

Abstract
Neutrophil chemotaxis is essential in responses to infection and underlies inflammation. In neutrophils, the small GTPase Rac1 has discrete functions at both the leading edge and in the retraction of the trailing structure at the cell’s rear (uropod), but how Rac1 is regulated at the uropod is unknown. Here, we identified a mechanism mediated by the trafficking protein synaptotagmin-like 1 (SYTL1 or JFC1) that controls Rac1-GTP recycling from the uropod and promotes directional migration of neutrophils. JFC1-null neutrophils displayed defective polarization and impaired directional migration to N-formyl-methionine-leucyl-phenylalanine in vitro, but chemoattractant-induced actin remodeling, calcium signaling and Erk activation were normal in these cells. Defective chemotaxis was not explained by impaired azurophilic granule exocytosis associated with JFC1 deficiency. Mechanistically, we show that active Rac1 localizes at dynamic vesicles where endogenous JFC1 colocalizes with Rac1-GTP. Super-resolution microscopy analysis shows adjacent distribution of JFC1 and Rac1-GTP, which increases upon activation. JFC1 interacts with Rac1-GTP in a Rab27a-independent manner to regulate Rac1-GTP trafficking. JFC1-null cells exhibited Rac1-GTP accumulation at the uropod and increased tail length, and Rac1-GTP uropod accumulation was recapitulated by inhibition of ROCK or by interference with microtubule remodeling. In vivo, neutrophil dynamic studies in mixed bone marrow chimeric mice show that JFC1−/− neutrophils are unable to move directionally towards the source of the chemoattractant, supporting the notion that JFC1 deficiency results in defective neutrophil migration. Our results suggest that defective Rac1-GTP recycling from the uropod affects directionality and highlight JFC1-mediated Rac1 trafficking as a potential target to regulate chemotaxis in inflammation and immunity.
Keywords: Neutrophils, chemotaxis, JFC1, Rac1, vesicular trafficking
Summary sentence : Neutrophil migration towards formyl-peptides is controlled by the vesicular trafficking-mediated removal of Rac1-GTP from the uropod which is regulated by JFC1 in a Rab27a-independent manner.
Graphical Abstract
INTRODUCTION:
Chemotaxis is an essential mechanism in cell development, metastasis, host response to infection and inflammation. Neutrophils (polymorphonuclear leukocytes) respond to chemical gradients of endogenous and exogenous molecular cues by undergoing cellular and molecular changes that include cell elongation, polarization and Ras-GTPase segregation, thus facilitating adaptation to the microenvironment, gradient recognition and motion (1). Cell migration is a cyclic process, involving the extension of protrusions at the cell front (lamellipodia) and retraction of the trailing structure at the rear of the cell (uropod). This process requires dynamic spatial and temporal activation of Ras GTPases. As a part of the polarization process, migrating cells segregate the Ras-GTPases CDC42, Rac1, Rac2 and RhoA, which play fundamental roles in the regulation of chemotaxis in all migrating cells (2). In neutrophils, low concentrations of the bacteria-derived mimetic peptide fMLF (formyl-methyl-leucyl-phenylalanine) induce shape change (polarization), formation of pseudopod extensions and ultimately facilitate chemotaxis of neutrophils (3). The process involves the activation of the fMLF receptor and downstream signaling pathways including Ras GTPase-regulated mechanisms and subsequent cytoskeletal rearrangements. Exposure of neutrophils to higher concentrations of fMLF induces motility arrest, and favors degranulation and the generation of oxygen-derived reactive oxygen species (4–7). The molecular mechanisms mediating the switch from the pro-motility to the pro-secretory neutrophil phenotypes are currently unknown.
The spatiotemporal distribution of Ras GTPases at discrete cellular poles where they associate with specific signaling hubs and downstream effectors is a fundamental characteristic of migrating cells. Thus, the concept of a central and single role for the canonical Ras GTPases has been recently challenged and is now accepted that instead, Ras GTPases play multiple functions defined by their localization in the chemotaxing cell (2). Furthermore, the distribution of Ras modulators in migrating cells, including guanine nucleotide exchange factors (GEFs) and GTPase-activating protein (GAPs), is also polarized and a similar pattern for Ras-downstream effectors has been proposed (2,8,9). Thus, the particular functions played by Rac, RhoA and Cdc42 during chemotaxis is defined by their dynamic transient interactions with strategically localized regulators and effectors suggesting that GTPase redistribution is important during migration.
Different from fibroblasts where Rac activation is confined to the leading lamellae and peripheral ruffles of migrating cells (10,11), in neutrophils, Rac1 is also known to function at the rear end (uropod) (12), where it is proposed to play a role in the organization of the Rho-myosin backness program (13). Rapid changes in the activity of Rac at the uropod that appear to match tail retraction and attachment/detachment to the surface has been reported supporting a cyclic activation of Rac1 at the rear end (12). However, many of the known Rac1 regulatory proteins including the GEFs Tiam1 and p-Rex1 (9,14) localize at the cell front. Active mitochondria, the source of ATP needed to reconstitute the GTP pool by nucleotide-diphosphate kinases have also been shown to localize at the cell front in migrating neutrophils and anterior localization of mitochondria correlates with faster migration velocities and increased directional persistence (15,16). Thus, it is unclear how cyclic loops of Rac1 activation-inactivation occurs at the uropod during chemotaxis in neutrophils given the different localization of Rac1, its accessory proteins and the needed metabolites. Based on the differential localization of Rac1 GEFs at the cell front, we hypothesize that Rac1 trafficking would be necessary to support Ras activation cycling at the rear end to maintain recurrent events of tail elongation and tail detachment, but no such mechanism has been described so far.
JFC1/Slp1, is a trafficking molecule first identified in leukocytes by our group, and subsequently characterized as a Rab27a-effector by three independent groups (17–19). The binding of JFC1 to Rab27a is mediated by its synaptotagmin-homology domain located in the amino terminus of the Rab effector, where the conserved residue tryptophan 83 is known to be essential for this interaction (20). JFC1 regulates exocytosis through the regulation of vesicular docking mediated by the interaction of its C2A domain with plasma membrane phosphoinositides (21). JFC1 also regulates vesicular trafficking through the recruitment of the RhoA GTPase-activating-protein (GMIP, Gem-interacting protein) and through the subsequent remodeling (depolymerization) of actin filaments surrounding neutrophil granules, a process that facilitates granule movement through cortical actin (6). Although JFC1 is essential for the secretion of azurophilic granules in neutrophils (22), its participation in the regulation of chemotaxis has not been explored so far.
In response to bacterial infections or sterile inflammation neutrophils migrate from the blood stream into tissues, a process that is important to combat infections but also induces systemic inflammation. Neutrophil transmigration is a multistep mechanism that involves rolling at and adhesion to the activated endothelium, endothelial transmigration and basal membrane degradation through the release of granule cargoes including MMP-9 and elastase, and the subsequent migration in the direction of a chemoattractant gradient towards the infectious or inflammatory foci (23,24). Neutrophil chemotaxis is essential for this last migration step and therefore, a better understanding of the molecular cues controlling chemotaxis is essential for both increasing responses to infection and decreasing inflammation. Previous studies from Sperandio’s and our laboratory elucidated a novel mechanism of neutrophil transmigration, characterized by the control of the mobilization of integrins VLA3 and VLA6 by Mst1 (25), but the molecular mechanisms regulating neutrophil directional migration are not completely elucidated.
Here, we identify a novel regulatory mechanism of neutrophil chemotaxis and migration controlled by JFC1. We show that JFC1 regulates vesicular trafficking, controls the uropod localization of Rac1-GTP and modulates neutrophil directional migration. The process is specifically controlled by the direct interaction of the trafficking molecule JFC1 with Rac1-GTP. We present evidence that JFC1 is a novel regulator of neutrophil directional movement both in vitro and in vivo and identify a novel mechanism that involves the regulation of Rac1 trafficking by JFC1.
RESULTS
JFC1 mediates neutrophil directional migration at low chemoattractant concentration
To study whether JFC1 modulates the chemotactic response in neutrophils, we performed migration assays using bone-marrow derived neutrophils from WT and JFC1 knockout mice (JFC1−/−). Lack of JFC1 expression in JFC1−/− leukocytes was confirmed by Western blot (Supplementary Fig. 1). Using a μ-slide chemotaxis chamber, we analyzed time-lapse and trajectory of chemotaxing neutrophils in spatially well-defined chemotactic gradients of the bacterial-derived peptide fMLF. We studied neutrophil chemotaxis using gradients generated by varying fMLF concentrations known to engage diverse molecular regulators(26). Similar to previous reports(27–29), gradients were generated using 10μM fMLF at the chemoattractant reservoir, which based on the calculated diffusion coefficient (30) produces a 0 to 10 μM fMLF gradient starting from the farthest end of the cell chamber (2×1 mm2) to the chemoattractant chamber at 30 minutes, when image collection begins. Alternatively, we used 2.5X fMLF (25μM at the chemoattractant chamber), to assess chemotaxis to a higher chemoattractant concentration (Fig. 1). When neutrophil migration was evaluated in response to 10 μM fMLF, directional migration was significantly impaired in the JFC1−/− neutrophils (Fig. 1a), despite showing normal velocity, migrated distance and persistence (Fig. 1b-d). Tracks of cell migration from one representative experiment are shown in Figs. 1e and 1f. Interestingly, the migration defect was not observed at 25 μM fMLF (Figs. 1g-l), suggesting that JFC1 regulates directionality of neutrophils specifically at initial low concentration gradients of the chemoattractant. The migration of wild type and JFC1−/− neutrophils at 1× and 2.5× fMLF gradients neutrophils is presented in Supplementary movies S1-S4.
FIGURE 1: Directional migration is defective in JFC1−/− neutrophils.
