sequences invade donor templates and are recognized by XPF for cleavage. Notably, redundant pathways besides XPF are also involved in clipping and processing DNA secondary structures at DSB ends. For instance, CtIP and MRE11 play important roles in processing DNA secondary structures formed at DSB ends, likely before the strand invasion step (Wang et al., 2014). In this aspect, we showed that inactivation of MRE11 or CtIP also leads to PDS sensitivity (Figure S9A). Co-depletion of MRE11 or CtIP with XPF has stronger effect than single depletion in repairing DSBs generated by I-SceI at Flex1, which contain DNA secondary structures (Figure S9B). More in-depth study will be needed in the future to understand how XPF-dependent and XPF-independent pathways function together to process DNA secondary structures at DSB ends to promote HR.
Unlike those with deficiencies in other NER genes, patients with XPF and mice deficient in ERCC1 and XPF show phenotypes of neurological deterioration and accelerated aging (Gregg et al., 2011). G4-forming sequences and repetitive sequences at CFSs have been shown to be linked to neurological diseases (Thys et al., 2015, Bose et al., 2014). Our findings reveal that XPF has an important role in repairing DSBs at sites with DNA secondary structures, including G4s and CFSs, suggesting a possible mechanism that contributes to the unique clinical symptoms of neurodegeneration stemming from XPF deficiency, which are not shared by other XP genes.
XPF Is Important for Repairing DSBs at DNA Secondary Structures that Have Accumulated in FANCM-Deficient Cells
DNA secondary structures often induce replication fork stalling, leading to fork collapse and DSB formation (Figure 7C). We showed that XPF is required for spontaneous mitotic recombination induced by structure-prone sequence Flex1 derived from CFS FRA16D (Zhang and Freudenreich, 2007, Wang et al., 2014, Wang et al., 2018). Decreased mitotic recombination at Flex1 in XPF-deficient cells is largely due to a defect in removing DNA secondary structures after DSB formation. This is supported by the observations that XPF deficiency leads to DSB accumulation at Flex1, and that even after DSBs are generated by I-SceI XPF is still required for HR when DSB ends contain secondary structures.
XPF-deficient cells are sensitive to PDS, supporting the role of XPF in repairing DSBs at G4s. Upon PDS treatment, replication stress is substantially potentiated in XPF-deficient cells, and DNA fiber analysis showed replication slowdown in the presence of PDS when XPF is deficient. These data support the idea that XPF is required to avoid problems at G4s on replication forks by processing them.
At stalled replication forks, MUS81 cleaves forks to promote HR-mediated repair (Hanada et al., 2007). Our study supports the idea that XPF functions downstream of MUS81 to repair DSBs that are generated by MUS81-mediated cleavage of stalled replication forks. We showed that increased DSB formation in XPF-deficient cells after both HU and PDS treatment is dependent on MUS81. Loss of XPF induces replication stress and reduces replication track length in WT cells upon PDS treatment, but no further increase of replication stress and reduction of fork track length are observed when XPF is depleted in MUS81 KO cells. Although it has been shown that ERCC1/XPF functions together with MUS81 to cleave CFSs in mitosis (Naim et al., 2013) and is involved in cleaving special DNA structures such as ICLs, R-loops, and DNA cruciforms (Lu et al., 2015, Sollier et al., 2014), XPF does not seem to play an essential role as MUS81 in cleaving stalled replication forks for DSB formation in S-phase.
FANCM promotes fork reversal to remove DNA secondary structures formed at Flex1 (Wang et al., 2018). We showed that increased mitotic recombination at Flex1 caused by FANCM deficiency depends on XPF and both FANCM and XPF are important in preventing DSB formation after PDS treatment. Thus, XPF plays an important role in repairing DSBs that have accumulated at structure-prone DNA sequences when FANCM is deficient. As DNA secondary structures such as G4s are very abundant in the human genome, we propose that the concerted roles of FANCM in protecting genomic loci containing DNA secondary structures and XPF in repairing DSBs arising at these sequences underlie the synthetic lethal phenotype of FANCM and XPF deficiencies.
