

Abstract
Cisplatin is an effective chemotherapeutic agent, but significant nephrotoxicity limits its clinical use. Despite extensive investigation of the acute cellular and molecular responses to cisplatin, the mechanisms of progression from acute to chronic kidney injury have not been explored. We used functional and morphological metrics to establish a time-point when the transition from acute and reversible kidney injury to chronic and irreparable kidney disease is clearly established. In mice administered 1 or 2 doses of intraperitoneal cisplatin separated by 2 weeks, kidney function returned toward baseline two weeks after the first dose, but failed to return to normal two weeks following a second dose. Multiphoton microscopy revealed increased glomerular epithelial and proximal tubular damage in kidneys exposed to two doses of cisplatin compared with those exposed to a single dose. In contrast, there was no evidence of fibrosis, macrophage invasion, or decrease in endothelial cell mass in chronically diseased kidneys. Pathway analysis of microarray data revealed regulated necrosis as a key determinant in the development of chronic kidney disease after cisplatin administration. Western blot analysis demonstrated activation of proteins involved in necroptosis and increased expression of kidney injury markers, cellular stress response regulators, and upstream activators of regulated necrosis, including Toll-like receptors 2 and 4. These data suggest that unresolved injury and sustained activation of regulated necrosis pathways, rather than fibrosis, promote the progression of cisplatin-induced acute kidney injury to chronic kidney disease.
Keywords: Regulated Necrosis, Failed Repair, Cisplatin CKD
Graphical Abstract
Introduction
Cisplatin, though an effective chemotherapeutic agent, is limited by its nephrotoxicity. Administration of multiple cycles of cisplatin leads to permanent loss of kidney function, with severe and life-limited chronic kidney disease (CKD) emerging after successful cisplatin therapy.1 There is no known preventive therapy, and avoidance of additional doses limits the chances of successful treatment of otherwise responsive tumors. Thus, there is considerable motivation to identify molecular targets implicated in cisplatin-induced CKD.
The development of CKD, defined as a fixed loss of glomerular filtration that is often progressive, is associated with loss of renal mass and the accumulation of fibrous tissue. Often, but not always, this loss of function is associated with glomerulosclerosis. Thus a long held theory of progression states that the progressive accumulation of fibrous tissue destroys the entire nephron so that whole nephrons are lost to filtration.2 Co-incidental to this mounting fibrosis is the stimulation of a proinflammatory state characterized by macrophage infiltration and generation of profibrotic chemokines and growth factors including TGFß.2
Although interstitial fibrosis is a clinical hallmark of CKD, its role in the progression of CKD is increasingly questioned.3 Instead, growing interest in another explanation for CKD in both humans and animals centers on the development of atubular glomeruli that emerge during the course of fixed renal failure in animal models and in humans.4 Specifically, atubular glomeruli have been implicated in the progression of renal insufficiency in animal models of unilateral ureteral obstruction and in human patients with glomerular diseases (IgA nephropathy, congenital nephrotic syndrome, diabetic nephropathy) and tubular diseases (ischemic, nephrotoxin-induced and chronic pyelonephritis).3-7 Marcussen et al. have demonstrated a correlation between atubular glomeruli and reduced renal function in animal models of lithium and cisplatin-induced CKD, but in the nearly three decades since these studies were published, little work has been done to determine the natural history of chronic cisplatin nephrotoxicity, much less its mechanism.8,9
Murine models of cisplatin-induced kidney injury show that multiple administrations of cisplatin are necessary to confer chronic disease. We have established a murine model of cisplatin-induced CKD using two 15mg/kg doses of cisplatin administered two weeks apart and have shown a 50% fixed loss in GFR that extended beyond twenty-five weeks.10 Chronic renal failure in these mice was confirmed by progressive loss of renal mass that was associated with the development of reduced proximal epithelial extension into the glomerular capsule, as well as the development of atubular glomeruli and loss of proximal tubule mass. There was no detectable glomerulosclerosis histologically, and two-dimensional images of collagen distribution did not show marked changes in collagen distribution in diseased kidneys compared with controls. Therefore, fibrosis does not appear to play an obvious role in the evolution of CKD. Rather, the morphologic hallmark of CKD in this model was the “uncapped glomerular lesion,” characterized by a loss of visceral epithelial cells in the glomerular capsule. Severe lesions resulted in atubular glomeruli with collapsed proximal tubules.10
The molecular mechanism for the observed morphologic and functional changes in the transition from acute kidney injury (AKI) to CKD following cisplatin re-exposure has yet to be elucidated. Exposure of renal tubule cells to cisplatin has been shown to activate complex signaling pathways involved in apoptosis, cell proliferation and differentiation, and inflammatory and stress responses.11,12 After single exposure of the kidney to cisplatin, proximal tubule cells die by necrosis through a relatively newly discovered pathway of regulated necrosis, while apoptosis plays a minor role in vivo.13-15 Regulated necrosis consists of several distinct pathways that end in the death of cells and release of damage associated molecular patterns (DAMPs) that may perpetuate injury and activate inflammatory pathways. Regulated death may emerge from injury in the nucleus, endoplasmic reticulum, and mitochondria all of which are damaged by CP. Both necroptosis, mediated by the proteins Receptor Interacting Serine/Threonine Kinase (RIPK) 1 and 3, and Mixed Lineage Kinase Domain-Like (MLKL) that may emerge from signals formed after nuclear damage, and the activation of the mitochondrial permeability transition pore, mediated by Cyclophilin D (CypD), have been shown to be responsible for the necrosis observed in acute cisplatin toxicity.16 Whether these pathways of regulated necrosis engaged in acute toxicity are involved in the generation of chronic cisplatin nephrotoxicity is unknown.
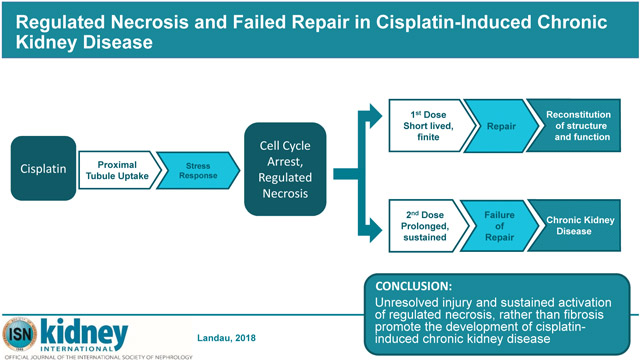
In this study, we established a time-point, two weeks after the second dose (15 mg/kg) of cisplatin, when the transition between AKI and CKD is complete. We then analyzed the kidney’s molecular profile at this time-point to compare with an earlier state, two weeks after a single-dose of cisplatin, where renal function is still recoverable. Two weeks after a single dose, GFR is near normal, plasma creatinine levels and kidney mass are normal, and there is minimal morphologic evidence of injury, indicating near completeness of repair. However, two weeks after a second cisplatin dose, or four weeks after the first dose, treated mice demonstrate elevated plasma creatinine levels, reduced kidney mass and GFR and significant glomerular epithelial injury that persists, indicating established CKD. To identify potential biomolecules and processes implicated in the progression of cisplatin induced AKI to CKD, we compared molecular and histologic markers at these stages. The data suggest that following the second dose of cisplatin, unresolved injury and increased rates of regulated necrosis, rather than fibrosis, drive disease progression.
