

SUMMARY
JC polyomavirus (JCPyV) is a ubiquitous human pathogen that causes progressive multifocal leukoencephalopathy (PML). The entry receptors for JCPyV belong to the 5-hydroxytryptamine 2 receptor (5-HT2R) family, but how individual members of the family function to facilitate infection is not known. We used proximity ligation assay (PLA) to determine that JCPyV interacts with each of the 5-HT2 receptors (5-HT2Rs) in a narrow window of time during entry. We used CRISPR-Cas9 to randomly introduce stop codons in the gene for each receptor and discovered that the second intracellular loop of each was necessary for infection. This loop contains a motif possibly involved in receptor internalization by β-arrestin. Mutation of this motif and small interfering RNA (siRNA) knockdown of β-arrestin recapitulated the results of our CRISPR-Cas9 screen, showing that this motif is critical. Our results have implications for the role these receptors play in virus infection and for their normal functioning as receptors for serotonin.
Graphical Abstract
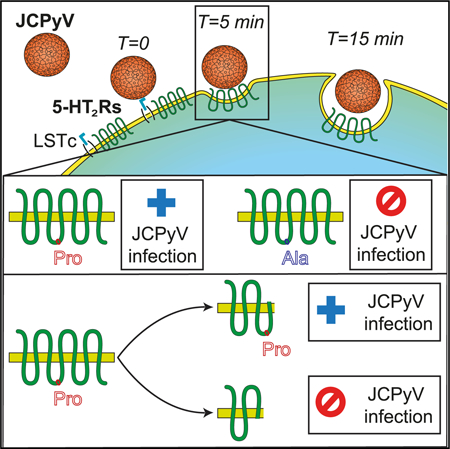
In Brief
5-HT2 receptors are important for infection of cells by JC virus (JCPyV). Assetta et al. show that JCPyV interacts transiently with each of three 5-HT2 receptors during entry and pinpoint a critical role for a proline in the second intracellular loop of each receptor in facilitating virus infection.
INTRODUCTION
Progressive multifocal leukoencephalopathy (PML) is a fatal neurodegenerative disease characterized by lytic JCPyV infection of oligodendrocytes and astrocytes in the CNS (Assetta and Atwood, 2017; Haley and Atwood, 2017). PML occurs in ~3% of patients with HIV, and the mortality rate in AIDS-associated PML cases is approximately 50% (Cinque et al., 2003; Garvey et al., 2011; Khanna et al., 2009; Major, 2010). Individuals undergoing immunomodulatory therapy for diseases such as multiple sclerosis (MS) or Crohn’s disease are also at risk of developing PML for which there is no treatment (Carson et al., 2009; Haley and Atwood, 2017; Kleinschmidt-DeMasters and Tyler, 2005; Neu et al., 2010). The only option is to restore immune surveillance in these patients.
JCPyV attachment to host cells is mediated by recognition of the receptor motif α2,6-linked glycan lactoseries tetrasaccharide c (LSTc) (Neu et al., 2010). JCPyV also requires 5-HT2 receptors(5-HT2AR, 5-HT2BR, and 5-HT2CR) to infect cells (Assetta et al., 2013; Elphick et al., 2004; Maginnis et al., 2010). The 5-HT2Rs are Gαq/11-coupled receptors and are composed of seven transmembrane domains, a glycosylated extracellular N-terminal domain, three extracellular loops (ECL1–3), three intracellular loops (ICL1–3), and one intracellular C-terminal tail. The second intracellular loop of all three receptors contains an important structural domain characterized by a DRY motif and by the presence of a proline 6 amino acids downstream of the DRY motif (proline 6). It was previously reported that proline 6 in 5-HT2CR is involved in β-arrestin binding (Marion et al., 2006). β-arrestin binding to the 5-HT2Rs is crucial to initiate internalization because it acts as a scaffold for AP2 and clathrin (Bohn and Schmid, 2010; Shenoy and Lefkowitz, 2011). Transfection of HEK293A cells, a poorly permissive cell line, with human 5-HT2Rs confers susceptibility to infection by facilitating viral entry into host cells, and a function-blocking antibody directed against 5-HT2AR inhibits JCPyV infection of glial cells (Assetta et al., 2013; Elphick et al., 2004). Drugs targeting one isoform or multiple isoforms of the 5-HT2Rs showed different degrees of inhibition to JCPyV infection, suggesting that these receptors may have a cooperative role in JCPyV entry (Elphick et al., 2004 ; O’Hara and Atwood, 2008). JCPyV does not seem to interact with 5-HT2Rs at the plasma membrane because JCPyV binding to cells overexpressing the 5-HT2Rs is not enhanced (Assetta et al., 2013). JCPyV enters host cells via clathrin-mediated endocytosis, and the 5-HT2Rs are also internalized by the same mechanism (Mayberry et al., 2019; Pho et al., 2000; Querbes et al., 2004). It is not yet known whether there is an interaction between JCPyV and the 5-HT2Rs during entry, and studies to clarify whether there is a redundant role for each individual isoform in the context of JCPyV infection of glial cells have not been performed. Additionally, it is not known what structural domains of the 5-HT2Rs are crucial for JCPyV infection, although recently a motif in the C terminus of the 5HT2A receptor was shown to be important for virus internalization and infection (Mayberry et al., 2019). In this study, mutagenesis of an ASK (Ala-Ser-Lys) motif in the C-terminal tail of 5HT2AR and small interfering RNA(siRNA) knockdown of beta-arrestin reduced JCPyV infection (Mayberry et al., 2019).
In the present work, we exploit the ability of the guide RNA/ caspase 9 (gRNA/Cas9) complex to cause double-strand breaks (DSBs) that are randomly repaired through the non-homologous end joining (NHEJ) pathway (Ran et al., 2013). Using CRISPR/Cas9, we generated genetically modified human glial cell lines to investigate the exact role of the three 5-HT2R isoforms in JCPyV infection. We isolated single cells and performed clonal expansion and deep sequencing of each clone to ascertain the nature of the gene modifications. This approach allowed us to isolate different mutants for analyses and to compare their susceptibilities to infection by JCPyV. By analyzing a panel of CRISPR mutants, we were able to identify critical motifs in the second intracellular loop of each receptor that were responsible for conferring susceptibility of the cells to infection by JCPyV.
RESULTS
JCPyV Transiently Interacts with 5-HT2AR, 5-HT2BR, and 5-HT2CR
SVG-A cells were exposed to JCPyV, and a proximity ligation assay (PLA) was used to test for an interaction between the 5-HT2Rs and JCPyV VP1 at 0, 5, and 15 min post-infection (Figures 1A–1C). A significant increase in signal was detected at 5 min post-entry when compared to uninfected cells. There was not an observable increase in signal at 0 min when the virus initially binds to the plasma membrane, supporting our previous observation that JCPyV does not interact with the 5-HT2Rs at the cell surface (Assetta et al., 2013). The interaction between JCPyV and the 5-HT2Rs is transient because, at 15 min post-entry, the PLA signal returned to baseline levels. To test JCPyV specificity, the experiment was repeated at 5 min post-infection, targeting JCPyV VP1 and the transferrin receptor (TFR) (Figure 1D). No increase in the PLA signal was detected.
Figure 1. JCPyV Transiently Interacts with 5-HT2AR, 5-HT2BR, and 5-HT2CR during Entry.