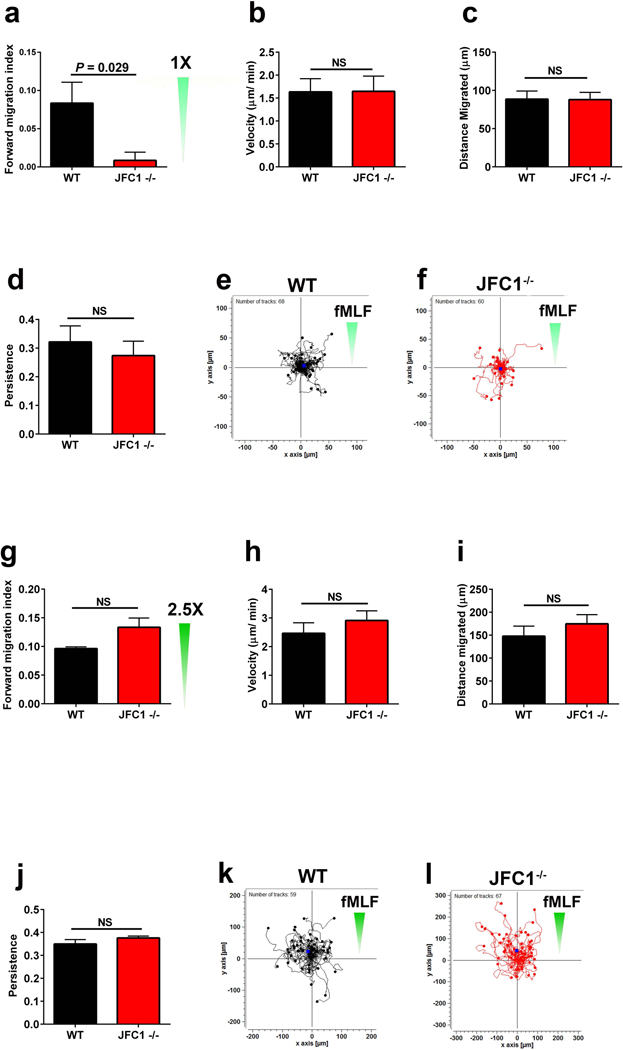
(a-l) WT or JFC1−/− mouse bone marrow neutrophils were analyzed in chemotaxis using collagen-coated ibidi μ-slide chemotaxis chambers. Gradients were generated using 10 μM fMLF (a-f) or 25 μM fMLF (g-l) at the chemoattractant reservoir, which produces 0 to 10 or 25 μM fMLF gradients at 30 minutes (see methods and results sections). Cell movement was recorded at 2 min intervals for 1 hour and tracks for the cells were mapped using the Manual Tracking plug-in of ImageJ software. The forward migration index (efficiency of directed cell migration) (a and g), mean velocity (b and h), distance migrated (c and i) and persistence (d and j) were calculated using the Chemotaxis and Migration Tool software (Ibidi). The results are expressed as mean ± SEM from at least 3 independent experiments (n=6 for a-d and n=3 for g-j), *p<0.05; NS, not significant. (e, f, k and l) Data showing tracks of cell migration from one representative experiment. Distance from the origin is indicated on x and y axes in μm. The direction of the chemotactic gradient is indicated with green triangles.
JFC1−/− neutrophils exhibit decreased polarization index upon fMLF stimulation
Cell shape polarization provides the necessary morphological and molecular modifications to induce the acquisition of functional and spatial asymmetry to facilitate chemotaxis. Given the similarity in neutrophil chemotactic patterns between fields of uniform fMLF and fMLF gradients (26), we tested neutrophil polarization by treating cells to uniform concentrations of fMLF ranging from low nM to 1 μM (Fig. 2). We show that at the nanomolar range, granulocytes undergo molecular mechanisms that prepare them for the process of cell motion (cell elongation), while micromolar fMLF concentrations induce molecular modifications to halt chemotaxis and to prepare the cells for the process of exocytosis (Figs. 2b and c). The transition to a migratory phenotype induced by fMLF was quantified using a measurement of cell shape polarization calculated as an increase in the length-to-width ratio of the cell (Figs. 2a and b). Representative images showing cell polarization at different fMLF concentrations are shown in Fig. 2b and larger fields in Supplementary Figure S2a. In these studies, although both wild type and JFC1−/− neutrophils show shape polarization at 10–100 nM fMLF, JFC1−/− neutrophils exhibit a significant decrease in polarization index as compared to the wild type neutrophils at both 10 and 100 nM fMLF concentrations (Fig. 2b). At 1 μM fMLF concentration, both the wild type and JFC1−/− neutrophils were less elongated and more rounded, as this is a higher than optimal concentration of fMLF for polarization that instead prepares the cells for the process of exocytosis. The cell polarization- defective phenotype was observed both after 2 or 10 min exposure to the chemoattractant (Supplementary Fig. S2b and Figure 2b), suggesting that the defect is caused by a persistent molecular deficiency rather than a delay in signaling pathways.
FIGURE 2: JFC1−/− neutrophils exhibit decreased polarization index upon fMLF stimulation and the defect is independent of impaired exocytosis.
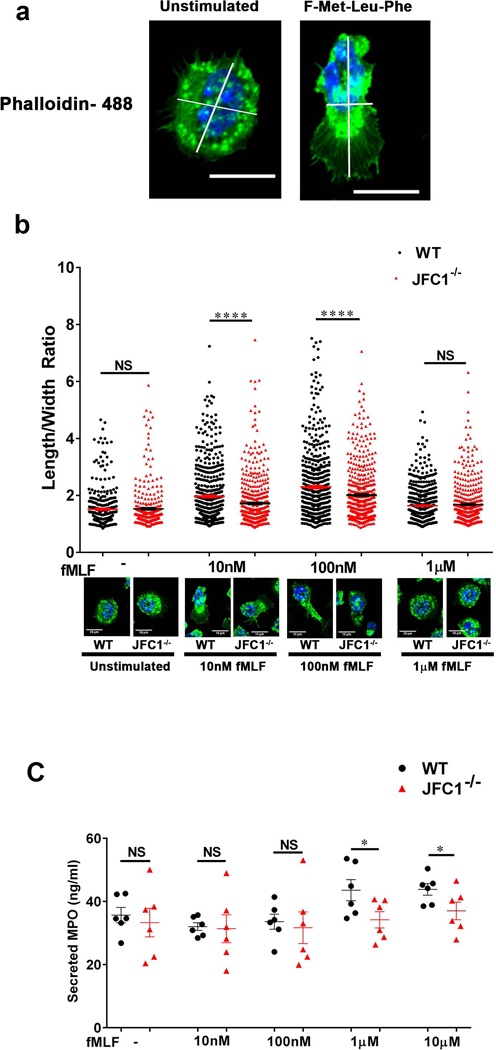
Mouse bone marrow neutrophils from WT and JFC1−/− mice were left unstimulated or stimulated with either 10nM, 100nM or 1μM fMLF for 10 min at 37° C, followed by fixation and staining with phalloidin-488. The length to width ratio of the cells were quantified using ImageJ software. (a) Representative images of unstimulated cells or cells stimulated with 100nM fMLF for 10 min. The scale bar represents 10μm. (Larger fields are shown in Supplementary Fig. S2a). (b) The elongation index of WT vs JFC1−/− cells calculated as the length to width ratio of the cell is indicated as mean ± SEM and was determined from 6 WT and 6 JFC1−/− mice. A minimum of 50 cells were measured from each mouse. A representative cell for WT and JFC1−/− neutrophils for each condition is shown. Larger fields can be seen in supplementary figure 2. **** p< 0.0001; NS, not significant. (c) Mouse bone marrow neutrophils from WT (shown in black) or JFC1−/− (shown in red) neutrophils were isolated and stimulated with the either 10nM, 100nM, 1μM or 10μM fMLF for 10 min at 37 °C, and myeloperoxidase (MPO) in the cell supernatants was determined by ELISA. Data are represented as mean ± SEM, (n=6). Each symbol represents an individual mouse. *p<0.05; NS, not significant.
The trafficking molecules synaptotagmin VII, the JFC1 binding partner Rab27a, and the Rab27a effector Slp5 have been associated with the regulation of chemotaxis in neutrophils through the localized secretion of proteases, a mechanism proposed to facilitate uropod detachment (31–33). Since neutrophil chemoattractants are able to induce both neutrophil migration and exocytosis and JFC1 regulates exocytosis, we evaluated whether the function of JFC1 in chemotaxis was, at least in part, explained by its role in regulated secretion. To test this hypothesis, we analyzed exocytosis in WT and JFC1−/− neutrophils stimulated with fMLF in a dose-dependent manner and correlated the results to those triggered by the same concentration of chemoattractant during cell polarization and exocytosis. In Figure 2c, we show that JFC1 regulates azurophilic granule exocytosis in mouse neutrophils. In agreement with previous reports, high concentrations of chemoattractant (≥1 μM) were necessary to trigger exocytosis of azurophilic granules, the main source of neutrophil proteases (Fig. 2c) (34). Although JFC1-deficient neutrophils show defective azurophilic granule exocytosis, their cargo is released at concentrations of chemoattractant that are too high to induce the morphological changes that precede the chemotactic response, suggesting that defective chemotaxis is not explained by impaired azurophilic granule exocytosis in the JFC1−/− neutrophil.
fMLF-induced actin polymerization, calcium signaling and Erk signaling are unaffected in JFC1−/− neutrophils
Actin polymerization is required for effective directional migration (35) and sequential actin assembly and disassembly is necessary for neutrophil chemotaxis (36). To determine whether a differential actin polymerization pattern in JFC1−/− neutrophils is responsible for their impaired chemotaxis, we monitored the dynamic changes observed in actin polymerization and depolymerization induced by a wide concentration range of fMLF using phalloidin. Despite slightly increased basal levels of F-actin in JFC1−/− neutrophils compared to WT cells (Fig. 3a-b), no significant differences in the dynamics of actin remodeling were observed between WT and JFC1−/− cells for the wide range of fMLF concentrations and time points evaluated (Figs. 3c-f), indicating normal actin remodeling and response to fMLF in JFC1−/− neutrophils. A transient increase in intracellular free calcium is a hallmark of chemoattractant receptor activation (37,38) as indicated by neutrophils lacking the transient receptor potential cation channel TRPM2, which display diminished calcium elevation and impaired migration in response to fMLF (39). Here, using a cell membrane-permeable calcium sensor (Fluo-8) to monitor the dynamic changes in intracellular calcium, we show that JFC1−/− neutrophils have normal calcium signaling upon treatment with a wide range of fMLF concentration, suggesting again that the activation of the formyl-peptide receptor downstream signaling pathways is intact in JFC1−/− neutrophils (Figs. 3g-k). Next, we analyzed whether the fMLF-induced downstream signaling was impaired in JFC1−/− neutrophils. Since Erk activation was recently shown to be required for neutrophil migration in response to fMLF (40), we focused on Erk phosphorylation. In Figure 3l and m, we show that the activation of Erk in response to fMLF is normal in JFC1−/− neutropils, thus ruling out signaling defects as the cause of defective neutrophil migration in JFC1-deficiency.
Figure 3: F-actin dynamics and calcium release upon fMLF stimulation are unaffected in JFC1−/− neutrophils.