Results
Quantification of Physiologic and Morphologic Kidney Measurements
We developed a CKD model using two intraperitoneal doses of cisplatin (15 mg/kg) administered two weeks apart (Figure 1). Mice were sacrificed at two weeks after the first cisplatin dose, four weeks after the first dose, and two weeks after the second dose (four weeks after the first dose). At each time point, we measured plasma creatinine and GFR, and collected kidney tissue for histological examination (Figure 1). Previously we reported on the characteristics of the induction of CKD at nine weeks following the first dose using this two-dose model. We now focus on an earlier time at four weeks following the first dose where both the fall in GFR and the appearance of tubular injury are evident. Two weeks after the second dose of cisplatin (four weeks after the first dose) the animals lost weight and their GFR and kidney weight were reduced as compared to control and single dosed animals (Figure 2). Plasma creatinine (PCr) reflected this lower GFR in these animals. Two and four weeks following a single dose of cisplatin, PCr had returned to normal, although GFR was still somewhat reduced (Figure 2). Kidney weight was not reduced at this time point. However, two weeks after the second dose increased PCr and reduced GFR and decreased kidney weight were evident and indicated the establishment of CKD. Previously we have shown that this reduction in renal mass and GFR is sustained for several months after two doses and indeed the reduction in renal mass progresses.10
Figure 1.

Mouse Model of Chronic Kidney Disease. One or two doses of cisplatin (15 mg/kg) were administered intraperitoneally to mice aged 8-12 weeks. Groups were comprised of 3 mice. Blood was sampled by retro-orbital puncture under brief isoflurane anesthesia at 2-week intervals beginning 2 weeks after the first dose to measure serum creatinine. Mice were sacrificed at 0 weeks, 2 weeks after the 1st dose, 4 weeks after the 1st dose, 2 weeks after the second dose (4 weeks after the 1st dose), and 4 weeks after the 2nd dose (6 weeks after the first dose). At the time of sacrifice mice were anesthetized, GFR was measured by inulin clearance and a final blood sample was obtained. The left kidney was surgically removed and processed for mRNA and protein analysis. The animal was then perfused intravascualarly with buffered 4% paraformaldehyde for several minutes and the right kidney was harvested for preparation of frozen sections and MPM analysis.
Figure 2.

Second Dose of Cisplatin Results in Sustained Renal Injury (A) Glomerular filtration rate (GFR) following a single dose of cisplatin recovers nearly completely by four weeks after the first dose. However, GFR two weeks following the second dose is significantly (p < 0.05) reduced by approximately 50%. (B) Perfused kidney weight was unaffected by a single dose of cisplatin, but kidney weight decreased by approximately 30% following the second dose of cisplatin.
Multiphoton microscopy of cisplatin treated kidneys revealed variable degrees of both capsular and tubular damage distributed throughout the cortical region (Figure 3A-B). Capsular and tubular damage in diseased kidneys was manifested by reduced protein staining, enlarged nuclei, reduced cytoplasm with increased nuclear/cytoplasmic ratio, and thinned tubular walls (Figure 3B). A subset of tubules remains morphologically normal after the two doses of cisplatin, but most tubules show at least portions of damaged tubular cells. Glomeruli that show damage at the glomerular-tubular junction and complete flattening (uncapping) of the proximal cell extensions at this junction are associated with abnormal proximal tubule cells in later segments of these nephrons often associated with collapsed lumens (Figure 3-C). Kidneys exposed to a single dose of cisplatin show far less capsular and tubular damage with the majority of tubules appearing morphologically normal, when compared with kidneys exposed to two doses of cisplatin (Figure 3B). Rare tubules in 2 wk −1 dose kidneys display marked damage throughout the tubule, and an even smaller subset of these damaged tubules are atubular. The damage visualized at this timepoint is predominantly restricted to individual nephrons with surrounding normal tubules. After two additional weeks, without additional cisplatin (4 wk – 1 dose), there is no statistical difference in damage, although there is a trend for reduction in damaged capsules (Figure 3C). If a second cisplatin dose is given (4 wk – 2 dose), there is a statistically significant increase in capsular damage and atubular glomeruli, corresponding to the reduction in GFR (Figure 3C).
Figure 3.

Multiphoton Microscopy Reveals Increased Morphological Damage 2 weeks After a 2nd Dose of Cisplatin. Patterns of proximal tubular cell damage at capsule and glomerulo-tubular junction, 2 weeks after second dose of cisplatin. (A) 3D reconstruction of glomeruli based on MPM imaging. I: Normal cuboidal tubular cells of the glomerular capsule enclose a large proportion of the surface of a glomerulus. II: Partial glomerular capsule cuboidal cell loss is visible in 3D reconstruction, leading to a proximal tubule with minor changes to nuclear morphology (prominent nucleoli). III: Complete cuboidal cell loss was associated with visually detectable proximal tubule damage, characterized by reduced nuclear spacing, and reduced protein staining. IV: Glomerulus without detectabe proximal tubule connection showing small glomerular size and adjacent irregular cellular aggregate without visible lumen.
(B) Nuclear stain (DAPI) green, protein stain (eosin) red. I: Low power view of kidney two weeks after a single dose of cisplatin (2 wk – 1 dose) had normal overall appearance. II: High power view of 2 wk – 1 dose nephron shows many glomerular capsule cuboidal cells and associated normal proximal tubule showing homogeneously bright protein staining (red), clear brush border, and round nuclei with single nucleoli. III: High resolution 3D reconstructions of normal glomerular capsule shows smooth surface from cuboidal glomerular capsule cells near the tubular junction. IV: Low power view 2 weeks after 2nd dose of cisplatin (4 wk – 2 dose) shows variable protein staining with areas of increased nuclear density corresponding to damaged nephrons. V: High power view of damaged nephron unit (demarcated by blue line) in kidney 2 weeks after second dose of cisplatin (4 wk – 2 dose). Changes include vacuoles in proximal tubule cells at glomerulo-tubular junction, reduction of cuboidal cells of glomerular capsule (tubular epithelial cells extending into capsule), markedly reduced protein staining in proximal tubule, swollen, irregular nuclei with marginalized chromatin, loss of brush border, increased nucleus/cytoplasm ratio, and high nuclear density. VI: High resolution 3D reconstructions of damaged glomerular capsule shows “uncapping” of capsule with associated disorganization of the underlying nuclei.
