

Abstract
The immune-mediated tissue destruction of graft-vs-host disease (GvHD) remains a major barrier to greater use of hematopoietic stem cell transplantation (HSCT). Mesenchymal stem cells (MSCs) have intrinsic immunosuppressive qualities and are being actively investigated as a therapeutic strategy for treating GvHD. We characterized Cymerus™ MSCs, which are derived from adult, induced pluripotent stem cells (iPSCs), and show they display surface markers and tri-lineage differentiation consistent with MSCs isolated from bone marrow (BM). Administering iPSC-MSCs altered phosphorylation and cellular localization of the T cell-specific kinase, Protein Kinase C theta (PKCθ), attenuated disease severity, and prolonged survival in a humanized mouse model of GvHD. Finally, we sevaluated a constellation of pro-inflammatory molecules on circulating PBMCs that correlated closely with disease progression and which may serve as biomarkers to monitor therapeutic response. Altogether, our data suggest Cymerus iPSC-MSCs offer the potential for an off-the-shelf, cell-based therapy to treat GvHD.
Keywords: PKCθ, Graft-vs-host disease, Mesenchymal stem cell, Induced pluripotent stem cell, Humanized mouse model
1. Introduction
Hematopoietic stem cell transplantation (HSCT) can provide full hematopoietic reconstitution after myeloablative therapy commonly used to treat hematologic malignancies, solid tumors, or immunemediated bone marrow (BM) failure diseases, such as aplastic anemia (van den Brink et al. 2015; Ratajczak and Suszynska, 2016; Dietz et al. 2016). Graft-versus-host disease (GvHD) develops when immune-competent cells in the stem cell graft are activated and damage host tissues (Shlomchik 2007). Affecting up to 80% of allogeneic HSCT recipients (Garnett et al. 2013), GvHD remains a significant barrier to the broader use of HSCT in the clinic.
T cells contribute prominently to GvHD pathophysiology. Skin, gut, and liver are major organs targeted by T cells during GvHD, but damage to hematopoietic tissues also occurs (Ramadam and Paczesny, 2015). T cells are activated by antigen-specific signals delivered through the T cell receptor, coupled with antigen non-specific signals conveyed through CD28 co-stimulatory receptors. The T cell-specific kinase, Protein Kinase C theta (PKCθ), is phosphorylated on multiple residues, downstream of CD28 signaling. Functional PKCθ is essential for mediating GvHD responses (Valenzuela et al. 2009), including through its nuclear regulation of pro-inflammatory gene expression (Sutcliffe et al. 2011). Reducing PKCθ activity in alloreactive T cells; therefore, constitutes an attractive approach to limiting GvHD.
Strategies to prevent or treat GvHD include prophylactic or therapeutic administration of immunosuppressive agents, such as cyclosporine, although prognosis is poor for patients who progress to a steroid-refractory state (Westin et al. 2011). More recently, MSC-based therapies are being rigorously investigated. While administering MSCs for steroid-refractory GvHD is promising (Fernández Vallone et al., 2013), there remains a critical need both for more effective treatments for acute GvHD, in general, and for improving the performance of MSC-based therapies, specifically.
Induced pluripotent stem cells (iPSCs) can proliferate almost indefinitely without losing pluripotency, making it feasible to generate a nearly limitless supply of iPSCs from a single blood or tissue donation (Lei and Schaffer 2013). Harnessing the expansion potential of iPSCs prior to differentiation, allows for producing vast numbers of iPSC-derived MSCs, without excessively expanding the MSCs themselves. This enables ongoing production of commercial quantities of minimally-expanded MSCs from a single iPSC line.
Here we report on the characterization and pre-clinical efficacy of the Cymerus™ MSCs, derived from iPSCs through the mesenchy-moangioblast pathway (Vodyanik et al., 2010). Immunophenotyping and in vitro functional studies show iPSC-MSCs exhibit a typical MSC phenotype and a normal karyotype. Using a humanized mouse model of GvHD, we demonstrate single- or dual-dose infusions of iPSC-MSCs, given under therapeutically relevant conditions, attenuate GvHD severity and provide a significant survival benefit. We demonstrate that the immunosuppressive effects of iPSC-MSCs result from their modulating PKCθ phosphorylation and cellular localization in T cells. Finally, we show a reduced expression of pro-inflammatory molecules correlate highly with clinical response to iPSC-MSC administration, suggesting these may serve as biomarkers to monitor therapeutic response.
2. Materials and methods
2.1. Animals
Animal studies were approved by the Institutional Animal Care and Use Committee, University of Massachusetts Amherst. Six-weeks-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were rested for one week prior to use and housed under pathogen-free conditions in microisolator cages with acidified, antibiotic water throughout the experimental procedures.
2.2. iPSC-MSC derivation
The iPSCs used to generate CYP-001 were manufactured on Cynata’s behalf by Cellular Dynamics International, Inc. (Madison, WI). The iPSCs were derived from CD34-enriched peripheral blood mononuclear cells using an episomal plasmid-based, transgene-free, viral-free and feeder layer-free reprogramming procedure. Manufacture of the iPSC-MSCs commenced with the expansion of iPSCs in Essential-8™ Complete Medium and standard plastic tissue culture dishes coated with recombinant vitronectin. The expanded iPSCs were harvested as single cells and plated on 6-well plates coated with Collagen IV, and the medium changed to differentiation medium (Iscove’s modified Dulbeccos Medium) supplemented with Ham’s F-12 Nutrient Mix, BMP-4, and Activin A. The cells were subsequently transferred to semi-solid mesenchymal-Colony Forming Medium containing Stem-Span™ Serum Free Expansion Medium, ES-Cult™ M3120, Human Endothelial Serum-Free Medium (ESFM), and human fibroblast growth factor-2 (FGF-2). Cells were incubated until spherical mesenchymoangioblast colonies formed. The resulting colonies were harvested using a 100 μM strainer and resupended in Mesenchymal Serum-Free Expansion Medium containing StemLine II, Human ESFM, and FGF-2, grown in adherent culture on fibronectin/collagen-coated flasks. The final harvested iPSC-MSCs were cryopreserved and stored in the vapor phase of liquid nitrogen until use.
2.3. Assessing IDO and PD-L1 expression by IFNγ-licensed iPSC-MSCs
iPSC-MSCs were seeded on fibronectin- and collagen-coated plates. The next day they were licensed with 50 ng/mL of Recombinant Human IFNγ (BioLegend, San Diego, CA) for 24 and 48 h in M-SFEM, at 37 °C in 5% CO2. Cells were split and treated as follows: RNA was isolated using the QuickRNA miniprep kit (Zymo Research, Irvine, CA) and qPCR was performed using the SYBR Green Master Mix (BioTools, Jupiter, FL) to assess indoleamine 2,3-dioxygenase (IDO) mRNA levels. β-Actin was the reference gene used for normalization. Cells were also stained with antibodies specific for surface markers, as indicated. Data were acquired on a BD LSR Fortessa Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ) and analyzed using FlowJo (version 10.0, Treestar, Ashland, OR).
2.4. iPSC-MSC labeling and in vivo trafficking
For cell labeling, iPSC-derived MSCs and BM-MSCs were labeled using Celsense (Pittsburgh, PA) 19F reagents, under Celsense protocols. Nuclear Magnetic Resonance (NMR) analysis was performed to confirm 19F integration into the cells. For ex vivo tissue detection confirmation, three animals each were dosed through the tail vein with low (8 × 105) or high (3 × 106) doses of 19F-labeled iPSC-MSCs or low or high doses of BM-MSCs. After 24 h, animals were sacrificed, and selected tissues were harvested. Tissues were analyzed for the presence of cells by both NMR and immunohistochemistry, to confirm that 19F signals were representative of the cells’ location. Alexa Fluor® 488 dual fluorescent tag (ThermoFisher Scientific, Waltham, MA) and STEM101 mouse monoclonal antibody specific for the human cell nucleus marker, Ku80 (StemCells, Newark, CA) were used to facilitate IHC. Some animals were followed up to 28 days for long-term biodistribution studies, in vivo, of labeled cells.
2.5. Graft-vs-host disease induction
Human PBMCs (StemCell Technologies, Vancouver, BC, Canada) were thawed and rested overnight at 37 °C in 5% CO2. NSG mice were conditioned with 2 Gy of total body irradiation from a 137Cs source, then rested for 4 h. PBMCs were washed with sterile PBS and 10 × 106 cells in 150 μL of sterile PBS were delivered to NSG mice via the tail vein.
2.6. iPSC-MSC administration
iPSC-MSCs were thawed at 37 °C, washed in sterile PBS, 2 × 106 cells/mL were resuspended in 150 μL sterile PBS. For single- and dual-dose treatments, 2 × 106 iPSC-MSCs were administered into NSG mice via tail vein injection on day +14 only, or on days +14 and +18, respectively, after GvHD induction.
2.7. Biomarker analysis
BM, spleen, and peripheral blood were collected on day +19 to determine percent PBMCs [positive human CD45 cells % / (positive human CD45 cells % + positive mouse CD45 cells %)] and infiltration of human CD4 and CD8 T cells. Human CD4 and CD8 T cells were stained with antibodies specific for CD25, pPKCθ (Thr538), NOTCH1, and T-BET. Data were acquired on a BD LSR Fortessa Flow Cytometer (Becton Dickinson) and analyzed using FACSDiva Software (version 8.0, Becton Dickinson) and FlowJo (version 10.0, Treestar).
