

Abstract
Anthropogenic copper pollution of environmental waters from sources such as acid mine drainage, antifouling paints, and industrial waste discharge is a major threat to our environment and human health. This study presents an optical sensing system that combines self-assembled glutaraldehyde-cross-linked double-layered poly-ethylenimine (PEI-GA-PEI)-modified nanoporous anodic alumina (NAA) interferometers with reflectometric interference spectroscopy (RIfS) for label-free, selective monitoring of ionic copper in environmental waters. Calibration of the sensing system with analytical solutions of copper shows a linear working range between 1 and 100 mg L–1, and a low limit of detection of 0.007 ± 0.001 mg L–1 (i.e., ~0.007 ppm). Changes in the effective optical thickness (ΔOTeff) of PEI-GA-PEI-functionalized NAA interferometers are monitored in real-time by RIfS, and correlated with the amount of ionic copper present in aqueous solutions. The system performance is validated through X-ray photoelectron spectroscopy (XPS) and the spatial distribution of copper within the nanoporous films is characterized by time-of-flight-secondary ion mass spectroscopy (TOF-SIMS). The specificity and chemical selectivity of the PEI-GA-PEI-NAA sensor to Cu2+ ions is verified by screening six different metal ion solutions containing potentially interfering ions such as Al3+, Cd2+, Fe3+, Pb2+, Ni2+, and Zn2+. Finally, the performance of the PEI-GA-PEI-NAA sensor for real-life applications is demonstrated using legacy acid mine drainage liquid and tap water for qualitative and quantitative detection of copper ions. This study provides new opportunities to develop portable, cost-competitive, and ultrasensitive sensing systems for real-life environmental applications.
Graphical Abstract
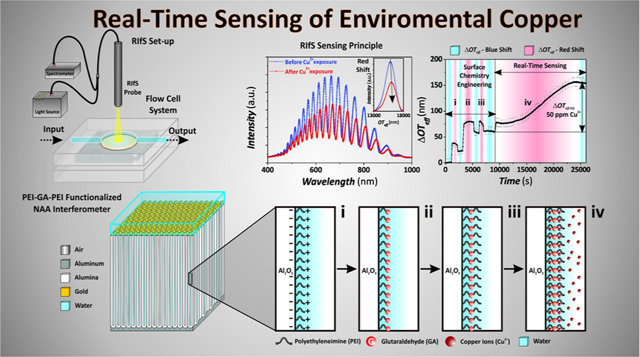
The use of copper is constantly increasing in materials and products of commercial importance such as cosmeceuticals,1 agriculture,2 construction,3 chemical industries,4 and electronics.3,5 This rapid diversification and expansion in the use of copper is dramatically increasing its impact on natural environments. Copper can be released into the environment during its mining and also from copper-based products such as metal-based biocides in agriculture, antifouling paints in marine systems,6,7 and domestic and industrial waste emissions.8–10 Once released into the environment, copper becomes highly soluble and percolates into soil and water in its various toxic forms.11,12 Copper is a broad spectrum biocide, being free ionic (Cu2+) and inorganic complexes (Cu(OH)+) in its most toxic forms.13 The maximum permissible limit of Cu2+ ions in drinking water cannot exceed 2 mg L–1 (i.e., 2 ppm) and 1.3 mg L–1 (i.e., 1.3 ppm), as established by the World Health Organization (WHO) and the U.S. Environmental Protection Agency (EPA), respectively.14 Therefore, there is an urgent need to develop monitoring systems that can perform highly sensitive, selective, cost-competitive, user-friendly, and reliable detection of copper ions in environmental waters. Current benchmark techniques used to detect copper in aqueous solutions include inductively coupled plasma-optical emission spectroscopy/mass spectroscopy (ICP-OES/MS),15 atomic absorption spectroscopy (AAS),16 anodic stripping voltammetry (ASV),17 UV–visible18 and fluorescence spectroscopies.19 Though these methods offer good detection limits and broad linear working ranges, they require significant capital and maintenance investments, laborious sample preparation processes, and highly trained personnel and cannot be miniaturized into portable sensing systems for in situ analysis applications.
Current progress in nanotechnology is enabling development of advanced analytical tools for heavy metal ions sensing. An outstanding example of this is the combination of thin nanoporous films with optical techniques such as fluid imbibition-coupled laser interferometry,20 interferogram average over wavelength,21 and reflectometric interference spectroscopy (RIfS).22–28 These systems provide novel approaches for developing label-free optical sensors able to monitor binding events in real-time. The nanoporous structure of sensing platforms such as porous silicon and nanoporous anodic alumina (NAA) enables enhanced sensitivities due to their high surface area, which provides more ligand sites for binding interactions. Chemical functionalization of these nanoporous substrates enables high chemical selectivity toward a broad range of analyte species such as proteins,29 small molecules30 and ions,31 nucleotides,32,33 and whole cells.34,35
This study presents an innovative optical sensing system combining chemically modified NAA optical interferometers with RIfS for sensitive and highly selective detection of copper ions (Figure 1a,b). The novelty stems from our identification that the modification of the surface chemistry of NAA interferometers with layers of glutaraldehyde cross-linked polyethylenimine (PEI-GA-PEI) gives this system chemical selectivity to specifically detect copper ions in aqueous solutions. The interaction between copper ions and PEI-GA- PEI-modified NAA interferometers is translated into quantifiable changes in the effective optical thickness of these nanoporous films (i.e., sensing principle) (Figure 1c,d). PEI-GA-PEI chemical functional layers provide excellent selectivity toward copper ions in complex real-life environmental solutions containing interfering organic and inorganic impurities (Figure 1e,f).36–38 The performance of this copper sensing system is systematically assessed in terms of working range, sensitivity, linearity, low limit of detection, chemical selectivity, and real-life application. Our study provides new opportunities to develop ultrasensitive, highly selective, low-cost, portable sensing systems able to monitor trace levels of copper ions in environmental waters.
Figure 1.