(C) Quantification of injured capsules during development of cisplatin-CKD revealed that single dose damage was not statistically increased compared to untreated controls but some atubular glomeruli evolved. There was no detectable change at 4 weeks after a single dose, but a second dose of cisplatin induced increased percentage of injured tubules and accumulation of atubular glomeruli compared to controls, 2 week – 1 dose, and 4 week – 1 dose kidneys (Chi-squared, p < 0.01).
Notably, second harmonic generation imaging showed that none of these changes were associated with fibrosis in the regions of tubular damage and that the mild increases in interstitial collagen at two weeks after the second dose (4 wk – 2 dose) were not spatially correlated to damaged tubules (Figure 4).
Figure 4.

Nephron Damage was not Associated with Collagen Fibrosis by Second Harmonic Generation Second harmonic generation sections of 2 week – 1 dose kidney showing minimal collagen (green). Low-power views show collagen is predominantly perivascular in both 2 week – 1 dose and 4 week – 2 dose kidneys. High power views show some interstitial peritubular collagen (green), but the collagen was not associated with abnormal tubules or "atubular" glomeruli. Scale bars=500 μm (left panel), 50 μm (right panel).
We performed additional staining to evaluate whether other cellular elements indicative of chronicity was present (Figure 5). Following the second dose of cisplatin, we found no increase in macrophage numbers (F4/80 staining – data not shown) or decrease in endothelial cell mass (CD34 staining). However, there was a sustained increase in activated myofibroblasts (SMA staining). Remarkably there was a reduction in Ki67 staining two weeks after the second dose in marked contrast to the increase seen two weeks after a single dose (Figure 5I).
Figure 5.


Histological Evaluation of Cellular Elements Indicative of Chronicity. All images are original magnification of 40X, scale bars = 100 μm. (A) Tunel staining for apoptosis. (B) No statistically significant difference in Tunel staining was observed in kidney sections at 2 weeks or 4 weeks after the 1st dose and 2nd doses of cisplatin. (C) Smooth muscle actin (SMA) staining for activated myelofibroblasts. (D) SMA staining was significantly increased at 2 and 4 weeks following the 2nd dose of cisplatin compared with similar time points following the 1st dose of cisplatin. (E) CD34 staining for endothelial cells. (F) CD34 staining was significantly reduced at 2 weeks following the 2nd dose of cisplatin, but recovered by 4 weeks following the 2nd dose. Histological Evaluation of Cellular Elements Indicative of Chronicity
All images are original magnification of 40X, scale bars = 100 μm. (G) Ki67 staining of the tubule compartment increased through the 4 week period following 1 dose of cisplatin (triangle). By contrast, Ki67 staining does not increase at this time point in the 2 dose model (circle) and is no different from untreated control levels. Ki67 staining also was not increased through the 2 week period following 1 (triangle) or 2 (circle) doses of cisplatin when compared with untreated control levels.
Reduced renal mass mostly confined to the proximal tubule, without significant involvement of invading macrophages or reduction in endothelial cell mass, suggested to us that an analysis of the renal transcriptome in the two-week interval between cisplatin doses would reveal the important processes arising from the nephron that are responsible for the establishment of CKD. We therefore generated whole kidney chip arrays to fully describe the state of the renal transcriptome prior to cisplatin administration, two weeks after a single dose, and two weeks after the second dose.
Microarray analysis reveals distinct patterns of gene expression after one or two doses of cisplatin.
Principal Component Analysis (PCA) and unsupervised hierarchical clustering analysis demonstrated that each treatment group of samples clusters separately (Figure 6). Multigroup comparison (ANOVA, p < 0.05; Benjamini-Hochberg FDR < 0.01) identified 1679 differentially expressed genes across the treatment groups of samples (q < 0.01). When analyzed in a kidney-specific database of IPA, these differentially expressed genes were significantly associated with damage and injury to renal tubules (Table 1). In a non-kidney specific database, notably enriched networks included those related to cell signaling and movement, cell death and survival, and renal necrosis (Table 2).
Figure 6.

Changes in Kidney Transcriptional Profile Following Cisplatin Administraiton (A) Principal component analysis (PCA) of the multi-group ANOVA comparison, illustrating the significant variation between the gene expression profiles of the control (blue, WK0), one dose (yellow, WK2), and two dose (pink, WK4) animals
Table 1 ∣.
Top diseases and functions from kidney-specific database. Top 15 diseases and functions (P < 0.001) identified after applying a kidney filter to the 1689 transcripts from ANOVA
| Top Diseases or Functions Annotation | p-Value |
|---|---|
| Damage of renal tubule | 8.67E-08 |
| Damage of kidney | 3.33E-06 |
| Thrombus | 5.50E-06 |
| Crescentic glomerulonephritis | 3.48E-05 |
| Injury of renal tubule | 3.48E-05 |
| Nephritis | 6.83E-05 |
| Acute phase crescentic glomerulonephritis | 7.53E-05 |
| Thrombosis of renal glomerulus | 9.24E-05 |
| Injury of kidney | 1.31E-04 |
| Cell death | 6.75E-04 |
| Damage of tubulointerstitium | 7.65E-04 |
| Accumulation of phagocytes | 1.29E-03 |
| Urological disorder of kidney | 1.36E-03 |
| Abnormal morphology of renal glomerulus | 1.52E-03 |
| Glomerulonephritis | 1.71E-03 |
Table 2 ∣.