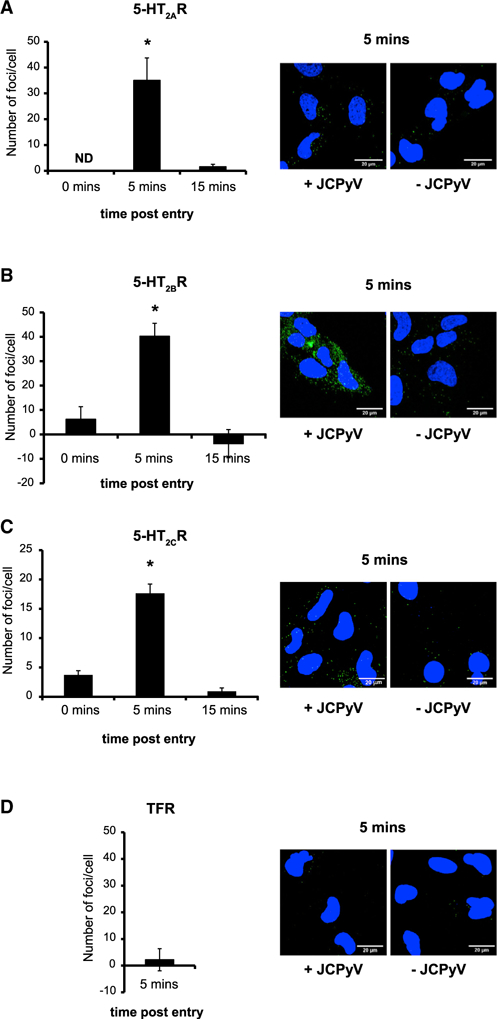
Close proximity between the major capsid protein VP1 and either 5-HT2AR (A), 5-HT2BR (B), 5-HT2CR (C), or transferrin receptor (TFR) (D) was quantified by PLA assay. The number of green fluorescent foci per cell was quantified using the BlobFinder software. The average number of foci per cell from samples that were not incubated with JCPyV was subtracted from the average number of foci per cell obtained from samples that were incubated with JCPyV. Experiments were performed in triplicate, and at least 50 cells per replicate were analyzed. Error bars represent SD. *p < 0.05. Representative images at 5 min post-entry in the presence (+JCPyV) or absence of JCPyV (–JCPyV) are included in each panel.
Deletion of the Second Intracellular Loop of the 5-HT2C Receptor Correlates with Reduced Susceptibility to Infection
CRISPR-Cas9 was used to directly modify the coding sequence of 5-HT2AR, 5-HT2BR, and 5-HT2CR in SVG-A cells. Briefly, we selected the gRNA sequences from the GeCKOv2 library published by Sanjana et al. (2014) and used a single vector lentiviral system to deliver them into SVG-A cells. After lentiviral infection and drug selection, single cells were isolated on rafts and expanded to generate clonal populations. The edited genomes were characterized using next-generation sequencing. Clones displaying nonsynonymous mutations leading to a premature stop codon were selected for further analysis. The gRNA directed against the human 5-HT2AR targeted the second intracellular loop coding sequence. One CRISPR-modified clonally expanded cell line was selected (5-HT2AR∆1-SVG-A cells). Two sequences carrying different nonsynonymous mutations were identified (Figures 2A and S1A). The DRY motif is disrupted in the first sequence, and it is preserved in the second. After the nonsynonymous mutations, there are 16 amino acids that differ from the wild-type(WT) sequence and they precede a premature stop codon. JCPyV infection of 5-HT2AR∆1-SVG-A cells was not inhibited when compared to WT-SVG-A cells (Figure 2B). The gRNA directed against the human 5-HT2BR targeted the third intracellular loop. One CRISPR-modified clonally expanded cell line was selected (5-HT2BR∆1-SVG-A cells), and it is characterized by four sequences carrying different 5-HT2BR nonsynonymous mutations (Figures 2A and S1A). JCPyV infection of 5-HT2BR∆1-SVG-A cells was not inhibited when compared to WT-SVG-A cells (Figure 2B). The gRNA directed against the human5-HT2CR targeted the coding sequence of the second intracellular loop, and the CRISPR-modified sequence obtained from one clone (5-HT2CR∆1-SVG-A cells) is shown in Figures 2A and S1A. Interestingly, the core sequence of the second intracellular loop starting with the DRY motif was completely disrupted, and JCPyV infection of the 5-HT2CR∆1-SVG-A cells was significantly inhibited when compared to WT SVG-A cells(Figure 2).
Figure 2. Deletion of the Second Intracellular Loop of the 5-HT2CR Correlates with Reduced Susceptibility to Infection.
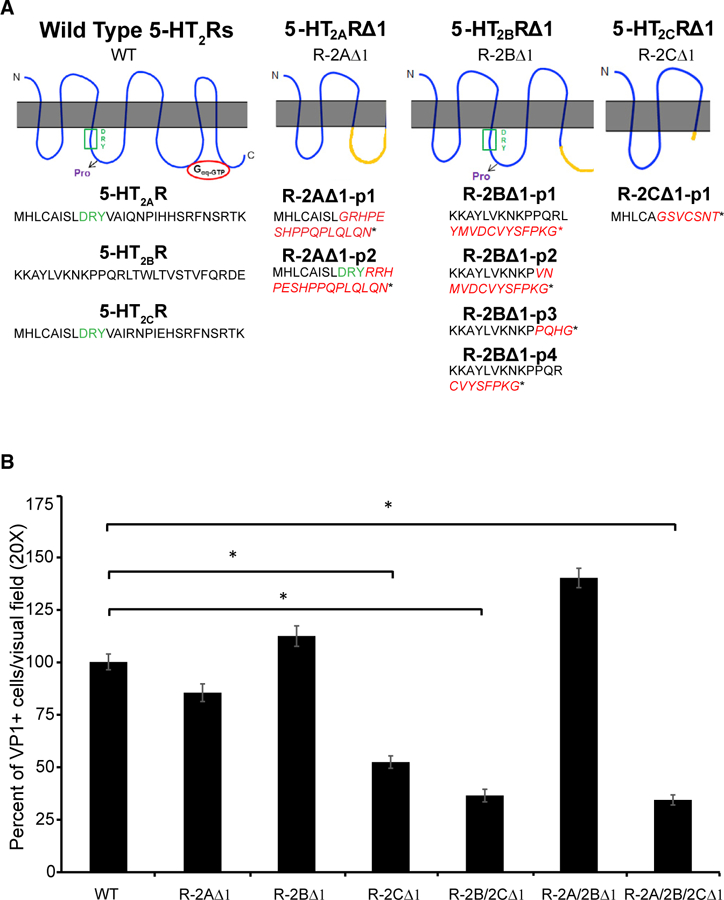
(A) The protein sequence of the 5-HT2AR∆1 (R-2A∆1), 5-HT2BR∆1 (R-2B∆1), and 5-HT2CR∆1 (R-2C∆1) compared to the WT is shown. Red letters highlight where the frameshift occurred, and the star indicates the stop codon.
(B) WT and genetically modified SVG-A cell lines were infected with JCPyV for 1 h at 37°c, and cells were stained for VP1. VP1-and DAPI-positive nuclei per visual field were quantified. Percent infection in WT SVG-A cells was set to 100% and the results normalized to this value. The results represent the average of three independent experiments performed in triplicate; at least 15 fields of view were imaged per replicate. Error bars represent SEM. *p < 0.05.
See also Figure S1.
Double and Triple Mutants that Include the 5-HT2CR Modification Show Reduced Susceptibility to Infection
5-HT2BR∆1-SVG-A cells were infected with lentiviral particles encoding for a gRNA targeting 5-HT2CR to generate a double mutant 5-HT2B/2CR∆1 -SVG-A cell line. The gRNA was the same used to generate 5-HT2CR∆1-SVG-A cells. The edited genome of a selected clonal population was characterized by next generation sequencing, and the nonsynonymous mutation was identical to the one described in Figures 2A and S1A.JCPyV infection of 5-HT2B/2CR∆1-SVG-A cells was reduced by ~60% when compared to WT-SVG-A cells, similar to 5-HT2CR∆1-SVG-A single mutant(Figure 2B). We proceeded to infect 5-HT2B/2CR∆1-SVG-A cells with lentiviral particles encoding for a gRNA targeting 5-HT2AR to generate a triple 5-HT2A/2B/2CR∆1-SVG-A cell line. The gRNA was the same used to generate 5-HT2AR∆1-SVG-A cells. The sequencing results are shown in Figure S1C. Again, JCPyV infection of 5-HT2A/2B/2CR∆1-SVG-A cells was 60% decreased when compared to WT-SVG-A cells similar to 5-HT2CR∆-SVG-A cells and 5-HT2B/2CR∆1-SVG-A cells (Figure 2). Finally, we generated 5-HT2A/2BR∆1-SVG-A cell to WT-SVG-A cells (Figures 2B andS1B). Our results indicate that the 5-HT2CR receptor was dominant in the presence of truncated 5HT2AR and 5HT2BR that kept the second intracellular loop intact.