Inhibiting XPF Is a Promising Strategy for Targeted Treatment of FANCM-Deficient Tumors
FANCM deficiency is associated with breast and ovarian cancers, with a particularly strong predisposition toward hard-to-treat triple-negative breast cancer (Neidhardt et al., 2017, Kiiski et al., 2014, Kiiski et al., 2017, Dicks et al., 2017). Reduced FANCM expression is also commonly found in sporadic head and neck squamous cell carcinoma (Wreesmann et al., 2007). Our study identifies a synthetic lethality interaction between XPF and FANCM, providing a targeted therapeutic strategy for treating FANCM-deficient tumors by inactivating XPF.
It has been shown that G4-stabilizing compounds can selectively eliminate HR-compromised tumors, including those with a BRCA1 or BRCA2 deficiency (Zimmer et al., 2016). Based on our findings that FANCM and XPF are involved in preventing DNA secondary structure formation and in repairing DSBs at G4s, respectively, compromised XPF or FANCM function would cause cell sensitivity to PDS, and inhibition of XPF would enhance toxicity of FANCM-deficient cells to PDS. We also demonstrated a synergistic effect of PDS and suppression of XPF expression in killing FANCM-deficient cells. This combined treatment strategy could allow us to reduce the dose of PDS and XPF inhibition to a level that is not toxic to normal cells but can effectively kill FANCM-deficient tumor cells. Small-molecule inhibitors for XPF at nanomolar range with good specificity have been identified, with potential for development into potent anti-cancer drugs (Arora et al., 2016). Our study provides a rationale for using these inhibitors in the effective treatment of FANCM-deficient tumors.
Limitations of the Study
In this study, we identified an important role of ERCC1/XPF in removing DNA secondary structures at DSB ends to facilitate HR. A step-by-step in vitro biochemistry analysis will be needed to further validate the model that the cleavage by ERCC1/XPF of DNA secondary structures occurs after invasion of the 3′ single-strand tail to the template in the D loop. We also showed synthetic lethal interaction of FANCM and XPF in cells, which offers a novel targeted treatment strategy for FANCM-deficient tumors, but mouse study will be needed for proof of principle in vivo.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Orlando D. Schärer for valuable suggestions and comments on this work.
The HCT116 FANCM KO cell line was kindly provided by Dr. Lei Li (University of Texas MD Anderson Cancer Center). XPF-WT cDNA was kindly provided by Dr. Stephen C. West (The Francis Crick Institute, UK). The shRNA vector pLKO.1-blast (#26655) and gRNA/Cas9 vector pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988) are from Addgene.
This work is supported by NIH grants CA187052, CA197995, and GM080677 to X.W.
Author Contributions
Conceptualization, X.W. and S.L.; Investigation, S.L., H.L., Z.W., Q.H., and H.W.; Writing – Original Draft, X.W. and S.L.; Writing – Review & Editing, X.W., S.L., H.L., and Z.W., Supervision, X.W., R.X., and T.C.; Funding Acquisition: X.W.
Declaration of Interests
The authors declare no competing interests.
Published: June 28, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.05.017.
Supplemental Information
References
- Adair G.M., Rolig R.L., Moore-Faver D., Zabelshansky M., Wilson J.H., Nairn R.S. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000;19:5552–5561. doi: 10.1093/emboj/19.20.5552. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adair, G.M., Rolig, R.L., Moore-Faver, D., Zabelshansky, M., Wilson, J.H. & Nairn, R.S.. 2000. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J., 19, 5552-5561. [DOI] [PMC free article] [PubMed]
- Ahmad A., Robinson A.R., Duensing A., Van Drunen E., Beverloo H.B., Weisberg D.B., Hasty P., Hoeijmakers J.H., Niedernhofer L.J. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008;28:5082–5092. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ahmad, A., Robinson, A.R., Duensing, A., Van Drunen, E., Beverloo, H.B., Weisberg, D.B., Hasty, P., Hoeijmakers, J.H. & Niedernhofer, L.J.. 2008. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol., 28, 5082-5092. [DOI] [PMC free article] [PubMed]