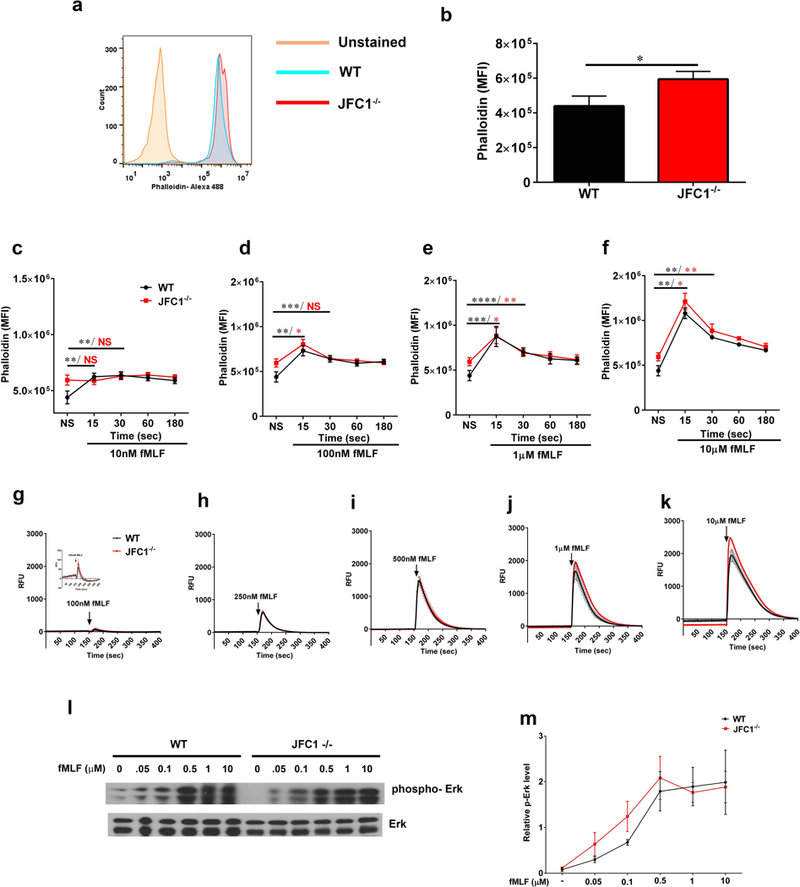
(a-f) Quantitative analysis of actin remodeling. Mouse bone marrow neutrophils from WT or JFC1−/− mice were left unstimulated (a, b) or were stimulated with either 10nM (c), 100nM (d), 1μM (e) or 10μM (f) fMLF for the indicated times at 37° C. The cells were then fixed, actin stained using Phalloidin-488 and the cells were analyzed by flow cytometry. The mean fluorescence intensity was calculated using FlowJo software. a. Representative histogram showing the basal F-actin levels in WT and JFC1−/− neutrophils. b. Quantitative analysis of the basal F-actin levels in WT vs JFC1−/− neutrophils. Mean ± SEM from 8 mice, * p<0.05. (c-f) Time course of actin polymerization at the indicated fMLF concentration. Data are indicated as mean ± SEM from 3 WT (shown in black) and 3 JFC1−/− (shown in red) mice. * p<0.05, ** p<0.01, *** p<0.001, **** p< 0.0001. (g-k) Quantitative analysis of calcium flux. Mouse bone marrow neutrophils were seeded onto poly D-lysine coated plates and loaded with Fluo8, a fluorescent calcium sensitive indicator for 30 min. Calcium levels were measured by monitoring the fluorescence intensity at Ex/Em = 490/525 nm using the FLIPR-384 system over time. fMLF was added at the 150 sec time point after beginning the read. Mean ± SEM from 3 WT (shown in black) and 3 JFC1−/− (shown in red) mice. (l) Western blot analysis of neutrophil signaling in response to fMLF activation. Mouse bone marrow neutrophils from WT or JFC1−/− mice were left unstimulated or were stimulated with either 50 nM, 100 nM, 500 nM, 1 μM or 10 μM fMLF for 2 minutes at 37° C. Immunoblots are representative of four independent experiments. (m) Quantification of Erk phosphorylation was performed by densitometry. The intensity of phosphorylated Erk was normalized to that of total Erk to calculate relative p-Erk levels. Data are represented as mean ± SEM from an n=4 mice.
Active Rac1-GTP colocalizes with JFC1-positive vesicles and interacts with JFC1
Rac1−/− neutrophils exhibit a chemotactic defect similar to that observed in the JFC1−/− neutrophils, where the speed of migration is unaffected while directional migration towards the source of the chemoattractant is defective (41). Furthermore, the neutrophil migration defect in the absence of Rac1 was shown to be specific for low chemoattractant concentrations (26). Given these similarities in phenotypes, we hypothesized that JFC1 could regulate neutrophil chemotaxis through Rac1. Furthermore, the role of JFC1 in the regulation of vesicular trafficking raises the question as to whether JFC1 directly regulates Rac1-GTP localization during chemotaxis by controlling vesicular trafficking. To address this, we studied the dynamic distribution of active Rac1 (Rac1-Q61L) in WT and JFC1−/− neutrophils using pseudo-TIRFM (p-TIRFM), a technique that facilitates the analysis of both organelles that are adjacent to the plasma membrane and vesicles located deeper into the cell, while maintaining the excellent signal to background ratio of conventional TIRFM (42). Primary neutrophils were transfected with the expression vector Rac1-Q61L-GFP and the distribution of the expressed chimera was analyzed by p-TIRFM. We detected Rac1-Q61L-GFP as dynamic puncta resembling small vesicles, which based on size and motility, were different from neutrophil granules (Fig. 4a and supplementary movies S5 and S6), but were similar to other vesicles of endocytic origin previously observed in neutrophils (43). Importantly, JFC1−/− neutrophils stimulated with fMLF showed increased accumulation of Q61L-GFP-positive vesicles in the uropod, which in addition appeared elongated when compared to the uropods present in wild type cells (Fig. 4a, fMLF).
FIGURE 4: Active Rac1 colocalizes with JFC1 in a fMLF-dependent manner.
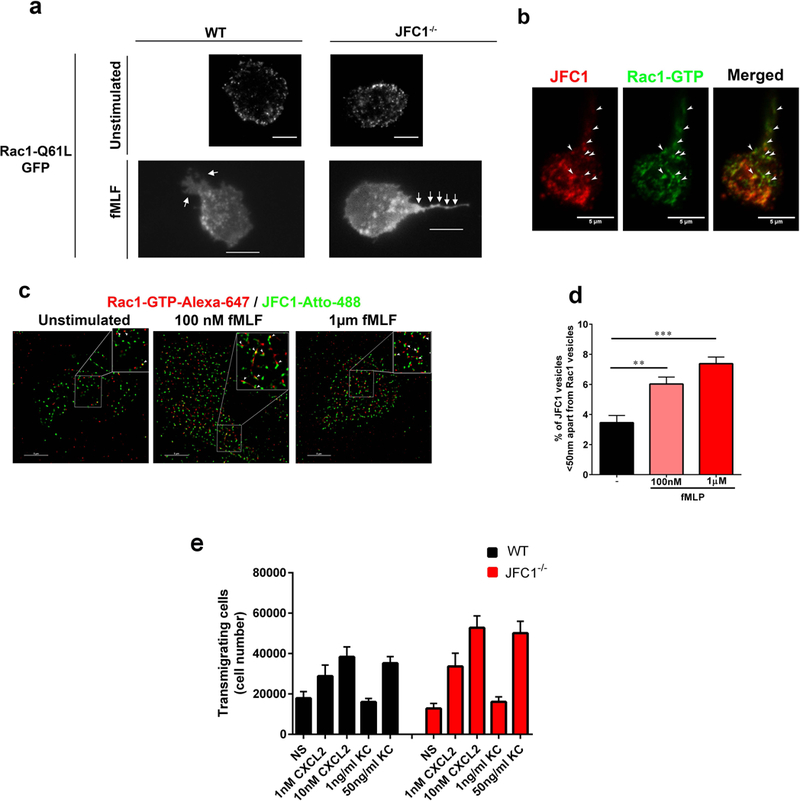
(a) The distribution of Rac1-Q61L transfected into primary WT or JFC1−/− neutrophils was analyzed by TIRF microscopy. Upper panels, representative images of unstimulated neutrophils are shown and representative dynamic studies are presented in supplementary movies S5 and S6. Lower panels, representative images of fMLF-stimulated neutrophils are shown. The white arrows point to the uropods of these cells showing accumulation of Rac1-Q61L-GFP in JFC1−/− but not in wild type neutrophils. Scale bars: 5 μm. (b) Immunofluorescence analysis of endogenous JFC1 (red) and Rac1-GTP (green) in neutrophilic-differentiated HL60 cells. Arrowheads indicate examples of vesicles that are positive for both JFC1 and Rac1 at the uropod and back body. Scale bar, 5 μm. (c) Super-resolution microscopy analysis (STORM) of the localization of endogenous JFC1 and Rac1-GTP in neutrophilic-differentialted HL60 cells. JFC1 and Rac1-GTP are detected adjacent to each other (arrowheads <50 nm apart) compatible with putative, in situ, protein-protein interaction; Scale bar, 5 μm. (d) Quantification of the super-resolution microscopy analysis showing the distance between JFC1 and Rac1-GTP centroids was performed as described under “Materials and Methods” and results are expressed as a percentage of total pairs at <50nm distance for each cell. A total of at least 10,000 JFC1 and Rac1-GTP pairs were analyzed from at least 4 individual cells. Mean ± S.E.M, **, p < 0.01, ***, p<0.001.(e) Chemotactic responses of wild type (WT, black bars) or JFC1−/− (red bars) neutrophils in response to the CXC chemokines CXCL2 and KC. Mean ± SEM, n=6 mice analyzed independently in two independent experiments. NS, non-stimulated.
Colocalization of endogenous JFC1 and active Rac1 (Rac1-GTP) at neutrophil vesicles localized at the uropod of polarized cells was demonstrated by confocal microscopy analysis (Figure 4b and Supplementary Figure S3), further suggesting that Rac1-GTP is transported by JFC1 through vesicular trafficking. Next, to study whether JFC1 and active Rac1 are distributed in close proximity, thus putatively supporting their in situ interaction, we analyzed the localization of JFC1 and active Rac1 in neutrophils using super-resolution microscopy (STORM), (Fig. 4c). Because Rac1-biosensors rely on the intrinsic interaction of Rac1-binding domains of Rac effectors with the active Rac, and this would interfere with the interaction of the active Rac1 moiety in the biosensor with JFC1, and therefore block the putative JFC1-dependent localization of the Rac1 biosensor, we analyzed active Rac1 localization using a validated antibody that detects the endogenous active form of Rac1 (Rac1-GTP) but not Rac1-GDP(44–48). First, we confirmed the specificity of the antibody for endogenous Rac1-GTP showing: a) that the anti-Rac1-GTP antibody efficiently pulls down GTP-loaded Rac1 (Supplementary Figure S4); b) that the amount of Rac1-GTP pulled-down from human neutrophils is markly increased upon neutrophil activation (Supplementary Figure S4); c) that Rac1-GTP localizes not only at the uropod but also at cellular ruffles in activated neutrophils (Supplementary Figure S5a); d) that stimulated neutrophils show significant increase in Rac1-GTP labeling thus validating its use in immunofluorescence methods (Supplementary Figure S5b), and e) importantly, that anti-Rac1-GTP detects active Rac1-Q61L but not the GDP-bound inactive mutant Rac1T17N, also by immunofluorescence (Supplementary Figure S6). Finally, GTP-Rac1 staining is blocked by a specific Rac1 inhibitor (48). Here, we show that endogenous Rac1-GTP and JFC1 localize adjacent to each other at distances that are compatible with putative protein-protein interaction (<50nm). Importantly, quantitative analysis of super-resolution studies shows that the percentage of JFC1-Rac1-GTP molecular pairs detected in close proximity is significantly increased upon fMLF stimulation, further supporting an activation-dependent mechanism of Rac1-GTP recruitment by JFC1 (Fig. 4d). Next, to determine whether this mechanism also operates upon neutrophil stimulation with other chemoattractants such as CXC chemokines, we analyzed the neutrophil response to CXCL2 and KC. We show that chemotaxis to these chemokines is not defective in JFC1−/− neutrophils at any of the concentrations tested (Figure 4e), supporting that the JFC1-dependent activation of Rac1-GTP may operate as a mechanism to discriminate between the different chemotactic factors.