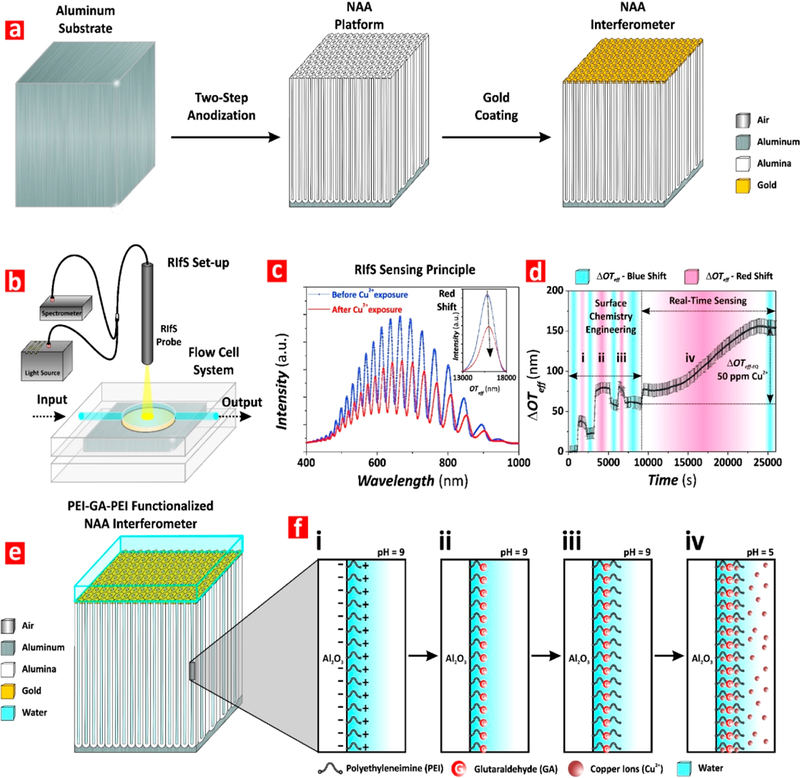
Production of PEI-GA-PEI-functionalized NAA interferometers and assessment of binding affinity for detection of copper ions using RIfS. (a) Illustration describing the two-step anodization process used to produce NAA interferometers (left, aluminum substrate; center, NAA interferometer; right, gold-coated NAA interferometer). (b) Schematic showing the RIfS setup used to monitor binding interactions between PEI-GA-PEI-modified NAA interferometers and copper ions in real-time under dynamic flow conditions. (c) Representative RIfS spectra of PEI-GA-PEI-functionalized NAA interferometers before and after exposure to Cu2+ ions (inset showing the characteristic fast Fourier transform (FFT) spectra used to estimate the effective optical thickness (OTeff) of NAA interferometers associated with the different stages of the process: surface chemistry engineering (i–iii) and real-time sensing (iv)). (d) Real-time effective optical thickness changes (ΔOTeff) associated with the surface chemistry engineering and sensing stages: (i) electrostatic functionalization of the inner surface of the nanopores of NAA interferometers with PEI molecules; (ii) cross-linking of immobilized PEI molecules with glutaraldehyde (GA); (iii) immobilization of second PEI functional layer; and (iv) binding to Cu2+ ions. (e) Schematic showing the structure of PEI-GA-PEI-functionalized NAA interferometers. (f) Illustration showing details of the inner surface chemistry of gold-coated PEI-GA-PEI-functionalized NAA interferometers during the different stages of the sensing process (i–iv).
EXPERIMENTAL SECTION
Materials.
High purity (99.9997%) aluminum (Al) foils of thickness 0.32 mm were purchased from Goodfellow Cambridge Ltd. (U.K.). Oxalic acid (H2C2O4), perchloric acid (HClO4), chromic acid (H2CrO4), lead(II) nitrate (Pb-(NO3)2), nickel(II) sulfate (NiSO4), zinc chloride (ZnCl2), aluminum chloride hexahydrate (AlCl3 6H2O), cadmium nitrate tetrahydrate (Cd(NO3)2 4H2O), iron chloride (FeCl3), hydrochloric acid (HCl), nitric acid (HNO3), and glutaraldehyde (GA) were acquired from Sigma-Aldrich (Australia). Ethanol (C2H5OH), phosphoric acid (H3PO4), sodium chloride (NaCl), and copper(II) sulfate pentahydrate (CuSO4 5H2O) were purchased from ChemSupply (Australia). Branched polyethylenimines (PEI) Lupasol G20 (50 wt % in H2O, MW 1300 g mol–1), Lupasol HF (56 wt % in H2O, MW 25000 g mol–1), and Lupasol P (50 wt % in H2O, MW 750000 g mol–1) were provided by BASF (Germany) and stored under N2 until use. Real legacy acid mine drainage solution was kindly provided by Copper Mines of Tasmania (Australia). Ultrapure water 18.2 MΩ cm from a Milli-Q Advantage A10 water purification system was used to prepare all the aqueous solutions used in this study. pH adjustments were performed using an ION 700 m (Eutech Instruments, Singapore).
Fabrication of NAA Interferometers.
Al substrates were sonicated in EtOH and ultrapure water for 15 min each and dried under air stream. Then Al chips were electropolished in a mixture of HClO4 and EtOH 1:4 (v:v) at 20 V and 5 °C for 3 min in an electrochemical reactor with a circular window of ~1 cm in diameter. The first anodization step was performed in 0.3 M oxalic acid electrolyte at 40 V and 6 °C for 20 h. The resulting NAA layer was chemically removed by wet etching in 0.2 M H2CrO4 and 0.4 M H3PO4 at 70 °C for 3 h. The second anodization step was performed using the same conditions than those used during the first step but for 2 h. Finally, the NAA films were pore-widened by wet chemical etching in H3PO4 5 wt % at 35 °C for 15 min.39–42
Optical Characterization.
Details of the flow system and RIfS setup used in this study are provided in the Supporting Information. RIfS spectra (Figure 1b–d) were acquired in the wavelength range 400–1000 nm and processed by applying fast Fourier transform (FFT) to estimate the effective optical thickness (OTeff) of NAA interferometers according to eq 1:
| (1) |
where OTeff, neff, andLp are the effective optical thickness, the effective refractive index, and the physical thickness of the NAA platform, respectively, whereas θ is the angle of incidence of light (i.e., θ = 0° in this case).
Chemical Modification of NAA Interferometers.
As-prepared NAA interferometers were coated with an ultrathin film of gold ~4–5 nm thick using a sputter coater equipped with a film thickness monitor (sputter coater 108 Auto, Cressington) to enhance the intensity of spectral fringes.43–45 Prior to sensing, the inner surface of NAA interferometers was chemically functionalized with GA-cross-linked double PEI layers through a three-step procedure monitored in real-time by RIfS. This process was performed in a flow cell system (Supporting Information) using effective optical thickness changes (ΔOTeff) as sensing parameter. A stable baseline in water was obtained. Then, NAA interferometers were exposed to a PEI functional solution (0.2 wt %, 0.1 mol L–1 NaCl, pH 9) until complete saturation of the inner surface by PEI functional groups, denoted by a plateau in (ΔOTeff) (Figure 1d-i). Milli-Q water was injected into the system to remove loosely bound PEI molecules from the inner surface of NAA interferometers. Self-assembled PEI molecules immobilized onto the inner surface of the NAA interferometers by electrostatic interaction were cross-linked by exposing the nanoporous films to 2.5 vol % GA solution for a given time, followed by a thorough washing with Milli-Q water (Figure 1d-ii). The sensing platforms were then exposed to fresh PEI functional solution (0.2 wt %, 0.1 mol L–1 NaCl, pH 9) as in the first step to cross-link a secondary layer of PEI molecules (Figure 1d-iii). Finally, Milli-Q water was flowed through the system to remove unbounded PEI molecules.
Calibration and Detection of Cu2+ Ions.