- Al-Minawi A.Z., Saleh-Gohari N., Helleday T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 2008;36:1–9. doi: 10.1093/nar/gkm888. [DOI] [PMC free article] [PubMed] [Google Scholar]; Al-Minawi, A.Z., Saleh-Gohari, N. & Helleday, T.. 2008. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res., 36, 1-9. [DOI] [PMC free article] [PubMed]
- Arora S., Heyza J., Zhang H., Kalman-Maltese V., Tillison K., Floyd A.M., Chalfin E.M., Bepler G., Patrick S.M. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget. 2016;7:75104–75117. doi: 10.18632/oncotarget.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]; Arora, S., Heyza, J., Zhang, H., Kalman-Maltese, V., Tillison, K., Floyd, A.M., Chalfin, E.M., Bepler, G. & Patrick, S.M.. 2016. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget, 7, 75104-75117. [DOI] [PMC free article] [PubMed]
- Bardwell A.J., Bardwell L., Tomkinson A.E., Friedberg E.C. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science. 1994;265:2082–2085. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]; Bardwell, A.J., Bardwell, L., Tomkinson, A.E. & Friedberg, E.C.. 1994. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science, 265, 2082-2085. [DOI] [PubMed]
- Bennardo N., Cheng A., Huang N., Stark J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bennardo, N., Cheng, A., Huang, N. & Stark, J.M.. 2008. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet., 4, e1000110. [DOI] [PMC free article] [PubMed]
- Bose P., Hermetz K.E., Conneely K.N., Rudd M.K. Tandem repeats and G-rich sequences are enriched at human CNV breakpoints. PLoS One. 2014;9:e101607. doi: 10.1371/journal.pone.0101607. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bose, P., Hermetz, K.E., Conneely, K.N. & Rudd, M.K.. 2014. Tandem repeats and G-rich sequences are enriched at human CNV breakpoints. PLoS One, 9, e101607. [DOI] [PMC free article] [PubMed]
- De Silva I.U., Mchugh P.J., Clingen P.H., Hartley J.A. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; De Silva, I.U., Mchugh, P.J., Clingen, P.H. & Hartley, J.A.. 2000. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol., 20, 7980-7990. [DOI] [PMC free article] [PubMed]
- Dicks E., Song H., Ramus S.J., Oudenhove E.V., Tyrer J.P., Intermaggio M.P., Kar S., Harrington P., Bowtell D.D., Group A.S. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget. 2017;8:50930–50940. doi: 10.18632/oncotarget.15871. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dicks, E., Song, H., Ramus, S.J., Oudenhove, E.V., Tyrer, J.P., Intermaggio, M.P., Kar, S., Harrington, P., Bowtell, D.D., Group, A.S., et al, 2017. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget, 8, 50930-50940. [DOI] [PMC free article] [PubMed]
- Durkin S.G., Glover T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]; Durkin, S.G. & Glover, T.W.. 2007. Chromosome fragile sites. Annu. Rev. Genet., 41, 169-192. [DOI] [PubMed]
- Enzlin J.H., Scharer O.D. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 2002;21:2045–2053. doi: 10.1093/emboj/21.8.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]; Enzlin, J.H. & Scharer, O.D.. 2002. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J., 21, 2045-2053. [DOI] [PMC free article] [PubMed]
- Fishman-Lobell J., Haber J.E. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science. 1992;258:480–484. doi: 10.1126/science.1411547. [DOI] [PubMed] [Google Scholar]; Fishman-Lobell, J. & Haber, J.E.. 1992. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science, 258, 480-484. [DOI] [PubMed]
- Gregg S.Q., Robinson A.R., Niedernhofer L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair (Amst.) 2011;10:781–791. doi: 10.1016/j.dnarep.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gregg, S.Q., Robinson, A.R. & Niedernhofer, L.J.. 2011. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair (Amst.), 10, 781-791. [DOI] [PMC free article] [PubMed]