2.8. LEGENDPlex™ Bead-based immunoassay
Peripheral blood for cytokine analysis was obtained on day +19 from animals via cardiac puncture, immediately following humane euthanasia. The LEGENDPlex™ Human Th1/Th2 panel (8-plex; BioLegend) was used to determine IFNγ. Data were acquired on a BD LSR Fortessa Flow Cytometer (Becton Dickinson) and analyzed using LEGENDPlex™ Software, Version 7.0 (BioLegend).
2.9. Protein subcellular localization
BM, spleen, and peripheral blood were collected on day +19. Single cell suspensions were prepared and surface stained for CD4 and CD8 T cells. Samples were fixed and permeabilized using the Foxp3 Staining Buffer Kit (BD Biosciences) and stained with fluorescently-conjugated antibodies specific for pPKCθ (Thr538), NOTCH1, and T-BET. Nuclei were stained using cell-permeable DRAQ5™ Fluorescent Probe (ThermoFisher Scientific). Cells were visualized and quantified using an ImageStream®X Mark II Imaging Flow Cytometer (EMD Millipore, Billerica, MA). Subcellular localization of pPKCθ (Thr538), NOTCH1, and T-BET were determined using the Nuclear Localization Wizard, IDEAS® Software, upon masking of nuclear and non-nuclear regions to quantify proteins localized in and out of the nucleus.
2.10. Statistical analyses
Data are the mean ± SEM; all in vitro experiments were repeated at least three times. Unpaired, two-tailed Student’s t-test using (Prism5; GraphPad Software, San Diego, CA) was used for statistical comparison of two groups, with Welch’s correction applied when variances were significantly different. Two-way ANOVA (Prism 5; GraphPad Software) was used for the comparison of variables, which are influenced by two different categories and followed by Bonferroni post-test. For in vivo experiments, survival benefit was determined using Kaplan–Meier analysis with an applied log-rank test. P values of ≤0.05 were considered significantly different.
3. Results
3.1. iPSC-derived MSCs phenotypically resemble native MSCs, respond to IFNγ licensing, and dampen PBMC activation potential
MSCs are identified by a constellation of criteria: adherence to the tissue culture dish, tri-lineage differentiation potential, and expression of several distinct surface markers together with the absence of others (Dominici et al. 2008). MSCs also acquire immunosuppressive functions following exposure to pro-inflammatory cytokines, such as interferon gamma (IFNγ); a process referred to as IFNγ-licensing.
Cymerus™ iPSC-MSCs were derived from CD34-enriched peripheral blood mononuclear cells using an episomal plasmid-based, transgene-free, viral-free, feeder-layer-free process, prior to differentiating and expanding in culture (Supplemental Fig. S1). iPSCs are pluripotent and possess indefinite growth potential. This characteristic makes them especially attractive for in vitro expansion, without undergoing senescence, prior to differentiation. However, this intrinsic growth potential may also predispose these cells to genetic instability and putative tumor formation. Therefore, we examined the genetic stability of the iPSC-MSCs by karyotyping. As shown in Supplemental Fig. S2, we confirmed that the iPSC-MSCs we generated for use in this study are genetically stable, with no clonal abnormalities detected at the applied band resolution of 400–450 bands.
We further characterized the fully-differentiated iPSC-MSCs by examining their surface markers and whether their expression changed after exposure to IFNγ. We found that molecules expressed on iPSC-MSCs are consistent with an MSC phenotype (Supplemental Fig. S3 A–I). iPSC-MSCs exhibit tri-lineage differentiation (Supplemental Fig. S4 A–C), also in agreement with their characterization as MSCs (Rebelatto et al. 2008). We assessed post-thaw senescence of iPSC-MSCs, because replicative senescence in cryopreserved cells, following ex vivo expansion, may reduce in vivo potency (Galipeau 2013; Turinetto et al. 2016; de Witte et al. 2017). Our results suggest that up to one week following cell thawing, iPSC-MSCs cultured in vitro do not exhibit signs of functional senescence, as measured by β-galactosidase staining (Supplemental Fig. S4 D).
In response to IFNγ exposure, native MSCs can acquire immunosuppressive capabilities. MSCs use two well-characterized mechanisms to curtail immune cell activation: through the Programmed Cell Death Protein (PD)-1-Programmed Cell Death Protein-Ligand (PD-L)1 signaling axis (Yan et al. 2014) and through immune-modulating indoleamine 2,3-dioxygenase (IDO; Shi et al. 2010). MSCs licensed by IFNγ upregulate and can secrete soluble PD-L1, an immune checkpoint inhibitor (Davies et al. 2017). We found that iPSC-MSCs express moderate amounts of PD-L1 that were further increased in response to IFNγ (Fig. 1A). At the protein level, intracellular IDO in IFNγ-licensed iPSC-MSCs, increased approximately 3-fold over baseline expression after 24 h of exposure to IFNγ, and nearly 5-fold after 48 h in culture with IFNγ (Fig. 1B). We noted very high IDO transcript levels in iPSC-MSCs cultured in the presence of IFNγ for 24 h, which increased further after 48 h of IFNγ exposure (Fig. 1C).
Fig. 1.
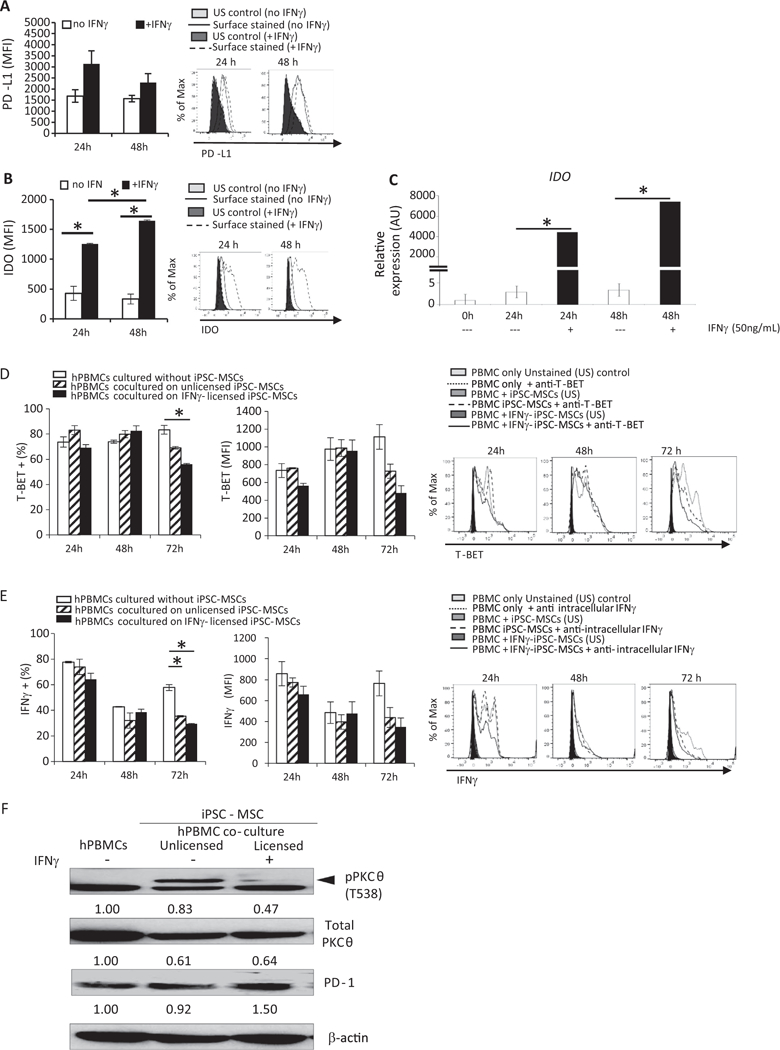
iPSC-derived MSCs phenotypically resemble native MSCs, respond to IFNγ licensing, and dampen PBMC activation potential. iPSC-MSCs were plated as described; IFNγ was added to some cells. 24 or 48 h later, cells were harvested and stained with antibodies specific for (A) PD-L1 or (B) IDO. Cells were permeabilized prior to IDO staining. Data were acquired on a BD LSR Fortessa Flow Cytometer and analyzed using FlowJo software. For (C) IDO gene expression, cells were harvested after 24 or 48 h of culture with or without IFNγ.
Total RNA was reverse transcribed, and IDO expression determined by quantitative real-time PCR using specific forward and reverse primers. For co-culture experiments, stimulated PBMCs were added to iPSC-MSCs and cultured an additional 24, 48, or 72 h. PBMCs were harvested and stained with antibodies specific for (D) T-BET. Percent positive cells and amount of protein expressed, indicated by median fluorescence intensity (MFI), was determined using flow cytometry. For some cultures, golgi plug was added during the last 6 h and (E) IFNγ levels were determined by intracellular staining and flow cytometric analysis. iPSC-MSCs were cultured without or with INFγ for 48 h. Stimulated PBMCs were added to iPSC-MSCs and cultured an additional 96 h. Expression of (F) pPKCθ was determined by immunoblotting. Loading control was β-actin. Data are the mean + SEM of three independent experiments or, for immunoblotting, are representative of two independent replicates that showed similar results. *P < .05; unpaired Student’s t-test.