JFC1 interacts with Rac1 in its active form
To test if JFC1 interacts with Rac1 we performed co-immunoprecipitation analysis. We found that JFC1 interacts strongly with constitutively active Rac1 (Rac1-Q61L) but very weakly with dominant negative Rac1 (Rac1 T17N) (Fig. 5a). The interaction of JFC1 with Rac1-GTP was more than 2.5 fold higher than with Rac1-WT and 4 fold higher than with Rac1-T17N (Fig. 5b). To determine whether the JFC1-interacting GTPase Rab27a regulates this interaction, we analyzed the ability of the JFC1 point mutant JFC1-W83S, which lacks affinity for Rab27a (20), to pull-down Rac1-GTP. Here we show that JFC1-W83S pulls down active Rac1 as efficiently as wild type JFC1 (Fig. 5c), indicating that tryptophan 83 is dispensable for the JFC1-Rac1-GTP interaction. In a similar assay, we show that the point mutation W83S completely abolishes the interaction between JFC1 and Rab27a (Fig. 5d). Altogether, our data indicates that JFC1 binds to Rac1-GTP at intracellular vesicles in a Rab27a-independent manner.
FIGURE 5: JFC1 interacts with active Rac1 in a Rab27a-independent manner.
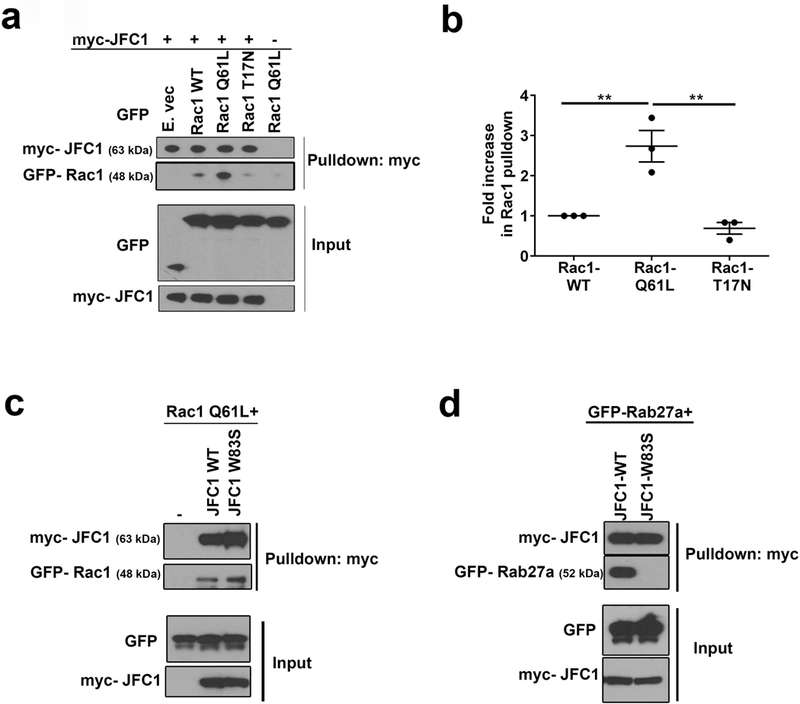
(a) Coimmunoprecipitation analysis of the JFC1-Rac1 interaction. Cells were transfected with myc-JFC1 and with either WT Rac1-GFP, the constitutively active Rac1 Q61L-GFP or the dominant negative Rac1 T17N-GFP. Cell lysates were used in pulldown assays, carried out using anti-myc antibodies and magnetic beads. Western blots are representative of at least three experiments with similar results. (b) Densitometric quantification of the immunoprecipitated bands from 3 independent experiments using the ImageJ software. The data is represented as mean ± SEM. ** p< 0.01. (c) Pulldown experiments were performed in cells transfected with GFP-Rac1Q61L with either wild type myc-JFC1 or with the point mutant myc-JFC1-W83S, which lacks binding to Rab27a. (d) Pulldown experiments were performed in cells transfected with EGFP-Rab27a with either myc-JFC1 WT or with the myc-JFC1-W83S mutant.
JFC1−/− neutrophils show increased tail length and accumulation of Rac1-GTP at the uropod
In a chemotaxing neutrophil, active Rac1 not only localizes at the lamellipodia but also at the uropod (12), where Rac1 regulates uropod function at the trailing edge through the Rho and myosin axis (13). To better understand the molecular mechanism regulating active Rac1 localization at the uropod, we analyzed the subcellular localization of endogenous, active, Rac1-GTP in neutrophils from WT and JFC1−/− mice. In these assays, the cells were stimulated with fMLF (100 nM) and labeled using the anti-Rac1-GTP antibody. The measured Rac1-GTP fluorescence intensity in whole cells showed similar global Rac1-GTP levels in WT and JFC1−/− neutrophils, indicating no defects in global Rac1 activation in the JFC1−/− cells upon fMLF stimulation (Fig. 6a). Rac1 positive uropods were observed as early as 2 minutes post-fMLF stimulation (data not shown) and the accumulation of Rac1-GTP in the tail increased significantly upon 10 minutes of fMLF treatment. Importantly, JFC1−/− neutrophils exhibit significantly longer Rac1-GTP-positive tails as compared to the WT neutrophils at the 10-minute time point (Figs. 6b and c). This characteristic phenocopies the defect observed in the Rac1−/− neutrophils where the Rac1−/− neutrophils exhibit increased uropod lengths (41). The longer Rac1 tails in JFC1−/− neutrophils correlated with an increase in the percentage of total Rac1-GTP present at the uropod (Fig. 6d) and an increase in Rac1-GTP tail intensity per unit area (Fig. 6e), thus indicating a net increase in the accumulation of Rac1-GTP in the rear end of the polarized JFC1−/− neutrophil. Rab27a−/− neutrophils did not exhibit a similar increase in Rac1-GTP tail length (Supplementary Fig. S7), further suggesting that this function of JFC1 is independent of Rab27a. Altogether, our data suggest that JFC1 is necessary to retrieve/recycle Rac1-GTP from the uropod through vesicular trafficking in a Rab27a-independent manner. Importantly, inhibition of microtubule remodeling significantly increased the percentage of cells with Rac1-GTP positive tails and Rac1-GTP accumulation at the uropod (Figs. 6f-h). These data support a cross-talk between vesicular trafficking and Rac1-GTP accumulation although other effects of Nocodazole, including direct modulation of Rac1-GTP activity, cannot be ruled out. Inhbition of ROCK significantly increased the length of the Rac1-GTP tails in the WT neutrophils and caused accumulation of Rac1-GTP at the uropod (Figs. 6i-k), further supporting a cross-regulation between RhoA downstream pathways, Rac1 recycling and tail retraction. Interestingly, the activation of wild type neutrophils induced a net reduction of total RhoA activity (Fig. 6l). This differed from that observed in JFC1−/− neutrophils, which showed a defective response maintaining the levels of RhoA activity even under stimulated conditions. In principle, this suggests that both inhibition of RhoA downstream kinase (ROCK) or inability of neutrophils to engage in RhoA activity fluctuations alters chemotaxis. Although apparently conflicting, these results go hand-in-hand with previous observations showing that both inhibition of myosin light chain (MLC) phosphorylation cause by impaired Rac1 and RhoA activity (13) and excesive p-MLC phosphorylation, impair chemotaxis (49).
FIGURE 6: JFC1−/− neutrophils show increased Rac1-GTP tail length and accumulation of Rac1-GTP at the uropod.