Top diseases and functions associated with changes in kidney transcriptional profile following cisplatin administration. Top diseases and functions (P < 0.001) identified from a general (non–organ specific) database for 1689 transcripts identified by ANOVA. Significance was calculated from the p-score (p-score = −log [P value]) and focus molecules are the number of molecules that appear in network and in the dataset
| Top Diseases and Functions |
p-score | p-value | Focus Molecules |
Molecules in Network |
|---|---|---|---|---|
| Lipid Metabolism, Small Molecule Biochemistry, Vitamin and Mineral Metabolism | 12 | 1E-12 | 16 | AADAC,ABCG2,ACSS1,ALDH1A1,CDC42BPA,DPEP1,EIF3A,EMILIN1,FAM151A,IL17RB,MID1,NAPSA,Nrg1,PKD1,POU2F1,TTR,VWF |
| Cell-To-Cell Signaling and Interaction, Cellular Movement, Hematological System Development and Function | 9 | 0.000000001 | 21 | ABCC4,ALB,APOE,AQP2,C1QA,CASP3,CCL2,CCL5,CXCL10,CYP27B1,DBP,EGR1,ETS1,F3,FAT1,IL1B,INF2,KIRREL,LRP1,mir-193,MYD88,NCF1,NFKBIA,NOS2,NPHS1,NR1H4,PLAUR,RELA,SCNN1A,SLC14A1,SLC51A,TLR4,TNF,VDR,WT1 |
| Cellular Movement, Hematological System Development and Function, Immune Cell Trafficking | 6 | 0.000001 | 18 | ACE,CAV1,Ccl2,CCL2,CCL4,CCL5,CFB,CXCL3,F2RL1,folic acid,GBP4,HGF,ICAM1,IL17RD,IL1B,IRF4,MAPK14,MYD88,NOS3,NTN1,PROC,PROCR,RELA,S1PR2,SMAD4,SMAD7,SMURF1,THBD,TLR2,TLR4,TNF,TNFRSF12A,TNFRSF1B,TNFSF12,YBX1 |
| Cell Death and Survival, Organismal Injury and Abnormalities, Renal Necrosis/Cell Death | 6 | 0.000001 | 16 | AGT,CD68,CDKN1A,Clec2d (includes others),Cyp4a14,D-glucose,DKK3,EGF,GLIS2,Gsta4,HLA-DMA,ITGB5,JAG1,Kap,MAP2K3,MAPK13,MTOR,NLRP3,PPIB,RHOA,SMAD3,SPC25,TCIRG1,TFE3,TGFB1,TSC22D1,VEGFA,VIM,YBX1 |
| Organismal Injury and Abnormalities, Cellular Movement, Cardiovascular System Development and Function | 5 | 0.00001 | 16 | aldosterone,BDKRB1,BDKRB2,BGN,BIRC2,COL18A1,COL1A1,COL1A2,COL3A1,COL4A2,COL4A3,CR1L,CTGF,CXCR3,D-glucose,DCN,EDN1,FAS,FBN1,FN1,GLIS2,HGF,IKBKB,MAP2K1,MAPK1,MAPK3,MAPK14,MGP,MMP2,NUDT19,NUPR1,SMAD3,TGFB1,TSC22D3,WFDC2 |
| Cellular Development, Cellular Growth and Proliferation, Organ Development | 5 | 0.00001 | 13 | ANGPT2,BAG1,CAMK2A,CDH1,CTNNB1,CYBA,DBNL,EGFR,FOXI1,ID2,KPNA2,MIR17HG,MMP7,MMP14,MMP23B,NDE1,PKD1,PKD2,PRKCSH,PROS1,PVALB,RHOB,SLC26A4,SLC4A1,TCF3 |
| Cellular Movement, Immune Cell Trafficking, Hematological System Development and Function | 4 | 0.0001 | 15 | Abcb1b,ATP1A1,AXL,CD9,Cdc42,COL4A3,D-glucose,EDNRB,FCER1G,FCGR2B,FN1,GAS6,HBEGF,IFNG,IGF1,IL18BP,IL1B,ILK,ITGA3,ITGB1,LAMA5,LAMB1,LAMB2,MMP9,NID1,NOS2,PDGFB,PLAT,SLC9A3,SOCS3,STAT3,THY1,TNF,TP53,VEGFA |
| Endocrine System Disorders, Gastrointestinal Disease, Metabolic Disease | 4 | 0.0001 | 11 | ABCA1,AGT,ARPC1B,BAX,BMP2,CA2,CALB1,D-glucose,EIF4EBP1,EIF4G1,FABP1,FOS,MLXIPL,nicotinic acid,PDCD4,PRKCA,SPP1,TGFB1,TRIM24,TRPV5,TSC2,VCAM1 |
| Cell Morphology, Organismal Injury and Abnormalities, Metabolic Disease | 3 | 0.001 | 12 | AGT,AGTR1,ANGPT2,APAF1,BMP4,BMPR1A,CP,CTNNB1,Cyp2c44,GAS2,HMOX1,HSPB1,IL1B,LCN2,MAPKAPK3,MGST1,MYC,NOX4,PTGS2,REN,SLC12A1,SLC2A1,SMAD1,SOX9,TCF4,TGFB1,TNF,TXNIP,VAMP3 |
Interactive networks between the differentially expressed genes and the downstream function “necrosis of kidney” revealed activation of necrosis acutely (3 days) following the first dose of cisplatin (unpublished data), inhibition of necrosis at two weeks after the first dose, and sustained reactivation of necrosis two weeks following the second dose (Figure 6). More detailed analysis of the microarray results can be found in the supplementary data.
Second dose of cisplatin administration results in activation of stress response and regulated necrosis proteins
Protein levels of KIM1, NGAL, p-RIPK1, p-RIPK3, p-MLKL, MLKL, TLR4 and TLR2 were examined by western blot analysis. In addition, levels of phosphorylated JNK and ERK proteins were evaluated because the MAPK genes, which were identified as differentially expressed by microarray analysis, influence the activity of these proteins. Western blot analysis of KIM1 showed similar band strength for control and 2 week – 1 dose samples, minimal increased band strength for 4 week – 1 dose samples, and markedly increased band strength for 2-dose samples (Figure 8A-B). NGAL showed significantly increased protein expression at two weeks following the first and second doses of cisplatin (Figure 8A). However, by four weeks after the first dose of cisplatin, NGAL protein expression returned to baseline (Figure 8B).
Figure 8.

Expression of NGAL and KIM1 as Marker of Unresolved Injury Following Cisplatin Administration
Protein isolated from control (n=3), 2 week-1 dose cisplatin (n=3), 4 week – 1 dose (n=3), and 4 week-2 dose cisplatin (n=3) mice. Western blot analysis of NGAL and KIM1 with GAPDH and beta-tubulin used as endogenous controls. Results are mean ± SE (n=3, compared with control, p < 0.05) from triplicate blotting. A) KIM1 protein expression significantly increased following the second dose of cisplatin, while NGAL protein expression increased following one and two doses of cisplatin. B) The increase in NGAL protein expression observed at two weeks following a single dose of cisplatin was resolved by four weeks. There was no observed increase in KIM-1 protein expression at two or four weeks after a single dose of cisplatin.
Phosphorylated ERK (T202, Y204) demonstrated increased band strength in 2-dose samples only, while phosphorylated JNK (T183, Y185) showed significantly increased expression in 1-dose and 2-dose samples compared with control samples (Figure 9). Proteins involved in regulated necrosis showed significantly increased expression (TLR2 and TLR4) and activation (p-RIPK1, p-RIPK3, p-MLKL) following one and two doses of cisplatin (Figure 10). We were unable to find an adequate antibody for CypD and thus were not able to include this protein in our western blot analysis, but chip arrays and PCR showed increased levels (supplemental data).
Figure 9.