- Hanada K., Budzowska M., Davies S.L., Van Drunen E., Onizawa H., Beverloo H.B., Maas A., Essers J., Hickson I.D., Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]; Hanada, K., Budzowska, M., Davies, S.L., Van Drunen, E., Onizawa, H., Beverloo, H.B., Maas, A., Essers, J., Hickson, I.D.. & Kanaar, R.. 2007. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol., 14, 1096-1104. [DOI] [PubMed]
- Hansel-Hertsch R., Di Antonio M., Balasubramanian S. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017;18:279–284. doi: 10.1038/nrm.2017.3. [DOI] [PubMed] [Google Scholar]; Hansel-Hertsch, R., Di Antonio, M. & Balasubramanian, S.. 2017. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol., 18, 279-284. [DOI] [PubMed]
- Hodskinson M.R., Silhan J., Crossan G.P., Garaycoechea J.I., Mukherjee S., Johnson C.M., Scharer O.D., Patel K.J. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell. 2014;54:472–484. doi: 10.1016/j.molcel.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hodskinson, M.R., Silhan, J., Crossan, G.P., Garaycoechea, J.I., Mukherjee, S., Johnson, C.M., Scharer, O.D. & Patel, K.J.. 2014. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell, 54, 472-484. [DOI] [PMC free article] [PubMed]
- Ivanov E.L., Haber J.E. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995;15:2245–2251. doi: 10.1128/mcb.15.4.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ivanov, E.L. & Haber, J.E.. 1995. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 2245-2251. [DOI] [PMC free article] [PubMed]
- Kiiski J.I., Pelttari L.M., Khan S., Freysteinsdottir E.S., Reynisdottir I., Hart S.N., Shimelis H., Vilske S., Kallioniemi A., Schleutker J., Leminen A. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. U S A. 2014;111:15172–15177. doi: 10.1073/pnas.1407909111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kiiski, J.I., Pelttari, L.M., Khan, S., Freysteinsdottir, E.S., Reynisdottir, I., Hart, S.N., Shimelis, H., Vilske, S., Kallioniemi, A., Schleutker, J., Leminen, A.., 2014. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. U S A, 111, 15172-15177. [DOI] [PMC free article] [PubMed]
- Kiiski J.I., Tervasmaki A., Pelttari L.M., Khan S., Mantere T., Pylkas K., Mannermaa A., Tengstrom M., Kvist A., Borg A. FANCM mutation c.5791C>T is a risk factor for triple-negative breast cancer in the Finnish population. Breast Cancer Res. Treat. 2017;166:217–226. doi: 10.1007/s10549-017-4388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kiiski, J.I., Tervasmaki, A., Pelttari, L.M., Khan, S., Mantere, T., Pylkas, K., Mannermaa, A., Tengstrom, M., Kvist, A., Borg, A., et al. 2017. FANCM mutation c.5791C>T is a risk factor for triple-negative breast cancer in the Finnish population. Breast Cancer Res. Treat., 166, 217-226. [DOI] [PMC free article] [PubMed]
- Klein Douwel D., Boonen R.A., Long D.T., Szypowska A.A., Raschle M., Walter J.C., Knipscheer P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014;54:460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; Klein Douwel, D., Boonen, R.A., Long, D.T., Szypowska, A.A., Raschle, M., Walter, J.C. & Knipscheer, P.. 2014. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell, 54, 460-471. [DOI] [PMC free article] [PubMed]
- Klein Douwel D., Hoogenboom W.S., Boonen R.A., Knipscheer P. Recruitment and positioning determine the specific role of the XPF-ERCC1 endonuclease in interstrand crosslink repair. EMBO J. 2017;36:2034–2046. doi: 10.15252/embj.201695223. [DOI] [PMC free article] [PubMed] [Google Scholar]; Klein Douwel, D., Hoogenboom, W.S., Boonen, R.A.. & Knipscheer, P.. 2017. Recruitment and positioning determine the specific role of the XPF-ERCC1 endonuclease in interstrand crosslink repair. EMBO J., 36, 2034-2046. [DOI] [PMC free article] [PubMed]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., Fitzhugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]; Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., Fitzhugh, W.., et al. 2001. Initial sequencing and analysis of the human genome. Nature, 409, 860-921. [DOI] [PubMed]