We next evaluated what effects iPSC-MSCs have on human T cell proliferation and differentiation potential in vitro. We determined the suppressive capacity of the iPSC-MSCs using a previously-described immunopotency assay (Bloom et al. 2015). A normalized immunopotency assay value (IPAv) was calculated by dividing the IPAv of each sample by the IPAv of the reference standard. This allowed us to compare the level of suppression conveyed by each sample relative to the reference standard. The IPAv for products used in this study (Supplemental Table S1) indicates that iPSC-MSCs provide a modest suppressive effect on CD4 T cell proliferation during in vitro co-culture.
After antigenic stimulation, human PBMCs upregulate signaling molecules that further facilitate their activation and differentiation potential. These include the high-affinity subunit of the IL-2 receptor, CD25, the transmembrane receptor, NOTCH1, the master transcriptional regulator of T helper type1 (Th1) cells, T-BET (T-box expressed in T cells), and the pro-inflammatory cytokine, IFNγ (Osborne and Minter 2007; Minter et al. 2005). Compared to PBMCs cultured alone, coculturing PBMCs with iPSC-MSCs significantly reduced the percentage of cells expressing T-BET (Fig. 1D) and IFNγ (Fig. 1E), suggesting iPSC-MSCs reduce the potential of activated T cells to adopt a Th1 cell fate. The amount of T-BET or IFNγ produced on a per cell basis was also lower in PBMCs co-cultured with iPSC-MSCs (Fig. 1D, E, respectively), although this downward trend did not reach statistical significance during the short culture period. Co-culturing PBMCs with iPSC-MSCs did not alter expression of CD25 or of NOTCH1 (Supplemental Fig. S5, A and B, respectively), although NOTCH1 levels also decreased by 72 h in co-culture.
The T cell-specific kinase, PKCθ, functions within a signal-amplifying cascade, to fully activate T cells (Isakov and Altman 2012) and promote tissue destruction in GvHD (Valenzuela et al. 2009). To assess iPSC-MSC influence on PKCθ phosphorylation, we asked whether co-culturing iPSC-MSCs with PBMCs altered pPKCθ expression. iPSC-MSCs were left unlicensed or licensed with IFNγ for 48 h then co-cultured with stimulated PBMCs for an additional 96 h. We observed reduced expression of pPKCθ in PBMCs co-cultured with IFNγ-licensed, but not with unlicensed, iPSC-MSCs (Fig. 1 F). In parallel, PD-1 levels also increased following co-culture with licensed, but not unlicensed iPSC-MSCs.
Collectively, these data show iPSC-MSCs phenotypically resemble native MSCs derived from BM and respond to IFNγ exposure in ways that are consistent with those of native MSCs, cultured under similar conditions. Furthermore, co-culturing with IFNγ-licensed iPSC-MSCs dampened the immune response of PBMCs, as measured by proliferation, expression of well-described activation and differentiation markers, and reduction of pro-inflammatory IFNγ, supporting the notion that iPSC-MSCs exert functional, immune-modulating actions, in vitro.
3.2. iPSC-MSC administration, in vivo, reduces cytokine production and weight loss in mice with GvHD
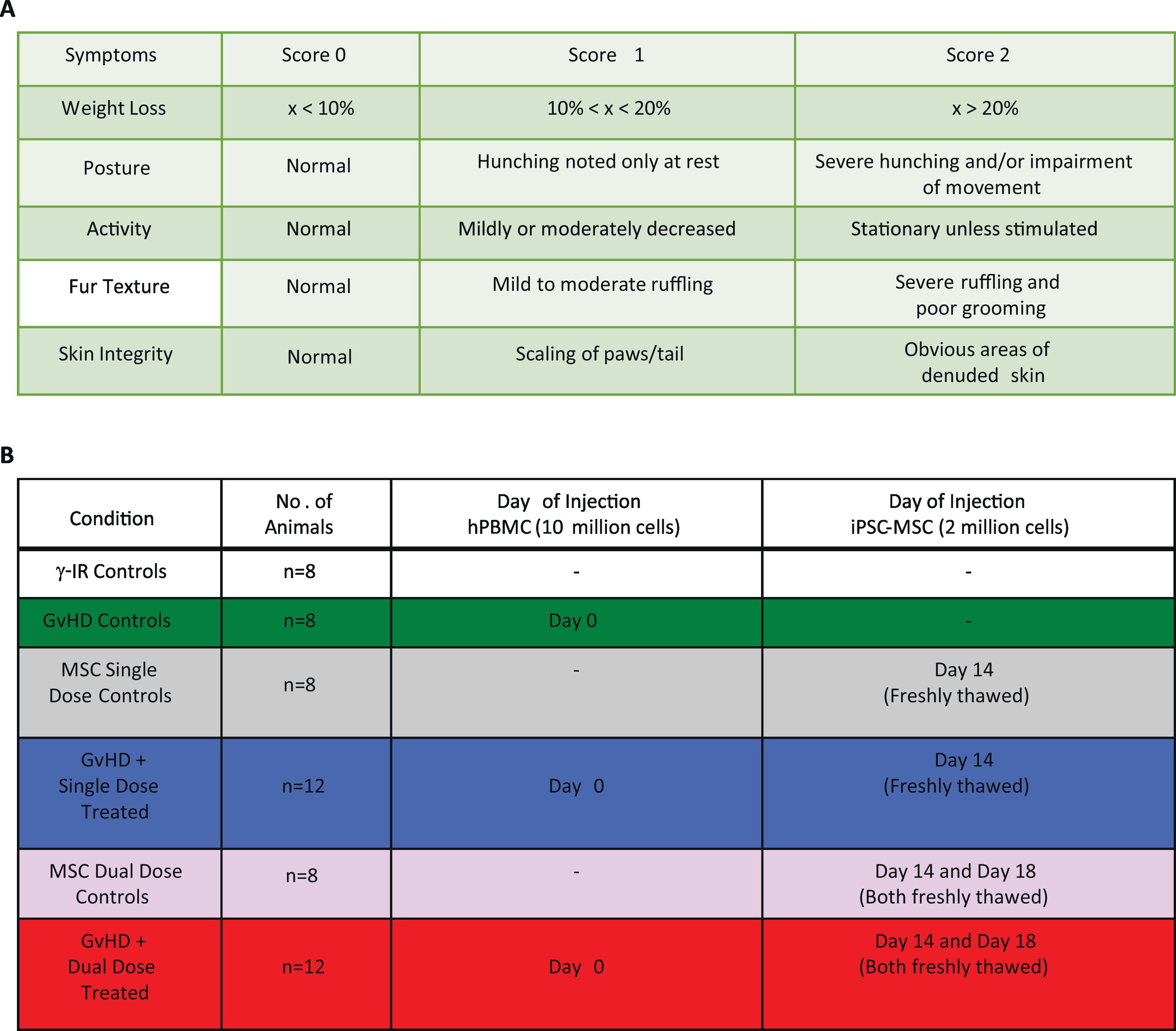
Humanized mouse models have been used to evaluate the efficacy of MSC therapy to diminish disease severity (Roemeling-van Rhijn et al. 2013). We utilized a lymphocyte transfer model of GvHD (Ozay et al. 2016), in which human PBMCs are transferred into transgenic NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice lacking T, B and NK cells, to investigate the therapeutic benefit of giving single or dual doses of iPSC-MSCs, under clinically-relevant conditions (Supplemental Fig. S6).
Before defining the in vivo therapeutic activity of iPSC-MSCs, we first verified that transferred PBMCs expanded to the same extent in iPSC-MSC-treated animals as they did in untreated GvHD controls. We found no differences in percentages of circulating PBMCs collected from untreated mice and from mice that received single- or dual-dose treatments of iPSC-MSCs (Fig. 2A). Furthermore, percentages of circulating CD4 and CD8 T cells also did not differ significantly between treated and untreated animals (Fig. 2, B and C, respectively). These data are consistent with in vitro suppression data which showed only modest effects in co-culture (Supplemental Table S1), and with a previous report using a similar mouse model, and which demonstrated the protective effects afforded by MSCs are not due to accelerated elimination of disease-inducing PBMCs (Tobin et al. 2013). We also confirmed that Cymerus iPSC-MSCs traffick, in vivo, in a manner consistent with BM-derived MSCs and what has been reported in the literature (Rustad and Gurtner 2012; Supplemental Fig. S7A–F). We labeled iPSC-MSCs and BM-derived MSCs with 19F or Alexa 488, administered both low (8 × 105) and high (3 × 106) doses of cells via injection through the tail vein, and assessed their biodistribution 24 h later using Nuclear Magnetic Resonance and immunofluorescence approaches. iPSC-MSCs and BM-derived MSCs migrated first to the lungs, then to the liver, although iPSC-MSCs appeared to exit from the lungs and infiltrate the liver with somewhat slower kinetics, compared to BM-derived MSCs (Supplemental Fig. S7A, B). We used histological and immunofluorescent staining of lung sections to visualize iPSC-MSCs as single cells (Alexa488-positive; Supplemental Fig. S7C, D, F), or as larger clusters of cells in which both Alexa488 and the human Ku80-specific antibody, STEM101, could be co-detected (Supplemental Fig. S7C, E, F). No MSCs were detected in animals beyond 7 days (data not shown), also consistent with previous reports (Sensebé and Fleury-Cappellesso 2013). Altogether, these data suggest that Cymerus iPSC-MSCs and BM-derived MSCs traffick and persist similarly in vivo, following intravascular administration.
Fig. 2.

