

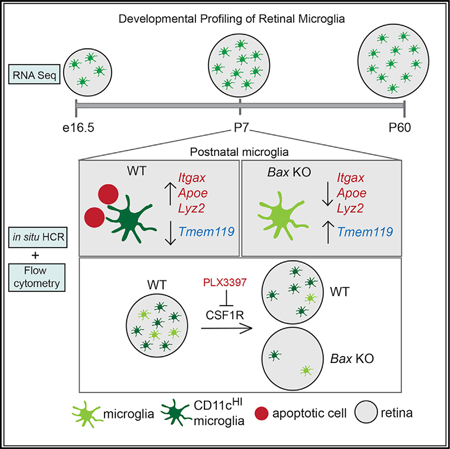
SUMMARY
Microglia have important remodeling functions in neurodevelopment, aging, and disease, with evidence for molecular diversity. However, the signaling pathways and environmental cues that drive diverse states of microglia are incompletely understood. We profiled microglia of a discrete developing CNS region, the murine retina. We found distinct transcriptional signatures for retinal microglia across development and peak postnatal density of a population that resembles aging and disease-associated microglia (DAM) and CD11c+ microglia of developing white matter. While TREM2 signaling modulates the expression of select genes, the DAM-related signature is significantly reduced in retinas lacking Bax, a proapoptotic factor required for neuronal death. Furthermore, we found postnatal retinal microglia highly expressing CD11c are resistant to loss or inhibition of colony stimulating factor 1 receptor (CSF1R), while most microglia can be eliminated in Bax knockout retina. Thus, developmental apoptosis promotes a microglia gene signature linked to CSF1R independence that shares features with microglia in developing white matter and in disease.
Graphical Abstract
In Brief
Microglia have essential remodeling functions in the CNS, especially in development. Anderson et al. profile retinal microglia across development and show that apoptosis in early postnatal retina promotes a microglia gene signature related to aging and disease, as well as independence from CSF1R signaling for survival.
INTRODUCTION
Microglia, the resident macrophages of the CNS, are dynamic cells with a spectrum of functions during development, aging, and disease (reviewed in Colonna and Butovsky, 2017 and Li and Barres, 2018). During development, brain microglia progress through stepwise changes in gene expression and chromatin accessibility (Matcovitch-Natanetal.,2016) and gradually acquire a homeostatic gene expression signature (Bennett et al., 2016; Butovsky et al., 2014; Zhang et al., 2014). This developmental program is partially governed by transcription factors required for microglial identity (Buttgereit et al., 2016; Holtman et al., 2017; Kierdorf et al., 2013; Matcovitch-Natan et al., 2016) and survival and differentiation signals, such as transforming growth factor-β (TGF-β), colony-stimulating factor 1 (CSF1) and interleukin 34 (IL-34) (Bohlen et al., 2017; Butovsky et al., 2014; Buttgereit et al., 2016; Chitu et al., 2016; Gosselin et al., 2014). The core identity of microglia, distinct from other macrophages, is intrinsic and dependent upon ontogeny (Bennett et al., 2018). However, environmental factors and functional demands of the CNS heavily influence microglial functional state and gene expression (Bennett et al., 2018; Gosselin et al., 2014; Lavin et al., 2014). This is strikingly apparent during development, where there is remarkable diversity of microglia phenotype (De et al., 2018; Hagemeyer et al., 2017; Hammond et al., 2019; Li et al., 2019; Wlodarczyk et al., 2017). But how specific developmental events drive changes in microglial states is largely unknown.
Interestingly, developmental microglia have important functions that parallel microglia in disease (Anderson and Vetter, 2019; Hammond et al., 2018). Comparisons of aging and disease models have identified a shared gene expression signature for rare disease-associated microglia (DAM), or microglial neurodegenerative phenotype (MGnD), which are characterized by the upregulation of genes involved in phagocytosis and lipid metabolism and concurrent downregulation of homeostatic genes (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017). Many DAM genes are also enriched in transient subsets of developmental microglia but are absent in healthy adult microglial cells (Butovsky et al., 2014; Hagemeyer et al., 2017 ; Hammond et al., 2019; Li et al., 2019; Wlodarczyk et al., 2017). These findings suggest the existence of shared mechanisms and roles for microglia in developmental and disease processes that have yet to be fully understood.
