

Abstract
Here, a folate-targeted pH-sensitive bortezomib conjugate was developed for cancer-specific drug delivery and therapy. The conjugate showed improved cellular uptake, penetration, and anticancer activity compared to free bortezomib, bortezomib-mannitol derivative, and PEGylated bortezomib conjugate in the folate receptor overexpressing cancer cells and their 3D spheroids.
The ubiquitin-proteasome pathway (UPP) involved in protein turnover is essential to cellular homeostasis and survival 1. Targeting the UPP by proteasome inhibitors is considered a new targeted approach for treating human cancer 2. Bortezomib (BTZ) is the first FDA-approved proteasome inhibitor anticancer drug for multiple myeloma and mantle cell lymphoma. Its major anticancer mechanisms include the upregulation of the proapoptotic protein, NOXA, and suppression of the NF-κB signaling pathway 3. BTZ has been also found to increase the sensitivity of chemotherapy 4 and stimulate autophagy 5. Recently, BTZ has been tested in treating solid tumors, such as ovarian cancers 6 and glioma 7, in clinical trials. However, its therapeutic outcome is compromised by poor solubility, insufficient tumor specificity, drug resistance, and unsatisfactory efficacy toward solid tumors. The commercial BTZ product (Velcade®) is a BTZ-mannitol derivative and only increases BTZ solubility. These difficulties have led to the development of new proteasome inhibitors 8. Unfortunately, these attempts focus mainly on improving drug bioavailability and fail to address other important issues. In addition, a few drug delivery systems, e.g. polymeric conjugates 9–11 and micelles 12, 13, are currently under development. However, these systems are still far from a drug-like molecule/system.
Targeting of cell surface receptors is a major strategy to improve the drug’s cancer specificity. Folate receptors (FRs) are overexpressed on many types of cancer cells in response to their rapid proliferation 14. Various FR targeting strategies have been developed for tumor-specific imaging and therapy 14. FRs are glycosylphosphatidylinositol-anchored proteins that bind to folic acid (FA)/folate with high affinity 15. Since its small size, facile chemistry, lack of immunogenicity, and low costs, folate is a mostly used small-molecule ligand for development of FR-targeted tumor-specific therapeutics 14. In this study, we reported a novel pH-sensitive FA-modified BTZ conjugate for cancer-specific drug delivery and therapy.
To improve the performance of BTZ, two pH-sensitive BTZ-polymer conjugates have been developed, the PEG-BTZ and dextran-BTZ conjugates 9, 10. Although the hydrophilic polymers increase BTZ’s solubility and blood circulation time, their cellular uptake and efficacy may be compromised by polymers’ “stealth” properties. In addition, the lack of ‘active’ targeting moieties may increase the risk of the off-tumor toxicity. However, these studies suggest that the formation of boronate ester may be a good strategy to prepare the BTZ conjugate, in which the ester bond serves as a pH-sensitive linker.
In our design, a facile method was used to synthesize the FA-modified BTZ conjugates, including both the small-molecule conjugate, FA-Cat-BTZ, and polymeric conjugate, FA-PEG-Cat-BTZ (Fig. 1 and S1–2). The FA was first modified by dopamine, which works as a catechol linker (Cat), and then coupled to the boronic acid of BTZ via the formation of the boronate ester bond. The structure - activity relationship revealed that the boronic acid group was a proteasome binding site and critical for BTZ’s proteasome inhibitory capability 3. The esterification of the boronic acid would lower the activity of the conjugated BTZ, and only after the FR-mediated endocytosis, BTZ could be liberated and kill cancer cells. Therefore, the strategy would decrease the BTZ’s off-target toxicity 9. The conjugate, FA-Cat-BTZ, showed the distinct optical properties compared to FA-Cat and BTZ, as evidenced by their maximum wavelengths (276nm vs. 284nm and 267nm) (Fig. 2A). In the 1H-NMR spectra, all characteristic peaks of the FA-Cat-BTZ conjugate were clearly shown (Fig. 2B). The peak integration results indicated that the FA/BTZ ratio was around 1/1. The mass spectra indicated that the molecular weight of the conjugate was 923.1 [M-H] in the APCI- mode (Fig. 2C). All these data indicated that the FA-Cat-BTZ conjugate was successfully synthesized.
Fig.1.