iPSC-MSC administration, in vivo, reduces cytokine production and weight loss in mice with GvHD. The percent of (A) human CD45 (B) human CD4 and (C) human CD8 positive PBMCs, was determined by flow cytometric analysis of peripheral blood samples from single- (n = 8) and dual-dose control (n = 8), GvHD control (n = 12), single- (n = 12) and dual-dose-treated mice (n = 12) harvested on day +19 after disease induction. (D) Circulating IFNγ was measured for cohorts of mice described in A, using standard ELISA techniques. (E) The percent weight change for cohorts of mice described in A was also determined. Data are the mean + SEM. *P ≤.05, **P < .01, ***P < .001; unpaired Student’s t-test.
Pro-inflammatory cytokines released into the circulation during GvHD can cause significant weight loss in patients (Pajak et al. 2008). In the pre-clinical model used here, high levels of circulating IFNγ correlate closely with disease severity (Ozay et al. 2016). We monitored IFNγ levels, as well as changes in weight on day +19, for mice that were left untreated or were treated with single- or dual-doses of iPSC-MSCs. We observed significantly lower IFNγ in mice that received dual-doses of iPSC-MSCs, compared to untreated and to single-dose treated mice (Fig. 2D). Reduced plasma cytokines correlated with significant relief from the cachexia-associated weight loss typically observed in this Th1-mediated model (Fig. 2E). Although administering single or dual doses of iPSC-MSCs did not affect percentages of circulating PBMCs, compared to untreated GvHD control mice, the in vivo capacity of transferred PBMCs to produce proinflammatory cytokines was greatly diminished in mice that received dual doses of iPSC-MSCs.
3.3. iPSC-MSC administration reduces BM-infiltration and expression of proinflammatory molecules in mice with GvHD
The mechanisms MSCs utilize to attenuate GvHD have not been fully elucidated, but reports suggest they influence immune cell activation and trafficking to target organs (Li et al. 2014). The in vivo efficacy of human BM-derived MSCs varies greatly, both by donor and by in vitro expansion protocols. For these reasons, making a direct and useful treatment comparison between Cymerus iPSC-MSCs and donor-derived native MSCs is both challenging and arbitrary. Therefore, we induced GvHD in lightly-irradiated NSG mice, harvested blood and tissue samples +19 days after GvHD-induction, at the peak of disease, and evaluated target-tissue infiltration and markers of immune activation in untreated and in iPSC-MSC-treated mice. When we examined the BM, the major target of immune destruction in this model, we noted the percentages of BM-infiltrating PBMCs were significantly lower in mice given iPSC-MSCs, regardless of whether they received single or dual doses, compared to untreated controls (Fig. 3A). We detected decreased BM-infiltration of CD4 (Fig. 3B) and CD8 T cells (Fig. 3C,) which corresponded to higher total BM cellularity in iPSC-MSC-treated mice, compared to untreated GvHD control animals (Fig. 3D). In contrast, percentages of human T cells in the spleen were less affected by iPSC-MSC-treatment (Supplemental Fig. S8 A–C). Thus, one means by which iPSC-MSCs may function in vivo in this model, is by protecting the BM from immune-mediated destruction caused by infiltrating PBMCs.
Fig. 3.
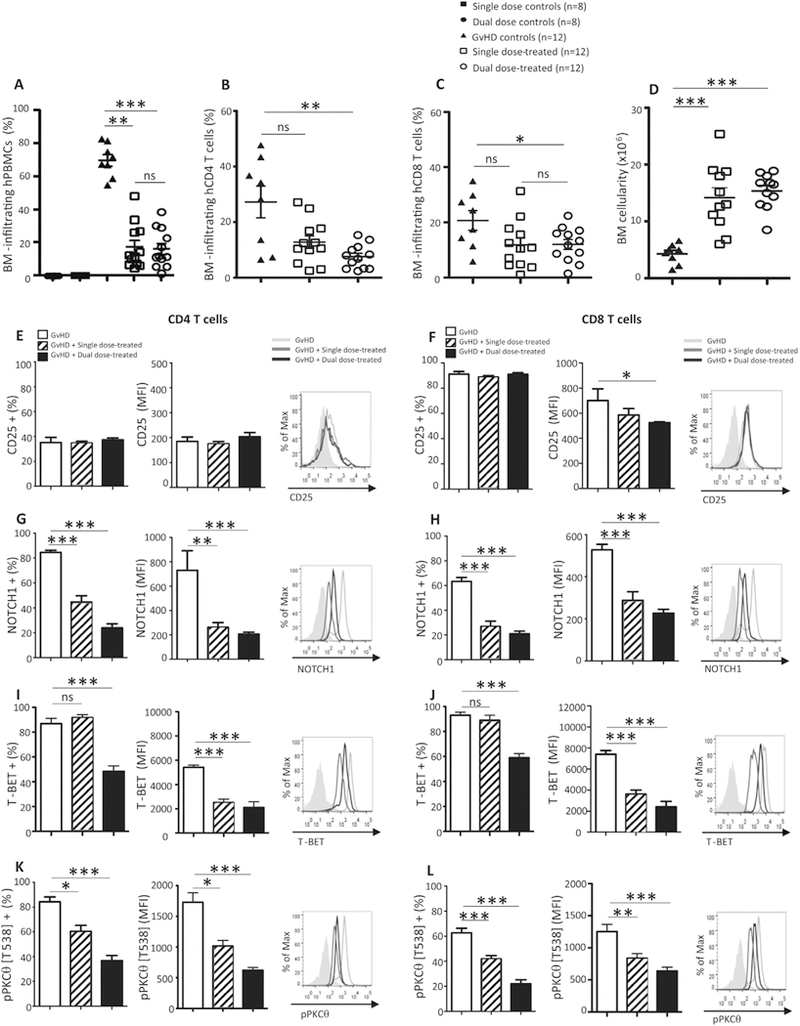
iPSC-MSC administration reduces BM-infiltration and expression of proinflammatory molecules in mice with GvHD. The percent of (A) total human PBMCs, (B) human CD4 and (C) human CD8 T cells infiltrating the BM was determined by flow cytometric analysis of BM samples from single-(n = 8) and dual-dose control (n = 8), GvHD control (n = 12), single- (n = 12) and dual-dose-treated mice (n = 12) harvested on day +19 after disease induction. (D) Total BM cellularity was assessed for each of the cohorts of mice described. Flow cytometry was used to determine expression of (E, F) CD25, (G, H) NOTCH1, (I, J) T-BET, and (K, L) pPKCθ for populations of human CD4 (E, G, I, K, respectively) and human CD8 T cells (F, H, J, L, respectively) retrieved from the BM of untreated and treated mice with GvHD. Data are the mean + SEM. *P ≤ .05, **P < .01, ***P < .001; unpaired Student’s t-test.
The lethal GvHD induced in this model is driven by CD4 Th1 and cytolytic CD8 T cells. We used flow cytometry to measure the expression of CD25, NOTCH1, and T-BET, to ask whether iPSC-MSC treatment attenuated this Th1-mediated response, in vivo. Except for CD25 expression on BM-infiltrating CD4 T cells, which did not differ between treatments (Fig. 3E), there was significantly lower expression of all the pro-inflammatory markers evaluated on day +19. Compared to the levels expressed in untreated GvHD control animals, CD25 expression was lower on CD8 T cells recovered from the BM of iPSC-MSC-treated animals (Fig. 3F). NOTCH1 (Fig. 3, G and H) and T-BET (Fig. 3, I and J), both of which are upregulated in Th1-mediated diseases (Minter et al. 2005; Roderick et al. 2013), were also significantly decreased in BM CD4 and CD8 T cells from mice treated with single- or dual-dose regimens of iPSC-MSCs. Consistent with our in vitro co-culture results, we observed that treating animals with single- or dual-doses of iPSC-MSCs also significantly reduced expression of pPKCθ in CD4 and CD8 T cells recovered from the BM (Fig. 3, K and L; respectively).
Collectively, our data suggest iPSC-MSCs decrease the activation and infiltration of pro-inflammatory CD4 and CD8 T cells in the BM. We conclude that the reduced expression on PBMCs of multiple markers of activation, coupled with the low level of PKCθ phosphorylation, in BM-infiltrating CD4 and CD8 T cells, result from immune modulation mediated by iPSC-MSCs.
3.4. iPSC-MSCs attenuate GvHD severity and prolong survival in mice
We assessed disease severity in our humanized model of GvHD, using a standardized scoring system (Ozay et al. 2016), after randomly assigning mice to one of six treatment cohorts (Supplemental Fig. S9 A, B). Mice were humanely euthanized when they reached a cumulative score of “8” which was recorded as the day of lethal GvHD induction. We determined the survival benefit of administering iPSC-MSCs using Kaplan–Meier analyses with an applied log-rank test, with a P value of < 0.05 considered significantly different.
Administering iPSC-MSCs significantly attenuated disease symptoms, compared to untreated mice (Fig. 4A), and we noted further significant differences in clinical scores between single- and dual-dose treatments when we evaluated mice on days +24 and + 25 post-GvHD-induction. During survival studies, single- and dual-dose treatments conferred significant survival benefits over untreated GvHD controls (P < .0001; Fig. 4B). Animals receiving dual-dose regimens survived slightly longer (median = 57 days, range = 31–82 days) than single-dose-treated animals (median = 48 days, range = 33–60 days), although this difference was not statistically significant (P = .0715). Utilizing this pre-clinical model of GvHD, we conclude that administering single or dual doses of iPSC-MSCs, under clinically relevant conditions, significantly attenuates disease severity and conveys a robust survival benefit.