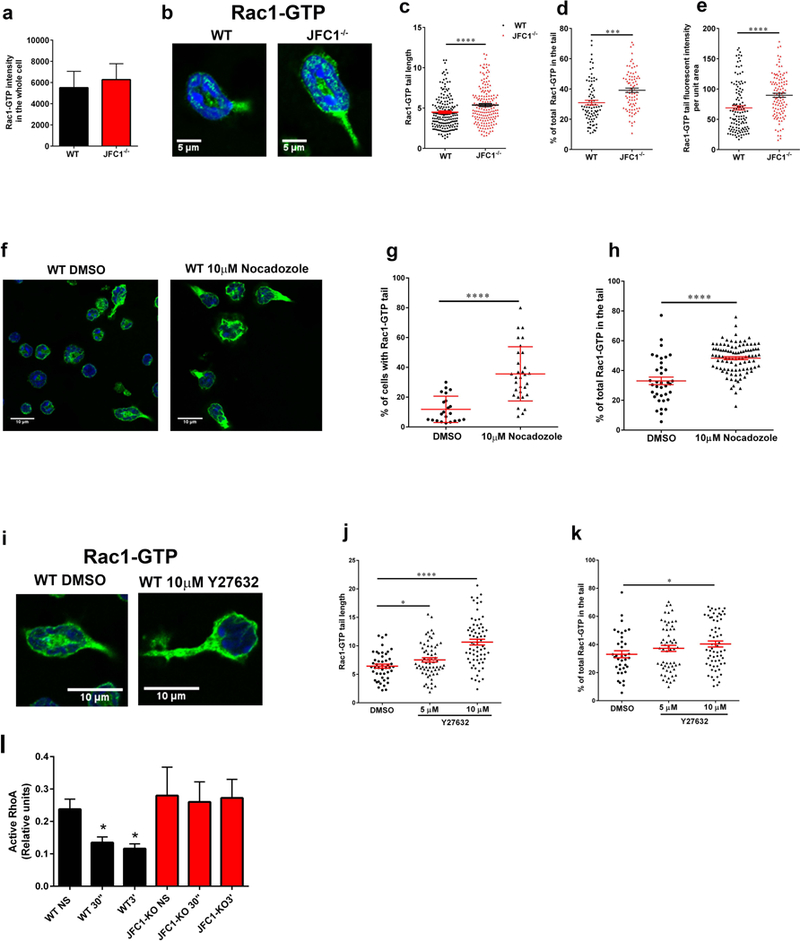
(a-e) Comparative quantitative analysis of the localization of endogenous active Rac1 in stimulated wild type and JFC1−/− neutrophils. (a) Quantitative analysis of total Rac1-GTP in neutrophils isolated from WT and JFC1−/− mice after stimulation with 100nM fMLF for 10 min at 37 °C. Active Rac1 was quantified using an antibody specific for the GTP bound form of Rac1 as described under “Material and Methods” and Rac1-GTP fluorescence intensity in the whole cell was analyzed using ImageJ. The data is indicated as mean± SEM of the arbitrary fluorescent units from an n=3 mice. (b) Representative images of WT and JFC1−/− cells showing Rac1-GTP accumulation in the tail upon fMLF stimulation for 10 min. (c) The length of Rac1-GTP uropods was analyzed using the ImageJ software. The data is expressed as mean ± SEM from a total of 180 cells from 3 independent experiments. (d) The % of total Rac1-GTP accumulated in the tail for each cell was calculated from the fluorescent intensity of Rac1-GTP in the uropod relative to the whole cell, measured using the ImageJ software. (e) The tail area was measured and Rac1 tail intensity per unit area was calculated using ImageJ. (c-e). WT (black circles); JFC1−/− (red triangles). Data are represented as mean ± SEM and were obtained from an n=4 mice. *** p<0.001, **** p<0.0001. (f-k) Effects of microtubule disruption (f-h) or ROCK inhibition (i-k) on the distribution of endogenous active Rac1 in WT neutrophils. Isolated neutrophils were seeded onto glass coverslips for 30 min, treated with the indicated inhibitors and stimulated with 100nM fMLF for 10 min at 37 °C. Active Rac1 was quantitatively analyzed by confocal microscopy as described above. (f) Representative image of WT and JFC1−/− cells showing Rac1-GTP accumulation in the tail upon treatment with 10μM Nocadozole followed by fMLF stimulation. (g) Percentage of total cells expressing Rac1-GTP-positive tails. (h) % of total (whole cell) Rac1-GTP accumulated in the tail for each Nocodazole- or vehicle-treated cell. (i) Representative images of WT and JFC1−/− cells showing Rac1-GTP accumulation in the tail upon treatment with 10μM Y27632 prior to fMLF stimulation. (j) The length of the Rac1-GTP tails in vehicle vs Y27632-treated cells. The data is expressed as mean ± SEM obtained from 3 independent experiments. (k) Percentage of total Rac1-GTP accumulated at the uropods of Y27632 or vehicle treated cells. n=3 mice. Mean ± SEM. (l) Quantitative analysis of RhoA activity in wild type (WT) and JFC1−/− neutrophils that have been left untreated (NS) or stimulated with fMLF for 30 seconds (30”) or for 3 minutes (3’). Mean ± SEM, n=3. *, p<0.05.
JFC1−/− neutrophils show decreased in vivo migration towards chemotactic stimuli
Neutrophil migration is a multistep mechanism that involves endothelial and basal membrane degradation through the release of granule cargo, and the subsequent transmigration in the direction of a chemoattractant gradient towards the infectious or inflammatory foci (23,24). To determine if JFC1 regulates migration in vivo, we performed neutrophil dynamics studies in mice. To this end, we generated a 1:1 mixed bone marrow chimera of JFC1−/− mice and DsRed WT mice and transplanted it into an irradiated WT recipient mouse (Fig. 7a). Using the chimeric mice, we analyzed neutrophil migration in response to fMLF, in vivo, using the cremaster muscle model and adapting it to a microinjection system, in which neutrophil migration is induced by a single adjacent dose of chemotactic peptide (50). We found that while wild type cells migrated directionally towards the chemoattractant gradient, the JFC1−/− neutrophils were unable to move directionally towards the source of the chemoattractant thus recapitulating the in vitro defective phenotype (Fig. 7b and c, and movie S7). Quantitative analysis of cell displacement shows that JFC1−/− neutrophils move defectively towards the chemoattractant in vivo (Fig. 7c). In agreement with the in vitro data, the velocity and the distance migrated were not impaired in the JFC1−/− neutrophils (Fig. 7d and 7e). We also observed increased number of JFC1−/− neutrophils in circulation in the chimeric mouse (Fig. 7f). The phenotype was confirmed in JFC1−/− mice, which similar to other mice with defective neutrophil recruitment (51), displayed neutrophilia (Fig. 7g). In principle, since in vivo experiments were performed in a wild type background, the results rule out possible JFC1-deficient phenotypes associated with secretion/production of intermediate pro-inflammatory molecules and instead support a defective migratory phenotype caused by neutrophil intrinsic defects in JFC1 deficiency. Altogether, our data supports a JFC1-mediated trafficking mechanism for Rac1-GTP retrieval from the uropod and demonstrate that JFC1 is required for effective directional neutrophil migration both in vitro and in vivo.
Figure 7: In vivo directional migration is defective in JFC1−/−chimeric mice.
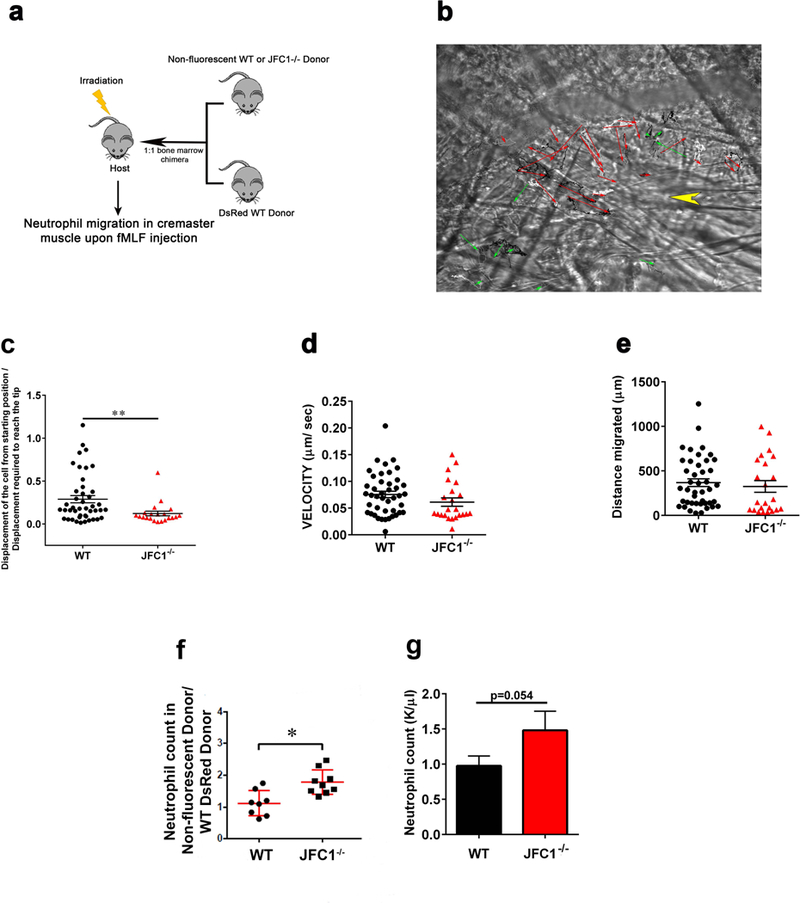
(a) Schematic depiction showing the technique used for the generation of the mixed bone-marrow chimera mice. Bone marrow chimera mice with 1:1 ratio of DsRed labeled wild type and either non-fluorescent WT or JFC1−/− bone marrow were prepared. The host CD45.1 male mice were irradiated and then retro-orbitally injected with 1:1 mixture of wild type and knock out bone marrow isolated under sterile conditions from leg bones of CD45.2 male donor mice. To compare the fMLF induced chemotaxis of wild type and JFC1−/− neutrophils, cremaster muscle imaging was performed on the chimera mice. At about 50 μm distance from a venule 1 μl of 10 μM fMLF was microinjected into the muscle with a glass canule. WT and JFC1−/− leukocytes were imaged using far red oblique illumination and RFP epifluorescence imaging was used to identify the wild type neutrophils. Cell tracks were traced using the Manual Tracking plugin in ImageJ. (b) Representative image showing the migration vectors of the WT cells in red arrows and the JFC1−/− cells in green arrows. The point of fMLF injection is indicated by the yellow arrowhead (movie S7) (c) The ratio of the displacement of the cell from the starting position to the displacement required to reach the glass canule tip where the fMLF was injected is shown. The velocity of migration (d) and distance migrated (e) were calculated using the Chemotaxis and Migration Tool software (Ibidi). Each symbol represents an individual cell and the data was obtained from 2 independent experiments. All values are expressed as mean ± SEM. (f) Neutrophil counts in the chimeric mice were measured by FACS analysis. The data is expressed as mean ± SEM from an n of at least 8 mice. (g) Peripheral blood was collected from WT and JFC1−/− mice in EDTA-coated capillary tubes. Complete blood cell counts were obtained using a Hemavet Hematology analyzer. Blood neutrophil counts are expressed as mean ± SEM from 7 WT and 7 JFC1−/− mice.
DISCUSSION
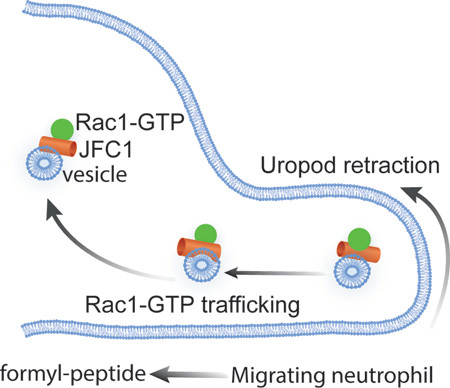
In this study, we show that the vesicular transport effector JFC1 is a novel regulator of active Rac1 trafficking and neutrophil chemotaxis. Our data support the idea that Rac1-GTP subcellular distribution during chemotaxis is regulated by JFC1 through vesicular trafficking, and that Rac1-GTP transport and removal from the uropod is important for maintaining directional movement (Fig. 8). Altogether, we have identified a novel mechanism that requires the dynamic distribution of intracellular Rac1-GTP by JFC1 to favor the maintenance of cell polarity and migration.
Figure 8: Model for JFC1-mediated control of neutrophil chemotaxis through the regulation of Rac1-GTP trafficking.