The retina is a discrete CNS region with well-defined developmental processes. To link microglial transcriptional states to key developmental events, we profiled retinal microglia from embryonic age to adulthood. We found that microglial developmental gene expression in the retina shares similarities with microglia that are associated with aging, disease, and developing white matter. We provide evidence that this gene expression profile is largely driven by developmental apoptosis and coupled with independence from CSF1 receptor (CSF1R) signaling. In addition, we found that TREM2 modulates the expression of select, but not all, DAM-related genes. Thus, we identify and characterize a population of microglia in the developing retina that may broaden our understanding of microglial function in development aging and disease.
RESULTS
Retinal Microglia Have Distinct Transcriptional Signatures across Development
Microglia are present in the mouse retina as early as e11.5 (Santos et al., 2008) and have important developmental functions (Anderson et al., 2019; Checchin et al., 2006; Frade and Barde, 1998; Huang et al., 2012; Jobling et al., 2018). They are branched at e12.5 with increasing morphological complexity over time, and a more amoeboid appearance at postnatal day (P)7, (Figure 1A). To determine whether retinal microglia follow a stepwise developmental program, we isolated GFP+CD45+CCR2− cells from CX3CR1-GFP mouse (Jung et al., 2000) retinas at embryonic day (e)16.5, P7, and adult (P60) by fluorescence-activated cell sorting (FACS) (Figures 1B and 1C). We performed highthroughput RNA sequencing on purified microglia and whole retinas at each time point (Table S1). Principal component analysis verified that gene expression of microglia and whole retina samples varied primarily by age (PC1) and sample type (PC2) (Figure 1D). Gene expression was highly correlated between samples of the same type, regardless of age (Figure S1A). The sorted microglia samples were enriched for 25 highly expressed or specific microglial genes (Butovsky et al., 2014) compared with whole retina (Figure S1C), demonstrating efficient isolation.
Figure 1. Retinal Microglia Express Core Transcription Factors and Adult Brain Microglial Genes but Have Unique Developmental Transcriptional States.
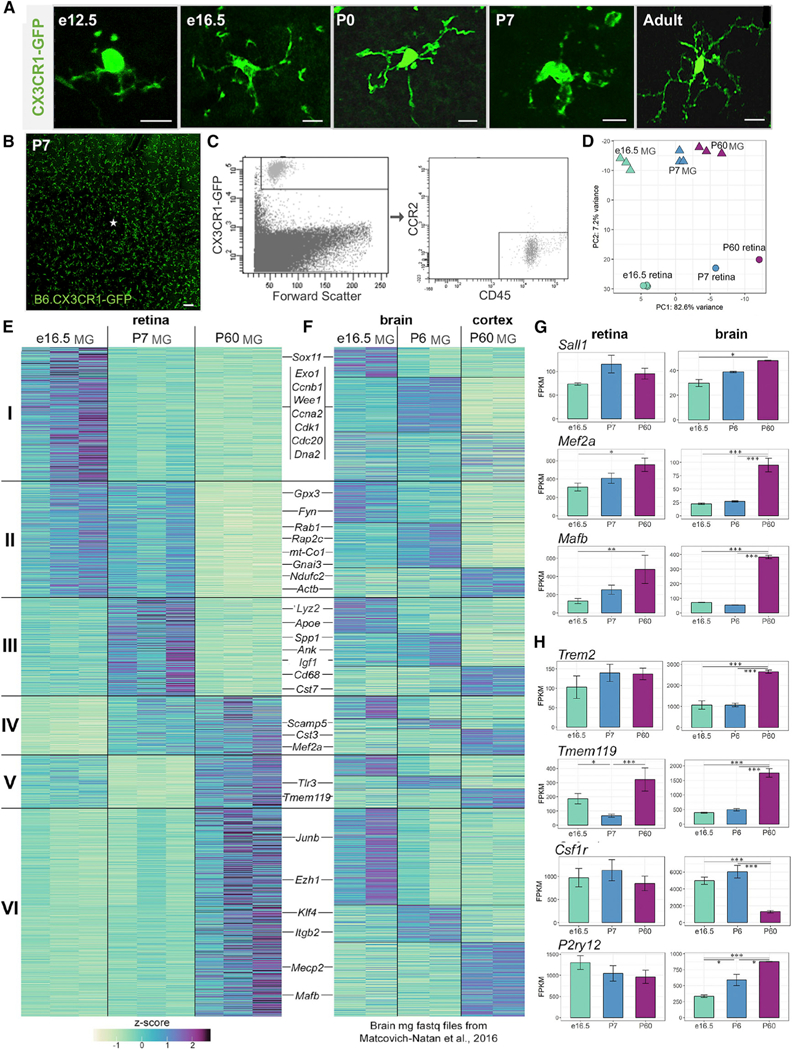
(A) Confocal images of individual microglia in retinal whole mounts. CX3CR1-GFP (green). Scale bar, 10 µm.