Fig. 4.
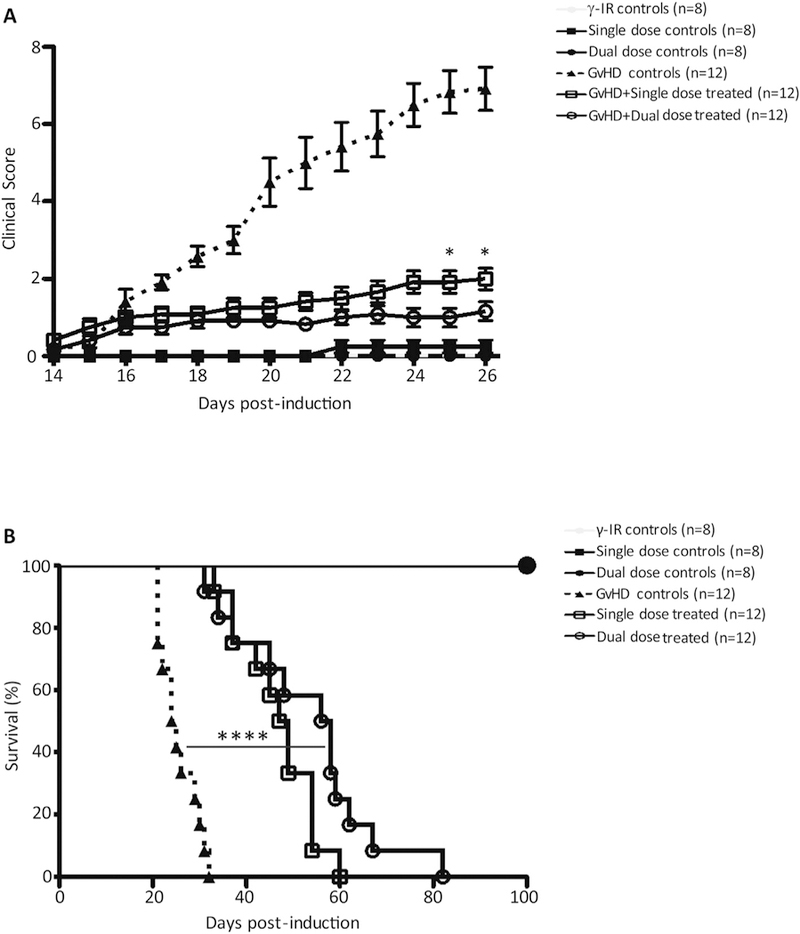
iPSC-MSCs attenuate GvHD severity and prolong survival in mice. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were conditioned with 2Gy of γ-irradiation from a 137Cs source. Control mice received only γ-irradiation (n = 8), a single (n = 8), or a dual dose of iPSC-MSCs (n = 8) without GvHD induction. Four hours after irradiation, GvHD was induced and mice were stratified into GvHD control, single- and dual-dose groups. The GvHD control group received no treatment (n = 12). For single-dose treatment (n = 12), mice received 2 × 106 freshly-thawed iPSC-MSCs on day +14 after induction; for dual-dose treatment (n = 12), mice received 2 × 106 freshly-thawed iPSC-MSCs on day +14 and on day +18 after induction. Clinical scores were generated based on standard criteria and mice were removed from the study when they reached a total score of “8”. (A) Cumulative scores of mice in all treatment groups for the first 26 days of the study are shown. (B) Survival of mice with GvHD left untreated or treated was followed until they reached a score of “8” and were removed from the study. Significant differences between clinical scores were determined using two-way ANOVA and followed by Bonferroni post-test. Survival benefit of iPSC-MSC treatments was determined using Kaplan–Meier analysis with an applied log-rank test. *P ≤.05, ****P < .0001.
3.5. iPSC-MSC-treatment alters subcellular localization of pPKCθ in BM-infiltrating T cells
One means by which protein activity can be regulated is through changes in subcellular compartmentalization (Bauer et al. 2015). Dynamic redistribution of proteins between the cytosol and the nucleus may increase, decrease, or completely alter protein-protein interactions to differentially modulate biological outcomes (Cyert 2001). Indeed, a nuclear role for PKCθ has also been reported in human CD4 T cells, functioning as part of a complex that facilitates transcription of proinflammatory genes, including IFNG (Sutcliffe et al. 2011). To further understand, on a molecular level, the effects of treating mice with iPSC-MSCs, we asked whether the nuclear localization of pro-inflammatory proteins was altered following iPSC-MSC administration.
Using imaging flow cytometry, we analyzed the subcellular distribution of NOTCH1, T-BET, and pPKCθ, in BM-infiltrating CD4 and CD8 T cells from animals induced with GvHD and given single or dual doses of iPSC-MSC. Nuclear expression of NOTCH1 and T-BET in CD4 and CD8 T cells recovered from the BM was not significantly altered by iPSC-MSC treatment (Supplemental Fig. S10, A–D). In contrast, BM samples collected on day +19 from untreated animals with GvHD contained high percentages of nuclear pPKCθ-expressing CD4 and CD8 T cells. This nuclear localization of pPKCθ was confirmed by high positive nuclear similarity scores (Fig. 5). However, the percentages of cells expressing nuclear pPKCθ were significantly lower, both in CD4 and CD8 T cells, in BM samples from mice treated with iPSC-MSCs (Fig. 5, A, B, D, E). Furthermore, the amount of pPKCθ detected in the nucleus, was also significantly reduced following treatment with iPSC-MSCs, both in CD4 and CD8 T cells (Fig. 5, C and F).
Fig. 5.

iPSC-MSC-treatment alters subcellular localization of pPKCθ in BM-infiltrating T cells. BM samples were collected on day +19 and single cell suspensions prepared. Cells were surface-stained for CD4 and CD8 expression with fluorescently-conjugated antibodies, fixed, permeabilized and stained intracellularly with fluorescently-conjugated antibodies specific for pPKCθ (Thr538). Nuclei were stained using cell-permeable DRAQ5™ fluorescent probe. Cells were visualized, nuclear pPKCθ was quantified, and nuclear similarity score was determined using an ImageStream®X Mark II Imaging Flow Cytometer for BM-infiltrating (A–C) CD4 and (D–F) CD8 T cells. Subcellular localization of pPKCθ was determined using the Nuclear Localization Wizard and IDEAS® Software following masking of nuclear and non-nuclear regions. Images are representative of 12 mice analyzed for each cohort. Data are the mean + SEM. *P ≤ .05, **P < .01, ***P < .001; unpaired Student’s t-test.
Altogether, these data indicate that, by day +19, a high percentage of T cells found in the BM express elevated levels of nuclear pPKCθ. Moreover, treating mice with iPSC-MSCs reduced the infiltration of nuclear pPKCθ-expressing T cells and, further, acted to diminish the amount nuclear pPKCθ within these CD4 and CD8 T cells.
3.6. Pro-inflammatory molecules expressed by circulating PBMCs correlate with therapeutic response to iPSC-MSC administration
A minimally-invasive means to identify biomarkers to predict or monitor therapeutic responses to iPSC-MSC administration would be of great clinical value. Therefore, we asked whether utilizing flow cytometry to analyze expression of pro-inflammatory markers on peripheral blood CD4 and CD8 T cells, found to be significantly different on BM-infiltrating immune cells, also correlated with disease severity. We measured expression of CD25 (Fig. 6A, B), NOTCH1 (Fig. 6C, D), T-BET (Fig. 6E, F), and pPKCθ (Fig. 6G, H) in circulating PBMCs from untreated and iPSC-MSC-treated mice, collected on day +19 after disease induction. Compared to PBMCs from untreated mice, the expression of all these pro-inflammatory molecules was significantly reduced as measured by the percent positive cells, the level of the proteins expressed, or both, following therapeutic administration of iPSC-MSCs.
Fig. 6.