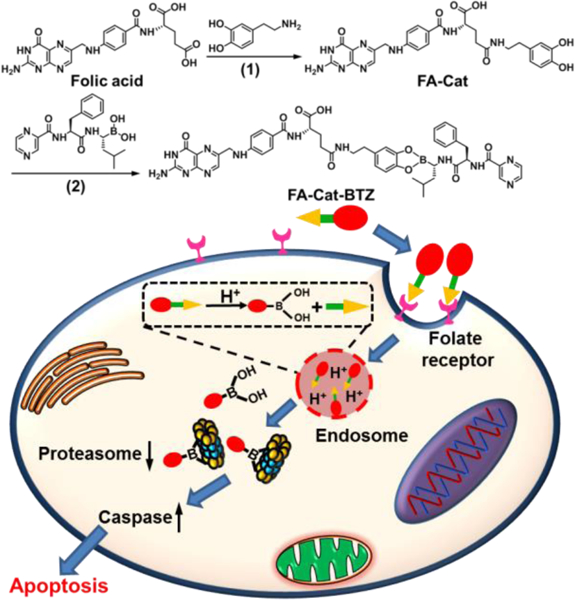
Synthesis and drug delivery mechanism of FA-Cat-BTZ. (1) DMSO, DCC/NHS, 50 ºC; (2) DMSO, room temperature.
Fig. 2.
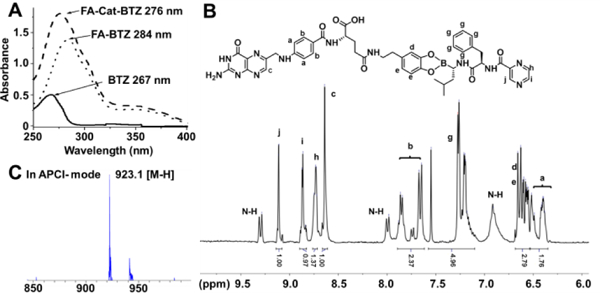
Chemical characterization of FA-Cat-BTZ. (A) UV-Vis absorbance. (B) 1H NMR in DMSO-d6. (C) Mass spectrometry in negative atmospheric pressure chemical ionization (APCI-) mode.
The pH sensitivity and in vitro drug release were evaluated by three methods. In the TLC, FA-Cat-BTZ stayed at the starting point due to its high polarity, while after the incubation with the acid, a new spot was generated, which had the similar position to that of free BTZ (low polarity) (Fig. 3A). The spot at the starting point didn’t disappear because of the generation of the polar FA-Cat residue after acidic hydrolysis. The mass of two spots were detected in situ as BTZ (367.2 [M-H2O+H]) and FA-Cat (575.2 [M-H]) by the TLC/MS (Fig. S3). In the 1H-NMR spectra, the peak of phenolic hydroxyl groups of FA-Cat-BTZ could not be seen because of the esterification, while after incubation of FA-Cat-BTZ with the acid for 1 h, the phenolic hydroxyl peak came back at 8.3 ppm, confirmed by the spectrum of FA-Cat with the exposed phenolic hydroxyl groups (Fig. 3B). The cleavage kinetic of the boronate ester was studied using Cat-BTZ 16 at various pHs (5.5–8.5) which might simulate the normal tissue, tumor extracellular, and endosomal pHs and in the 100% mouse plasma to estimate the drug stability in the blood. The released BTZ was monitored by the RP-HPLC over 120min. The boronate ester between Cat and BTZ could be quickly and completely cleaved at pH 5.5, while it only showed a mild cleavage at pH 7.4 or 8.5 and in the plasma, indicating that the drug release from the BTZ conjugate was pH-dependent (Fig. 3C). These data confirmed that the BTZ was conjugated to the FA-Cat via the pH-sensitive boronate ester linker. Once cleaved, the BTZ could be liberated from the conjugate, which would facilitate the cytosolic drug release from endosomes upon the FR-mediated endocytosis. Furthermore, the released BTZ with the intact boronic acids (Fig. 3B) ensured its proteasome binding affinity.
Fig. 3.
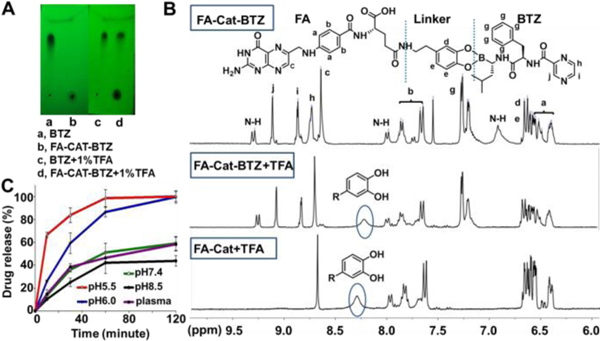
pH sensitivity and drug release of the BTZ conjugate. (A) TLC under UV254nm. (B) 1H NMR in DMSO-d6. (C) Drug dissociation determined by RP-HPLC. Panels A and B: the samples were treated with 1% TFA. Panel C: the samples were incubated at the indicated pHs.