After functionalization (sections i–iii, Figure 1d), the sensing performance of PEI-GA-PEI-modified NAA interferometers was assessed by measuring ΔOTeff upon exposure to six different concentrations of Cu2+, ranging from 1 to 100 mg L–1 at pH 5. These copper analytical solutions were prepared by diluting 0.1 M stock solution of Cu2SO4 5H2O in Milli-Q water. PEI-GA-PEI-modified NAA interferometers were packed in a flow cell system, through which the copper analytical solutions were flowed at an optimized rate of 100 μL min–1 using a peristaltic pump (LongerPump, Thermoline Scientific, Australia). A stable baseline was first established in Milli-Q water at pH 5 for ~15 min before injection of ionic copper solutions into the flow cell. Binding of copper ions to PEI-GA-PEI-functionalized NAA interferometers was monitored in real-time through changes in ΔOTeff by RIfS. This process continued until all the available ligand sites in the inner surface of NAA were saturated with Cu2+ ions. Control experiments were performed with nonfunctionalized NAA interferometers using 10 and 100 mg L–1 analytical solutions of Cu2+.
Assessment of Chemical Selectivity.
The chemical selectivity of the system toward copper ions was assessed by exposing a set of freshly prepared PEI-GA-PEI-modified NAA sensing platforms to 25 mg L–1 individual aqueous solutions of Cd2+, Ni2+, Fe3+, Al3+, Pb2+, and Zn2+ ions. Effective optical thickness changes upon exposure to these analytical solutions were compared against those obtained for copper ions for the same concentration.
Real-Life Environmental Application.
The performance of PEI-GA-PEI-modified NAA interferometers to detect copper ions in complex matrixes was evaluated using AMD and tap water solutions with known Cu2+ concentration and benchmarked against ICP-OES. A volume of 100 mL of acid mine drainage (AMD) liquid was poured into a 250 mL glass beaker, and the pH was adjusted to 5 (i.e., initial pH ~2.2). A 50 mL aliquot of supernatant containing dissolved metal ions was separated from the precipitate and used for detection of copper content through RIfS in PEI-GA-PEI-modified NAA interferometers. The ion metal content of the AMD solution was established by inductively coupled plasma optical emission spectroscopy (ICP-OES). Duplicates of the diluted samples were acidified with 3–4 drops of HNO3 to prevent bacterial growth. All the results were processed with Multicomponent Spectral Fitting. Calibration standards and QC standards were prepared in 1% HNO3.
Structural Characterization of NAA.
The structural features of the NAA interferometers were established by field emission gun-scanning electron microscopy (FEG-SEM FEI Quanta 450). FEG-SEM images were processed using ImageJ.
RESULTS AND DISCUSSION
Structural Characterization of NAA Interferometers.
The geometric features of NAA interferometers (i.e., nanopore diameter, dp; nanopore length, Lp; and interpore distance, dint; Figure 2a) were established by FEG-SEM image analysis. Figure 2b–d compiles a set of representative FEG-SEM images showing the cross-sectional (Figure 2b,c) and top (Figure 2d) views of NAA interferometers fabricated by a two-step anodization process.39–42 Figure 2b shows a cross-sectional FEG-SEM image of a NAA interferometer featuring straight cylindrical nanopores from top to bottom, which grow perpendicularly to the underlying aluminum substrate during anodization. These nanopores have a closed oxide barrier layer at the bottom (Figure 2c), with an average nanopore length of Lp = 5.5 ± 0.2 μm (i.e., physical thickness of the optical film). The top surface of NAA shows an array of nanopores of uniform size and distribution arranged in a self-organized hexagonal pattern (Figure 2d). The average nanopore diameter was dp = 65 ± 4 nm, with an average interpore distance of dint = 105 ± 5 nm. NAA interferometers with these geometric features display well-resolved and intense Fabry-Pérot interference fringes in their RIfS spectra that are suitable for sensing applications.
Figure 2.
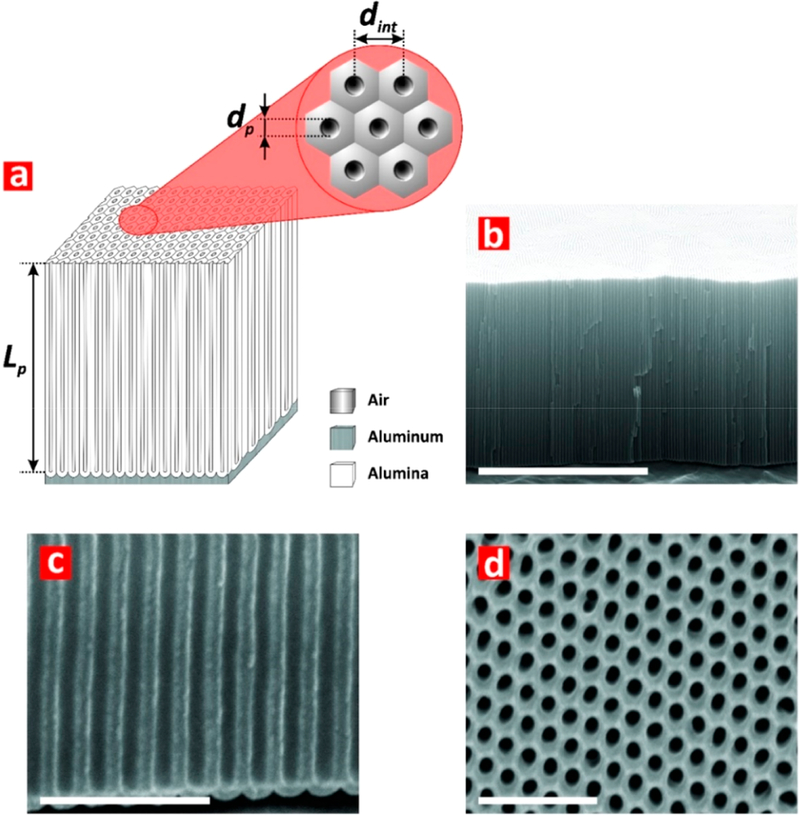
Structural features of NAA interferometers produced by two-step anodization. (a) Schematic of a NAA interferometer with details of geometric features (i.e., nanopore diameter, dp; nanopore length, Lp; and interpore distance, dint). (b) General cross-sectional FEG-SEM image view of a NAA interferometer featuring straight cylindrical nanopores from top to bottom (scale bar = 5 μm). (c) Magnified cross-sectional FEG-SEM image view showing details of the oxide barrier layer (scale bar = 500 nm). (d) Top FEG-SEM view of hexagonally arranged cylindrical nanopores in NAA interferometers (scale bar = 500 nm).
Preliminary Optimization of Sensing Features of NAA Interferometers.
Preliminary experiments were carried out to optimize the sensing performance of PEI-GA-PEI-functionalized NAA interferometers toward copper ions. Three sensing features were optimized: (i) molecular weight of PEI functional molecules, (ii) flow rate of the analytical solutions, and (iii) the surface chemistry architecture of PEI-GA-PEI functional layers. These three parameters were selected for their effect on the sensing performance of this system, as demonstrated in previous studies.46,47 Freshly prepared PEI-GA-PEI-modified NAA interferometers were exposed to a 100 mg L–1 Cu2+ analytical solution at pH 5. Effective optical thickness changes (ΔOTeff) in these NAA sensing platforms upon modification of these three working parameters were measured by RIfS to establish the most optimal conditions for Cu2+ sensing. The obtained results, described in detail in the Supporting Information and summarized in Figure 3, established that the best performing combination of these working parameters was PEI molecules of 750 000 g mol–1 molecular weight (Figure 3a), a flow rate of 100 μL min–1 (Figure 3b), and a sandwiched PEI-GA-PEI surface chemistry architecture (Figure 3c).