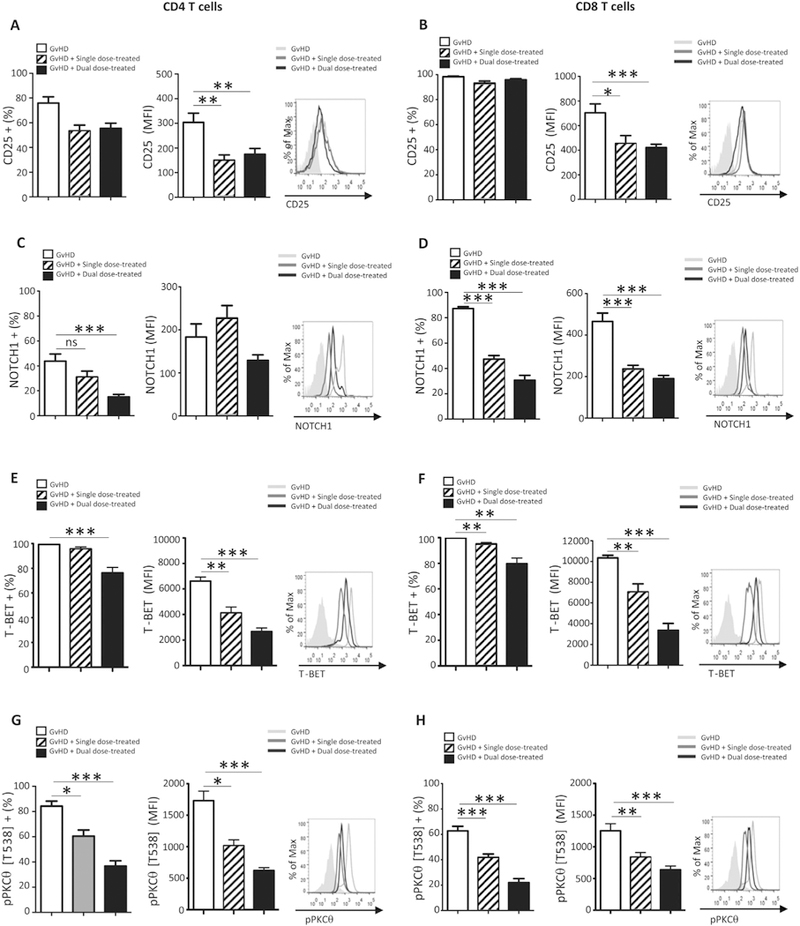
Pro-inflammatory molecules expressed by circulating PBMCs correlate with therapeutic response to iPSC-MSC administration. Peripheral blood samples from GvHD control (n = 12), single- (n = 12) and dual-dose-treated (n = 12) mice were harvested on day +19 after disease induction. Flow cytometry was used to determine expression of (A, B) CD25, (C, D) NOTCH1, (E, F) T-BET, and (G, H) pPKCθ was determined for populations of human CD4 (A, C, E, G, respectively) and human CD8 T cells (B, D, F, H, respectively) in the circulation of untreated and treated mice with GvHD. Data are the mean + SEM. *P ≤.05, **P < .01, ***P < .001; unpaired Student’s t-test.
Collectively, we demonstrate administering Cymerus iPSC-MSCs as a cell-based therapy provides relief from acute symptoms and significantly prolongs survival in a pre-clinical model of GvHD. Our data suggest iPSC-MSCs modulate PKCθ phosphorylation and subcellular localization. Furthermore, flow cytometric analysis of pro-inflammatory molecules may provide a set of biomarkers on circulating PBMCs that correlate closely with therapeutic response to iPSC-MSC administration.
4. Discussion
Since the first experimental use of MSCs to successfully treat steroid-resistant GvHD (Le Blanc et al. 2004) administering MSCs as a cell-based therapy has become an area of intensive investigation. However, inconsistent results in the clinic underscores all we do not yet understand about the precise mechanisms of action, the longevity of MSCs post-transfer, or even what contributes to the vast variability in performance between MSCs derived from different donors (Galipeau 2013), all of which make a meaningful, direct comparison between Cymerus iPSC-MSCs and human BM-derived native MSCs difficult, if not impossible. While there are some indications cryopreserving MSCs adversely affects their immunosuppressive capacity (Pollock et al. 2015; François et al. 2012), other reports suggest MSCs can undergo multiple passages or repeated freezing without exhibiting any diminished function (Mamidi et al. 2012). Extensive ex vivo expansion has been linked to telomere shortening in cultured MSCs (Bernardo et al. 2012), but whether this contributes to functional senescence and impaired efficacy, in vivo, is not known. Additionally, in vitro studies indicate senescence can be reversed by priming with IFNγ (Chinnadurai et al. 2017), but further studies are required to determine whether this phenomenon also occurs in vivo. One means to bypassing replicative senescence in MSCs may be to derive them from iPSCs (Sabapathy and Kumar, 2016). We show that iPSC-MSCs can be generated and expanded using a standardized protocol and Good Manufacturing Proce-dures, maintain their genetic stability, and display low levels of senescence immediately after thawing, which decreased over 48 h in culture, even without the addition of IFNγ. Furthermore, IFNγ licensing significantly increased IDO expression in iPSC-MSCs, consistent with the findings that IDO-mediated suppression is an important mechanism of immune modulation by MSCs (Chinnadurai et al. 2017). While we could not track the senescence of transferred iPSC-MSCs in vivo, the significant survival benefit noted following single- or dual-dose administration, indicate that iPSC-MSCs function for a sufficient length of time after infusion, to produce a durable immunomodulatory response.
Donor T cells traffic to secondary lymphoid organs within 24 h after HSCT (Anderson et al. 2008). Following antigenic stimulation there, alloreactive CD4 and CD8 T cells expand and migrate to target tissues. In the humanized model of GvHD used in this study, the BM is the primary target organ, and mice die of lethal BM failure approximately three weeks after PBMC infusion. The accumulation of activated T cells in the BM of untreated mice, and the pro-inflammatory markers they express, are consistent with a Th1-mediated immune response, and one that will also drive effector functions of CD8 cytolytic T cells. This includes increased CD25 expression, upregulation of NOTCH1 and T-BET, and production of IFNγ, as well as sustained phosphorylation of the T cell-specific kinase, PKCθ, which is recruited to membrane lipid rafts following co-stimulation through the T cell receptor and CD28 (Kong et al. 2011). The high-affinity IL-2 receptor, CD25, is upregulated in activated T cells in a NOTCH1-dependent manner (Palaga et al. 2003; Adler et al. 2003). Furthermore, physical association of NOTCH1 with PKCθ aids in assembly of the Carma1-BCL10-MALT1 supramolecular signaling complex in CD4 T cells, a prerequisite to activating the NF-κΒ (nuclear factor-κB) transcriptional complex (Shin et al. 2014).
PKCθ is one of nine members of the PKC family. It is required for mediating immune destruction in GvHD, identifying it as an attractive therapeutic target (Zhang et al. 2013). However, close structural homology between PKCs make designing small molecule inhibitors to single isoforms challenging (Xu et al. 2004). Long-believed to be a cytosolic-resident kinase, a seminal study by Sutcliffe et al. (2011) revealed a nuclear function for PKCθ in CD4 T cells, including positively regulating transcription of pro-inflammatory genes such as IFNG. We utilized imaging flow cytometry to evaluate the cellular localization of pPKCθ in BM-infiltrating CD4 and CD8 T cells. We found nuclear pPKCθ highly expressed in > 80% of CD4 and CD8 T cells in the BM of untreated mice. By contrast, both the percentage of cells expressing nuclear pPKCθ, as well as the quantity of nuclear pPKCθ detected, was significantly reduced in CD4 and CD8 T cells collected from the BM of dual dose-treated animals. MSCs migrate to sites of inflammation, where the local concentration of IFNγ or TNF may be sufficient to license their immunomodulatory functions. iPSC-MSCs may be similarly recruited to the BM in this model of GvHD and exert immune suppressive effects on infiltrating T cells by reducing nuclear pPKCθ expression.
Alternatively, our data show that iPSC-MSCs may exit the lungs with slower kinetics than BM-derived MSCs. Recent reports suggest that interactions between MSCs and lung macrophages can modulate macrophage polarization, promoting an anti-inflammatory M2 phenotype, over the pro-inflammatory M1 state. This redirection of macrophage polarization may occur either as a result of macrophages directly engulfing MSCs (Braza et al. 2016) or through the paracrine effects of anti-inflammatory components secreted by the MSCs themselves (Morrison et al., 2017). Macrophages are known to migrate to sites of inflammation. Therefore, it is possible that increased iPSC-MSC residence in the lungs may facilitate M2 polarization of lung macrophages, which then migrate to and infiltrate the BM, thus dampening immune destruction. However, further experimentation will be necessary to test this hypothesis.
Mechanistically, it is not entirely clear how iPSC-MSCs affect cytosolic versus nuclear distribution of pPKCθ. Surface expression of PD-L1 on iPSC-MSCs has the potential to interact with its cognate receptor PD-1, on activated CD4 and CD8 T cells. This interaction negatively regulates T cell activity by recruiting the phosphatase, SHP2, to surface micro-aggregates (Yokosuka et al. 2012). Furthermore, it was recently demonstrated that SHP2 preferentially dephosphorylates CD28, within T cell receptor-CD28 signaling clusters (Hui et al. 2017). Given that PKCθ functions downstream of CD28 engagement (Huang et al. 2002), one means by which iPSC-MSCs may downregulate pPKCθ activity is through diminished CD28 signaling.
The immune modulator, IDO, also suppresses effector T cell functions, acting as the rate-limiting enzyme in the tryptophan catabolism pathway. IDO upregulation can suppress local tissue destruction during active GvHD (Jasperson et al. 2008). Mechanistically, limiting tryptophan availability has been shown to negatively regulate the AKT-mTOR pathway, as well as PKCθ phosphorylation (Metz et al. 2012). Gluco-kinase1 (GLK1), an amino acid-sensing molecule, phosphorylates PKCθ on threonine residue 538 following stimulation through the T cell receptor (Chuang et al. 2011). In the absence of tryptophan, its kinase activity is limited, potentially impacting PKCθ phosphorylation. We noted robust expression of IDO by unlicensed iPSC-MSCs. This was further increased following exposure to IFNγ and co-culturing PBMCs with licensed iPSC-MSCs reduced PKCθ phosphorylation. Although numerous reports have correlated high IDO expression with potent MSC-mediated immune suppression, at least one report suggests that immune suppression can proceed in an IDO-independent manner, through PD-L1-PD1 signaling (Chinnadurai et al. 2014). Therefore, a distinct, or perhaps redundant, means by which licensed iPSC-MSCs may act to suppress T cells is through IDO-mediated regulation of tryptophan availability.