- Li W.H., Gu Z., Wang H., Nekrutenko A. Evolutionary analyses of the human genome. Nature. 2001;409:847–849. doi: 10.1038/35057039. [DOI] [PubMed] [Google Scholar]; Li, W.H., Gu, Z., Wang, H. & Nekrutenko, A.. 2001. Evolutionary analyses of the human genome. Nature, 409, 847-849. [DOI] [PubMed]
- Lu S., Wang G., Bacolla A., Zhao J., Spitser S., Vasquez K.M. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 2015;10:1674–1680. doi: 10.1016/j.celrep.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lu, S., Wang, G., Bacolla, A., Zhao, J., Spitser, S. & Vasquez, K.M.. 2015. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep.,10, 1674-1680. [DOI] [PMC free article] [PubMed]
- Lukusa T., Fryns J.P. Human chromosome fragility. Biochim. Biophys. Acta. 2008;1779:3–16. doi: 10.1016/j.bbagrm.2007.10.005. [DOI] [PubMed] [Google Scholar]; Lukusa, T. & Fryns, J.P.. 2008. Human chromosome fragility. Biochim. Biophys. Acta, 1779, 3-16. [DOI] [PubMed]
- Lyndaker A.M., Goldfarb T., Alani E. Mutants defective in Rad1-Rad10-Slx4 exhibit a unique pattern of viability during mating-type switching in Saccharomyces cerevisiae. Genetics. 2008;179:1807–1821. doi: 10.1534/genetics.108.090654. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lyndaker, A.M., Goldfarb, T. & Alani, E.. 2008. Mutants defective in Rad1-Rad10-Slx4 exhibit a unique pattern of viability during mating-type switching in Saccharomyces cerevisiae. Genetics, 179, 1807-1821. [DOI] [PMC free article] [PubMed]
- Manandhar M., Boulware K.S., Wood R.D. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene. 2015;569:153–161. doi: 10.1016/j.gene.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; Manandhar, M., Boulware, K.S. & Wood, R.D.. 2015. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene, 569, 153-161. [DOI] [PMC free article] [PubMed]
- McLuckie K.I., Di Antonio M., Zecchini H., Xian J., Caldas C., Krippendorff B.F., Tannahill D., Lowe C., Balasubramanian S. G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J. Am. Chem. Soc. 2013;135:9640–9643. doi: 10.1021/ja404868t. [DOI] [PMC free article] [PubMed] [Google Scholar]; McLuckie, K.I., Di Antonio, M., Zecchini, H., Xian, J., Caldas, C., Krippendorff, B.F., Tannahill, D., Lowe, C. & Balasubramanian, S.. 2013. G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J. Am. Chem. Soc., 135, 9640-9643. [DOI] [PMC free article] [PubMed]
- Mehta A., Haber J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mehta, A. & Haber, J.E.. 2014. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol., 6, a016428. [DOI] [PMC free article] [PubMed]
- Melton D.W., Ketchen A.M., Nunez F., Bonatti-Abbondandolo S., Abbondandolo A., Squires S., Johnson R.T. Cells from ERCC1-deficient mice show increased genome instability and a reduced frequency of S-phase-dependent illegitimate chromosome exchange but a normal frequency of homologous recombination. J. Cell Sci. 1998;111(Pt 3):395–404. doi: 10.1242/jcs.111.3.395. [DOI] [PubMed] [Google Scholar]; Melton, D.W., Ketchen, A.M., Nunez, F., Bonatti-Abbondandolo, S., Abbondandolo, A., Squires, S. & Johnson, R.T.. 1998. Cells from ERCC1-deficient mice show increased genome instability and a reduced frequency of S-phase-dependent illegitimate chromosome exchange but a normal frequency of homologous recombination. J. Cell Sci., 111 (Pt 3), 395-404. [DOI] [PubMed]
- Mu D., Park C.H., Matsunaga T., Hsu D.S., Reardon J.T., Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]; Mu, D., Park, C.H., Matsunaga, T., Hsu, D.S., Reardon, J.T. & Sancar, A.. 1995. Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem., 270, 2415-2418. [DOI] [PubMed]
- Naim V., Wilhelm T., Debatisse M., Rosselli F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013;15:1008–1015. doi: 10.1038/ncb2793. [DOI] [PubMed] [Google Scholar]; Naim, V., Wilhelm, T., Debatisse, M. & Rosselli, F.. 2013. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol., 15, 1008-1015. [DOI] [PubMed]
- Neidhardt G., Hauke J., Ramser J., Gross E., Gehrig A., Muller C.R., Kahlert A.K., Hackmann K., Honisch E., Niederacher D. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol. 2017;3:1245–1248. doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]; Neidhardt, G., Hauke, J., Ramser, J., Gross, E., Gehrig, A., Muller, C.R., Kahlert, A.K., Hackmann, K., Honisch, E., Niederacher, D., et al, 2017. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol., 3, 1245-1248. [DOI] [PMC free article] [PubMed]