Figure 3.
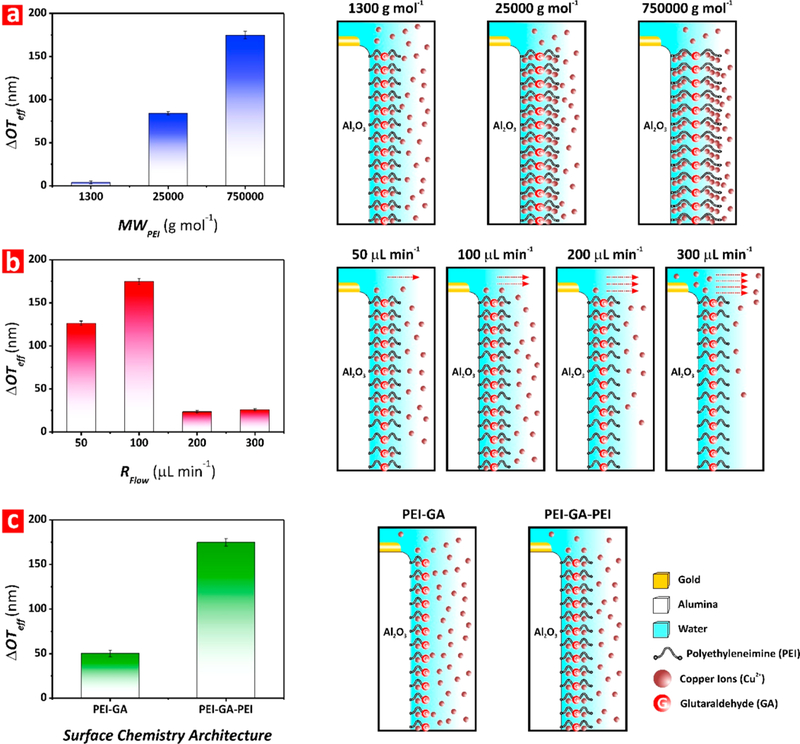
Optimization of working parameters to maximize sensing of Cu2+ ions in NAA interferometers (note: error bars denote standard deviation from average measurements obtained from n = 3 independent experiments). (a) Bar chart showing the ΔOTeff associated with each molecular weight of PEI assessed in this study (i.e., MWPEI = 1 300, 25 000, and 750 000 g mol–1) (left) and illustration showing the effect of this working parameter on the sensing performance of PEI-GA-PEI-modified NAA interferometers (right). (b) Bar chart showing the ΔOTeff associated with each flow rate of analytical solution assessed in this study (i.e., RFlow = 50, 100, 200, and 300 μL min–1) (left) and schematic showing the effect of this working parameter on the sensing performance of PEI-GA-PEI-modified NAA interferometers (right). (c) Bar chart showing the ΔOTeff associated with each surface chemistry architecture assessed in this study (i.e., PEI-GA and PEI-GA-PEI) (left) and illustration depicting the effect of this working parameter on the sensing performance of PEI-GA-PEI-modified NAA interferometers (right).
Real-Time Monitoring of Copper Ions.
The surface chemistry engineering and real-time sensing (sections i–iii and iv in Figure 1d, respectively) were monitored in real-time by RIfS. First, a ΔOTeff baseline is established in Milli-Q water and 0.1 M NaCl in Milli-Q water at pH 9 for ~15 min each. No significant change in ΔOTeff is observed during the transition from Milli-Q water to NaCl solution. After this step, 0.2 wt % PEI solution in 0.1 M NaCl at pH 9 is injected into the flow system. The exposure of the NAA interferometers to the PEI solution leads to a sharp and rapid increase in ΔOTeff, which stabilizes at a value of ~35 nm. The surface alumina (i.e., Al2O3) is negatively charged at slightly basic pH (i.e., pH = 9). As a result, positively charged PEI molecules are immobilized onto the inner surface of alumina by strong electrostatic interactions.48,49 The adsorption of the PEI functional layer onto the inner surface of the nanopores increases the effective refractive index of the NAA interferometers, red-shifting the OTeff of the optical film (Figure 1d-i).
After achieving a plateau (i.e., saturation of the inner surface of NAA with PEI molecules), fresh 0.1 M NaCl solution at pH 9 and Milli-Q water are sequentially flowed through the system. ΔOTeff slightly decreases to a new equilibrium value of ~23 nm during this stage, confirming a stable adsorption of PEI functional layers. This slight blue shift in ΔOTeff is collectively attributable to lower refractive index of water and removal of loosely bound PEI molecules from the inner surface of NAA. Next, PEI-modified NAA interferometers are exposed to a 2.5 vol % GA solution in Milli-Q water, resulting in a red shift in ΔOTeff of ~79 nm (Figure 1d-ii). The chemical cross-linking of PEI with GA enhances the stability of the PEI functional layer by creating intramolecular bonds between PEI molecules immobilized onto the inner surface of NAA. Once stabilized, Milli-Q water and 0.1 M NaCl at pH 9 solutions are flowed through the system in a sequential fashion to remove physisorbed GA molecules and to establish a new ΔOTeff baseline prior to creating the second PEI functional layer. During this process, ΔOTeff blue-shifts and achieves a new stable baseline at ~58 nm. Fresh PEI solution (0.2 wt %, 0.1 mol L–1 NaCl, pH 9) is flowed again through the system to create a double PEI functional layer cross-linked to the primary PEI layer through GA (i.e., PEI-GA-PEI surface chemistry architecture) (Figure 1d-iii). Finally, 0.1 M NaCl at pH 9 and Milli-Q water solutions are sequentially flowed through the system. A total increment of ~4 nm in ΔOTeff is observed after deposition of the second PEI functional layer, with the final equilibrium baseline of ΔOTeff achieved at ~62 nm. Then, PEI-GA-PEI-modified NAA interferometers were exposed to different analytical solutions of Cu2+ ions with controlled concentrations (i.e., real-time sensing stage, Figure 1d-iv). Figure 4a shows a representative example of real-time monitoring of ΔOTeff upon exposure to [Cu2+] = 75 ppm solution (Figure S1 in the Supporting Information summarizes the obtained result for all the Cu2+ ions concentrations). As this graph reveals, ΔOTeff increases sharply upon exposure to the analytical solution containing Cu2+ until the binding groups present in the PEI functional layers are completely saturated with Cu2+ ions (i.e., plateau in ΔOTeff). The chemical binding between Cu2+ ions and the functional groups of GA-cross-linked PEI layers red-shifts the RIfS interference pattern. For instance, as Figure 4a shows, when PEI-GA-PEI-modified NAA platforms are exposed to a 75 mg L–1 solution of Cu2+ ions, ΔOTeff increases progressively up to ~130 nm, from the previously established baseline in Milli-Q water, until it achieves a stable value. This indicates that the PEI functional layers inside the nanopores of NAA are saturated with Cu2+ ions (i.e equilibrium state). Once the binding equilibrium state is achieved, Milli-Q water at pH 5 is flowed through the system to remove unbound Cu2+ ions and to establish the total ΔOTeff associated with 75 mg L–1 of Cu2+ ions, which was measured to be ~124 nm with respect to the previous baseline obtained in Milli-Q water. The kinetics of this binding reaction for each [Cu2+] is also characterized by the saturation time (tsat), defined as the time at which the equilibrium state is reached, as indicated in Figure 4a. Figure 4b summarizes the average ΔOTeff values for each surface chemistry engineering stage (i.e., first PEI functional layer, GA cross-linking, and second PEI functional layer) and real-time sensing (i.e., for [Cu2+] = 1–100 ppm).