CRISPR-Cas9-Mediated N-Terminal Targeting of5-HT2AR and 5-HT2BR Significantly Decreases JCPyV Infection
To confirm that the second intracellular loop of the 5-HT2Rs could have a crucial role in facilitating JCPyV entry, gRNA stargeting the N-terminal coding region of 5-HT2AR and 5-HT2BR were designed. The web-based tool called CRISPR design (http://zlab.bio/guide-design-resources) was used, cell lines from single cell clones were generated, and the genome editing was characterized as described previously (Figure S2A). JCPyV infection of 5-HT2AR∆2-SVG-A cells and 5-HT2BR∆2-SVG-A cells was reduced compared to WT and had the same phenotype as the 5-HT2CR∆-SVG-A cells (Figure 3A). Transfection of the mutant cell lines with plasmids expressing the WT versions of 5-HT2AR, 5-HT2BR, and 5-HT2CR partially rescued infection, confirming that our gRNAs were specific for the chosen targets (Figure 3B). To determine whether JCPyV infection could be completely abolished by targeting all three receptors at the same time, triple mutant cell lines were generated using the custom design gRNAs to target 5-HT2AR and 5-HT2BR and the gRNA obtained from the GeCKOv2 library to target 5-HT2CR, and sequencing results are shown in Figure S2B (5-HT2A/2B/2CR∆2-SVG-A cell line). JCPyV infection of the triple mutant was modestly reduced when compared to single mutants. The transfection of plasmids expressing the WT form of either 5-HT2AR, 5-HT2BR, or 5-HT2CR partially rescued JCPyV infection of 5-HT2A/2B/2CR∆2-SVG-A cell line (Figure 3C). As a control, we infected 5-HT2A/2B/2CR∆2-SVG-A cells with SV40 and saw no difference in susceptibility to infection compared to WT cells (Figure 3D). We then tested JCPyV binding and entry in 5-HT2A/2B/2CR∆2-SVG-A cells compared to WT SVG-A. Virus binding was measured by flow cytometry using labeled JCPyV-488. The binding of labeled virus to the triple knockout was not reduced compared to WT (Figure 3E). We then tested for JCPyV entry by JC pseudovirus (JCPsV) transduction. JCPsV has been shown to mirror JCPyV attachment and entry, and it does not have the ability to replicate making it a good tool to test for viral entry defects (Gee et al., 2013). We found that JCPsV transduction of 5-HT2A/2B/2CR∆2-SVG-A cells was significantly decreased when compared to WT SVG-A cells (Figure 3F).Finally, we treated 5-HT2A/2B/2CR∆2-SVG-A cells with ritanserin, a 5-HT2R antagonist, at a 25 µM concentration and found that JCPyV infection could be further reduced (Figure 3G).
Figure 3. CRISPR-Cas9-Mediated N-Terminal Targeting of 5-HT2AR and 5-HT2BR Significantly Decreases JCPyV Infection.
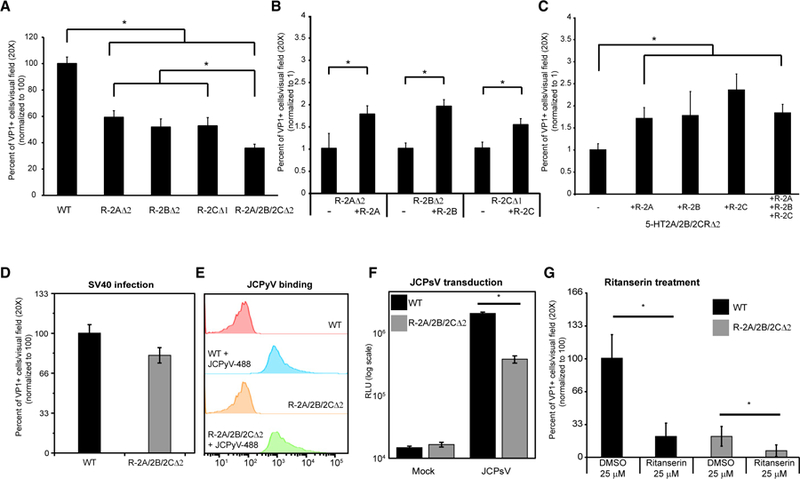
5-HT2AR∆2-SVG-A (R-2A∆2), 5-HT2BR∆2-SVG-A (R-2B∆2), and 5-HT2A/2B/2CR∆2-SVG-A (R-2A/2B/2C∆2) cells were generated.
(A) Mutant cell lines were challenged with JCPyV and infection scored.
(B and C) 5-HT2AR∆2-SVG-A (R-2AD2), 5-HT2BR∆2-SVG-A (R-2B∆2), and 5-HT2CR∆1-SVG-A (R-2B∆1) cells (B) and the triple mutant cells (C) were rescued by transfecting the cells with plasmids expressing WT receptors.
(D) SV40 infection was scored, and it was not reduced in triple mutant SVG-A cells compared to WT.
(E) JCPyV-488 binding was tested by flow cytometry and was not reduced in triple-mutant SVG-A cells compared to WT.
(F) WT or triple-mutant cells were transduced with JC pseudoviruses (JCPsV) and luciferase activity measured as relative light units (RLUs) on a log scale. JCPsV entry was significantly reduced in triple-mutant cells.
(G) The 5HT2Rs antagonist ritanserin inhibits JCPyV infection in the triple-mutant SVG-A cells.
(A, B, C, D, and G) VP1-and DAPI-positive nuclei were quantified, and results are expressed as percent of infected cells. Numbers were normalized to WT SVG-A cellsinfection level (A, D, and G) or to mock transfected cells (B and C), set to either 100% (A, D, and G) or 1% (B and C). All results represent the average of three independent experiments performed in triplicate. At least 15 fields of view were imaged per replicate for the infection experiments. At least 10,000 events wererecorded per replicate in (E). Error bars represent SEM. *p < 0.05.
See also Figure S2.
The Second Intracellular Loop of the 5-HT2Rs Is Important for JCPyV Infection
We hypothesized that the difference in the infection phenotypes between 5-HT2CR∆1-SVG-A cells and in 5-HT2AR∆1 and 5-HT2BR∆1-SVG-A cells could be due to incomplete disruption of the second intracellular loop (ICL-2). We generated truncation mutants of the three receptors by inserting a stop codon either before the beginning of ICL-2 or at the end of the fourth trans-membrane domain (Figure 4A). We transfected HEK293A cells with WT or the truncation mutants and challenged with JCPyV. Overexpression of WT receptors supported JCPyV infection as previously reported (Assetta et al., 2013). The receptors truncated at the end of the fourth transmembrane domain showed a significant increase in JCPyV infection when compared to cells transfected with the empty vector. In contrast, receptors that were truncated before the beginning of ICL-2 did not support JCPyV infection. WT and truncation mutant receptors were equally expressed at the cell surface (Figures S3A and S3B). The second intracellular loop of all three WT receptors contains an important structural domain characterized by a DRY motif and by the presence of a proline, 6 amino acids downstream of the DRY motif (proline 6; Figure 4B). It was previously reported that proline 6 in 5-HT2CR is involved in barrestin binding, which initiates internalization and could trigger specific signaling cascades (Marion et al., 2006). We mutated proline 6 to an alanine in 5-HT2AR-YFP, 5-HT2BR-YFP, and 5HT2CR-YFP fusion constructs and transfected HEK293A cells with either the WT or the mutated version of the receptors (Figure 4c). Overexpression of the proline to alanine mutant receptors impaired JCPyV infection. The proline-to-alanine mutant receptors were equally expressed at the cell surface when compared to WT receptors (Figures S3C and S3D). To determine whether there is a link between β-arrestin and 5-HT2R-mediated JCPyV infection, we treated WT and 5-HT2A/2B/2CR∆2-SVG-A cells with siRNA targeting β-arrestin (β-arrestin KD) or a non-targeting oligonucleotide (neg). Efficient knockdown was tested by western blot. Knockdown of β-arrestin in WT SVG-A cells causes a significant reduction in JCPyV infection, although it does not reduce infection further in 5-HT2A/2B/2CR∆2-SVG-A cells. Strikingly, the infection in 5-HT2A/2B/2CR∆2-SVG-A cells is similar to WT SVG-A cells treated with β-arrestinsiRNA (Figure 4D).