- Niedernhofer L.J., Essers J., Weeda G., Beverloo B., De Wit J., Muijtjens M., Odijk H., Hoeijmakers J.H., Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 2001;20:6540–6549. doi: 10.1093/emboj/20.22.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]; Niedernhofer, L.J., Essers, J., Weeda, G., Beverloo, B., De Wit, J., Muijtjens, M., Odijk, H., Hoeijmakers, J.H. & Kanaar, R.. 2001. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J., 20, 6540-6549. [DOI] [PMC free article] [PubMed]
- Niedernhofer L.J., Odijk H., Budzowska M., Van Drunen E., Maas A., Theil A.F., De Wit J., Jaspers N.G., Beverloo H.B., Hoeijmakers J.H., Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; Niedernhofer, L.J., Odijk, H., Budzowska, M., Van Drunen, E., Maas, A., Theil, A.F., De Wit, J., Jaspers, N.G., Beverloo, H.B., Hoeijmakers, J.H. & Kanaar, R.. 2004. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol., 24, 5776-5787. [DOI] [PMC free article] [PubMed]
- Regairaz M., Zhang Y.W., Fu H., Agama K.K., Tata N., Agrawal S., Aladjem M.I., Pommier Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 2011;195:739–749. doi: 10.1083/jcb.201104003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Regairaz, M., Zhang, Y.W., Fu, H., Agama, K.K., Tata, N., Agrawal, S., Aladjem, M.I.. & Pommier, Y.. 2011. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol., 195, 739-749. [DOI] [PMC free article] [PubMed]
- Rhodes D., Lipps H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015;43:8627–8637. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rhodes, D. & Lipps, H.J.. 2015. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res., 43, 8627-8637. [DOI] [PMC free article] [PubMed]
- Rodriguez R., Miller K.M., Forment J.V., Bradshaw C.R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S.P. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012;8:301–310. doi: 10.1038/nchembio.780. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rodriguez, R., Miller, K.M., Forment, J.V., Bradshaw, C.R., Nikan, M., Britton, S., Oelschlaegel, T., Xhemalce, B., Balasubramanian, S. & Jackson, S.P.. 2012. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol., 8, 301-310. [DOI] [PMC free article] [PubMed]
- Sargent R.G., Meservy J.L., Perkins B.D., Kilburn A.E., Intody Z., Adair G.M., Nairn R.S., Wilson J.H. Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res. 2000;28:3771–3778. doi: 10.1093/nar/28.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sargent, R.G., Meservy, J.L., Perkins, B.D., Kilburn, A.E., Intody, Z., Adair, G.M., Nairn, R.S. & Wilson, J.H.. 2000. Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res., 28, 3771-3778. [DOI] [PMC free article] [PubMed]
- Sargent R.G., Rolig R.L., Kilburn A.E., Adair G.M., Wilson J.H., Nairn R.S. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc. Natl. Acad. Sci. U S A. 1997;94:13122–13127. doi: 10.1073/pnas.94.24.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sargent, R.G., Rolig, R.L., Kilburn, A.E., Adair, G.M., Wilson, J.H. & Nairn, R.S.. 1997. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc. Natl. Acad. Sci. U S A, 94, 13122-13127. [DOI] [PMC free article] [PubMed]
- Sijbers A.M., De Laat W.L., Ariza R.R., Biggerstaff M., Wei Y.F., Moggs J.G., Carter K.C., Shell B.K., Evans E., De Jong M.C. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]; Sijbers, A.M., De Laat, W.L., Ariza, R.R., Biggerstaff, M., Wei, Y.F., Moggs, J.G., Carter, K.C., Shell, B.K., Evans, E., De Jong, M.C., et al., 1996. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell, 86, 811-822. [DOI] [PubMed]
- Sollier J., Stork C.T., Garcia-Rubio M.L., Paulsen R.D., Aguilera A., Cimprich K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell. 2014;56:777–785. doi: 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sollier, J., Stork, C.T., Garcia-Rubio, M.L., Paulsen, R.D., Aguilera, A. & Cimprich, K.A.. 2014. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell, 56, 777-785. [DOI] [PMC free article] [PubMed]