The benefits of using MSCs as a therapeutic modality are being explored for a variety of conditions, including end-stage chronic heart disease (Butler et al., 2017), ischemic stroke (Toyoshima et al. 2017), acute myocardial infarct (Kanelidis et al. 2017), kidney failure (Urt-Filho et al. 2016), and critical limb ischemia (Liew and O’Brien, 2012). Developing an efficacious MSC product to treat GvHD has been an important industry focus for several years. Products such as MSC-100-IV™ (Mesoblast, LLC), its Japanese-approved counterpart, Temcell® (JCR Pharmaceutical Co.; Locatelli et al. 2017), and Multistem® (Athersys, Inc.; Maziarz et al. 2015) are being evaluated in clinical trials. Common denominators for these products are that adult MSCs are collected from donors, pooled and expanded in culture, then cryopreserved until use. Cymerus iPSC-MSCs are unique in that they are derived from mesenchymoangioblasts, which are in turn derived from iPSCs. In addition to the fact that iPSCs can undergo nearly indefinite ex vivo expansion without losing pluripotency, mesenchymoangioblasts and their progeny also have enormous ex vivo expansion potential: cultures derived from a single mesenchymoangioblast have been shown to accumulate up to 1022 MSCs in total (Vodyanik et al. 2010). Nonetheless, by primarily relying on expansion at the iPSC-level, extensive expansion of Cymerus iPSC-MSCs, post-differentiation, can be avoided. This approach facilitates the production of large numbers of Cymerus iPSC-MSCs from the same starting material, apparently without acquiring functional senescence or diminished immunosuppressive capacity in vivo.
5. Conclusion
A thorough characterization of Cymeryus iPSC-MSCs reveals they mitigate signaling through the PKCθ pathway to attenuate GvHD progression. The exact mechanism by which this occurs will require further experimentation and is currently under active investigation in our lab. Additionally, having the ability to monitor patient response to iPSC-MSCs administration in real time, using a straight-forward, minimally-invasive flow cytometric analysis of pro-inflammatory markers expressed by circulating PBMCs, provides clinicians with valuable feedback upon which to make informed treatment decisions. Altogether, our data strongly support the use of Cymerus iPSC-MSCs as a treatment for GvHD, and its clinical efficacy is currently being studied in the context of a small, first-in-human trial for the treatment of steroid refractory GvHD.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank Dr. Amy Burnside and the Flow Cytometry Core Facility, for use of and assistance with the Amnis ImageStream® imaging flow cytometer and University of Massachusetts Amherst Animal Care staff for excellent care of research animals. This work was performed in association with the Models2Medicine Center, Institute for Applied Life Sciences, University of Massachusetts, Amherst. The authors also thank Waisman Biomanufacturing, Madison, WI, for undertaking the immunopotency and karyotyping assays. HS is supported by an NIH Training Fellowship (T32 GM108556). BAO and LMM are supported by NIH 5P01CA166009; EIO, JV, and LMM were funded, in part, by Cynata Therapeutics, Limited.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scr.2019.101401.
Disclosure of conflicts of interest
EIO, JV, and LMM were supported, in part, by Cynata Therapeutics, Limited, manufacturer of the proprietary iPSC-MSC product, Cymerus™. RAM and KK are employees of Cynata Therapeutics Limited and IIS is a consultant to the company. RAM, IIS, and KK also hold stock in Cynata Therapeutics, Limited. The authors have no additional competing financial interests.
References
- Adler SH, Chiffoleau E, Xu L, Dalton NM, Burg JM, Wells AD, Wolfe MS, Turka LA, Pear WS, 2003. Notch signaling augments T cell responsiveness by enhancing CD25 expression. J. Immunol 171, 2896–2903. [DOI] [PubMed] [Google Scholar]
- Anderson BE, Taylor PA, McNiff JM, Jain D, Demetris AJ, Panoskaltsis-Mortari A, Ager A, Blazar BR, Shlomchik WD, Shlomchik MJ, 2008. Effects of donor T-cell trafficking and priming site on graft-versus-host disease induction by naive and memory phenotype CD4 T cells. Blood 111, 5242–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer NC, Doetsch PW, Corbett AH, 2015. Mechanisms regulating protein localization. Traffic 16, 1039–1061. [DOI] [PubMed] [Google Scholar]
- Bernardo ME, Zaffaroni N, Novara F, Cometa AM, Avanzini MA, Moretta A, Montagna D, Maccario R, Villa R, Daidone MG, Zuffardi O, Locatelli F, 2012. Human BM derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res 67, 9142–9149. [DOI] [PubMed] [Google Scholar]
- Bloom DD, Centanni JM, Bhatia N, Emler CA, Drier D, Leverson GE, McKenna DH Jr., Gee AP, Lindblad R, Hei DJ, Hematti P, 2015. A reproducible immunopotency assay to measure mesenchymal stromal cell-mediated T-cell suppression. Cytotherapy 17, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braza F, Dirou S, Forest V, Sauzeau V, Hassoun D, Chesné J, Cheminant-Muller MAA, Sagan C, Magnan A, Lemarchand P, 2016. Mesenchymal stem cells induce suppressive macrophages through phagocytosis in a mouse model of asthma. Stem Cells 34, 1836–1845. [DOI] [PubMed] [Google Scholar]
- Butler J, Epstein SE, Greene SJ, Quyyumi AA, Sikora S, Kim RJ, Anderson AS, Wilcox JE, Tankovich NI, Lipinski MJ, Ko YA, Margulies KB, Cole RT, Skopicki HA, Gheorghiade M, 2017. Intravenous allogeneic mesenchymal stem cells for nonischemic cardiomyopathy: safety and efficacy results of a Phase II-A randomized trial. Circ. Res 120, 332–340. [DOI] [PubMed] [Google Scholar]
- Chinnadurai R, Copland IB, Patel SR, Galipeau J, 2014. IDO-independent suppression of T cell effector function by IFN-γ-licensed human mesenchymal stromal cells. J. Immunol 192, 1491–1501. [DOI] [PubMed] [Google Scholar]
- Chinnadurai R, Rajan D, Ng S, McCullough K, Arafat D, Waller EK, Anderson LJ, Gibson G, Galipeau J, 2017. Immune dysfunctionality of replicative senescent mesenchymal stromal cells is corrected by IFNγ priming. Blood Adv 1, 628–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HC, Lan JL, Chen DY, Yang CY, Chen YM, Li JP, Huang CY, Liu PE, Wang X, Tan TH, 2011. The kinase GLK controls autoimmunity and NF-κB signaling by activating the kinase PKC-θ in T cells. Nat. Immunol 12, 1113–1118. [DOI] [PubMed] [Google Scholar]
- Cyert MS, 2001. Regulation of nuclear localization during signaling. J. Biol. Chem 276, 20805–20808. [DOI] [PubMed] [Google Scholar]
- Davies LC, Heldring N, Kadri N, Le Blanc K, 2017. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immuno-suppression. Stem Cells 35, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink MR, Velardi E, Perales MA, 2015. Immune reconstitution following stem cell transplantation. Hematol. Am. Soc. Hematol. Educ. Program 215–219. [DOI] [PubMed]
- Dietz AC, Lucchini G, Samarasinghe S, Pulsipher MA, 2016. Evolving hematopoietic stem cell transplantation strategies in severe aplastic anemia. Curr. Opin. Pediatr 28, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop DJ, Horwitz E, 2008. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. [DOI] [PubMed] [Google Scholar]
- Fernández Vallone VB, Romaniuk MA, Choi H, Labovsky V, Otaegui J, Chasseing NA, 2013. Mesenchymal stem cells and their use in therapy: what has been achieved? Differentiation 85, 1–10. [DOI] [PubMed] [Google Scholar]
- François M, Copland IB, Yuan S, Romieu-Mourez R, Waller EK, Galipeau J, 2012. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-γ licensing. Cytotherapy 14, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau J, 2013. The mesenchymal stromal cells dilemma–does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy 15, 2–8. [DOI] [PubMed] [Google Scholar]
- Garnett C, Apperley JF, Pavlů J, 2013. Treatment and management of graft-versushost disease: improving response and survival. Ther. Adv. Hematol 4, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Lo PF, Zal T, Gascoigne NR, Smith BA, Levin SD, Grey HM, 2002. CD28 plays a critical role in the segregation of PKC theta within the immunologic synapse. Proc. Natl. Acad. Sci 99, 9369–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD, 2017. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakov N, Altman A, 2012. PKC-theta-mediated signal delivery from the TCR/CD28 surface receptors. Front. Immunol 3, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH, Blazar BR, 2008. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood 111, 3257–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanelidis AJ, Premer C, Lopez J, Balkan W, Hare JM, 2017. Route of delivery modulates the efficacy of mesenchymal stem cell therapy for myocardial infarction: a meta-analysis of preclinical studies and clinical trials. Circ. Res 120, 1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong KF, Yokosuka T, Canonigo-Balancio AJ, Isakov N, Saito T, Altman A, 2011. A motif in the V3 domain of the kinase PKC-θ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol 12, 1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, Ringdén O, 2004. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 363, 1439–1441. [DOI] [PubMed] [Google Scholar]