(B) Retinal whole mount of B6.CX3CR1-GFP at P7. Scale bar, 100 µm.
(C) Gating strategy for sorting retinal microglia.
(D) Principal component analysis of RNA-seq datasets for whole retina samples (circles) and sorted microglia (MG) samples (triangles) at e16.5 (green), P7 (blue), and P60 (purple).
(E) Heatmap of scaled fragments per kilobase of transcript per million mapped reads (FPKM) values for differentially expressed genes, organized into 6 clusters.
(F) Brain microglia gene expression compared with the clusters in 1E, organized into 18 sub-clusters.
(G and H) Mean FPKM ± SEM for microglial transcription factors (G) and transmembrane proteins (H). *padj ≤ 0.05, **padj≤ 0.01, ***padj ≤ 0.001.
We identified 2,872 microglia-enriched genes (≥20-fold above whole retina) that were differentially expressed (adjusted p value [padj] ≤ 0.05) between any two time points by DESeq2. We then defined 6 gene clusters with varied expression kinetics by K-means clustering (Figures 1E and S1B). Cluster I genes with high embryonic expression were involved in cell division and DNA repair. Cluster II genes included those associated with cytoskeletal remodeling and exocytosis. Genes with peak expression postnatally (cluster III) included those involved with lipid metabolism, as well as lysosomal and phagocytic function. We saw an emergence of microglia-specific markers postnatally (cluster IV). Genes transiently decreased at P7 (cluster V) included genes involved in apoptosis as well as the microglia specific gene Tmem119. Cluster VI contained adult brain microglia genes, including Mafb, which is important for adult microglial gene expression in brain (Matcovitch-Natan et al., 2016). Thus, retinal microglia have distinct transcriptional states from embryonic age to adulthood.
To compare with the developmental expression patterns of brain microglia, we analyzed published brain datasets at age-matched time points (Matcovitch-Natan et al., 2016). Starting with raw sequencing data, we selected the same 2,872 genes, organized them by retinal microglia clusters, and further subdivided those clusters by K-means (Figure 1F). While some genes had similar developmental expression patterns in retina and brain, the clustering patterns were mostly distinct (Figure 1F). We next asked whether retinal microglia express key genes required for brain microglia development and identity. Transcription factors essential for adult homeostatic brain microglia, Mef2a and Mafb, increased from embryonic to adulthood, similar to brain microglia. Other important transcription factors, including Irf8, Runx1, Spi1 (PU.1), and Sall1, were highly expressed by e16.5 and not differentially expressed over time (Figures 1G and Table S1) (Buttgereit et al., 2016; Holtman et al., 2017; Kierdorf et al., 2013). Several core microglial genes, including Tmem119 and P2ry12, were also expressed (Figure 1H). Therefore, retinal microglial identity and development likely requires the same lineage-specific factors as brain microglia, but the varied expression of genes in retina may be due to the influence of local cues.