Figure 4.
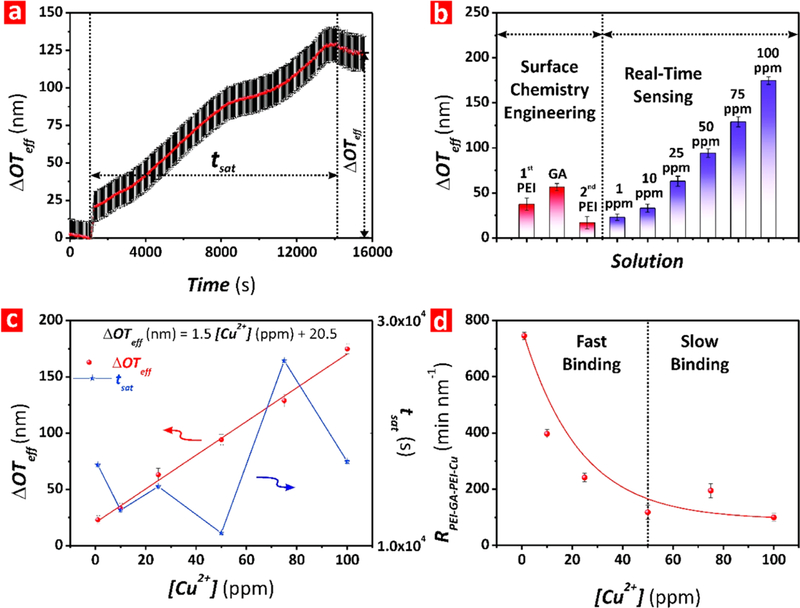
Assessment of the binding interaction between Cu2+ ions and PEI-GA-PEI-functionalized NAA interferometers for different concentrations of Cu2+ ions (note: error bars denote standard deviation from average measurements obtained from n = 3 independent experiments). (a) Real-time Cu2+ binding stage for [Cu2+] = 75 ppm, where the arrows indicate ΔOTeff and tsat for the binding reaction performed under dynamic flow conditions (note: the dotted line shown at the left of the graph indicates the time point at which the Cu2+ analytical solution was injected into the flow cell system, OTeff and time baselines). (b) Average values of ΔOTeff for each surface chemistry engineering stage and real-time sensing. (c) Correlation between ΔOTeff (left scale) and tsat (right scale) with [Cu2+] for PEI-GA-PEI-functionalized NAA interferometers. (d) Kinetic rate (RPEI-GA-PEI-Cu) for the binding reaction between Cu2+ ions and PEI-GA-PEI functional layers for the range of [Cu2+] (i.e., 1–100 ppm).
Calibration of PEI-GA-PEI-Modified NAA Interferometers for Cu2+ Sensing.
The sensing performance of our RIfS system was assessed by flowing analytical solutions of Cu2+ ions with controlled concentrations, from 1 to 100 mg L–1. The sensing parameters characterizing the performance of this system (i.e., linear working range, sensitivity, saturation time, and low limit of detection) were estimated by correlating ΔOTeff and tsat values for each Cu2+ ions concentration, as summarized in Figure 4c. This graph shows a strong linear correlation between ΔOTeff and [Cu2+] for the entire range of concentrations (1–100 mg L–1). A linear fitting between these parameters establishes the sensitivity (i.e., slope (S) = 1.55 ± 0.11 nm (mg L–1)–1) and the low limit of detection (i.e., LOD = 0.007 ± 0.001 mg L–1), calculated as the slope of the fitting line and 3σ =3 times the standard deviation of the lowest concentration of copper ions divided by the slope of the fitting line, respectively, with a linear working range from 1 to 100 mg L–1 and a linearity of R2 = 0.9926. As Figure 4c shows, tsat is relatively constant for [Cu2+] < 75 ppm. However, the saturation time increases above [Cu2+] = 75 ppm and decreases moderately for [Cu2+] = 100 ppm, with an average tsat = 4.7 ± 1.5 h, tsat-max = 7.3 h, and tsat-min = 3.1 h. The response time achieved by PEI-GA-PEI-functionalized NAA interferometers under the conditions of study is comparable to that reported in previous studies using NAA interferometers of similar nanoporous geometry and dynamic flow conditions.27,32,46,48,50 The main factor establishing the kinetics of copper ions is the binding mechanism to PEI-GA-PEI functional layers. PEI is a polymer with a branched structure and high content of amine-nitrogen functional groups with repeating C2H5N units that donate electrons and chelate metal ions.51 Nitrogen atoms in PEI chelate Cu2+ ions by coordination interaction, in which four nitrogen atoms bind one Cu2+ ion. The branched structure of the PEI-GA-PEI functional layer prevents the direct exposure of all the functional groups in the PEI molecules immobilized onto the inner surface of NAA interferometers. The progressive binding of copper ions leads to conformational changes in PEI molecules so four nitrogen atoms can chelate one Cu2+ ion. These conformational changes expose more functional binding sites in the PEI molecules, creating new binding interactions with Cu2+ ions and a progressive increment of ΔOTeff.
To gain a better insight into the kinetics mechanism of the binding interaction between Cu2+ ions and PEI-GA-PEI functional layers, we estimated the binding rate RPEI-GA-PEI-Cu, calculated as the ratio between ΔOTeff and tsat for each [Cu2+] (Figure 4d). RPEI-GA-PEI-Cu follows an exponential decay trend with the concentration of copper ions, revealing that, at low concentrations of copper ions (i.e., [Cu2+] < 50 ppm), the increasing concentration of analyte molecules accelerates the binding reaction due to enhancement of the frequency of interactions between Cu2+ ions and the functional groups in the PEI-GA-PEI layer since more ions are available for binding events inside the nanopores. However, for [Cu2+] ≥ 50 ppm, the reaction rate becomes almost constant and practically independent of [Cu2+], indicating that the reaction is rate-limited by the binding affinity between PEI-GA-PEI and Cu2+ ions and the conformational changes of PEI molecules upon initial exposure to Cu2+ ions.52 Note that control experiments with NAA interferometers without PEI-GA-PEI functional layers were performed to verify that red-shifts in the ΔOTeff of PEI-GA-PEI-functionalized NAA interferometers upon exposure to copper ions are exclusively due to selective surface chemistry interactions. Bare NAA interferometers were exposed to 10 and 100 mg L–1 analytical solutions of Cu2+ ions at pH 5. The measured ΔOTeff for nonmodified NAA interferometers upon exposure were ~8 and ~11 nm, respectively (Figure S2, Supporting Information). This result demonstrates that nonspecific adsorption of positively charged Cu2+ ions to the negatively charged surface of NAA due to electrostatic interactions is almost negligible as compared to ΔOTeff values achieved in PEI-GA-PEI-functionalized NAA interferometers.