To study the FR-mediated internalization, FITC was conjugated with FA, as a fluorescent probe. Unlike the anti-FR antibodies which are specific to individual FR isomers, FA-FITC can bind to most FR isomers and therefore, may better mimic the FA-modified drugs 17. Compared to the unmodified FITC, FA-FITC was more efficient to enter the FR overexpressing (FR+) cells (HeLa), while in the FR low-expressing (FR-) cells (A549) and retinoic acid (RA) pretreated HeLa cells (with the FR down-regulation 18, 19), no difference in the cellular uptake was observed between FA-FITC and FITC (Fig. 4A–B and S4–5). The FA preincubation significantly decreased the uptake of FA-FITC, but had no effects on the FITC uptake in FR+ cells (Fig. 4C), suggesting that FA-FITC was internalized mainly via the FR-mediated endocytosis. High level of cellular FRs also resulted in efficient uptake of FA-Cat-BTZ in HeLa cells, about 65% higher than that of BTZ, while in A549 cells, no significant difference was observed between BTZ and its conjugate (Fig. 4D).
Fig. 4.
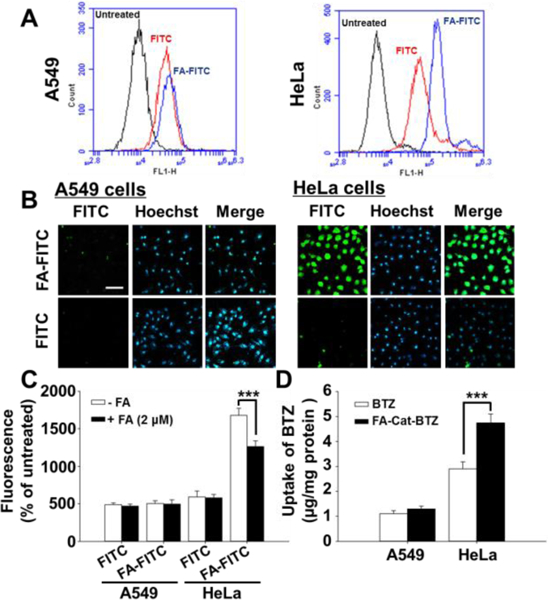
Folate-mediated cellular uptake. (A) Cellular uptake determined by FACS. (B) Cellar uptake determined by fluorescence microscopy. (C) FA competition study, determined by FACS. The cells were incubated with FA-FITC or FITC for 1.5 h. (D) Drug uptake, quantitated by RP-HPLC. The cells were incubated with BTZ or FA-Cat-BTZ for 4 h. Scale bar: 50 μm.
The cytotoxicity of the BTZ conjugates was evaluated after 2-day treatments (Fig. 5 and S6). FA-Cat-BTZ showed much higher cytotoxicity than BTZ and its commercial formulation (BTZ-mannitol derivative) in the FR+ cells (HeLa and MDA-MB-231), due to the FR-mediated endocytosis. The BTZ-mannitol derivative could only mildly increase the drug efficacy because of the increased drug solubility. In the FR- cells (A549) and FR down-regulated cells (RA-HeLa), all small molecules (FA-Cat-BTZ, BTZ, and BTZ-mannitol) had similar cytotoxicity probably because the FA modification could not benefit the drug uptake and the passive diffusion was the major driving force of the drug transport. The drug response data were confirmed by the IC50 values (Table S1). The non-drug components of the conjugate (FA-Cat) didn’t show any toxicity (Fig. S7). The data suggested that the FR-mediated endocytosis enhanced the uptake of FA-Cat-BTZ, resulting in its high efficacy in the FR+ cells. The drug efficacy of FA-Cat-BTZ was well correlated with the cellular FR levels (HeLa > MDA-MB-231 > A549) (Fig. S4). We also found that the PEGylation lowered the efficacy of the conjugated BTZ because the PEG inhibited the drug uptake 20, 21 although the ligand (FA) might increase the molecule’s interaction with the cell membrane. But this might be compensated via the multivalent conjugation of FA11 or BTZ 9 to the PEG.