Activation of Stress Response Following Cisplatin Administration. Western blot analysis of phospho-Thr183/Tyr185-JNK, phospho-Thr202/Tyr204-ERK, and Total ERK; GAPDH was used as an endogenous control. Results are mean ± SE (n=3, *: compared with control, p < 0.05) from triplicate blotting. Western Blots revealed significantly increased protein expression following the first cisplatin dose (pJNK) and second cisplatin dose (pJNK, pERK).
Figure 10.

Activation of Regulated Necrosis Following Cisplatin Administration. (A) Western blot analysis of p-RIPK1, p-RIPK3, p-MLKL, t-MLKL, TLR2, and TLR4; GAPDH and β-tubulin used as endogenous controls. Protein isolated from whole kidney lysate. (B) Results are mean ± SE (n=3, *: compared with control, p < 0.05) from triplicate blotting. Western Blots showed increased protein expression for p-RIPK1, pRIPK3, and TLR4 following the first and second doses of cisplatin.
Discussion
Previously we have shown that two 15mg/kg doses of cisplatin caused CKD characterized by sustained reduction in GFR and reduced kidney mass. We now identify a time point, two weeks after the second dose of cisplatin, at which time both features of CKD are present, indicating that chronicity has been established. Importantly, these indicators of chronicity are nearly completely resolved at a time point of two weeks after a single dose. We also note that these functional and morphologic characteristics of chronicity occur before invasion of macrophages and the depletion of endothelial cells, but when fibroblasts are already activated. Additionally, we observed progressive increase in atubular glomeruli, which coupled with our observation of reduced replicative response to injury following the second dose, we interpret to mean that tubular injury is unresolved and indicative of unsuccessful repair. Analysis of the renal transcriptome at this time should reveal the genes that are responsible for this failure to repair as well as those that are determinant of the transition between acute and chronic renal failure.
The analysis of the microarrays identified a set of genes and proteins that demonstrate sustained activation following the second dose of cisplatin. Markers of renal injury (NGAL, KIM1), cell cycle regulation and stress responsiveness (pERK, pJNK,), as well as regulated necrosis (MLKL, p-RIPK1, p-RIPK3, TLR2, TLR4) were sustained or reactivated after the second dose. The data suggest that following the first dose of CP there is short-lived activation of cell injury and regulated necrosis pathways, followed by nearly complete repair and regeneration, while following the second dose, there is prolonged expression of cell stress molecules and sustained activation of regulated necrosis, resulting in chronic disease.
The results of this study expand upon previous studies in two important ways. First, this study examines molecular responses to CKD at the transition between AKI and CKD. Second, this study identified a set of molecular candidates for further exploration of mechanisms of the progression from AKI to CKD following multiple doses of cisplatin.
Previously proposed as biomarkers for AKI, NGAL and KIM1 were identified in this study as molecular candidates in the progression from AKI to CKD. NGAL, which belongs to the lipocalin superfamily, promotes differentiation of kidney progenitors into epithelia, tubules, and complete nephrons. Beyond its diagnostic and prognostic function in AKI, exogenous administration of NGAL has been shown to be renoprotective, rescuing the kidney from ischemic-reperfusion injury.17-20 In the present study, NGAL protein levels were increased following one and two doses of cisplatin, which may suggest the kidney’s attempt to repair damage and protect against further injury. However, four weeks following a single dose of cisplatin, when renal function has nearly completely recovered, NGAL protein levels return to baseline, supporting the role of NGAL as a marker of kidney injury. Because NGAL protein levels in 2wk-1dose and 4wk-2dose animals did not differ significantly, it is likely that NGAL is a marker of ongoing injury and unsuccessful attempts at repair.
On the other hand, our data supports the use of KIM1 as a marker for CKD given that elevated levels of KIM1 during acute toxicity fall to pre-exposure levels two weeks after the first dose, show an only small increase at four weeks after the first dose, but rise to very high levels following the second dose of cisplatin when CKD is established. KIM1, a co-stimulatory T-cell transmembrane glycoprotein, is highly upregulated in AKI.21 Acutely, KIM1 appears to play a protective role, but the acute protective role of KIM1 may not mirror the chronic effects of KIM1 expression.21 As chronic expression of KIM1 is localized to the proximal tubule, which is the site of many of the morphological changes that impair nephron function, its reactivation may reflect unresolved injury.10,21 Moreover, KIM1 has been shown to activate the IL6 pathway, which, in other tissues, plays a protective role acutely, but impairs regeneration when sustained.22,23
Sustained activation of inflammatory pathways has traditionally been conceived to promote CKD development via fibrosis. Recently though, studies focusing on the relationship between fibrosis and functional decline have raised questions about fibrosis as a key process in CKD progression. It is well known that cisplatin causes chronic interstitial nephritis and interstitial fibrosis, however the role of these processes in the progression of renal disease is unclear.24 To elucidate the impact of fibrosis on renal function, Kawai et al. administered the antioxidant N, N’-diphenyl-p-phenylenediamine to block fibrosis following cisplatin administration. They found that inhibiting the expansion of interstitial fibrosis, increase in type III collagen, and α-SMA overexpression had no effect on acute renal injury or macrophage infiltration in the renal interstitium.24 More convincing evidence for the dissociation of fibrosis from functional decline was provided by the study of Maarouf et al., who demonstrate that stromal cells are the source of fibrosis and despite intervening in the activation of stromal cells and reducing interstitial fibrosis, they observed no effect on leukocyte recruitment, inflammation, functional decline or epithelial injury.25 Thus there is strong evidence that CKD may ensue regardless of the presence of renal fibrosis, suggesting that interstitial fibrosis itself does not promote renal dysfunction, but rather ongoing epithelial injury drives fibrosis as a secondary event in the development of CKD.24-26
Our data further support the notion that fibrosis is a secondary event in the development of CKD. Although our data show increased transcription of collagen genes and activation of myelofibroblasts following the second dose of cisplatin, we found no evidence for widening of the interstitial space, nor did we find evidence for increased macrophage infiltration. Using multiphoton microscopy and second harmonic generation to visualize collagen deposition in renal tissue from cisplatin-induced CKD we show that there is no spatial correlation between increased collagen fibrosis and defective nephrons (i.e., cap lesion).10 The lack of correlation between fibrosis and proximal tubule injury in our model furthers the hypothesis that sustained epithelial injury drives renal dysfunction and points to continued proximal tubule injury as the initiating determinant of the transition of AKI to CKD.3,26
We speculate that the unrelieved epithelial injury and the induction of regulated necrosis is a key event in the development of CKD. Regulated necrosis is the process by which injury induces the loss of membrane integrity and subsequent release of cell death associated molecular patterns, or DAMPS. One pathway leading to such loss of membrane integrity involves the induction of necroptosis, which is a kinase-dependent process of regulated necrosis mediated by the RIPK1-RIPK3-MLKL necroptosome. This process has been described in models of AKI and predominates in acute cisplatin nephrotoxicity.15 Regulated necrosis can activate necroinflammation, a process characterized by the auto amplification loop between cell necrosis and inflammatory tissue response.27-29 Of the regulated necrosis processes, necroptosis has been described as the “least inflammatory.”30 Enhanced necroptosis is thus consistent with our findings that F4/80 staining, and thus macrophage infiltration, does not increase following the 2nd dose of cisplatin (data not shown).