Figure 4. The Second Intracellular Loop of the 5-HT2Rs Is Important for JCPyV Infection.
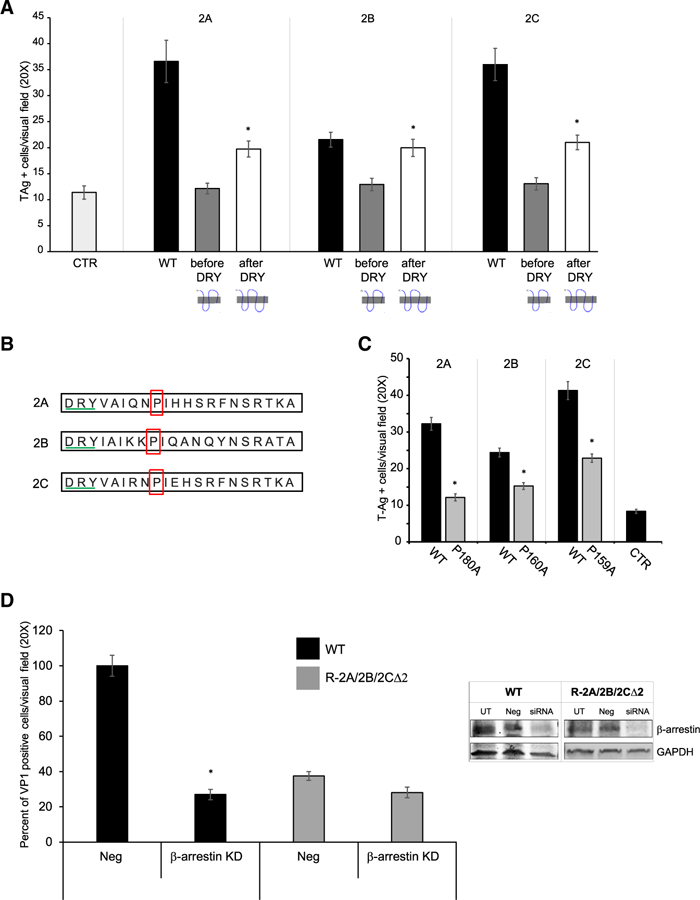
(A) HEK293A cells were transfected with WT or truncated constructs, challenged with JCPyV, and stained for TAg.
(B and C) The proline located 6 amino acids downstream of the DRY motif was mutated to an alanine in three constructs expressing 5-HT2AR, 5-HT2BR, or 5-HT2CR-YFP (B). HEK293A cells were transfected with WT or mutated constructs, challenged with JCPyV, and stained for Tag (C). Results are expressed as Tag-positive cells per visual field.
(D) WT and 5-HT2A/2B/2CR∆2-SVG-A (R-2A/2B/ 2C∆2) were treated with either a non-targeting or β-arrestin1-specific siRNA for 3 days and then challenged with JCPyV. VP1-and DAPI-positive nuclei were quantified, and results are expressed as percent of infected cells.
Numbers were normalized to WT SVG-A cells infection level, set to 100%. The results represent the average of either three or two independent experiments performed in triplicate, and at least 15 fields of view were counted per replicate. Error bars represent SEM. *p < 0.05. See also Figure S3.
DISCUSSION
JCPyV requires the 5-HT2Rs to facilitate entry into host cells, and here, we demonstrated that the virus transiently interacts with each of the 5HT2Rs during entry. Our approach using CRISPRCas9 allowed us to uncover an important role for the second intracellular loop of the 5-HT2Rs specifically of a proline located 6 amino acids downstream of the DRY motif. The first set of gRNAs used in this study generated premature stop codons at the end of the second intracellular loop of 5-HT2AR, in the middle of the third intracellular loop of 5-HT2BR, and at the beginning of the second intracellular loop of 5-HT2CR. JCPyV infection was significantly decreased in 5-HT2CR∆1-SVG-A cells but was not in 5-HT2AR∆1 and 5-HT2BR∆1-SVG-A cells. The generation of a triple mutant cell line containing these modifications did not further decrease JCPyV infection when compared to 5-HT2CR∆1-SVG-A cells, indicating that the missense mutations in 5-HT2AR and 5-HT2BR were not affecting JCPyV infection (Figure 2). However, when clonal cell lines were generated using gRNAs targeting the extreme N terminus of 5-HT2AR and 5-HT2BR, JCPyV infection was significantly decreased to levels equal to those observed in 5-HT2CR∆1-SVG-A cells (Figure 3). These data suggested that the second intracellular loop of the 5-HT2Rs plays a critical role in facilitating virus infection. The second intracellular loop of the 5HT2Rs contains a contiguous stretch of 10 amino acids beginning with the DRY motif and followed by a proline 6 amino acids downstream. This residue has been shown to be important for β-arrestin binding in the 5-HT2CR (Marion et al., 2006). When we mutated the proline to an alanine in all three constructs encoding the 5-HT2Rs, JCPyV infection was significantly decreased, but not eliminated (Figure 4B). β-arrestin has also been shown to bind to the third intracellular loop and to the C-terminal tail of the 5-HT2Rs (Gelberet al., 1999; Marion et al., 2006; McCorvy and Roth, 2015). Hence, this would explain the residual infection of HEK293A cells transfected with proline to alanine mutant receptors. This may also explain why the infection in cells transfected with the receptors truncated after the DRY motif was not as efficient as infection of WT cells (Figure 4A). We next confirmed a role for β-arrestin in JC virus infection by knocking it down with siRNA, and our results were consistent with that reported by Mayberry et al. (2019). siRNA treatment of WT cells decreased infection to the same extent as what we observed in the of 5-HT2A/2B/2CR∆2-SVG-A triple mutant cells. The fact that siRNA knockdown of β-arrestin did not reduce infection further in the triple mutant cells suggests that β-arrestin is exerting its main effect on infection by interacting with the 5HT2Rs. The residual infectivity in these experiments could be due to the presence of minor splice variants of the 5-HT2AR and 5-HT2BR not targeted by the custom designed gRNAs or by different and unknown entry receptors that do not rely on β-arrestin. The ritanserin experiment suggests that unknown minor splice variants could be the culprit; however, we used a non-toxic but relatively high concentration of the drug (25 µM) and it has been reported that, at such concentrations, ritanserin could also inhibit histamine-H1, dopamine-D2, adrenergic α1, and α2 receptors (Leysen et al., 1985).
The most prominent effect on JCPyV infection was obtained by individual receptor mutations, suggesting that the 5-HT2Rs do not have a redundant role (Figure 3A). It has been reported that 5-HT2Rs form homodimers and heterodimers in vitro and in vivo (Moutkine et al., 2017). Based on our findings, we hypothesize that JCPyV may favor 5-HT2 receptor heterodimers over homodimers to initiate infection. Nonetheless, when we overexpressed one 5-HT2 receptor isoform at a time in the triple mutant cell line, we were able to partially rescue the JCPyV infection phenotype (Figure 3C). This suggests either that JCPyV uses homodimers of 5-HT2 receptors as well or that there are unknown entry receptors that function in synergy with the 5-HT2 receptors. Further experiments are needed to investigate these hypotheses.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Walter J. Atwood (walter_atwood@brown.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
SVG-A cells, HEK293A (CRL-1573), HEK293T (ATCC CRL-11268), Lenti-X HEK293T cells (takara, cat# 632180) and African green monkey (CV-1) (ATCC, CCL-70) cells were used. SVG-A cells are a third-generation sub clone of the original SVG cell line (Major et al., 1985) and express the astrocyte marker GFAP. SVG-A cells and CV-1 were cultured in MEM supplemented with 10% fetal bovine serum and 1% of Amphotericin B, Penicillin, Streptomycin solution. HEK293A and HEK293T were cultured in DMEM supplemented with 10% fetal bovine serum and 1% of Amphotericin B, Penicillin, Streptomycin solution. All cells were grown in an incubator at 37°c with 5% CO2. SVG-A cells and HEK293A cells were authenticated using the ATCC cell line authentication service (STR profile). They have a 55% match with SVG p.12 (human fetal glial cells, CRL-8621) from ATCC and they are of male origin. HEK293A cells have an 81% match with the parental line and are of female origin. We are in the process of authenticating the other cell lines used in the study.