- Staresincic L., Fagbemi A.F., Enzlin J.H., Gourdin A.M., Wijgers N., Dunand-Sauthier I., Giglia-Mari G., Clarkson S.G., Vermeulen W., Scharer O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]; Staresincic, L., Fagbemi, A.F., Enzlin, J.H., Gourdin, A.M., Wijgers, N., Dunand-Sauthier, I., Giglia-Mari, G., Clarkson, S.G., Vermeulen, W. & Scharer, O.D.. 2009. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J., 28, 1111-1120. [DOI] [PMC free article] [PubMed]
- Thys R.G., Lehman C.E., Pierce L.C., Wang Y.H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics. 2015;16:60–70. doi: 10.2174/1389202916666150114223205. [DOI] [PMC free article] [PubMed] [Google Scholar]; Thys, R.G., Lehman, C.E., Pierce, L.C. & Wang, Y.H.. 2015. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics, 16, 60-70. [DOI] [PMC free article] [PubMed]
- Tsantoulis P.K., Kotsinas A., Sfikakis P.P., Evangelou K., Sideridou M., Levy B., Mo L., Kittas C., Wu X.R., Papavassiliou A.G., Gorgoulis V.G. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]; Tsantoulis, P.K., Kotsinas, A., Sfikakis, P.P., Evangelou, K., Sideridou, M., Levy, B., Mo, L., Kittas, C., Wu, X.R., Papavassiliou, A.G. & Gorgoulis, V.G.. 2008. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene, 27, 3256-3264. [DOI] [PubMed]
- Wang H., Li S., Oaks J., Ren J., Li L., Wu X. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 2018;9:2791. doi: 10.1038/s41467-018-05066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, H., Li, S., Oaks, J., Ren, J., Li, L. & Wu, X.. 2018. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun., 9, 2791. [DOI] [PMC free article] [PubMed]
- Wang H., Li Y., Truong L.N., Shi L.Z., Hwang P.Y., He J., Do J., Cho M.J., Li H., Negrete A. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell. 2014;54:1012–1021. doi: 10.1016/j.molcel.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, H., Li, Y., Truong, L.N., Shi, L.Z., Hwang, P.Y., He, J.., Do, J., Cho, M.J., Li, H., Negrete, A., et al 2014. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell, 54, 1012-1021. [DOI] [PMC free article] [PubMed]
- Wood R.D. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagen. 2010;51:520–526. doi: 10.1002/em.20569. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wood, R.D.. 2010. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagen, 51, 520-526. [DOI] [PMC free article] [PubMed]
- Wreesmann V.B., Estilo C., Eisele D.W., Singh B., Wang S.J. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J. Otorhinolaryngol. Relat. Spec. 2007;69:218–225. doi: 10.1159/000101542. [DOI] [PubMed] [Google Scholar]; Wreesmann, V.B., Estilo, C., Eisele, D.W., Singh, B. & Wang, S.J.. 2007. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J. Otorhinolaryngol. Relat. Spec., 69, 218-225. [DOI] [PubMed]
- Wu Y., Brosh R.M., Jr. G-quadruplex nucleic acids and human disease. FEBS J. 2010;277:3470–3488. doi: 10.1111/j.1742-4658.2010.07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu, Y. & Brosh, R.M., Jr.. 2010. G-quadruplex nucleic acids and human disease. FEBS J., 277, 3470-3488. [DOI] [PMC free article] [PubMed]
- Zhang H., Freudenreich C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell. 2007;27:367–379. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang, H. & Freudenreich, C.H.. 2007. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell, 27, 367-379. [DOI] [PMC free article] [PubMed]
- Zimmer J., Tacconi E.M.C., Folio C., Badie S., Porru M., Klare K., Tumiati M., Markkanen E., Halder S., Ryan A. Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Mol. Cell. 2016;61:449–460. doi: 10.1016/j.molcel.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zimmer, J., Tacconi, E.M.C., Folio, C., Badie, S., Porru, M., Klare, K., Tumiati, M., Markkanen, E., Halder, S., Ryan, A., et al. 2016. Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Mol. Cell, 61, 449-460. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.