Upstream regulation of necroptosis can occur through TNF and TLR4, which are also important mediators in acute cisplatin nephrotoxicity.13,16 Cisplatin-induced injury causes a release of DAMPs that activate TLR4 to stimulate the production of various chemokines and cytokines, including TNFα, which further exacerbate cisplatin nephrotoxicity.31-33 Reeves et al. demonstrate that inhibiting TNFα and TLR4 protects against acute cisplatin nephrotoxicity, and reduces renal dysfunction, tubule injury, and inflammation following cisplatin treatment.31,33 Although TLR2 has not been studied for its role in chronic cisplatin nephrotoxicity, it has been implicated in AKI following ischemia-reperfusion injury, as TLR2 knockout mice are protected from DAMP-driven inflammation in this model. 28,34
Our data supports and extends this work by suggesting a role for regulated necrosis in the progression of cisplatin-induced CKD. We observed significantly increased protein levels for TLR2 and TLR4, along with enhanced activation of RIPK3, RIPK1, and MLKL as evidenced by increased expression of p-RIPK1, p-RIPK3, and p-MLKL following cisplatin administration. CypD mRNA levels rose as well and may also initiate regulated necrosis (supplemental data). Coupled with our data on the functional and morphologic impact of cisplatin, the activation of these proteins following cisplatin administration points to regulated necrosis as an important mechanism for sustained injury and failure to repair, resulting in reduced renal mass and function, that begins after the first dose, but reaches the point of chronicity after the second dose.
Experiments are still needed to elucidate the mechanism driving increased regulated necrosis in the cisplatin-CKD model. Given the somewhat surprising TLR2 finding, one attractive future direction of study examines the role of TLR2 in regulated necrosis activation. In macrophages, stimulation of TL2, 3, 4, 5 or 9 coupled with inhibition of caspase 8 results in RIPK3 dependent regulated necrosis that occurs through TRIF.35 Though studies have shown that TLR3 and TLR4 can drive RIPK3 activation through RHIM association with TRIF, there is no current evidence for a role of TLR2 in driving this regulated necrosis mechanism.36 We are currently developing the TLR2 knockout mice and plan to evaluate the mechanism by which TLR2 contributes to regulated necrosis in the cisplatin-CKD model.
Cisplatin-induced models of AKI have focused on the involvement of cell stress and cell cycle pathways. DNA damaging agents, cisplatin among them, have been shown to activate the immediate early gene response.11 This response involves the MAPK pathway and subsequent activation of the transcription factors c-fos and c-Jun that then activate and repress a large number of genes involved in the cellular stress response.1 Two specific downstream components of the MAPK pathway, ERK and JNK, have been shown to be important in determining the outcome of acute injury. Specifically, ERK has been shown to promote cell death in cisplatin-induced AKI. 1,11 Inhibition of JNK and upregulation of ERK ameliorated necrosis in the ischemia/reperfusion model, demonstrating the complexities of these pathways under different stresses.11 Similar to the situation observed with sustained KIM1 expression, brief ERK activation has been shown to play a protective role in acute injury but sustained activation of ERK resulted in adverse cellular outcomes.37 Moreover, Murphy et al. postulated that protein products of the immediate early gene response function as molecular sensors for differences in signal duration, and increased signal duration alters the functional activity of the immediate early gene products, thus controlling biological outcome.37 Sustained activation of ERK and JNK following the second dose of cisplatin correlates with morphological and functional indications of failed tissue repair, suggesting a role for these immediate early gene products in modulating injury and repair processes in cisplatin nephrotoxicity. This failure to resolve brief activation of the immediate early gene response seems to mark the conversion of AKI to CKD in cisplatin-induced CKD.
The model developed here offers the potential to identify pathways and processes implicated in the progression to CKD. The observed progressive increase in atubular glomeruli suggests that once tubular damage progresses to the point where there is no connection between glomerulus and proximal tubule, there is no expected recovery. To confirm this, we suggest future studies perform sequential observation of the evolution of glomerular and proximal tubule lesions, which may be distinct in different species given the differences in the extent to which proximal tubules extend into the glomerulus.38 Capsule lesions in the mouse were always associated with diseased proximal tubule further down the nephron and reconstruction of nephrons would indicate the extent of damage and points of no return in species with limited extension into the glomerulus. The use of MPM would certainly facilitate this.
Beyond morphology, there is great interest in elucidating the molecular switch marking the “point of no return,” past which the kidney fails to recover. One potential switch activating further necroptosis is TWEAK, which has been shown to promote a second wave of renal damage in models of folic acid and IR models of AKI.13,39 Downstream TWEAK signaling through cIAP1/2 can activate RIPK1 and thus open the gate to further necroptosis and progression to CKD.40
Another important direction of future studies looks at the role of regulated necrosis and its mediators as it appears to be a central process involved in the progression from acute to chronic disease. The recent detection of p-MLKL in human kidneys after transplantation suggests that regulated necrosis is relevant to human disease and thus supports further investigation of necroptosis in human models of nephrotoxicity.41 However, pMLKL may be a difficult marker to use for necroptosis, given its ability to both promote and protect against necroptotic cell death. Phosphorylation of MLKL by RIPK3 triggers necroptotic cell death, Independent of RIPK3, MLKL participates in endosomal trafficking and production of extracellular vesicles, and pMLKL release in these vesicles has been shown to prevent MLKL-mediated cell death.42 Subsequent studies must assess the balance between the pro-necroptotic and anti-necroptotic functions of pMLKL in the progression of cisplatin-CKD. Future studies on the precise role of necroptosis in the development of cisplatin-induced CKD will have to take into account both the protective and the destructive role of necroptosis in the development of the disease.
Following the 2nd dose of CP, we observed increased CypD mRNA expression, which may be associated with CypD’s role in promoting mitochondrial permeability transition and necrotic cell death, while inhibiting apoptotic cell death.13,43 As mitochondrial injury plays a key role in acute cisplatin toxicity, this pathway is a highly promising one to pursue.1,44
Other potential targets for future investigation are KIM1 and NGAL, which demonstrated significant levels of sustained expression following the second dose of cisplatin, at a time when KIM1 and NGAL returned to near baseline or baseline after a single dose. Currently, KIM1 and NGAL are primarily recognized as urinary biomarkers for acute renal injury, though their functional role in chronic disease has yet to be elucidated.19,21 While other investigators have suggested the use of KIM1 and NGAL as markers for sustained renal injury,45 KIM1’s dramatic activation following the second cisplatin dose makes it a particularly attractive reporter for chronic disease.