Virus and pseudovirus
For JCPyV infection we used a lab adapted strain referred to as Mad-1/SVED described previously (Vacante et al., 1989). JCPyV was grown in SVG-A cells using 1700 cm2 roller bottles at 5% CO2 and at 37°c. Cells were cultured for 14 days, with the cell culture media replaced at 7 days. Viral lysates were harvested by scraping cells in the presence of cell culture media. This lysate was frozen at 80C and thawed in a 37°c water bath 3 times. Deoxycholate was added to obtain a final concentration of 0.25%. Samples were spun at 17000 x g for 30 mins and supernatant was used for infections. Simian virus 40 (SV40) strain 776 was propagated in the African green monkey kidney cell line CV-1 as previously described following the same protocol described for JCPyV (Norkin and Ouellette, 1976). JCPyV lysates, when needed, were purified as previously described (Liu and Atwood, 2001; Nelson et al., 2013). Briefly, lysates were sonicated three times on ice (50% amplitude 50% duty cycle, power 4, 1 min) and treated with Neuraminidase type II (Sigma, Cat# N6514) at 37°c for 1 hr to release virus from membranes. Samples were subjected to two rounds of Vertrel XF (Fisher scientific NC9715008) to extract lipids. The viral supernatant was pelleted through a 20% sucrose cushion in a Beckman SW40ti rotor at 150,000 x g at 4°c for 3 h. The viral pellet was resuspended into buffer A (10mM Tris-HCl, 50 mMNaCl, 0.1 mM CaCl2) and sonicated 3 times (30% amplitude 50% duty cycle, power 3, 1 min). The resuspended pellet was loaded onto a CsCl step gradient (1.29–1.35 g/ml) and spun at 115,000 x g at 4°C for 18 h in a Beckman SW55ti rotor. The band corresponding to DNA-containing virions was isolated and dialyzed extensively against buffer A. If needed JCPyV was directly labeled with Alexa Fluor 488 following to the manufacturer protocol (ThermoFisher Scientific, cat# A20000). JC pseudovirus (JCPsV) was generated as described previously (Gee et al., 2013). Briefly, pseudoviruses were produced by transfection of the VP1, VP2, VP3 and pSV40-Cluc plasmids into HEK293FT cells using the FuGENE® 6 Transfection Reagent (PromegaMadison, WI USA cat # E2692) in a 3:1:1:2 ratio. Mock pseudovirus controls were generated by transfecting 293FT cells with non-specific DNA (ns-DNA) and the pSV40-Cluc (NEB # N0318) reporter plasmid in a 5:1 ratio and following the same production/purification protocol as pseudovirus. As the viral structural proteins are expressed, they assemble into pseudovirus particles and package available plasmid DNA. The media is changed on the day following transfection to remove any extracellular transfection reagent. Cells were harvested 6 days post transfection and pseudovirus was purified as described above. Secreted luciferase was measured with the BioLuxCypridina Luciferase Assay kit (NEB E3309L).
METHOD DETAILS
Antibodies
A mouse monoclonal antibody obtained from the supernatant of a hybridoma was used to detect JCPyV VP1 (PAB597). Rabbit polyclonal primary antibodies were used to recognize the 5-HT2Rs and TFR in the proximity ligation assay. Anti-5-HT2AR and anti 5HT2CR were obtained from LS Biosciences, Inc cat# LS-A1106 and LS-A1119 respectively. Anti-5-HT2AR recognizes the N terminus while anti-5-HT2CR the C terminus. Anti-5-HT2BR was obtained from Novus Biologicals cat# NLS1111 and recognizes the second extracellular loop. Anti-TFR was purchased from Novus Biologicals cat# NB100–92243. For Western Blot analysis a rabbit monoclonal anti-β-arrestin½ was purchased from Cell Signaling Technologies (#4674) and a mouse monoclonal anti-GAPDH was purchased from Sigma-Aldrich (G8795). A Mouse monoclonal anti-pan Cadherin antibody was used as cell surface marker (abcam, ab6528) and Rabbit polyclonal anti-6X His tag was used to stain for truncated receptors (abcam, ab9108).
Indirect immunofluorescence assay
Three days (for SVG-A cells) or five days (for HEK293A cells) following viral challenge, cells were stained and counted by eye using epifluorescence microscopy (Nikon E800) as previously described (Assetta et al., 2013). Briefly, plates were fixed using ice-cold 100% methanol at 20C for at least 20 mins. They were treated with PBS containing 1% Triton X-100 for 5 mins and blocked with 10% goat serum for 30 mins at Room Temperature (RT). Samples were incubated for 1 hr at 37°C with PAB 597 (1:50) followed by staining with secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific) (1:500) and DAPI (Thermo Fisher Scientific). The amount of DAPI and VP1 stained nuclei per visual field was counted using Cell Profiler (Carpenter et al., 2006) and data are expressed as percentage of infected cells/visual field. Cells plated on coverslips and transfected with all mutant receptors were fixed with freshly made 4% PFA for 20 mins, permeabilized with Triton-X 1% for 15 mins, blocked with 10% goat serum for 30 mins. Cells transfected with truncated mutant receptors were incubated O/N with anti-6X His tag at 1/100 dilution in 2% goat serum. The next day cells were washed with PBS 1X and incubated with anti-pan Cadherin for 2 hr at 37°C in 2% goat serum. Cells were washed and incubated with anti-rabbit 488 and anti-mouse 594 at 1/500 dilution in PBS 1X at 37°C for 1 hr. Cells were washed with PBS 1 X and stained with 1/1000 DAPI solution (ThermoFisher Scientific, Cat# 62248) for 10 mins then mounted on slides.
Proximity ligation assays
To visualize close proximity between JCPyV VP1 and the 5-HT2Rs, WT-SVG-A cells were pre-chilled at 4°C for 20 mins and infected with purified JCPyV on ice for 1 hr. Three time points were analyzed, 0, 5, and 15 mins post infection. For the 0 mins time point cells were washed and fixed with 4% PFA immediately after infection. For the 5 and 15 mins time points, the virus was aspirated off, warm complete media was added, and the cells were shifted at 37°C for the respective time before fixation. Cells were permeabilized with PBS containing 0.5% Triton X-100 and were then blocked in 5% Donkey Serum. Cells were then stained for VP1 (1:2000 dilution) and 5-HT2Rs (1:25 dilution) or TFR (1:50 dilution) by overnight incubation at 4°C. These cells were then immunostained using the proximity ligation assay, following manufacturer’s instructions (Bethyl Labs). Cells were washed, and the cell nuclei were counterstained using DAPI. Fluorescence micrographs were collected by confocal microscopy. Fluorescent foci were quantified using the Blobfinder program (http://www.cb.uu.se/amin/BlobFinder/). As negative control uninfected samples were stained and processed at the same time. Blobfinder analysis was performed using a 7 by 7 blob size and at least 50 cells per sample were analyzed. The average number of blobs counted in the uninfected cells was subtracted from the infected samples. Experiments were performed in triplicate and error bars denote standard error.