Fig. 5.
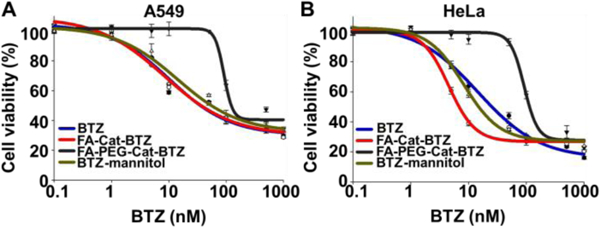
Cytotoxicity of the BTZ and its conjugates analyzed by the MTT Assay. (A) A549 cells. (B) HeLa cells. Incubation time: 48h.
To further understand the anticancer activity of the BTZ conjugates, the proteasome inhibition effect, caspase activity, and apoptosis were analyzed. Due to high drug uptake, even at low drug concentration (1 nM), FA-Cat-BTZ could dramatically inhibited the proteasome activity in HeLa cells, while the free BTZ and PEGylated BTZ conjugate didn’t show any inhibitory effects at 1 nM. At high drug concentrations, both BTZ and its conjugates showed similar inhibition effects, probably because of the saturation of the binding sites on the proteasomes. The non-drug component, FA-Cat, had no proteasome inhibitory capability. Compared to FA-Cat-BTZ and BTZ, the PEGylated BTZ conjugate had a much lower proteasome inhibitory capability (Fig. 6A). Here, although the boronic acid was initially blocked, the FR-mediated enhanced endocytosis (vs. the passive diffusion of BTZ) and the rapid pH-sensitive drug release (Fig. 3) ensured the high proteasome inhibition effect of FA-Cat-BTZ. All BTZ treatments elevated the activity of caspase 3/7 in a dose-dependent manner in both A549 and HeLa cells. Among them, FA-Cat-BTZ induced a significantly higher caspase activity than BTZ in HeLa cells, while no difference in the caspase activity was observed between BTZ and FA-Cat-BTZ in A549 cells (Fig. 6B). The cell apoptosis and death were further analyzed by the annexin V and PI double staining (Fig. 6C and S8). In HeLa cells, at 12 h, the number of apoptotic cells (Annexin V+ /PI-) upon the FA-Cat-BTZ treatment was significantly increased compared to that of the BTZ treatment (41.3% vs. 18.8%). At 24h, both treatments caused substantial cell death (Annexin V+ /PI+). But FA-Cat-BTZ was more efficient to kill cells than BTZ (75.6% vs. 51.2%). In A549 cells, BTZ and its conjugate didn’t show significant difference in their capabilities of inducing cell apoptosis and death. All data suggested that BTZ and FA-Cat-BTZ might exert the anticancer activity by the same caspase-dependent apoptotic pathway 22. However, the FA-mediated endocytosis and pH sensitivity of FA-Cat-BTZ enhanced the drug efficacy.
Fig. 6.
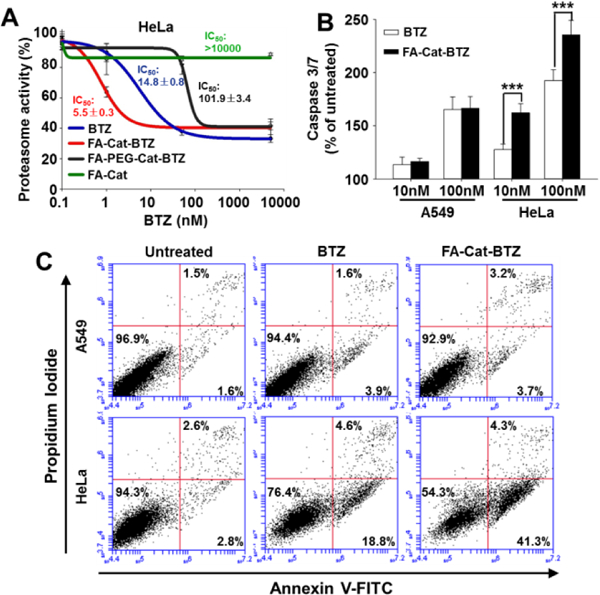
Proteasome activity, caspase 3/7 activity, and cell apoptosis/death. (A) Proteasome activity assay was carried out after 6 h treatment in Hela cells. (B) Caspase 3/7 activity was measured after 12 h treatment in Hela cells. (C) At 12 h after the treatments, cell apoptosis and death were analyzed by the annexin V and PI double staining.