Postnatal Retinal Microglia Resemble Microglia Associated with Aging, Disease, and Developing White Matter
To link retinal microglial gene expression with developmental events, we focused on the postnatal time period, which is characterized by significant remodeling (Silverman and Wong, 2018). We observed similarities between cluster III, containing genes that peaked at P7, and published datasets of microglia associated with aging and disease (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017) and developing white matter (Wlodarczyk et al., 2017). We compared each of our clusters with the top 500 upregulated genes from these published studies: microglia from developing white matter (CD11c+) (Wlodarczyk et al., 2017),DAM genes (Keren-Shaul et al., 2017), multiple models of aging and disease, and microglia exposed to lipopolysaccharide (LPS) (Holtman et al., 2015). Cluster III was highly enriched for DAM, aging and disease, and CD11c+signatures but not acute activation signature (LPS) by hypergeometric test (Figure 2A). To corroborate this, we performed gene set enrichment analysis via GAGE (generally applicable gene-set enrichment) for all genes expressed in the retina factoring in the level of gene expression (Luo et al., 2009) (Figure 2B). We found that CD11c+, DAM, and aging and disease microglia genes were significantly increased in our dataset from e16.5 to P7 and significantly decreased from P7 to P60. Many DAM-related genes were highly and selectively expressed at P7 (Figures 2C and S2A). There were 42 genes from cluster III that overlapped with both the top 500 genes altered in aging and disease and the top 500 genes upregulated in CD11c+microglial profiles (Figure 2D; Table S2).
Figure 2. Postnatal Retinal Microglia Resemble Aging and Disease-Associated Microglia and CD11c+ Microglia of the Developing White Matter.
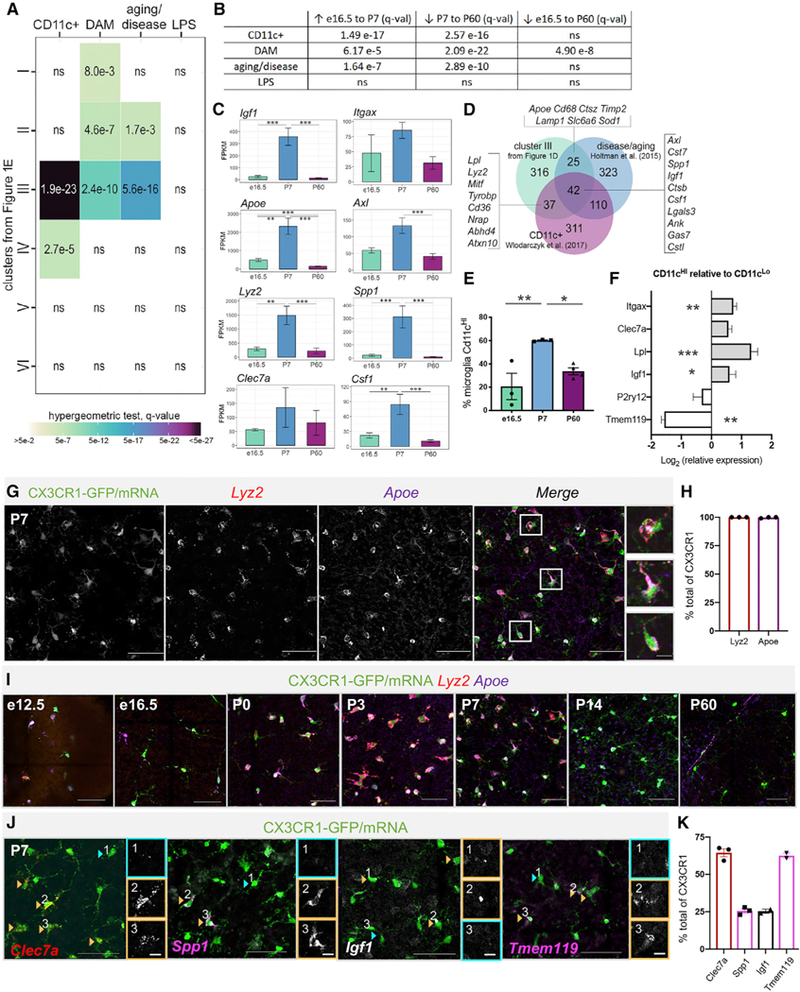
(A) Comparison by hypergeometric test of retinal microglia clusters (Figure 1E) with upregulated genes from published datasets.
(B) GAGE of top 500 upregulated genes from published datasets to all retinal microglia gene counts. ns= not significant.
(C) Mean FPKM ± SEM for genes overlapping with disease and aging datasets. *padj≤ 0.05, **padj ≤ 0.01,***padj≤ 0.001.
(D) Venn diagram of overlap between cluster III genes from Figure 1E, aging and disease genes, and CD11c+microglia of developing white matter.