Benchmark Validation of PEI-GA-PEI-Modified NAA Interferometers for Cu2+ Sensing.
PEI-GA-PEI-functionalized NAA interferometers were analyzed by XPS after exposure to 1, 25, 50, and 100 mg L–1 analytical solutions of Cu2+ ions. Figure S3a (Supporting Information) shows the ratio of copper to nitrogen (Cu/N) established by XPS, demonstrating that, at equilibrium, the amount of copper binding to the chelator (nitrogen) in PEI molecules is linearly dependent on the amount of copper ions present in the analytical solution. Copper binding increases linearly with increasing concentration of Cu2+ ions, as indicated by the linear fitting shown in Figure S3a. To further validate the selective binding of copper ions, the spatial distribution of Cu in PEI-GA-PEI-modified NAA interferometers was assessed by time-of-flight secondary ion mass spectrometry (TOF-SIMS) and 13C NMR analysis (Figures S3b and S4, Supporting Information).
Chemical Selectivity of PEI-GA-PEI-Modified NAA Interferometers toward Cu2+ Ions.
Label-free optical detection systems suffer from nonspecific binding interactions, which can lead to false positives or inaccurate quantification of analytes. PEI-GA-PEI-functionalized NAA interferometers were exposed to ion solutions spiked with 25 mg L–1 of potentially interfering ions such as Al3+, Fe3+, Cd2+, Ni2+, Pb2+, and Zn2+ to demonstrate the chemical selectivity toward Cu2+ ions. Changes in the effective optical thickness of these films upon exposure to each metal ion solution were compared with those obtained for a 25 mg L–1 Cu2+ ions solution. All of these analytical solutions were prepared in Milli-Q water with pH adjusted to 5. As Figure 5a shows, the ΔOTeff of PEI-GA-PEI-modified NAA interferometers underwent negligible changes upon exposure to Al3+, Fe3+, Ni2+, Pb2+, and Zn2+ ions. The most significant nonspecific change in ΔOTeff was observed for Cd2+ (i.e., 0.24 ± 0.4 nm), which is practically negligible as compared to that measured for the same concentration of Cu2+ ions (i.e., 63 ± 1 nm, ~262 times higher). Fourier transform- infrared spectroscopy (FT-IR) and X-ray absorption spectroscopy (XAS) analyses in our previous study indicate that GA cross-linking of PEI molecules generates structural changes that lead to the formation of a PEI network containing a high content of Schiff bases (i.e., imine nitrogens), with strong affinity and selectivity toward copper ions.38 These results demonstrate that PEI-GA-PEI-functionalized NAA interferometers feature high sensitivity and selectivity toward Cu2+ ions due to their functional surface chemistry architecture.
Figure 5.
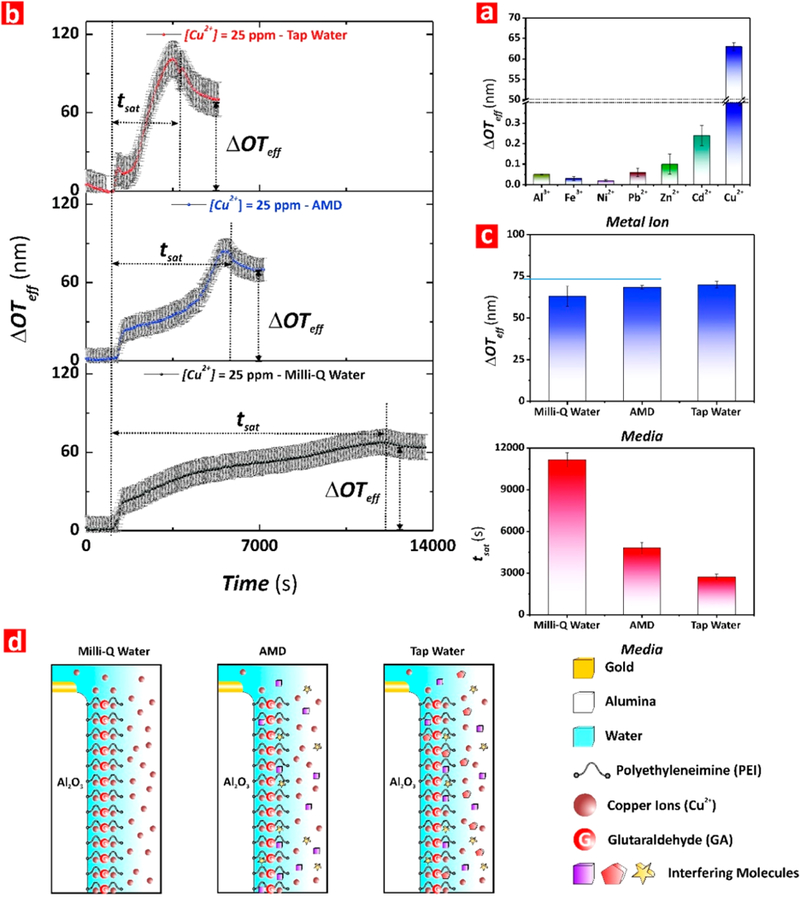
Assessment of chemical selectivity of PEI-GA-PEI-functionalized NAA interferometers for Cu2+ ions and performance assessment in complex matrices (note: error bars denote standard deviation from average measurements obtained from n = 3 independent experiments). (a) Bar chart showing the ΔOTeff measured upon exposure to analytical solutions of Al3+, Fe3+, Cd2+, Ni2+, Pb2+, Zn2+, and Cu2+ (i.e., [metal ion] = 25 ppm). (b) Real-time Cu2+ binding stage for each media (i.e., Milli-Q water, AMD, and tap water for [Cu2+] = 25 ppm), where the arrows indicate ΔOTff and tsat for each of these binding reactions performed under dynamic flow conditions (note: the dotted line shown at the left of the graphs indicates the time point at which the analytical solutions were injected into the flow cell system, OTeff and time baselines). (c) Bar chart showing the ΔOTeff (top) and tsat (bottom) measured in PEI-GA-PEI-functionalized NAA interferometers upon exposure to analytical and real-life solutions of Cu2+ (i.e., [Cu2+] = 25 ppm). (d) Schematic representation illustrating the effect of the media complexity (i.e., interfering molecules) on the sensing performance of PEI-GA-PEI-modified NAA interferometers.
Real-Life Application of PEI-GA-PEI-Modified NAA Interferometers for Cu2+ Ion Sensing.