The tumor architecture and microenvironment are known to influence the outcome of anticancer drugs. The cells embedded deeply in the tumor mass usually require a long distance for drugs to travel, which are likely to be drug-resistant. In addition, the dense tumor stroma increases the interstitial fluid pressure, which limits the convection/ penetration of therapeutics through the tumor tissue 23. The heterogeneity as well as cell-cell/cell-microenvironment communications pose significant barriers to drug delivery and effectiveness 24. We have successfully developed the tumor cell spheroids for the in vitro drug evaluation 21. Here, both the HeLa (FR+) and A549 (FR-) spheroids were developed. In the HeLa spheroids, the unmodified FITC showed weak fluorescence, indicating its limited penetration although it is a small molecule. In contrast, the FA modification substantially increased the penetration of FITC, indicating that the folate targeting strategy could enhance not only the drug uptake in cell monolayers (Fig. 4) but also the drug penetration in the 3D tumor tissues (Fig. 7A–B).
Fig. 7.
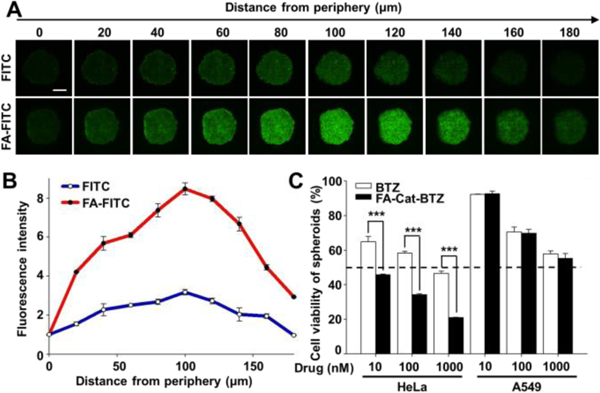
FA-mediated penetration and cytotoxicity in 3D cancer cell spheroids. (A) Spheroid penetration by FITC and FA-FITC, determined by confocal microscopy. Incubation time: 1.5 h. (B) Quantitation of the fluorescence intensity of the images by the ImageJ software. (C) Cell viability of the BTZ conjugate in the spheroids, determined by the CellTiter-Blue® Cell Viability Assay. Incubation time: 48 h. Scale bar: 200 μm.
Although BTZ was used to enhance sensitivity of chemotherapy drugs 4, the resistance to BTZ has been widely reported 25, 26. In our study, a BTZ resistance was observed upon the spheroid formation, as evidenced by the increased IC50 of BTZ (around 1000nM in spheroids in Fig. 7C vs. 31 nM in monolayers in Table S1). Surprisingly, unlike free BTZ, FA-Cat-BTZ could efficiently kill the HeLa cells in the 3D spheroids without a significant compromise (IC50 <10nM for both monolayers and spheroids), implying its capability of overcoming the BTZ resistance. In the A549 spheroids, no difference in cytotoxicity between BTZ and FA-Cat-BTZ was observed (Fig. 7C), suggesting that the FA-mediated FR targeting improved the drug penetration and anticancer activity. Here, the enhanced drug uptake and penetration increased the BTZ’s intracellular accumulation, which would overcome the BTZ resistance coming from the cancer’s 3D structure/microenvironment and upregulated efflux pumps, such as P-glycoprotein 25, 26.
In summary, we reported a novel small-molecule folate-targeted pH-sensitive BTZ conjugate for cancer treatment. The FA-Cat-BTZ conjugate was well characterized in terms of chemical structure, pH sensitivity and drug release. Due to the FR-mediated endocytosis and endosomal pH-triggered rapid drug release, the proteasome inhibitory and apoptosis-inducing capabilities of FA-Cat-BTZ were significantly increased. Compared to the BTZ, BTZ-mannitol derivative, and FA-PEG-Cat-BTZ, FA-Cat-BTZ showed the improved cellular uptake, penetration, and anticancer activity in FR+ cells in both 2D monolayer and 3D spheroid cancer models. The FA-Cat-BTZ conjugate might have great potential as a tumor-targeted proteasome inhibitor for tumor-specific drug delivery and effective cancer treatment.