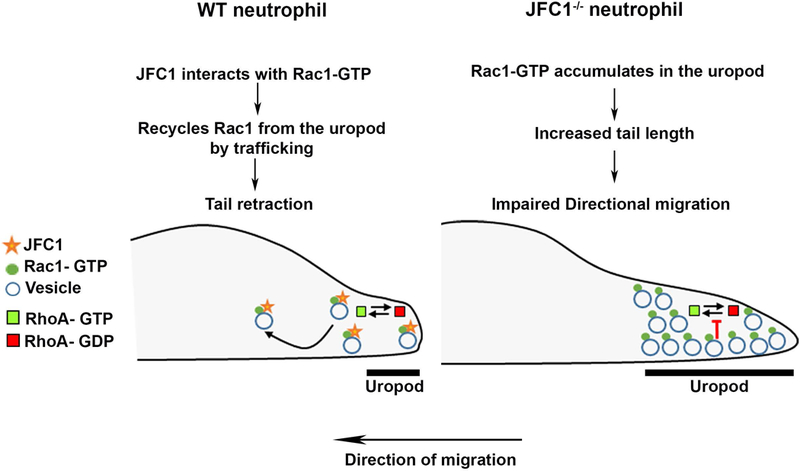
A schematic representation showing a proposed model for the sequence of events in a WT vs a JFC1−/− neutrophil. In a WT neutrophil the interaction of JFC1 with Rac1-GTP allows trafficking of Rac1-GTP, thus preventing its accumulation in the uropod and allowing for RhoA cyclic activation and subsequent tail retraction (left panel). In contrast, in a JFC1−/− neutrophil, Rac1-GTP, a proposed negative regulator of RhoA (52) but also suggested to positively regulate uropod-localized RhoA activity (13), accumulates in the uropod, affecting RhoA-activation-deactivation cycling, and causing increased uropod length and thus impaired directional migration (right panel).
Because Rac1 GEFs including Tiam1 and p-Rex1 are preferentially localized at the leading edge in migrating cells (9,14) our data showing that that JFC1 contributes to the removal of active Rac1 from the uropod suggest that JFC1-mediated Rac1-GTP trafficking contributes to the spatiotemporal regulation of Rac1 function at this cellular location where GEFs and GAPs maybe scarce. This mechanism could facilitate the reutilization of Rac1-GTP. Rac1-GTP is proposed to counteract RhoA (52); however, at the uropod of migrating neutrophils, Rac1-GTP positively regulates RhoA (13). JFC1, which is also known to decrease RhoA activity through recruitment of GMIP (Ref. (6) and Figure 6l), by physically retrieving Rac1-GTP from the uropod, may facilitate the transient cycles of RhoA activation-deactivation observed at the back of the cell during tail-retraction events (2). Thus, our data suggest that Rac1-GTP trafficking from the uropod by JFC1 mediates Rac1-GTP recycling. We propose that this mechanisms is used by neutrophils to facilitate transient RhoA activity oscillations during elongation and tail retraction, a mechanism known to require cyclic bursts of RhoA activity at the uropod in neutrophils(2,53).
Neutrophils respond to repetitive stimulation of increased concentrations of fMLF by modulating transient increases in F-actin formation triggered by increasing chemoattractant concentrations (54). We speculate that the morphological changes induced by repetitive stimulation with increasing fMLF concentrations are equally reversible once the concentrations are high enough to arrest movement and to induce exocytosis. Under experimental conditions of fMLF concentrations that induce exocytosis, JFC1 regulates RhoA inactivation at the granule membranes through the RhoA-GAP GMIP, a mechanism that favors granule mobilization through cortical actin to facilitate secretion (6). Thus, it is possible that JFC1 acts as a molecular switch that controls the fMLF-induced concentration-dependent spatiotemporal regulation of Rac1 and RhoA to regulate the cell preparedness for degranulation through RhoA inactivation at secretory granules. How this process is regulated is currently unknown. One possible scenario is that JFC1 post-transcriptional modifications, for example differential phosphorylation induced by low and high fMLF concentrations, modifies the affinity of JFC1 for Rac1 and/or Rab27a thus regulating the recruitment of JFC1 to different subcellular compartments. This is supported by previous observations that JFC1 subcellular distribution is regulated by phosphorylation (20) (55), and by the observations that JFC1 binds to and regulates Rac1 localization in a Rab27a-independent manner. Differential membrane recruitment of JFC1 could also be mediated by PI(3)K activation and PI(3,4,5)P3 formation, a phosphoinositide regulated at the uropod by Rac1 activation (56) and known to recruit JFC1 to the plasma membrane through its PI(3,4,5)P3-binding domain. Taken together, these studies suggest that PI(3)K substrates and kinases may play a role in recruiting JFC1 to specific membrane subdomains to facilitate its interaction with Rac1-GTP to enable trafficking.
Neutrophil chemotaxis and transmigration are essential mechanisms for host defense during infections but excessive neutrophil infiltration contributes to deleterious inflammatory processes, in endotoxemia, sepsis, trauma, cancer and autoinflammatory syndrome. Thus, a better understanding of the molecular cues controlling neutrophil chemotaxis is essential for both increasing responses to infection and decreasing inflammation. Our study elucidates a new mechanism of chemotaxis and neutrophil migration that involves the regulation of Rac1-GTP trafficking by the effector JFC1, and demonstrates that JFC1 regulates neutrophil directional movement both in vitro and in vivo. Our in vivo data indicate that JFC1 regulates neutrophil infiltration and inflammation through the regulation of cell-intrinsic mechanisms, and together with our in vitro studies, highlight that the control of Rac1-GTP localization by JFC1 may constitute a potential target for new therapeutics to decrease systemic inflammation.
MATERIALS AND METHODS:
Animals:
C57BL/6 JFC1−/− mice and their parental strain, C57BL/6 (wild type), were used. The JFC1/Sytl1-knockout mice were generated as previously described (6). Mice (6–12 wk old) were maintained in a pathogen-free environment and had access to food and water ad libitum. All animal studies were performed in compliance with the Department of Health and Human Services Guide for the Care and Use of Laboratory Animals. All studies were conducted according to National Institutes of Health and institutional guidelines and with approval of the animal review boards at The Scripps Research Institute and at the La Jolla Institute of Allergy and Immunology.
Expression vectors:
GFP- Rac1 WT, GFP- Rac1 Q61L, GFP- Rac1 T17N were obtained from Addgene. Myc-JFC1, DsRed-JFC1, myc-JFC1-W83S and GFP-Rab27a were generated as described before (20,57).
Antibodies and staining reagents:
Primary antibodies used for Western blotting and immunofluorescent staining were as follows: anti-GFP (A6455; Life Technologies, Carlsbad, CA), anti-myc (2278; Cell Signaling Technology), anti-active Rac1 (26903, New East Biosciences), anti-Rac1 (ARC03, Cytoskeleton). JFC1 antibody was raised by inoculating rabbits with the N-terminal peptide (17). The following secondary antibodies were used: Alexa fluor (488)-conjugated Goat anti-mouse IgM secondary antibody (Life Technologies), Alexa fluor (647)- conjugated donkey anti- rabbit IgG (Life Technologies), Alexa fluor (647) Goat anti-mouse IgM (Abcam) and Goat anti-rabbit IgG Atto 488 (Rockland). Alexa fluor 488- Phalloidin (A12379) and Rhodamine- Phalloidin (R415) were from Thermo Fischer Scientific.
Western Blotting:
Proteins were separated by gel electrophoresis using NuPAGE gels and 3-(N-morpholino) propanesulfonic acid buffer (Life Technologies). Proteins were transferred onto nitrocellulose membranes for 60 min at 100 V at 4°C. The membranes were blocked with tris-buffered saline (TBS) containing 5% (wt/vol) blotting-grade nonfat dry milk blocker (Rockland, Limerick, PA) and 0.1% (wt/vol) Tween 20. Proteins were detected by probing the membranes with the indicated primary antibodies at appropriate dilutions and using a detection system consisting of horseradish peroxidase–conjugated secondary antibodies (Bio-Rad Laboratories, Hercules, CA) and the chemiluminescence substrates SuperSignal, WestPico, and WestFemto (Thermo Scientific) and then visualized using Hyperfilm (Denville Scientific, Holliston, MA).
Pulldown:
For pulldown assays, 293T cells were lysed using lysis buffer (25mM Tris, 0.15M NaCl, 1mM EDTA, 1% NP40, 5% glycerol; pH 7.4), the samples were cleared by centrifugation and the supernatants were incubated with c-myc magnetic beads (Pierce, Thermo Scientific) at 4°C with rotation overnight. Following three washes with TBS Tween buffer, (100mM Tris, 0.6M NaCl, 0.2% Tween-20 Detergent; pH 7.5) the pulldown samples were subjected to western blotting.
Mouse neutrophil isolation:
Bone marrow–derived neutrophils were isolated using a Percoll gradient fractionation system as described (6). A three-layer Percoll gradient was used (52, 64, and 72%), and neutrophils were isolated from the 64/72% interface, washed in RPMI without phenol red, and used in all the assays. For exocytosis assays, calcium flux and actin remodeling assays, neutrophils were isolated using the anti- Ly6G microbead neutrophil purification kit (Miltenyi Biotec).
Human neutrophil isolation:
Human neutrophils were isolated from normal donor’s blood by Ficoll density centrifugation as previously described (58). All procedures regarding human subjects have been reviewed and approved by the Human Subjects Committee at The Scripps Research Institute and were conducted in accordance with the requirements set forth by the mentioned Human Subjects Committee and in accordance to NIH guidelines. The studies abide by the Declaration of Helsinki principles.
Immunofluorescence and confocal microscopy:
Neutrophils were seeded on untreated coverglasses (Cole-Parmer, Vernon Hills, IL) and incubated at 37°C for 30 min, then fixed with 3.7% paraformaldehyde for 10 min, permeabilized with 0.01% saponin, and blocked with 1% BSA in PBS. Samples were labeled with the indicated primary antibodies overnight at 4°C in the presence of 0.01% saponin and 1% BSA. Samples were washed and subsequently incubated with appropriate secondary antibodies. Samples were analyzed with a Zeiss LSM 710 laser scanning confocal microscope attached to a Zeiss Observer Z1 microscope at 21 °C, using a 63× oil Plan Apo, 1.4 numerical aperture (NA) objective. Images were collected using ZEN-LSM software keeping the laser power and gain constant during all acquisitions for comparative analysis of wild type and knockout neutrophils or vehicle vs various inhibitor treatments. Images were then processed using ImageJ (National Institutes of Health, Bethesda, MD) and Photoshop CS4 (Adobe). The tail lengths and fluorescent intensities were quantified using the ImageJ software.
Nucleofection:
Mouse neutrophil nucleofection was carried out using the 4D-Nucleofector X Unit system (Lonza) following the manufacturer’s instructions. In brief, mouse neutrophils were counted, and 1 × 106 cells were resuspended in 20 μl of Lonza P3 solution with 1 μg of DNA and the solutions were transferred into the X Unit and then subjected to nucleofection in the 4D-Nucleofector using program EA-100. The cells were then resuspended in phenol red–free RPMI (Life Technologies) and seeded into four-chamber 35-mm glass-bottom dishes (No. 1.5 borosilicate coverglass; In Vitro Scientific). Cells were incubated at 37°C for at least 4 h before microscopy analysis.