Generation of CRISPR modified SVG-A clonal cell lines
The lentiCRISPRv2 plasmid with a puromycin resistance was obtained from Addgene (#52961). Two additional lentiCRISPRv2 plasmids were generated by replacing the puromycin resistance with neomycin or hygromycin resistance to generate double or triple mutants. The lentiCRISPRv2 plasmid was mutated to insert aHpaI site after the puromycin resistance gene using Agilent QuikChange II XL Site Directed Mutagenesis Kit (#200521). Primers were ordered from Integrated DNA Technologies (IDT): lentiCRISPRv2_ g9959c_F GTTGATTGTCGAGTTAACGCGTTCAGGCACCG and lentiCRISPRv2_ g9959c_R CGGTGCCTGAACG CGTTAACTCGACAATCAAC. The puromycin resistance gene was removed from lentiCRISPRv2 by digesting with BamHI and HpaI and sequences encoding neomycin or hygromycin resistance were cloned into the lentiCRISPRv2 vector. The DNA encoding for the neomycin and hygromycin resistance were ordered from Blue Heron Biotech, LLC. The gRNAsequences targeting IL2 or IL3 of the 5-HT2Rs were obtained from the GeCKOv2 human library published by Sanjana et al. (2014). 5-HT2AR: GATTCTGGATGGC GACGTAG 5-HT2BR: AGCCACCTCAACGCCTAACA 5-HT2CR: GCACCTCTGCGCTATATCGC. The gRNAs targeting the N terminus of 5-HT2AR and 5-HT2BR were designed using a web-based tool called CRISPR design (http://zlab.bio/guide-design-resources). 5-HT2AR: AAAGTCATTACTGTAGAGCC 5-HT2BR: GTTATCTCTTCTAACTGGTC. The protocol provided by the Zhang lab was used to clone specific gRNAs in the lentiCRISPRv2 plasmids with minor changes (https://media.addgene.org/data/plasmids/52/52961/52961-attachment_B3xTwla0bkYD.pdf). Lentiviral particles were packaged in Lenti-X HEK293T cells plated in T75 cm2 flasks using the pCMV-dR8.2 packaging plasmid (https://www.addgene.org/8455/) and the VSV-G envelope encoding plasmid (www.addgene.org/8454/) at a ratio of 3 (pCMV-dR8.91), 0.3 (VSV-G) and 3 (lentiCRISPRv2). Fugene HD (Promega) was used at a 3:1 DNA:Fugene ratio for transfection following manufacturer instructions. Cells were incubated for 18 hr then media was replaced with 10 mls of DMEM with 30% FBS and cells were incubated at 37°C for 24hrs. Lentivirus was harvested every 12 hr, for a total of three harvests. Supernatants were combined filtered using a 0.45 µm filter and either used to infect SVG-A cells immediately or frozen at 80°C. SVG-A cells 60% confluent in a T75 cm2 flask were infected three times every 24 hr with 10 mls of the filtered lentivirussupplemented with Polybrene (Santa Cruz Biotechnology) at a concentration of 8 μg/ml. Cells were then drug selected with the suitable antibiotic: puromycin was used at 5 μg/ml for 3 days, geneticin (G418) at 600 μg/ml for 1 week and Hygromycin was used at 250 μg/ml for 4 days. Single cell isolation and expansion was performed to obtain a clonal population using Cellraft System (Cell Microsystem). The genome editing was confirmed using the CRISPR deep sequencing service performed at the Center for Computational and Integrative Biology (CCIB) DNA core at the Massachusetts General Hospital (MGH).
Flow cytometry
Cells were washed with 1X PBS and removed from plates using a cell stripper (Cellgro). The cells were pelleted, washed, and incubated in 100 µl of 1X PBS with JCPyV labeled with Alexa Fluor 488 (JCPyV-488) for 1 h on ice with occasional agitation. The cells were pelleted and washed with 1X PBS twice, then fixed in 1% paraformaldehyde (PFA), and analyzed for virus binding using a BD FACSCalibur (BD Bioscience).
Treatment of cells with ritanserin
Treatment with ritanserin was performed as described previously (O’Hara and Atwood, 2008). Briefly, cells at 60% confluence were pretreated with ritanserin (Tocris Bioscience) at 25 µM concentration O/N in complete medium at 37°c. The cells were then prechilled for 30 mins and infected on ice with JCPyV for 1 h in plain MEM 1X. The cells were washed with 1X PBS and warmed MEM with 10% FBS, antibiotics and ritanserin at 25 µM was added. The cells were stained for VP1 after 72 hr as described above.
Transient transfection
CRISPR-modified SVG-A clonal cell lines were plated in 24-well plates in MEM with 10% FBS overnight without antibiotics. Cells at 80% confluence were transfected using Lipofectamine 3000 (Invitrogen) following the manufacturer instructions with 1–2 μg of DNA per well of human 5-HT2AR, 5-HT2BR or 5-HT2CR in pcDNA3.1 or pcDNA3.1 control vector. The cells were incubated at 37°C for 4 h, the medium was replaced with MEM containing 10% FBS and antibiotics and incubated at 37°C for 24–48 hr. Cells were then infected with JCPyV for 1 hr at 37°C and stained for VP1 72 hr later. HEK293A cells were plated on glass coverslips in 24 well plate in DMEM with 10% FBS overnight without antibiotics. Cells at 80% confluence were transfected using Lipofectamine 3000 (Invitrogen) following the manufacturer instructions with 1–2 μg of DNA per well of each mutant receptor construct (either with point mutation or truncated) as well as the appropriate WT control. The cells were incubated at 37°C for 4 h, the medium was replaced with DMEM containing 10% FBS and antibiotics and incubated at 37°C for 48 hr. Cells were fixed and stained as described in the ‘‘immunofluorescence assay’’ section.
Site directed mutagenesis of 5-HT2Rs constructs
5-HT2Rs-YFP fusion constructs were generated as described previously (Assetta et al., 2013; Maginnis et al., 2010). Proline180, proline 160 and proline 159 of 5-HT2AR, 5-HT2BR or 5-HT2CR respectively were mutated to an alanine using the QuikChange II Site directed mutagenesis kit by Agilent following manufacturer instructions. XL-1 Blue super competent bacterial cells were used. Mutagenesis primers were designed using the QuikChange primer design tool (https://www.chem.agilent.com/store/primerDesignProgram.jsp):
| 5-HT2AR P180A_F | TGGTGGATGGCATTCTGGATGGCGACGTAG |
| 5-HT2AR P180A_R | CTACGTCGCCATCCAGAATGCCATCCACCA |
| 5-HT2BR P160A_F | GATTGGCCTGGATTGCCTTTTTGATGGCTATGTAACG |
| 5-HT2BR P160A_R | CGTTACATAGCCATCAAAAAGGCAATCCAGGCCAATC |
| 5-HT2CR P159A_F | GAAACGGCTATGCTCAATAGCATTACGTATTGCTACATACC |
| 5-HT2CR P159A_R | GGTATGTAGCAATACGTAATGCTATTGAGCATAGCCGTTTC |
Human 5-HT2AR, 5-HT2BR or 5-HT2CR in pcDNA3.1 expression vectors were used to generate truncation mutant receptors. A His tag was added before the stop codon in every mutant, as well as a XhoI cut site and an extra T (CATCATCATCACCACCAC TAA CTCGAG T). Primers were designed using the NEBaseChanger tool (http://nebasechanger.neb.com/) and the Q5 Site-Directed Mutagenesis Kit by NEB was used following manufacturer instructions. 5-HT2AR was truncated at position 510 (before the DRY motif) and at position 648 (after the DRY motif, at the end of the fourth transmembrane domain). 5-HT2BR was truncated at position 450 (before the DRY motif) and at position 579 (after the DRY motif, at the end of the fourth transmembrane domain). 5-HT2CR was truncated at position 447 (before the DRY motif) and at position 582 (after the DRY motif, at the end of the fourth transmembrane domain). Human 5-HT2AR, 5-HT2BR, or 5-HT2CR in pcDNA3.1 expression vectors were modified with the addition of a His tag at the C-terminal end of the protein. A BamHI site was added at the very end to help screen colonies (CATCATCATCACCACCAC TAA GGA TCC). Primers were designed using the NEBaseChanger tool (http://nebasechanger.neb.com/) and the Q5 Site-Directed Mutagenesis Kit by NEB was used following manufacturer instructions. All Primers were synthesized by Integrated DNA Technologies (IDT).