Supplementary Material
Acknowledgments
The work was supported by the National Cancer Institute of the National Institutes of Health (R15CA213103) to Dr. Lin Zhu.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Glickman MH and Ciechanover A, Physiol. Rev., 2002, 82, 373–428. [DOI] [PubMed] [Google Scholar]
- 2.Adams J, Cancer Cell, 2004, 5, 417–421. [DOI] [PubMed] [Google Scholar]
- 3.Chen D, Frezza M, Schmitt S, Kanwar J and Dou QP, Curr. Cancer Drug Targets, 2011, 11, 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cusack JC Jr., Liu R, Houston M, Abendroth K, Elliott PJ, Adams J and Baldwin AS Jr., Cancer Res., 2001, 61, 3535–3540. [PubMed] [Google Scholar]
- 5.Kao C, Chao A, Tsai C, Chuang W, Huang W, Chen G, Lin C, Wang T, Wang H and Lai C, Cell Death Dis., 2014, 5, e1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aghajanian C, Dizon DS, Sabbatini P, Raizer JJ, Dupont J and Spriggs DR, J. Clin. Oncol., 2005, 23, 5943–5949. [DOI] [PubMed] [Google Scholar]
- 7.Phuphanich S, Supko JG, Carson KA, Grossman SA, Nabors LB, Mikkelsen T, Lesser G, Rosenfeld S, Desideri S and Olson JJ, J. Neurooncol., 2010, 100, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teicher BA and Tomaszewski JE, Biochem. Pharmacol., 2015, 96, 1–9. [DOI] [PubMed] [Google Scholar]
- 9.Su J, Chen F, Cryns VL and Messersmith PB, J. Am. Chem. Soc., 2011, 133, 11850–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu W, Ding J, Li L, Xiao C, Zhuang X and Chen X, Chem. Commun. (Camb.), 2015, 51, 6812–6815. [DOI] [PubMed] [Google Scholar]
- 11.Santos FM, Matos AI, Ventura AE, Gonçalves J, Veiros LF, Florindo HF and Gois PM, Angewandte Chemie International Edition, 2017, 56, 9346–9350. [DOI] [PubMed] [Google Scholar]
- 12.Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R and Kabanov AV, Mol. Pharmaceutics, 2012, 9, 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasegawa U, Moriyama M, Uyama H and van der Vlies A, Colloid Polym Sci, 2015, 293, 1887–1892. [Google Scholar]
- 14.Low PS, Henne WA and Doorneweerd DD, Acc. Chem. Res., 2007, 41, 120–129. [DOI] [PubMed] [Google Scholar]
- 15.Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J, Yong EL, Xu HE and Melcher K, Nature, 2013, 500, 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su J, Chen F, Cryns VL and Messersmith PB, Journal of the American Chemical Society, 2011, 133, 11850–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W, Sarantopoulos A, de Jong JS, Arts HJ, van der Zee AG, Bart J, Low PS and Ntziachristos V, Nat. Med, 2011, 17, 1315–1319. [DOI] [PubMed] [Google Scholar]
- 18.Orr RB, Kreisler AR and Kamen BA, JNCI: Journal of the National Cancer Institute, 1995, 87, 299–303. [DOI] [PubMed] [Google Scholar]
- 19.Kamen BA and Smith AK, Adv. Drug Delivery Rev., 2004, 56, 1085–1097. [DOI] [PubMed] [Google Scholar]
- 20.Tu Y and Zhu L, J. Controlled Release, 2015, 212, 94–102. [DOI] [PubMed] [Google Scholar]
- 21.Zhu L, Wang T, Perche F, Taigind A and Torchilin VP, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 17047–17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pérez-Galán P, Roué G, Villamor N, Montserrat E, Campo E and Colomer D, Blood, 2006, 107, 257–264. [DOI] [PubMed] [Google Scholar]
- 23.Minchinton AI and Tannock IF, Nature Reviews Cancer, 2006, 6, 583. [DOI] [PubMed] [Google Scholar]
- 24.Trédan O, Galmarini CM, Patel K and Tannock IF, J. Natl. Cancer Inst., 2007, 99, 1441–1454. [DOI] [PubMed] [Google Scholar]
- 25.Rumpold H, Salvador C, Wolf AM, Tilg H, Gastl G and Wolf D, Biochem. Biophys. Res. Commun., 2007, 361, 549–554. [DOI] [PubMed] [Google Scholar]
- 26.O’Connor R, Ooi MG, Meiller J, Jakubikova J, Klippel S, Delmore J, Richardson P, Anderson K, Clynes M, Mitsiades CS and O’Gorman P, Cancer Chemother. Pharmacol., 2013, 71, 1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.