PEI-GA-PEI-functionalized NAA interferometers were exposed to complex, real acid mine drainage liquid (AMD) and tap water spiked with Cu2+ ions for detection and quantification of copper ions in complex matrixes (Figure 5). Table S1 (Supporting Information) shows the concentration of dissolved metals present in the AMD solution analyzed in our study. Analysis of the AMD solution by ICP-OES revealed that Al (~130 mg L−1) and Fe (>500 mg L–1) were the most abundant metal ions in these samples, with more than 4-fold and 16-fold higher concentrations, respectively, than other metal ions such as copper (~31 mg L−1) and zinc (~20 mg L–1). Freshly prepared PEI-GA-PEI-functionalized NAA interferometers were exposed to the AMD solution and changes in the effective optical thickness were assessed in real-time by RIfS (Figure 5b). The PEI-GA-PEI surface chemistry on the inner surface of NAA interferometers was engineered following the protocol outlined in Real-Time Monitoring of Copper Ions. After the final functionalization step, Milli Q-water at pH 5 was flowed through the system for 15 min to obtain a stable baseline. Then, the AMD analyte solution was flowed through the flow cell system. The ΔOTeff of PEI-GA-PEI-functionalized NAA interferometers increased sharply as a result of Cu2+ ions binding. Once the equilibrium was achieved (i.e., plateau in ΔOTeff), Milli-Q water (pH 5) was flowed again through the system to establish the total ΔOTeff resulting from the selective binding of Cu2+ ions present in the AMD solution. This protocol was repeated to analyze the system’s performance using tap water spiked with 25 mg L–1 of Cu2+ ions. Real-time monitoring of these binding processes through ΔOTeff is shown in Figure 5b. The RIfS response in terms of ΔOTeff for these processes established values of 69 ± 1 nm and 70 ± 2 nm for AMD and tap water solutions, respectively (Figure 5c). Using the calibration line obtained in Figure 4c, such a change in effective optical thickness corresponds to a concentration of copper ions of 32 ± 1 mg L–1. Interestingly, the amount of copper quantified using ICP-OES from the same AMD analyte solution was ~30.8 mg L–1, which would correspond to a total ΔOTeff of ~66.7 nm.
Therefore, the sensing performance of our RIfS system only deviates ~4.5% from the concentration value provided by a benchmark technique such as ICP-OES. This system could provide a cost-competitive solution for in situ copper ions sensing at a significantly reduced price per analysis, with miniaturized features for portability. The saturation time (tsat) for the binding reaction between Cu2+ and PEI-GA-PEI functional layers decreases in the following order Milli-Q water (11152 ± 450 s) > AMD (4797 ± 350 s) > tap water (2716 ± 200 s). The higher response of our RIfS system in terms of ΔOTeff for the AMD and tap water solutions can be attributed to the ionic strength of the solution. We speculate that the presence of other interfering organic and inorganic ions and complexes in these matrixes modifies the ionic strength of the medium. This could influence the conformation of PEI functional molecules so more binding groups are exposed to copper ions in the nanopores, increasing the binding reaction rate and reducing the saturation time of the reaction (Figure 5d). The excellent agreement between the results obtained by RIfS and ICP-OES for the quantification of copper ions clearly demonstrates the suitability of our sensing system to detect copper ions in real-life environmental samples.
CONCLUSIONS
In summary, this study has demonstrated the development of a label-free, real-time sensing system for the detection and quantification of copper ions combining nanoporous anodic alumina interferometers functionalized with double-layered glutaraldehyde-cross-linked polyethylenimine and reflectometric interference spectroscopy. The sensing performance parameters were established using analytical solutions of Cu2+ ions, where changes in the effective optical thickness of PEI-GA-PEI-functionalized NAA interferometers upon exposure to Cu2+ ions were used as sensing principle. The linear detection range of this system spans from 1 to 100 mg L–1, with a sensitivity of 1.5 ± 0.1 nm (mg L–1)–1, a low limit of detection of 0.007 ± 0.001 mg L–1, and a linearity of 0.9926. The chemical selectivity of the sensing system was assessed by exposing PEI-GA-PEI-modified NAA platforms to analytical solutions containing controlled amounts of potentially interfering ions such as Fe3+, Cd2+, Al3+, Ni2+, Pb2+, and Zn2+. The surface chemistry of the system showed excellent selectivity toward Cu2+ ions, and the effective optical thickness changes associated with other interfering ions were negligible as compared to those obtained for Cu2+ ions. Finally, we evaluated the performance of the system for real-life applications, establishing concentration of copper ions present in real acid mine drainage liquid and spiked tap water. The obtained results only deviated ~4.5% from the value obtained using ICP-OES.
Supplementary Material
ACKNOWLEDGMENTS
Authors thank the support provided by the Australian Research Council (ARC) through Grant Numbers DE140100549 and CE140100003, the School of Chemical Engineering, the University of Adelaide (UoA), the Institute for Photonics and Advanced Sensing (IPAS), the ARC Centre of Excellence for Nanoscale BioPhotonics (CNBP), the National Health and Medical Research Council (NHMRC) through Grant Numbers GNT1143296 and GNT1146627, and The University of Queensland.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.8b04963.
Further details on the optical setup, 13C NMR spectra of PEI molecules, XPS and TOF-SIMS analyses, real-time monitoring of ΔOTeff for different concentrations of copper ions, control experiments in as-produced NAA interferometers without PEI-GA-PEI functional layers, and a table summarizing the metal concentration in acid mine drainage liquid by ICP-OES (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Huseinel Hadmed H; Castillo RF J. Cosmet. Dermatol. 2016, 15 (4), 514–519. [DOI] [PubMed] [Google Scholar]
- (2).Rai M; Ingle A Appl. Microbiol. Biotechnol. 2012, 94 (2), 287–293. [DOI] [PubMed] [Google Scholar]