| 2A_trunc_510_His_F | ccactaactcgagtCTGGACCGCTACGTCGCC |
| 2A_trunc_510_His_R | tggtgatgatgatgCGAGATGGCGCAGAGGTG |
| 2B_trunc_450_His_F | ccactaactcgagtGTGGATCGTTACATAGCC |
| 2B_trunc_450_His_R | tggtgatgatgatgTGAAATGGCACAGAGATG |
| 2C_trunc_447_His_F | ccactaactcgagtCTGGATCGGTATGTAGCAATAC |
| 2C_trunc_447_His_R | tggtgatgatgatgCGATATAGCGCAGAGGTG |
| 2A_trunc_648_His_F | ccactaactcgagtGACGATTCGAAGGTCTTTAAG |
| 2A_trunc_648_His_R | tggtgatgatgatgCTGTAGCCCAAAGACTGG |
| 2B_trunc_579_His_F | ccactaactcgagtGGGATAGAGACTGATGTG |
| 2B_trunc_579_His_R | tggtgatgatgatgTTTAATAGGGACTGGAATG |
| 2C_trunc_582_His_F | ccactaactcgagtAGGGACGAAGAAAAGGTG |
| 2C_trunc_582_His_R | tggtgatgatgatgCAGTCCAATCACAGGGATAG |
| 2AWT_His_F | ccactaaggatccCTCGAGTCTAGAGGGCCC |
| 2AWT_His_R | tggtgatgatgatgCACACAGCTCACCTTTTCATTC |
| 2BWT_His_F | ccactaaggatccCTCGAGTCTAGAGGGCCC |
| 2BWT_His_R | tggtgatgatgatgTACATAACTAACTTGCTCTTCAGTTTTG |
| 2CWT_His_F | ccactaaggatccCTCGAGTCTAGAGGGCCC |
| 2CWT_His_R | tggtgatgatgatgCACACTGCTAATCCTTTCGC |
siRNA knock-down of β-arrestin
β-arrestin specific siRNA and control siRNA were obtained from Cell Signaling Technologies (SignalSilenceβ-arrestin1 siRNA #6218 and SignalSilence Control siRNA (unconjugated #6568) and were used at a concentration of 100 nM. Following manufacturer instructions, reverse transfection was performed using 1.5 μl of lipofectamine RNAiMAX and 60,000 cells per well of a 24 well plate or 7.5 μl of lipofectamine RNAiMAX and 300,000 cells per well of a 6 well plate. Cells were incubated for 72hrs then infected with JCPyV (24 well plates) or lysates were harvested for Western Blot (6 well plates).
Western blot
One well of a 6 well plate treated with β-arrestin1 specific siRNA or control siRNA was washed with ice-cold PBS 1X, Cells were incubated for 5 mins on ice in 350 μl of pre-chilled RIPA buffer supplemented with a protease inhibitor cocktail (Complete ULTRA tablets, Mini by Sigma-Aldrich/Roche). Cells were scraped and transferred to a microcentrifuge tube. Cells were rocked at 4°C for 30 mins then centrifuged for 15 mins at 14,000 rpm at 4°C. Supernatants were mixed with sample buffer, boiled for 5 mins at 95°C and resolved by SDS-PAGE (4%–15% Tris-HCl polyacrylamide gel). Proteins were transferred to a nitrocellulose membrane by Semidry transfer, blocked with 1X TBS 1% casein blocker (BioRad #161–0782) for 1hr at RT then incubated with anti-β-arrestin1/ 2 (1:500) O/N at 4°C. The membrane was washed 4 times with 1X TBS-T then incubated with goat anti-rabbit Alexa 680 (1:5,000) at RT for 1 hr, washed thoroughly with 1X TBS-T and imaged using the ChemiDoc MP Image system (BioRad). The membrane was then probed with anti-GAPDH (1:10,000) O/N at 4°C, washed 4 times with 1X TBS-T then incubated with goat anti-rabbit IRDye CW 800 (1:5,000) for 1 h at RT, washed thoroughly with 1X TBS-T and imaged using the ChemiDoc MP Image system (BioRad).
QUANTIFICATION AND STATISTICAL ANALYSIS
Information about the data quantification and statistical analysis is described in the figure legends. A Student’s t test was used to calculate the p value of each experiment using Microsoft Excel.
Blobfinder program (http://www.cb.uu.se/~amin/BlobFinder/) was used to determine the number of fluorescent foci per cell in the proximity ligation assay quantification shown in Figure 1.
Fiji was used to determine the Mander’s coefficient showed in Figure S3 using the plug in Coloc 2.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SV40 VP1 (PAB597) | lab generated, cross-reacts with JCPyV VP1 | N/A |
| Rabbit polyclonal anti-5-HT2AR | LS Biosciences | cat#LS-A1106; RRID: AB_10645379 |
| Rabbit polyclonal anti-5-HT2BR | Novus Biologicals | cat#NLS1111; RRID: RRID:AB_343312 |
| Rabbit polyclonal anti-5-HT2CR | LS Biosciences | cat#LS-A1119; RRID: AB_592691 |
| Rabbit polyclonal anti-transferrin receptor | Novus Biologicals | cat#NB100–92243; RRID: AB_1216384 |
| Rabbit monoclonal anti-β-arrestin1/2 | Cell signaling Technologies | Cat#4674; RRID: AB_10547883 |
| mouse monoclonal anti-GAPDH | Sigma-Aldrich | Cat#G8795 RRID:AB_1078991 |
| Mouse monoclonal anti-pan Cadherin | Abcam | Cat#ab6528; RRID:AB_305544 |
| Rabbit polyclonal anti-6X His tag | Abcam | Cat#ab9108; RRID:AB_307016 |
| Virus Strains | ||
| JCPyV | Vacante et al., 1989 | N/A |
| SV40 | Norkin and Ouellette, 1976 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ritanserin | Tocris Bioscience | Cat#1955 |
| Hygromycin B | ThermoFisher Scientific | Cat#10687010 |
| Geneticin (G418) | ThermoFisher Scientific | Cat#10131035 |
| Puromycin | ThermoFisher Scientific | Cat#A1113803 |
| Neuraminidase type II | Sigma | Cat#N6514 |
| Critical Commercial Assays | ||
| QuikChange II Site directed mutagenesis kit | Agilent | Cat#200555 |
| QuikChange II XL Site Directed Mutagenesis Kit | Agilent | Cat#200521 |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat#E0554S |
| BioLuxCypridina Luciferase Assay kit | NEB | Cat#E3309L |
| Fugene HD | Promega | Cat#E2311 |
| LipofectamineRNAiMAX | ThermoFisher Scientific | Cat#13778075 |
| Alexa Fluor 488 NHS Ester (Succinimidyl Ester) | ThermoFisher Scientific | Cat#A200000 |
| Lipofectamine 3000 | ThermoFisher Scientific | Cat#L3000008 |
| Experimental Models: Cell Lines | ||
| SVG-A cells (human fetal glial cells) | Major et al., 1985 | N/A |
| HEK293A cells | ATCC | Cat#CRL-1573; RRID:CVCL_0045 |
| HEK293T cells | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| Lenti-X HEK293T cells | Takara | cat#632180 |
| CV-1 (African green monkey) | ATCC | Cat#CCL-70; RRID:CVCL_0229 |
| Oligonucleotides | ||
| SignalSilence β-arrestin1 siRNA | Cell Signaling Technologies | Cat#6218 |
| SignalSilence Control siRNA | Cell Signaling Technologies | Cat#6568 |
| Mutagenesis primers see tables | This paper | N/A |
| Recombinant DNA | ||
| lentiCRISPRv2 plasmid-puromycin | Addgene | Cat#52961; RRID: Addgene_52961 |
| lentiCRISPRv2 plasmid-neomycin | This paper | N/A |
| lentiCRISPRv2 plasmid-hygromycin | This paper | N/A |
| pCMV-dR8.2 | Addgene | Cat#8455; RRID: Addgene_8455 |
| pCMV-VSV-G | Addgene | Cat#8454; RRID: Addgene_8454 |
| 5-HT2AR-pcDNA 3.1 | UMR cDNA resource center, University of Missouri-Rolla | Cat#HTR02A0001 |
| 5-HT2BR-pcDNA 3.1 | UMR cDNA resource center, University of Missouri-Rolla | Cat#HTR02B0000 |
| 5-HT2CR-pcDNA 3.1 | UMR cDNA resource center, University of Missouri-Rolla | Cat#HTR02C0000 |
| 5-HT2AR-YFP | Assetta et al., 2013; Maginnis et al., 2010 | N/A |
| 5-HT2BR-YFP | Assetta et al., 2013; Maginnis et al., 2010 | N/A |
| 5-HT2CR-YFP | Assetta et al., 2013; Maginnis et al., 2010 | N/A |
| 5-HT2AR-YFP-P180A | This paper | N/A |
| 5-HT2BR-YFP-P160A | This paper | N/A |
| 5-HT2CR-YFP-P159A | This paper | N/A |
| 5-HT2AR-pcDNA 3.1-trunc before DRY-His | This paper | N/A |
| 5-HT2AR-pcDNA 3.1-trunc after DRY-His | This paper | N/A |
| 5-HT2BR-pcDNA 3.1-trunc before DRY-His | This paper | N/A |
| 5-HT2BR-pcDNA 3.1-trunc after DRY-His | This paper | N/A |
| 5-HT2CR-pcDNA 3.1-trunc before DRY-His | This paper | N/A |
| 5-HT2CR-pcDNA 3.1-trunc after DRY-His | This paper | N/A |
| 5-HT2AR-His-pcDNA 3.1 | This paper | N/A |
| 5-HT2BR-His-pcDNA 3.1 | This paper | N/A |
| 5-HT2CR-His-pcDNA 3.1 | This paper | N/A |
| VP1-pwP vector | Gee et al., 2013 | N/A |
| VP2-ph2p vector | Gee et al., 2013 | N/A |
| VP3-ph2p vector | Gee et al., 2013 | N/A |
| phSv40-Cluc | Gee et al., 2013 | N/A |
| Software and Algorithms | ||
| Blobfinder | Center for Image Analysis (Uppsala University) | http://www.cb.uu.se/amin/BlobFinder/ |
| Fiji | Schindelin et al., 2012 | https://fiji.sc/ |
| CRISPR design | Zhang Lab | http://zlab.bio/guide-design-resources |
| QuikChange primer design tool | Agilent | https://www.chem.agilent.com/store/primerDesignProgram.jsp |
| NEBaseChanger tool | NEB | http://nebasechanger.neb.com/ |
Highlights.