(E) Average percentage of Cd11cHImicroglia (CD45+GFP+) at e16.5, P7 and P60 by flow cytometry (±SEM; n = 3 each, 2 retinas/sample); One-way ANOVA F(2,7) = 9.714, p = 0.0096 and Tukey’s multiple comparisons. *p ≤ 0.05,**p ≤ 0.01.
(F) Log-transformed relative gene expression in CD11cHI versus CD11cLo microglia from the same samples by qPCR. (±SEM; n = 5). Two-tailed paired t test *p ≤ 0.05, **p≤ 0.01, ***p ≤ 0.001.
(G) Max projected confocal image of HCR wholemount P7 retina. Scale bar, 50 µm. Inset scale bar, 10 µm.
(H) Percent CX3CR1-GFP+ cells expressing Apoe and Lyz2 by HCR (±SEM; n = 3 each).
(I) Max projected confocal images of HCR at e12.5, e16.5, P0, P3, P7, P14, and P60. Scale bar, 50µm.
(J) Heterogeneous expression by HCR of Clec7a (red), Spp1 (pink), Igf1 (white), and Tmem119 (pink) in Cx3cr1 expressing cells (green) at P7. Orange arrowheads and boxes indicate high expression and blue, low or no expression. Scale bar, 50 µm. Insets are individual channels without Cx3cr1; scale bar, 10 µm.
(K) Percent of CX3CR1-GFP+ cells expressing each gene by HCR (±SEM; n ≥ 2).
Although we found that DAM-related gene expression was particularly high in early postnatal retinal microglia, it was also present, albeit at much lower levels, embryonically. Thus, we asked whether a subpopulation of microglia expresses DAM genes with changing proportions over time, or whether DAM-related genes are expressed in all retinal microglia with varying levels across development. Since CD11c is highly expressed in DAM, in microglia adjacent to Aβ plaques in Alzheimer’s disease, and in microglia in white matter of developmental, aged or diseased brains (Kamphuis et al., 2016; Raj et al., 2017; Wlodarczyk et al., 2017; Yin et al., 2017), we first quantified the proportion of microglia highly expressing CD11c (CD45+GFP+CD11cHI) using flow cytometry (Figure S2B). We found that 20% were CD11cHI at 16.5, 60% at P7 and 33% at P60 (Figure 2E). To determine whether CD11cHI microglia more highly expressed DAM-related genes, we performed qPCR on sorted CD11cHI versus CD11cLo microglia at P7. Relative to CD11cLo, CD11cHI were enriched for the DAM-related genes, Lpl and Igf1, and had low expression of Tmem119 (Figure 2F).
Previous findings in disease report that not all CD11c+ microglia are DAM (Keren-Shaul et al., 2017). Thus, to directly determine the proportion of microglia expressing key DAM genes, we performed in situ hybridization chain reaction (HCR) (Choi et al., 2018) on retinal whole mounts, selecting genes from cluster III that were intersecting with genes in aging and disease microglia (Apoe), in developing white matter CD11c+ microglia (Lyz2), or both (Spp1 and Igf1) (see Figure 2D). We found that Apoe and Lyz2, the most highly upregulated DAM-related genes in our dataset, were expressed in virtually all Cx3cr1+microglia at P7 (Figures 2G and 2H). They were only expressed in a subset of retinal microglia embryonically (e12.5 and e16.5), showed peak expression at P3, and were significantly reduced by P14 and P60, consistent with RNA sequencing (RNA-seq) (Figure 2I). Only a proportion of Cx3cr1+microglia expressed Clec7a (64%), Spp1 (25%), and Igf1 (25%) at P7 (Figures 2J and 2K). In adult retina, we observed that a small number of Cx3cr1+ cells expressed DAM-related genes (Lyz2, Apoe, Itgax, Clec7a) (Figures S2C and S2E), but many were perivascular (data not shown).