Identifying key biomolecules with increased and sustained expression in cisplatin-induced CKD will allow for the elucidation of the functional effects of such activation on kidney regeneration and repair. We suggest that a focus on understanding how to inhibit sustained activation of key mediators of the stress response and regulated necrosis will enable repair and prevent the development of CKD following multiple exposures to cisplatin treatment.
Methods
Murine Model of Cisplatin-Induced Chronic Kidney Disease
The murine model of cisplatin-induced CKD was prepared as previously described with the following modification: kidneys were harvested at two weeks after the first dose and two weeks after the second dose of cisplatin.10 Initially we selected a cisplatin dose of 20 mg/kg, however mice were unable to survive a second injection at this dose. The cisplatin dose of 15 mg/kg was ultimately chosen because animals would survive the first dose, which was almost completely resolved by the second dose, and could be repeated after two weeks, when chronic and irreversible injury was established and animals would survive in a state of chronic renal failure.
Multiphoton Microscopy (MPM) Studies
Tissue Clearing and Multiphoton Imaging and Processing
Mouse kidney specimens were prepared, processed, and imaged using a tissue clearing protocol and multiphoton microscopy setup as previously described with minor modifications (see supplemental methods).10 Imaging was performed on a custom-built microscopy setup employing a Ti:Sapphire laser for excitation and a high numerical aperture and long working distance objective (Leica 0.95NA 1.95 mm WD) or with a shorter working distance objective with higher numerical aperture (Olympus 1.35NA) for higher resolution reconstructions. Second harmonic generation was simultaneously collected in transmittance during imaging. Tiled raster images for large reconstructions were collected overnight. Images were processed using standard ImageJ tools available with the FIJI distribution. Three-dimensional reconstructions were rendered by built-in volume viewing tools in FIJI.
Capsular Damage Analysis
Glomeruli were randomly marked on large volume 3D stacks taken at step sizes of 1-2um by employing the overlay function of ImageJ. Using customized scripts written in python, a cubical volume surrounding the position of marked glomeruli was extracted and composed into a randomized 3D mosaic containing glomeruli from all samples. The randomization ensured that the pathologist evaluating the level of capsular damage was blind to the time-point a given image originated from, reducing bias. Pathologists scored glomerular capsule damage based on reduction or visible damage to the cuboidal cells and categorized as 1) atubular, 2) clearly reduced/damaged, 3) normal, or 4) increased, indicated by score placed directly on glomerulus. The use of an 'increased' category reflects the finding that some glomerular capsules show higher percentage of cuboidal cell coverage than others, even in control specimens. Marked values were then linked with specimen of origin based on position of glomerulus on grid. Three to four kidneys resulting in at least 100 glomeruli/glomerular capsules at each time point were evaluated. Statistical comparisons of damage at different time points were done using the chi-square test for independence at a 0.01 level of significance.
Imunohistochemistry (IHC) studies
Immunohistochemical staining and scoring was performed for Ki67, F4/80, Tunel, α-smooth muscle actin (SMA), and CD34, as previously described, with blinded scoring performed by an expert renal pathologist using a square grid technique.10
RNA and Protein Isolation
RNA was isolated from mouse kidney using RNeasy® Kit (Qiagen, Hilden, Germany) according to manufacturer’s protocol. RNA concentrations were determined by absorbance at 260, 280, and 320 nm using a DS-11 Spectrophotometer (DeNovix Inc., Wilmington, DE). Kidney was lysed and homogenized in ice-cold RIPA buffer (Sigma, St. Louis, MO) supplemented with complete proteinase inhibitor (Roche Diagnostics, Risch-Rotkreuz, Switzerland) and phosphatase inhibitor (Sigma-Aldrich) cocktails using Bead Bug microtube homogenizer (Denville Scientific Inc., Holliston, MA) at 4,000rpm for 90 seconds. The tissue homogenates were centrifuged (13,000 × g, 15 min, 4 °C), supernatants were collected, and protein concentrations were determined (Bradford protein assay reagent, Bio-Rad.
Microarray Analysis
Microarray analysis was performed commercially with OneArray® (Phalanx Biotech Group, San Diego, CA) with RNA samples isolated from control (N=3), 2 week-1 dose (N=3), and 4 week-2 dose mice(N=3). Qlucore Omics Explorer v 3.2 (Qlucore AB, Lund, Sweden) was used for identifying differentially-expressed genes (q < 0.01) using a one-way analysis of variance (ANOVA). Principal component analysis plots, unsupervised hierarchical clustering, and heat maps were also generated in Qlucore. Differentially expressed genes identified by Qlucore were subjected to further analysis by Ingenuity Pathway Analysis (Ingenuity Systems QIAGEN Build: 389077M, Content version: 27821452, Redwood City, CA, USA).
Western Blot Analysis
Equal amounts of protein lysate were separated by SDS-Page and western blotting was carried out using the following antibodies: NGAL, KIM1 (R&D Systems, Minneapolis, MN), phospho-JNK, phospho-ERK, total-ERK, RIPK1, and RIPK3 (Cell Signaling Technologies, Danvers, MA, USA). Proteins were detected using a chemiluminescent substrate (SuperSignal® West Femto; Thermo Scientific, Rockford, IL, USA) and signal quantification was performed with ImageJ software (National Institutes of Health, Bethesda, MD, USA). More detailed methods are provided in the supplementary methods.
Supplementary Material
Figure 7.


Variation in “Necrosis of Kidney” Pathway Activity Ingenuity Pathway Analysis (IPA) prediction of the interactive network between the differentially expressed genes and the downstream function “necrosis of kidney”. Red, genes overexpressed; green, genes underexpressed; orange, function predicted to be activated; blue, function predicted to be inhibited. (A) Three days following the first dose (B) Two weeks following the first dose (C) Two weeks following the second dose. Changes in expression of Lcn2 (NGAL), CKDN1A (p21), MLKL and TLR2 were of particular interest given their known roles in injury response, cell cycle arrest, and regulated necrosis, respectively.
Figure 11.

Proposed Mechanism for Kidney Response to First Versus Second Dose of Cisplatin. Following the first dose of cisplatin, there is acute injury that stimulates repair and regeneration mechanisms that enable near complete reconstitution of renal function and structure. However, following the second dose of cisplatin, there is a sustained injury response, which result in a failure to repair and the development of CKD.