JC virus interacts transiently with each of the 5-HT2 receptors(5-HT2Rs)
CRISPR-Cas9 genetic knockout of 5-HT2 receptors reduces infection
The second intracellular loop of each 5-HT2 receptor is sufficient for infection
A proline in each 5-HT2 receptor is critical to support infection
ACKNOWLEDGMENTS
Work in the Atwood lab was supported by grants from the National Institute of Neurological Disease and Stroke, R01NS043097 and P01NS065719. Work in the Maginnis lab was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the NIH under grant number P20GM103423 (M.S.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.067.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Assetta B, and Atwood WJ (2017). The biology of JC polyomavirus. Biol.Chem 398, 839–855. [DOI] [PubMed] [Google Scholar]
- Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O’Hara BA, Allen Ramdial SA, and Atwood WJ (2013).5-HT2 receptors facilitate JC polyomavirus entry. J. Virol 87, 13490–13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, and Schmid CL (2010). Serotonin receptor signaling and regulation viab-arrestins. Crit. Rev. Biochem. Mol. Biol 45, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, et al. (2009). Progressive multifocal leukoencephalopathy after rituximab therapy in HIVnegative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 113, 4834–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinque P, Bossolasco S, Brambilla AM, Boschini A, Mussini C, Pierotti C, Campi A, Casari S, Bertelli D, Mena M, and Lazzarin A (2003). The effect of highly active antiretroviral therapy-induced immune reconstitution on development and outcome of progressive multifocal leukoencephalopathy: study of 43 cases with review of the literature. J. Neurovirol 9(Suppl 1), 73–80. [DOI] [PubMed] [Google Scholar]
- Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, et al. (2004). The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306, 1380–1383. [DOI] [PubMed] [Google Scholar]
- Garvey L, Winston A, Walsh J, Post F, Porter K, Gazzard B, Fisher M, Leen C, Pillay D, Hill T, et al. ; UK Collaborative HIV Cohort (CHIC) Study Steering Committee (2011). HIV-associated central nervous system diseases in the recent combination antiretroviral therapy era. Eur. J. Neurol 18, 527–534. [DOI] [PubMed] [Google Scholar]
- Gee GV, O’Hara BA, Derdowski A, and Atwood WJ (2013). Pseudovirusmimics cell entry and trafficking of the human polyomavirus JCPyV. Virus Res 178, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelber EI, Kroeze WK, Willins DL, Gray JA, Sinar CA, Hyde EG, Gurevich V, Benovic J, and Roth BL (1999). Structure and function of the third intracellular loop of the 5-hydroxytryptamine2A receptor: the third intracellular loop isa-helical and binds purified arrestins. J. Neurochem 72, 2206–2214. [DOI] [PubMed] [Google Scholar]
- Haley SA, and Atwood WJ (2017). Progressive multifocal leukoencephalopathy: endemic viruses and lethal brain disease. Annu. Rev. Virol 4, 349–367. [DOI] [PubMed] [Google Scholar]
- Khanna N, Elzi L, Mueller NJ, Garzoni C, Cavassini M, Fux CA, Vernazza P, Bernasconi E, Battegay M, and Hirsch HH; Swiss HIV Cohort Study (2009). Incidence and outcome of progressive multifocal leukoencephalopathy over 20 years of the Swiss HIV Cohort Study. Clin. Infect. Dis 48, 1459–1466. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, and Tyler KL (2005). Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med 353, 369–374. [DOI] [PubMed] [Google Scholar]
- Leysen JE, Gommeren W, Van Gompel P, Wynants J, Janssen PF, and Laduron PM (1985). Receptor-binding properties in vitro and in vivo of ritanserin: A very potent and long acting serotonin-S2 antagonist. Mol. Pharmacol 27, 600–611. [PubMed] [Google Scholar]
- Liu CK, and Atwood WJ (2001). Propagation and assay of the JC virus.Methods Mol. Biol 165, 9–17. [DOI] [PubMed] [Google Scholar]
- Maginnis MS, Haley SA, Gee GV, and Atwood WJ (2010). Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J. Virol 84, 9677–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO (2010). Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med 61, 35–47. [DOI] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, and Sever J (1985). Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc. Natl. Acad. Sci. USA 82, 1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S, Oakley RH, Kim KM, Caron MG, and Barak LS (2006). A beta-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J. Biol. Chem 281, 2932–2938. [DOI] [PubMed] [Google Scholar]
- Mayberry CL, Soucy AN, Lajoie CR, DuShane JK, and Maginnis MS (2019). JC polyomavirus entry by clathrin-mediated endocytosis is driven by β-arrestin. J. Virol 93, e01948–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy JD, and Roth BL (2015). Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther 150, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutkine I, Quentin E, Guiard BP, Maroteaux L, and Doly S (2017). Heterodimers of serotonin receptor subtypes 2 are driven by 5-HT 2C protomers. J. Biol. Chem 292, 6352–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CD, Carney DW, Derdowski A, Lipovsky A, Gee GV, O’Hara B, Williard P, DiMaio D, Sello JK, and Atwood WJ (2013). A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. MBio 4, e00729–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U, Maginnis MS, Palma AS, StrÖh LJ, Nelson CD, Feizi T, Atwood WJ, and Stehle T (2010). Structure-function analysis of the human JC polyomavirus establishes the LSTcpentasaccharide as a functional receptor motif. Cell Host Microbe 8, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norkin LC, and Ouellette J (1976). Cell killing by simian virus 40: variation in the pattern of lysosomal enzyme release, cellular enzyme release, and cell death during productive infection of normal and simian virus 40-transformed simian cell lines. J. Virol 18, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara BA, and Atwood WJ (2008). Interferon beta1-a and selective anti5HT(2a) receptor antagonists inhibit infection of human glial cells by JC virus. Virus Res 132, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pho MT, Ashok A, and Atwood WJ (2000). JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol 74, 2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, Benmerah A, Tosoni D, Di Fiore PP, and Atwood WJ (2004). A JC virus-induced signal is required for infection of glial cells by a clathrin-and eps15-dependent pathway. J. Virol 78, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 28, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, and Lefkowitz RJ (2011).b-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci 32, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacante DA, Traub R, and Major EO (1989). Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 170, 353–361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.