In disease, microglia downregulate homeostatic genes, including Tmem119, Siglech, and P2ry12 (Keren-Shaul et al., 2017; Krasemann et al., 2017). Of these, only Tmem119 was downregulated at P7 according to our sequencing data (cluster V, Figures 1E and 1H). Only 60% of P7 microglia expressed Tmem119 by HCR (Figures 2J and 2K), as compared to almost 100% in adult (Figure S2E). HCR also showed that roughly 50%–60% of P7 microglia expressed Itgax, which encodes CD11c (Figures S2C and S2D), consistent with our flow cytometry analysis (Figure 2E). Itgax cells variably colocalized with Clec7a, Igf1, and Tmem119, but we noted that Tmem119+Itgax+ cells commonly had inverse levels of expression (Figures S2C and S2D). Therefore, developing retinal microglia are a heterogeneous population, with increased expression of DAM-related genes from embryonic to postnatal periods.
Developmental Apoptosis Promotes Microglial DAM-Related Gene Expression
We hypothesized that developmental death of retinal neurons might be an important driver of this signature since large waves of apoptosis occur during the first postnatal week (Farah and Easter, 2005), and exposure to apoptotic cells can trigger DAM-related microglial gene expression (Ayata et al., 2018; Krasemann et al., 2017). Therefore, we analyzed retinas of mice deficient in Bax, a pro-apoptotic gene that is essential for developmental death of neurons in the postnatal retina (Péquignot et al., 2003). In non-neuronal cells, Bax is redundant to Bak1 (Degenhardt et al., 2002; Lindsten et al., 2000; Wei et al., 2001), and Bax is not directly required for microglia activation (Mac Nair et al., 2016). qPCR on sorted microglia (CD45+CD11b+) from Bax knockout (KO) retinas compared with littermate controls revealed a pronounced reduction in expression of DAM genes and a robust increase of Tmem119 (Figure 3A). By HCR, we found that the levels of Apoe and Lyz2 were diminished and the proportion of Cx3cr1+microglia expressing either gene was significantly reduced (Figures 3B and 3D). Similarly, a reduced proportion of microglia expressed Itgax in Bax KO retinas (Figures 3C and 3D), consistent with flow cytometry results (Figure 3E). Concurrently, the proportion of Tmem119expressing microglia increased to 91% (Figure 3D). We noted a decrease in total density of microglia at P7 in the Bax KO compared with wild-type (WT) littermate controls by flow cytometry (data not shown; p = 0.001) and by HCR for Cx3cr1 (Figures 3B and 3C), but this was likely not due to reduced proliferation since previous reports find limited microglia proliferation in postnatal retina (Santos et al., 2008). Overall, these findings show that neuronal apoptosis has a significant influence onDAM-related gene expression in postnatal retinal microglia.
Figure 3. Loss of Bax Reduces DAM-Related Gene Expression in Microglia.
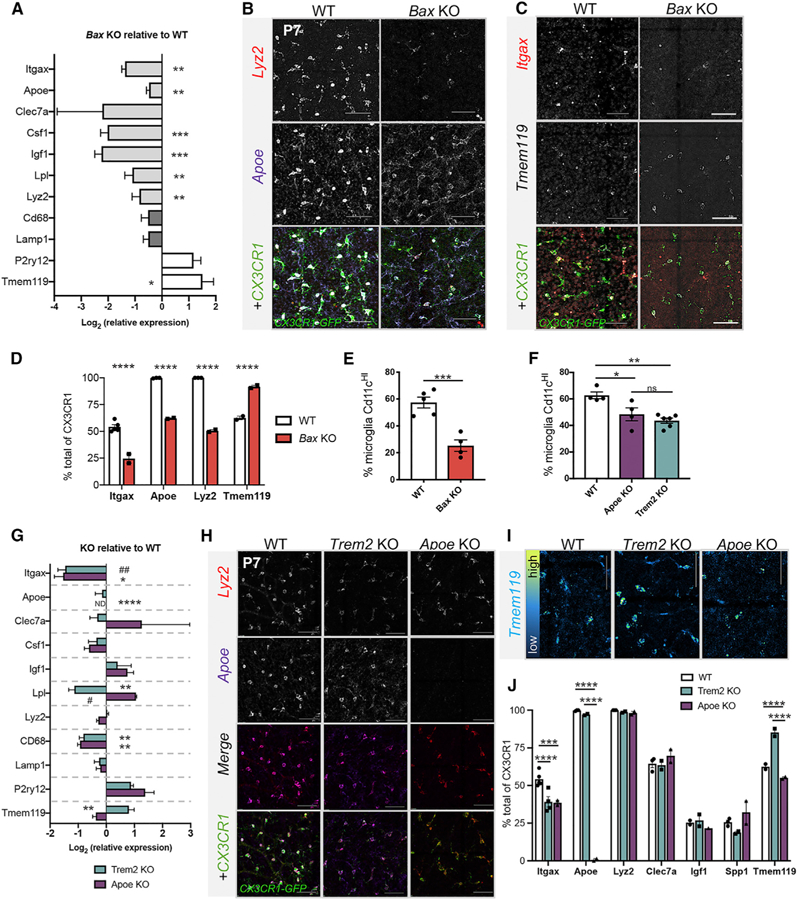
(A) Log-transformed relative gene expression by qPCR in sorted microglia from Bax KO relative to littermate WT controls. (±SEM; n = 4 KO except Lyz2 n = 2; n = 5 WT) Two-tailed unpaired t test *p ≤ 0.05, **p ≤ 0.01,***p ≤ 0.001.