Total Internal Reflection Fluorescence (TIRF) Microscopy:
TIRFM experiments were performed using a 100×/1.45 NA TIRF objective (Nikon Instruments, Melville, NY) on a Nikon TE2000U microscope custom modified with a TIRF illumination module as described (6). Images were acquired on a 14-bit, cooled charge-coupled device (CCD) camera (Hamamatsu) controlled through NIS-Elements software. The movies were recorded using 200 to 300-ms exposures, depending on the fluorescence intensity of the sample for 30 sec.
Super-resolution Microscopy:
Briefly, cells were labeled with anti-JFC1 and anti- active Rac1 primary antibodies and Alexa-647- and Atto-488-conjugated secondary antibodies. STORM imaging and analysis was performed exactly as described previously (43).
Three-dimensional STORM Data Quantification:
Images obtained on the Nikon N-storm (Nikon Inc) system were converted to high resolution images, fully calibrated, and imported into Imaris (Bitplane Inc) where they were analyzed using two well established modules: Spots, to mark the centroid location, and Colocalized Spots, to mark and score paired spots that lie within a defined distance from each other in three-dimensional space as previously described (43).
Neutrophil stimulation, ELISA and flow cytometry analysis:
Mouse neutrophils (1 × 106) isolated from the bone marrow were resuspended in phenol-red free RPMI and were stimulated with either 10nM, 100nM, 1μM or 10μM fMLP for 10min. The cells were spun down and the supernatants were collected for ELISA analysis. Active RhoA was determined using G-LISA activation assay (Cytoskeleton). MPO ELISA was performed using the R&D mouse MPO ELISA duo kit. To evaluate the plasma membrane expression of CD11b, the cells were blocked with PBS+ 1% BSA for 10 min and stained with anti–mouse-CD11b-Alexa 647 (clone M1/70; BD Biosciences, San Jose, CA). Ly-6G staining was used to gate the neutrophil population using anti–mouse-Ly6G-fluorescein isothiocyanate (clone 1A8; BD Biosciences). The cells were then washed, fixed with 1.5% paraformaldehyde and the samples were analyzed using the BD LSR II flow cytometer (BD Biosciences), and the data were processed using FlowJo (Ashland, OR) software. To evaluate F-actin polymerization, the cells were either left unstimulated or were stimulated with either 10nM, 100nM, 1μM or 10μM fMLF for 15 sec, 30 sec, 1 min and 3 min, followed by fixation with 2% paraformaldehyde for 15 min. The fixed cells were then stained with Phalloidin-488 in PBS containing 0.05% TX-100 for 30 min at room temperature. The cells were then washed, resuspended in PBS+ 1% BSA and analyzed using the BD LSR II flow cytometer as previously described.
Calcium flux assay:
Neutrophils (100,000) in HHBS medium (HBSS+ HEPES) were seeded onto 384-well poly-D-lysine coated black plates with a clear bottom. 25 μl of the Fluo-8 dye mixture was added to the cells and the cells were allowed to adhere for 30 min at 37 °C. The plate was loaded onto the FLIPPR machine, stimulant was added using the auto-dispense function and the fluorescence intensity was monitored at Ex/Em= 490/525 nm.
Neutrophil polarization assay:
Purified bone marrow neutrophils were seeded onto dishes with a glass coverslip for 30 min at 37 °C. The cells were then either left unstimulated or were stimulated with 10nM fMLF, 100nM fMLF or 1μM fMLF for 2 min or 10 min. Subsequently, cells were fixed with 3.7% paraformaldehyde for 10 min at room temperature. Fixed cells were blocked and permeabilized using PBS+ 1% BSA+ 0.01% saponin for 2 hours at room temperature, following which the cells were incubated with primary antibody diluted in PBS+ 1% BSA+ 0.01% saponin, overnight at 4 °C. The cells were washed and incubated with secondary antibody for 2 hours at room temperature. The cells were then stained with DAPI and mounted with Fluormount G and imaged using the Zeiss LSM 710 laser scanning confocal microscope attached to a Zeiss Observer Z1 microscope at 21 °C, using a 63× oil Plan Apo, 1.4 numerical aperture (NA) objective. Images were collected using ZEN-LSM software.
In vitro Chemotaxis assay:
Chemotaxis assay was performed using ibidi μ-slide chemotaxis chamber coated with collagen. Purified mouse bone-marrow neutrophils suspended in Hanks’ buffer containing 0.1% BSA were applied onto the slide and incubated at room temperature for 20 mins to allow cell attachment. Following cell attachment, chemoattractant chamber reservoirs were filled with HBSS containing 0.1% BSA and the chemoattractant was added to the upper reservoir. Gradients were generated using 10μM or 25 μM fMLF at the chemoattractant reservoir, which based on the calculated diffusion coefficient (30) produces a 0 to 10 μM fMLF gradient starting from the farthest end of the cell chamber (2×1 mm2) to the chemoattractant chamber at 30 minutes. Thus, cell migration imaging acquisition commenced after 30 min incubation to allow gradient formation. Cell migration was recorded at 1-minute intervals for 1 hour using the DIC setting on the Nikon TE2000U microscope. Tracks for each cell was plotted using the Manual Tracking plug-in of the ImageJ software. Velocity, distance migrated, forward migration index, persistence and track plots were obtained using the Chemotaxis and Migration tool V2.0 from Ibidi. Where indicated, chemotaxis was analyzed using Transwell Permeable Supports 3.0 μm polycarbonate membranes (Costar), using wild type or JFC1−/− neutrophils in response to the CXC chemokines CXCL2 and KC for 1 hour at 37°C following the manufacturer’s instructions. Migrating cells were quantified using flow cytometry.
In vivo chemotaxis assay:
Bone marrow chimera mice with 1:1 ratio of DsRed labeled wild type and non-fluorescent JFC1−/− bone marrow were prepared. The host CD45.1 male mice were irradiated and then retro orbitally injected with 1:1 mixture of bone marrow isolated under sterile conditions from leg bones of DsRed wild type (CD45.2) and JFC1−/− male donor mice. For two weeks after transplantation the mice were kept under sterile conditions on water supplemented with antibiotics. Six weeks after transplantation the reconstitution was confirmed with a FACS experiment. Peripheral blood samples contained less than 3% (average: 1.96%, standard deviation: 0.54) of host (CD45.1) leukocytes and within the donor (CD45.2) population the ratio between wild type and JFC1 knock out leukocytes was average 2.13 (standard deviation: 1.1), suggesting that JFC1 knock out bone marrow reconstituted at a lower level.
To compare the fMLF induced chemotaxis of wild type and JFC1−/− neutrophils, cremaster muscle imaging was performed on the chimera mice. The mice were anesthetized with isoflurane inhalation and were kept on a heating pad during the experiment. The cremaster muscle was exteriorized and cut open and pinned on top of a 0.75 cm wide 1 cm long, 15° tilted mirror of the surgical stage. The muscle was superfused with warmed bicarbonate buffered physiological salt solution (131.9 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl, 2 mM CaCl2, 18 mM NaHCO3, pH = 7.4). The testicle and its accessories were pulled out from the abdomen and were placed to the side. At about 50 μm distance from a venule 1 μl of 10 μM fMLF was microinjected into the muscle with a glass canule pulled from a capillary and grinded to a tip diameter of about 10 μm. The injected volume was confirmed with the help of grades painted on the canule with a marker pen (the distance between the grades was 2.3 mm, what corresponds to 1 μL volume at the internal diameter of the used capillary). The injection was confirmed by observation of tissue swelling around the canule tip. Far red oblique illumination adopted from the publication of Mempel et al. (59) was used to image every (wild type and knockout) leukocyte in the tissue and RFP epifluorescence imaging was used to identify the wild type neutrophils. For the oblique illumination the cremaster pinned on top of the tilted mirror was illuminated with 633 nm wavelength light (100 Watt HBO Nikon light source) through a Cy5 filter cube lacking an emission filter. The reflected light was detected with a Hamamatsu Orca R2 camera and an Olympus X91 upright microscope with an Olympus 20× objective was used. The fluorescent and the oblique illuminated images were collected sequentially.
Statistical analysis
Data are presented as mean, and error bars correspond to standard errors of the means (SEMs) unless otherwise indicated. Statistical significance was determined using the unpaired Student’s t-test or the ANOVA test using GraphPad InStat (version 3) or Excel software, and graphs were made using GraphPad Prism (version 4) software.
Supplementary Material
Acknowledgements:
This work was supported by National Institutes of Health Grants R01HL088256, R01AR070837 and R01DK110162 (to S. D. C.), and by fellowships from the American Heart Association (to M. R. and A.M.) and from the Cystinosis Research Foundation (to M.R. and J.Z.).