Translational Statement:
The two dose model of cisplatin-induced chronic kidney disease developed here mimics the development of chronic kidney disease (CKD) seen in patients undergoing treatment for cancer with cisplatin. The study also identifies the precise timing of the conversion of acute and reversible kidney injury to chronic and irreversibly injury and identifies molecular pathways associated with this conversion. It is suggested that by targeting the identified molecular pathways, cisplatin, an effective treatment for tumors that are otherwise resistant to chemotherapy, can be used more effectively (i.e., not limited by its nephrotoxicity) to treat such resistant tumors. As cisplatin causes chronic kidney injury not very different from other causes of tubolo-interstitial CKD, identifying such molecular therapeutic targets might be applicable to preventing other forms of CKD including ischemia reperfusion injury that follows cardiac surgery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
R Torres reports an ownership interest in Applikate Technologies, LLC, a startup developing technology for rapid tissue processing and imaging using multiphoton microscopy.
References
- 1.Pabla N & Dong Z Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney international 73, 994–1007 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Zeisberg M & Kalluri R Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. American journal of physiology. Cell physiology 304, C216–225, doi: 10.1152/ajpcell.00328.2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chevalier RL The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. American Journal of Physiology-Renal Physiology 311, F145–F161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forbes MS, Thornhill BA & Chevalier RL Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol 301, F110–117, doi: 10.1152/ajprenal.00022.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcussen N & Olsen T Atubular glomeruli in patients with chronic pyelonephritis. Laboratory investigation; a journal of technical methods and pathology 62, 467–473 (1990). [PubMed] [Google Scholar]
- 6.Thornhill BA, Forbes MS, Marcinko ES & Chevalier RL Glomerulotubular disconnection in neonatal mice after relief of partial ureteral obstruction. Kidney International 72, 1103–1112, doi: 10.1038/sj.ki.5002512 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Møller JC Dimensional changes of proximal tubules and cortical capillaries in chronic obstructive renal disease. Virchows Archiv 410, 153–158 (1987). [DOI] [PubMed] [Google Scholar]
- 8.Marcussen N, Ottosen P, Christensen S & Olsen T Atubular glomeruli in lithium-induced chronic nephropathy in rats. Laboratory investigation; a journal of technical methods and pathology 61, 295–302 (1989). [PubMed] [Google Scholar]
- 9.Marcussen N Atubular glomeruli in cisplatin-induced chronic interstitial nephropathy. APMIS 98, 1087–1097 (1990). [DOI] [PubMed] [Google Scholar]
- 10.Torres R et al. Three-Dimensional Morphology by Multiphoton Microscopy with Clearing in a Model of Cisplatin-Induced CKD. Journal of the American Society of Nephrology, ASN. 2015010079 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safirstein RL Acute renal failure: from renal physiology to the renal transcriptome. Kidney International 66, S62–S66 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Grigoryev DN et al. The local and systemic inflammatory transcriptome after acute kidney injury. Journal of the American Society of Nephrology 19, 547–558 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linkermann A et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A 110, 12024–12029, doi: 10.1073/pnas.1305538110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y et al. A Role for Tubular Necroptosis in Cisplatin-induced AKI. Journal of the American Society of Nephrology: JASN 26, 2647–2658, doi: 10.1681/ASN.2014080741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linkermann A et al. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. Journal of the American Society of Nephrology, ASN. 2012121169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kers J, Leemans JC & Linkermann A An overview of pathways of regulated necrosis in acute kidney injury. Seminars in nephrology 36, 139–152 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Mori K et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. Journal of Clinical Investigation 115, 610 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra J et al. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. Journal of the American Society of Nephrology 15, 3073–3082 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Mishra J et al. Neutrophil gelatinase-associated lipocalin: a novel early urinary biomarker for cisplatin nephrotoxicity. American journal of nephrology 24, 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Singer E et al. Neutrophil gelatinase-associated lipocalin: pathophysiology and clinical applications. Acta physiologica 207, 663–672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonventre JV Kidney injury molecule-1 (KIM-1): a urinary biomarker and much more. Nephrology Dialysis Transplantation 24, 3265–3268 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Jin X et al. Paradoxical effects of short-and long-term interleukin-6 exposure on liver injury and repair. Hepatology 43, 474–484 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Cuadros T et al. HAVCR/KIM-1 activates the IL-6/STAT-3 pathway in clear cell renal cell carcinoma and determines tumor progression and patient outcome. Cancer research 74, 1416–1428 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Kawai Y et al. The effect of antioxidant on development of fibrosis by cisplatin in rats. Journal of pharmacological sciences 111, 433–439 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Maarouf OH et al. Paracrine Wnt1 drives interstitial fibrosis without inflammation by tubulointerstitial cross-talk. Journal of the American Society of Nephrology 27, 781–790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kefaloyianni E et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI insight 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulay SR, Linkermann A & Anders HJ Necroinflammation in Kidney Disease. J Am Soc Nephrol 27, 27–39, doi: 10.1681/ASN.2015040405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulay SR, Kumar SV, Lech M, Desai J & Anders H-J How kidney cell death induces renal necroinflammation. Seminars in nephrology 36, 162–173 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Linkermann A, Stockwell BR, Krautwald S & Anders H-J Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nature Reviews Immunology 14, 759 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Sarhan M, Land WG, Tonnus W, Hugo CP & Linkermann A Origin and consequences of necroinflammation. Physiological reviews 98, 727–780 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Miller RP, Tadagavadi RK, Ramesh G & Reeves WB Mechanisms of cisplatin nephrotoxicity. Toxins 2, 2490–2518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramesh G & Reeves WB TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. American Journal of Physiology-Renal Physiology 285, F610–F618 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Zhang B, Ramesh G, Uematsu S, Akira S & Reeves WB TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. Journal of the American Society of Nephrology 19, 923–932 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leemans JC et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. The Journal of clinical investigation 115, 2894–2903 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He S, Liang Y, Shao F & Wang X Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proceedings of the National Academy of Sciences 108, 20054–20059 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser WJ et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3 and MLKL. Journal of Biological Chemistry, jbc. M113. 462341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy LO, Smith S, Chen R-H, Fingar DC & Blenis J Molecular interpretation of ERK signal duration by immediate early gene products. Nature cell biology 4, 556–564 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Chevalier RL & Forbes MS Generation and evolution of atubular glomeruli in the progression of renal disorders. Journal of the American Society of Nephrology 19, 197–206 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Martin-Sanchez D et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proceedings of the National Academy of Sciences 115, 4182–4187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brenner D, Blaser H & Mak TW Regulation of tumour necrosis factor signalling: live or let die. Nature Reviews Immunology 15, 362 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Gong Y-N et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300. e216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon S, Kovalenko A, Bogdanov K & Wallach D MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47, 51–65. e57 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Baines CP et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Brooks C, Wei Q, Cho S-G & Dong Z Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. The Journal of clinical investigation 119, 1275–1285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ko GJ et al. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. American Journal of Physiology-Renal Physiology 298, F1472–F1483 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.