- (3).Van Beers D; Graedel TS Afr. J. Sci. 2003, 99 (1–2), 61–69. [Google Scholar]
- (4).Richardson HW Handbook of Copper Compounds and Applications; CRC Press, 1997. [Google Scholar]
- (5).Pagett R Energy and Commodities In Building Global Resilience in the Aftermath of Sustainable Development: Planet, People and Politics; Springer International Publishing: Cham, Switzerland, 2018; pp 83–86. [Google Scholar]
- (6).Claisse D; Alzieu C Mar. Pollut. Bull. 1993, 26 (7), 395–397. [Google Scholar]
- (7).Castritsi-Catharios J; Neofitou N; Vorloou A Toxicol. Environ. Chem. 2015, 97 (1), 116–123. [Google Scholar]
- (8).Chung BY; Song CH; Park BJ; Cho JY Pedosphere 2011, 21 (5), 621–627. [Google Scholar]
- (9).Gunatilake SK, Methods of Removing Heavy Metals from Industrial Wastewater. J. Multidisciplinary Eng. Sci. Stud. 2015, 1 (1).1218 [Google Scholar]
- (10).Samsudeen N; Matheswaran M Bioremediation of Industrial Wastewater Using Bioelectrochemical Treatment In Bioremediation: Applications for Environmental Protection and Management; Springer, 2018; pp 115–126. [Google Scholar]
- (11).Cao ZH; Hu ZY Chemosphere 2000, 41 (1), 3–6. [DOI] [PubMed] [Google Scholar]
- (12).Minkina TM; Linnik VG; Nevidomskaya DG; Bauer TV; Mandzhieva SS; Khoroshavin VY J. Soils Sediments 2018, 18(6), 2217–2228. [Google Scholar]
- (13).Kaur S; Kempson I; Xu H; Nydén M; Larsson M RSC Adv. 2018, 8 (22), 12043–12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Flemming C; Trevors J ‘Water, Air, Soil Pollut. 1989, 44 (1–2), 143–158. [Google Scholar]
- (15).Al-Saydeh SA; El-Naas MH; Zaidi SJ J. Ind. Eng. Chem. 2017, 56, 35–44. [Google Scholar]
- (16).Cui C; He M; Hu BJ Hazard. Mater. 2011, 187 (1–3), 379–385. [DOI] [PubMed] [Google Scholar]
- (17).Freedman YE; Ronen D; Long GL Environ. Sci. Technol 1996, 30 (7), 2270–2277. [Google Scholar]
- (18).Nolan MA; Kounaves SP Anal. Chem. 1999, 71 (16), 3567–3573. [Google Scholar]
- (19).Wen T; Qu F; Li NB; Luo HQ Arabian J. Chem. 2017, 10, S1680–S1685. [Google Scholar]
- (20).Eckstein C; Acosta LK; Pol L; Xifré-Pérez E; Pallares J; Ferré-Borrull J; Marsal LF ACS Appl Mater. Interfaces 2018, 10 (12), 10571–10579. [DOI] [PubMed] [Google Scholar]
- (21).Mariani S; Strambini LM; Barillaro G Anal. Chem. 2016, 88 (17), 8502–8509. [DOI] [PubMed] [Google Scholar]
- (22).Möhrle BP; Kohler K; Jaehrling J; Brock R; Gauglitz G Anal. Bioanal. Chem. 2005, 384 (2), 407–413. [DOI] [PubMed] [Google Scholar]
- (23).Gauglitz G; Brecht A; Kraus G; Mahm W Sens. ActuatorsB 1993, 11 (1–3), 21–27. [Google Scholar]
- (24).Curtis CL; Doan VV; Credo GM; Sailor MJ J. Electrochem. Soc. 1993, 140 (12), 3492–3494. [Google Scholar]
- (25).Belge G; Beyerlein D; Betsch C; Eichhorn K-J; Gauglitz G; Grundke K; Voit B Anal. Bioanal Chem. 2002, 374 (3), 403–411. [DOI] [PubMed] [Google Scholar]
- (26).Santos A; Kumeria T; Losic D Anal. Chem. 2013, 85 (16), 7904–7911. [DOI] [PubMed] [Google Scholar]
- (27).Kumeria T; Rahman MM; Santos A; Ferré-Borrull J; Marsal LF; Losic D Anal. Chem. 2014, 86 (3), 1837–1844. [DOI] [PubMed] [Google Scholar]
- (28).Mariani S; Robbiano V; Strambini LM; Debrassi A; Egri G; Dähne L; Barillaro G Nat. Commun. 2018, 9, 5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kacmaz S; Ertekin K; Mercan D; Oter O; Cetinkaya E; Celik E Spectrochim. Acta, Part A 2015, 135, 551–559. [DOI] [PubMed] [Google Scholar]
- (30).Choi HW; Sakata Y; Kurihara Y; Ooya T; Takeuchi T Anal. Chim. Acta 2012, 728, 64–68. [DOI] [PubMed] [Google Scholar]
- (31).Piehler J; Brecht A; Gauglitz G Anal. Chem. 1996, 68 (1), 139–143. [DOI] [PubMed] [Google Scholar]
- (32).Chen Y; Santos A; Wang Y; Kumeria T; Wang C; Li J; Losic D Nanoscale 2015, 7 (17), 7770–7779. [DOI] [PubMed] [Google Scholar]
- (33).Lin VS-Y; Motesharei K; Dancil K-PS; Sailor MJ; Ghadiri MR Science 1997, 278 (5339), 840–843. [DOI] [PubMed] [Google Scholar]
- (34).Sauer M; Brecht A; Charisse K; Maier M; Gerster M; Stemmler I; Gauglitz G; Bayer E Anal. Chem. 1999, 71 (14), 2850–2857. [DOI] [PubMed] [Google Scholar]
- (35).Kumeria T; Kurkuri MD; Diener KR; Parkinson L; Losic D Biosens. Bioelectron. 2012, 35 (1), 167–173. [DOI] [PubMed] [Google Scholar]
- (36).Santos A; Balderrama VS; Alba M; Formentín P; Ferré-Borrull J; Pallarès J; Marsal LF Adv. Mater. 2012, 24 (8), 1050–1054. [DOI] [PubMed] [Google Scholar]
- (37).Lindén JB; Larsson M; Coad BR; Skinner WM; Nydén M RSC Adv. 2014, 4 (48), 25063–25066. [Google Scholar]
- (38).Lindén JB; Larsson M; Kaur S; Skinner WM; Miklavcic SJ; Nann T; Kempson IM; Nyden M RSC Adv. 2015, 5 (64), 51883–51890. [Google Scholar]
- (39).Masuda H; Fukuda K Science 1995, 268 (5216), 1466–1468. [DOI] [PubMed] [Google Scholar]
- (40).Nielsch K; Choi J; Schwirn K; Wehrspohn RB; Gösele U Nano Lett. 2002, 2 (7), 677–680. [Google Scholar]
- (41).Lee W; Park S-J Chem. Rev. 2014, 114 (15), 7487–7556. [DOI] [PubMed] [Google Scholar]
- (42).Santos A; Kumeria T; Losic D TrAC, Trends Anal. Chem. 2013, 44, 25–38. [Google Scholar]
- (43).Macias G; Hernández-Eguía LP; Ferré-Borrull J; Pallares J; Marsal LF ACS Appl. Mater. Interfaces 2013, 5 (16), 8093–8098. [DOI] [PubMed] [Google Scholar]
- (44).Kumeria T; Losic D Nanoscale Res. Lett. 2012, 7 (1), 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dronov R; Jane A; Shapter JG; Hodges A; Voelcker NH Nanoscale 2011, 3 (8), 3109–3114. [DOI] [PubMed] [Google Scholar]
- (46).Law CS; Sylvia GM; Nemati M; Yu J; Losic D; Abell AD; Santos A ACS Appl. Mater. Interfaces 2017, 9 (10), 8929–8940. [DOI] [PubMed] [Google Scholar]
- (47).Kumeria T; Gulati K; Santos A; Losic D ACS Appl. Mater. Interfaces 2013, 5 (12), 5436–5442. [DOI] [PubMed] [Google Scholar]
- (48).Lindén JB; Larsson M; Kaur S; Nosrati A; Nydén MJ Appl. Polym. Sci. 2016, 133, 43954. [Google Scholar]
- (49).Kaur S; Kempson IM; Linden JB; Larsson M; Nyden M Biofouling 2017, 33 (2), 184–194. [DOI] [PubMed] [Google Scholar]
- (50).Kumeria T; Rahman MM; Santos A; Ferré-Borrull J; Marsal LF; Losic D ACS Appl. Mater. Interfaces 2014, 6 (15), 12971–12978. [DOI] [PubMed] [Google Scholar]
- (51).Maketon W; Zenner CZ; Ogden KL Environ. Sci. Technol. 2008, 42 (6), 2124–2129. [DOI] [PubMed] [Google Scholar]
- (52).Law CS; Lim SY; Abell AD; Santos A Anal. Chem. 2018, 90 (16), 10039–10048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.