Abbreviations:
- fMLF
N-formyl-methionine-leucyl-phenylalanine
- MPO
myeloperoxidase
Footnotes
Competing financial interests: The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1.Wang F (2009) The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harbor perspectives in biology 1, a002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pertz O (2010) Spatio-temporal Rho GTPase signaling - where are we now? J Cell Sci 123, 1841–1850 [DOI] [PubMed] [Google Scholar]
- 3.Wong CH, Heit B, and Kubes P (2010) Molecular regulators of leucocyte chemotaxis during inflammation. Cardiovascular research 86, 183–191 [DOI] [PubMed] [Google Scholar]
- 4.Lakschevitz FS, Visser MB, Sun C, and Glogauer M (2015) Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cell Mol Immunol 12, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun R, Iribarren P, Zhang N, Zhou Y, Gong W, Cho EH, Lockett S, Chertov O, Bednar F, Rogers TJ, Oppenheim JJ, and Wang JM (2004) Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J Immunol 173, 428–436 [DOI] [PubMed] [Google Scholar]
- 6.Johnson JL, Monfregola J, Napolitano G, Kiosses WB, and Catz SD (2012) Vesicular trafficking through cortical actin during exocytosis is regulated by the Rab27a effector JFC1/Slp1 and the RhoA-GTPase-activating protein Gem-interacting protein. Mol Biol Cell 23, 1902–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monfregola J, Johnson JL, Meijler MM, Napolitano G, and Catz SD (2012) MUNC13–4 protein regulates the oxidative response and is essential for phagosomal maturation and bacterial killing in neutrophils. The Journal of biological chemistry 287, 44603–44618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly BA, Rice J, Feig LA, and Buchsbaum RJ (2005) Tiam1-IRSp53 complex formation directs specificity of rac-mediated actin cytoskeleton regulation. Mol Cell Biol 25, 4602–4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pegtel DM, Ellenbroek SI, Mertens AE, van der Kammen RA, de Rooij J, and Collard JG (2007) The Par-Tiam1 complex controls persistent migration by stabilizing microtubule-dependent front-rear polarity. Current biology : CB 17, 1623–1634 [DOI] [PubMed] [Google Scholar]
- 10.Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, and Matsuda M (2002) Activation of rac and cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol Cell Biol 22, 6582–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, and Hahn KM (2000) Localized Rac activation dynamics visualized in living cells. Science 290, 333–337 [DOI] [PubMed] [Google Scholar]
- 12.Gardiner EM, Pestonjamasp KN, Bohl BP, Chamberlain C, Hahn KM, and Bokoch GM (2002) Spatial and temporal analysis of Rac activation during live neutrophil chemotaxis. Current biology : CB 12, 2029–2034 [DOI] [PubMed] [Google Scholar]
- 13.Pestonjamasp KN, Forster C, Sun C, Gardiner EM, Bohl B, Weiner O, Bokoch GM, and Glogauer M (2006) Rac1 links leading edge and uropod events through Rho and myosin activation during chemotaxis. Blood 108, 2814–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen G, Dimitriou I, Milne L, Lang KS, Lang PA, Fine N, Ohashi PS, Kubes P, and Rottapel R (2012) The 3BP2 adapter protein is required for chemoattractant-mediated neutrophil activation. J Immunol 189, 2138–2150 [DOI] [PubMed] [Google Scholar]
- 15.Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, Zhang J, and Junger WG (2015) mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol 210, 1153–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai SP, Bhatia SN, Toner M, and Irimia D (2013) Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 104, 2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAdara Berkowitz JK, Catz SD, Johnson JL, Ruedi JM, Thon V, and Babior BM (2001) JFC1, a novel tandem C2 domain-containing protein associated with the leukocyte NADPH oxidase. The Journal of biological chemistry 276, 18855–18862 [DOI] [PubMed] [Google Scholar]
- 18.Fukuda M (2002) Synaptotagmin-like protein (Slp) homology domain 1 of Slac2-a/melanophilin is a critical determinant of GTP-dependent specific binding to Rab27A. The Journal of biological chemistry 277, 40118–40124 [DOI] [PubMed] [Google Scholar]
- 19.Strom M, Hume AN, Tarafder AK, Barkagianni E, and Seabra MC (2002) A family of Rab27-binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. The Journal of biological chemistry 277, 25423–25430 [DOI] [PubMed] [Google Scholar]
- 20.Johnson JL, Pacquelet S, Lane WS, Eam B, and Catz SD (2005) Akt regulates the subcellular localization of the Rab27a-binding protein JFC1 by phosphorylation. Traffic 6, 667–681 [DOI] [PubMed] [Google Scholar]
- 21.Catz SD, Johnson JL, and Babior BM (2002) The C2A domain of JFC1 binds to 3’-phosphorylated phosphoinositides and directs plasma membrane association in living cells. Proc Natl Acad Sci U S A 99, 11652–11657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brzezinska AA, Johnson JL, Munafo DB, Crozat K, Beutler B, Kiosses WB, Ellis BA, and Catz SD (2008) The Rab27a effectors JFC1/Slp1 and Munc13–4 regulate exocytosis of neutrophil granules. Traffic 9, 2151–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolaczkowska E, and Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nature reviews. Immunology 13, 159–175 [DOI] [PubMed] [Google Scholar]
- 24.Ley K, Laudanna C, Cybulsky MI, and Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature reviews. Immunology 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 25.Kurz AR, Pruenster M, Rohwedder I, Ramadass M, Schafer K, Harrison U, Gouveia G, Nussbaum C, Immler R, Wiessner JR, Margraf A, Lim DS, Walzog B, Dietzel S, Moser M, Klein C, Vestweber D, Haas R, Catz SD, and Sperandio M (2016) MST1-dependent vesicle trafficking regulates neutrophil transmigration through the vascular basement membrane. J Clin Invest 126, 4125–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Sun C, Glogauer M, and Bokoch GM (2009) Human neutrophils coordinate chemotaxis by differential activation of Rac1 and Rac2. J Immunol 183, 2718–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ittner A, Block H, Reichel CA, Varjosalo M, Gehart H, Sumara G, Gstaiger M, Krombach F, Zarbock A, and Ricci R (2012) Regulation of PTEN activity by p38delta-PKD1 signaling in neutrophils confers inflammatory responses in the lung. The Journal of experimental medicine 209, 2229–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamakura S, Nomura M, Hayase J, Iwakiri Y, Nishikimi A, Takayanagi R, Fukui Y, and Sumimoto H (2013) The cell polarity protein mInsc regulates neutrophil chemotaxis via a noncanonical G protein signaling pathway. Dev Cell 26, 292–302 [DOI] [PubMed] [Google Scholar]
- 29.Futosi K, Nemeth T, Pick R, Vantus T, Walzog B, and Mocsai A (2012) Dasatinib inhibits proinflammatory functions of mature human neutrophils. Blood 119, 4981–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebrahimzadeh PR, Hogfors C, and Braide M (2000) Neutrophil chemotaxis in moving gradients of fMLP. Journal of leukocyte biology 67, 651–661 [DOI] [PubMed] [Google Scholar]
- 31.Colvin RA, Means TK, Diefenbach TJ, Moita LF, Friday RP, Sever S, Campanella GS, Abrazinski T, Manice LA, Moita C, Andrews NW, Wu D, Hacohen N, and Luster AD (2010) Synaptotagmin-mediated vesicle fusion regulates cell migration. Nature immunology 11, 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh RK, Furze RC, Birrell MA, Rankin SM, Hume AN, and Seabra MC (2014) A role for Rab27 in neutrophil chemotaxis and lung recruitment. BMC Cell Biol 15, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh RK, Liao W, Tracey-White D, Recchi C, Tolmachova T, Rankin SM, Hume AN, and Seabra MC (2012) Rab27a-mediated protease release regulates neutrophil recruitment by allowing uropod detachment. J Cell Sci 125, 1652–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramadass M, and Catz SD (2016) Molecular mechanisms regulating secretory organelles and endosomes in neutrophils and their implications for inflammation. Immunol Rev 273, 249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manes S, Gomez-Mouton C, Lacalle RA, Jimenez-Baranda S, Mira E, and Martinez AC (2005) Mastering time and space: immune cell polarization and chemotaxis. Semin Immunol 17, 77–86 [DOI] [PubMed] [Google Scholar]
- 36.Pollard TD, and Borisy GG (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 37.Meshulam T, Proto P, Diamond RD, and Melnick DA (1986) Calcium modulation and chemotactic response: divergent stimulation of neutrophil chemotaxis and cytosolic calcium response by the chemotactic peptide receptor. J Immunol 137, 1954–1960 [PubMed] [Google Scholar]
- 38.Pettit EJ, and Fay FS (1998) Cytosolic free calcium and the cytoskeleton in the control of leukocyte chemotaxis. Physiol Rev 78, 949–967 [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, and Mori Y (2008) TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature medicine 14, 738–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang ER, Liu S, Wu LF, Altschuler SJ, and Cobb MH (2016) Chemoattractant concentration-dependent tuning of ERK signaling dynamics in migrating neutrophils. Sci Signal 9, ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun CX, Downey GP, Zhu F, Koh AL, Thang H, and Glogauer M (2004) Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood 104, 3758–3765 [DOI] [PubMed] [Google Scholar]
- 42.Mudrakola HV, Zhang K, and Cui B (2009) Optically resolving individual microtubules in live axons. Structure. 17, 1433–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson JL, He J, Ramadass M, Pestonjamasp K, Kiosses WB, Zhang J, and Catz SD (2016) Munc13–4 Is a Rab11-binding Protein That Regulates Rab11-positive Vesicle Trafficking and Docking at the Plasma Membrane. The Journal of biological chemistry 291, 3423–3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, and Shinohara ML (2012) Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal 5, ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dietz DM, Sun H, Lobo MK, Cahill ME, Chadwick B, Gao V, Koo JW, Mazei-Robison MS, Dias C, Maze I, Damez-Werno D, Dietz KC, Scobie KN, Ferguson D, Christoffel D, Ohnishi Y, Hodes GE, Zheng Y, Neve RL, Hahn KM, Russo SJ, and Nestler EJ (2012) Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat Neurosci 15, 891–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castillo-Lluva S, Tatham MH, Jones RC, Jaffray EG, Edmondson RD, Hay RT, and Malliri A (2010) SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nature cell biology 12, 1078–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Fotheringham L, Wittchen ES, and Hartnett ME (2015) Rap1 GTPase Inhibits Tumor Necrosis Factor-alpha-Induced Choroidal Endothelial Migration via NADPH Oxidase- and NF-kappaB-Dependent Activation of Rac1. The American journal of pathology 185, 3316–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long DL, Willey JS, and Loeser RF (2013) Rac1 is required for matrix metalloproteinase 13 production by chondrocytes in response to fibronectin fragments. Arthritis and rheumatism 65, 1561–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Qin W, Xu X, Xiong Y, Zhang Y, Zhang H, and Sun B (2017) Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc Natl Acad Sci U S A 114, 4483–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foy DS, and Ley K (2000) Intercellular adhesion molecule-1 is required for chemoattractant-induced leukocyte adhesion in resting, but not inflamed, venules in vivo. Microvasc Res 60, 249–260 [DOI] [PubMed] [Google Scholar]
- 51.Forlow SB, Schurr JR, Kolls JK, Bagby GJ, Schwarzenberger PO, and Ley K (2001) Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood 98, 3309–3314 [DOI] [PubMed] [Google Scholar]
- 52.Guilluy C, Garcia-Mata R, and Burridge K (2011) Rho protein crosstalk: another social network? Trends Cell Biol 21, 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong K, Pertz O, Hahn K, and Bourne H (2006) Neutrophil polarization: spatiotemporal dynamics of RhoA activity support a self-organizing mechanism. Proc Natl Acad Sci U S A 103, 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wallace PJ, Wersto RP, Packman CH, and Lichtman MA (1984) Chemotactic peptide-induced changes in neutrophil actin conformation. J Cell Biol 99, 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLeish KR, Merchant ML, Creed TM, Tandon S, Barati MT, Uriarte SM, and Ward RA (2017) Frontline Science: Tumor necrosis factor-alpha stimulation and priming of human neutrophil granule exocytosis. Journal of leukocyte biology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, and Huttenlocher A (2010) Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell 18, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson JL, Ellis BA, Noack D, Seabra MC, and Catz SD (2005) The Rab27a-binding protein, JFC1, regulates androgen-dependent secretion of prostate-specific antigen and prostatic-specific acid phosphatase. The Biochemical journal 391, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Markert M, Andrews PC, and Babior BM (1984) Measurement of O2- production by human neutrophils. The preparation and assay of NADPH oxidase-containing particles from human neutrophils. Methods Enzymol 105, 358–365 [DOI] [PubMed] [Google Scholar]
- 59.Mempel TR, Moser C, Hutter J, Kuebler WM, and Krombach F (2003) Visualization of Leukocyte Transendothelial and Interstitial Migration Using Reflected Light Oblique Transillumination in Intravital Video Microscopy. Journal of Vascular Research 40, 435–441 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.