- Lei Y, Schaffer DV, 2013. A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc. Natl. Acad. Sci. U. S. A 24 (110), E5039–E5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Jiang YM, Sun YF, et al. , 2014. CCR7 expressing mesenchymal stem cells potently inhibit graft-versus-host disease by spoiling the fourth supplemental Billingham’s tenet. PLoS One 9, e115720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew A, O’Brien T, 2012. Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Res. Ther 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli F, Algeri M, Trevisan V, et al. , 2017. Remestemcel-L for the treatment of graft versus host disease. Expert. Rev. Clin. Immunol 13, 43–56. [DOI] [PubMed] [Google Scholar]
- Mamidi MK, Nathan KG, Singh G, Thrichelvam ST, Mohd Yusof NA, Fakharuzi NA, Zakaria Z, Bhonde R, Das AK, Majumdar AS, 2012. Comparative cellular and molecular analyses of pooled BM multipotent mesenchymal stromal cells during continuous passaging and after successive cryopreservation. J. Cell. Biochem 113, 3153–3164. [DOI] [PubMed] [Google Scholar]
- Maziarz RT, Devos T, Bachier CR, Goldstein SC, Leis JF, Devine SM, Meyers G, Gajewski JL, Maertens J, Deans RJ, Van’t Hof W, Lazarus HM, 2015. Single and multiple dose MultiStem (multipotent adult progenitor cell) therapy prophylaxis of acute graft-versus-host disease in myeloablative allogeneic hematopoietic cell transplantation: a phase 1 trial. Biol. Blood Marrow Transplant 21, 720–728. [DOI] [PubMed] [Google Scholar]
- Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, Link CJ, Prendergast GC, 2012. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncol. Immunol 1, 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG, Cho OH, Palaga T, Gottipati S, Telfer JC, Kostura L, Fauq AH, Simpson K, Such KA, Miele L, Golde TE, Miller SD, Osborne BA, et al. , 2005. Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat. Immunol 6, 680–688. [PubMed] [Google Scholar]
- Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, Krasnodembskaya AD, 2017. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am. J. Respir. Crit. Care Med 196, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne BA, Minter LM, 2007. Notch signalling during peripheral T-cell activation and differentiation. Nat. Rev. Immunol 7, 64–75. [DOI] [PubMed] [Google Scholar]
- Ozay EI, Gonzalez-Perez G, Torres JA, Vijayaraghavan J, Lawlor R, Sherman HL, Garrigan DT, Burnside AS, Osborne BA, Tew GN, Minter LM, 2016. Intracellular delivery of Anti-pPKCθ (Thr538) via protein transduction domain mimics immunomodulation. Mol. Ther 24, 2118–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajak B, Orzechowska S, Pijet B, Pijet M, Pogorzelska A, Gajkowska B, Orzechowski A, 2008. Crossroads of cytokine signaling—the chase to stop muscle cachexia. J. Physiol. Pharmacol 59 (Suppl. 9), 251–264. [PubMed] [Google Scholar]
- Palaga T, Miele L, Golde TE, Osborne BA, 2003. TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J. Immunol 171, 3019–3024. [DOI] [PubMed] [Google Scholar]
- Pollock K, Sumstad D, Kadidlo D, McKenna DH, Hubel A, 2015. Clinical mesenchymal stromal cell products undergo functional changes in response to freezing. Cytotherapy 17, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan A, Paczesny S, 2015. Various forms of tissue damage and danger signals following hematopoietic stem-cell transplantation. Front. Immunol 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak MZ, Suszynska M, 2016. Emerging strategies to enhance homing and engraftment of hematopoietic stem cells. Stem Cell Rev 12, 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebelatto CK, Aguiar AM, Moretão MP, Senegaglia AC, Hansen P, Barchiki F, Oliveira J, Martins J, Kuligovski C, Mansur F, Christofis A, Amaral VF, Brofman PS, Goldenberg S, Nakao LS, Correa A, 2008. Dissimilardifferentiation of mesenchymal stem cells from BM, umbilical cord blood, and adipose tissue. Exp. Biol. Med 233, 901–913. [DOI] [PubMed] [Google Scholar]
- Roderick JE, Gonzalez-Perez G, Kuksin CA, Dongre A, Roberts ER, Srinivasan J, Andrzejewski C Jr., Fauq AH, Golde TE, Miele L, Minter LM, 2013. Therapeutic targeting of NOTCH signaling ameliorates immune-mediated BM failure of aplastic anemia. J. Exp. Med 210, 1311–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemeling-van Rhijn M, Khairoun M, Korevaar SS, Lievers E, Leuning DG, IJzermans JNM, Michiel GH, Betjes MGH, Genever PG, van Kooten C, de Fijter HJW, Ton J, Rabelink TJ, Baan CC, Weimar W, Roelofs H, Hoogduijn MJ, Reinders ME, 2013. Human Bone marrow- and adipose tissue-derived mesenchymal stromal cells are immunosuppressive in vitro and in a humanized allograft rejection model. J. Stem Cell Res. Ther. Suppl 6, 20780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad KC, Gurtner GC, 2012. Mesenchymal stem cells home to sites of injury and inflammation. Adv. Wound Care (New Rochelle) 1, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy V, Kumar S, 2016. hiPSC-derived iMSCs:NextGen MSCs as an advanced therapeutically active cell resource for regenerative medicine. J. Cell. Mol. Med 20, 1571–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensebé L, Fleury-Cappellesso S, 2013. Biodistribution of mesenchymal stem/stromal cells in a preclinical setting. Stem Cells Int 2013 (Article ID 678063, 5 pages). [DOI] [PMC free article] [PubMed]
- Shi Y, Hu G, Su J, et al. , 2010. Mesenchymal stem cells: a new strategy for immuno-suppression and tissue repair. Cell Res 20, 10–518. [DOI] [PubMed] [Google Scholar]
- Shin HM, Tilahun ME, Cho OH, Chandiran K, Kuksin CA, Keerthivasan S, Fauq AH, Golde TE, Miele L, Thome M, Osborne BA, Minter LM, 2014. NOTCH1 can initiate NF-κB activation via cytosolic interactions with components of the T cell signalosome. Front. Immunol 5, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik WD, 2007. Graft-versus-host disease. Nat. Rev. Immunol 7, 340–352. [DOI] [PubMed] [Google Scholar]
- Sutcliffe EL, Bunting KL, He YQ, Li J, Phetsouphanh C, Seddiki N, Zafar A, Hindmarsh EJ, Parish CR, Kelleher AD, McInnes RL, Taya T, Milburn PJ, Rao S, 2011. Chromatin-associated protein kinase C-θ regulates an inducible gene expression program and microRNAs in human T lymphocytes. Mol. Cell 41, 704–719. [DOI] [PubMed] [Google Scholar]
- Tobin LM, Healy ME, English K, Mahon BP, 2013. Human mesenchymal stem cells suppress donor CD4(+) T cell proliferation and reduce pathology in a humanized mouse model of acute graft-versus-host disease. Clin. Exp. Immunol 172, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima A, Yasuhara T, Date I, 2017. Mesenchymal stem cell therapy for ischemic stroke. Acta Med. Okayama 71, 263–268. [DOI] [PubMed] [Google Scholar]
- Turinetto V, Vitale E, Giachino C, 2016. Senescence in human mesenchymal stem cells: functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci 17 10.3390/ijms17071164. (7) pii: E1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urt-Filho A, Oliveira RJ, Hermeto LC, Pesarini JR, de David N, de Barros Cantero W, Falcão G, Marks G, Antoniolli-Silva ACMB, 2016. Mesenchymal stem cell therapy promotes the improvement and recovery of renal function in a preclinical model. Genet. Mol. Biol 39, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela JO, Iclozan C, Hossain MS, Prlic M, Hopewell E, Bronk CC, Wang J, Celis E, Engelman RW, Blazar BR, Bevan MJ, Waller EK, Yu XZ, Beg AA, 2009. PKCtheta is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice. J. Clin. Invest 119, 3774–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodyanik MA, Yu J, Zhang X, Tian S, Stewart R, Thomson JA, Slukvin II, 2010. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 7, 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westin JR, Saliba RM, De Lima M, Amin Alousi A, Hosing C, Qazilbash MH, Khouri IF, Shpall EJ, Anderlini P, Rondon G, Andersson BS, Champlin R, Couriel DR, 2011. Steroid-refractory acute GVHD: predictors and outcomes. Adv. Hematol 10.1155/2011/601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Witte SFH, Lambert EE, Merino A, Strini T, Douben HJCW, O’Flynn L, Elliman SJ, de Klein AJEMM, Newsome PN, Baan CC, Hoogduijn MJ, 2017. Aging of BM- and umbilical cord-derived mesenchymal stromal cells during expansion. Cytotherapy 19, 798–807. [DOI] [PubMed] [Google Scholar]
- Xu ZB, Chaudhary D, Olland S, Wolfrom S, Czerwinski R, Malakian K, Lin L, Stahl ML, Joseph-McCarthy D, Benander C, Fitz L, Greco R, Somers WS, Mosyak L, 2004. Catalytic domain crystal structure of protein kinase C-theta (PKCtheta). J. Biol. Chem 279, 50401–50409. [DOI] [PubMed] [Google Scholar]
- Yan Z, Zhuansun Y, Liu G, et al. , 2014. Mesenchymal stem cells suppress T cells by inducing apoptosis and through PD-1/B7-H1 interactions. Immunol. Lett 162, 248–255. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T, 2012. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med 209, 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EY, Kong KF, Altman A, 2013. The yin and yang of protein kinase C-theta (PKCθ): a novel drug target for selective immunosuppression. Adv. Pharmacol 66, 267–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.