(B and C) Confocal images of HCR retinal wholemounts of P7 WT (left) BaxKO (right) for Lyz2, Apoe, and Cx3cr1 (B) or Itgax, Tmem119, and Cx3cr1 (C). Scale bar, 50 µm.
(D) Percent of Cx3cr1+ cells expressing each gene by HCR (±SEM; n ≥ 2). Two-way ANOVA: interaction, F(3,13) = 122.4, p < 0.0001; gene F(3,13) = 190.2, p <0.0001; genotype F(1,13) = 215.5, p < 0.0001 and Sidak’s multiple comparison tests. ****p≤ 0.0001. WT data from Figures 2H, 2K, and S2D.
(E) Percent (±SEM) of CD11cHIof total CD45+CD11b+microglia from P7 WT (n = 5) and Bax KO (n = 4). Unpaired t test t(7) = 5.415 ***p = 0.001.
(F) Percent (±SEM) of CD11cHI of total CD45+GFP+(WT) or CD45+CD11b+ (KOs) microglia from P7 WT (n = 4), Apoe KO (n = 4), and P6/7 Trem2 KO (n = 6). Oneway ANOVA F(2,11) = 10.33 p = 0.003 and Tukey’s multiple comparisons *p≤ 0.05, +**p≤ 0.01.
(G) Log-transformed relative gene expression by qPCR (±SEM) in sorted microglia from KOs relative to WT controls. Apoe KO (n = 3); Trem2 KO (n = 6 except n = 3 for Lyz2, Cd68, Lamp1, and P2ry12); WT (n ≥ 6 except n ≥ 3 Itgax, Lyz2, and Lpl). Two-tailed unpaired t test *p ≤ 0.05, **p ≤ 0.01. ND, not detectable. #, Welch’s correction. (H)
(H and I) Max projected confocal images of HCR P7 retinal wholemounts from WT (left), Apoe KO (middle), and Trem2 KO (right) for Lyz2, Apoe, and Cx3cr1 (H) or Tmem119 (I). Tmem119 is pseudo-colored to reflect levels in (I). Scale bar, 50 µm.
(J) Percent of Cx3cr1+ cells expressing each gene by HCR (±SEM; n ≥ 2). Two-way ANOVA: interaction, F(12,29) = 59.01, p < 0.0001; gene F(6,29) = 248.0, p < 0.0001; genotype F(2,29) = 67.96, p < 0.0001 and Tukey’s multiple comparison tests. ***p ≤ 0.001, ****p ≤ 0.0001. WT data from Figures 2H, 2K, and S2D.
Next, we sought to identify the pathways driving this signature. Since TREM2 can mediate the phagocytosis of apoptotic cells (Takahashi et al., 2005) and is required for the full acquisition of the DAM signature (Keren-Shaul et al., 2017), we analyzed Trem2 KO retinas. We also examined retinas deficient in Apoe since ApoE is a ligand for TREM2 and is required for expression of select DAM genes in disease (Krasemann et al., 2017). The proportion of CD11cHI microglia at P7 was slightly, but significantly, decreased in both Apoe (~48%) and Trem2 KO (~43 %) retinas relative to WT(~60%) (Figure 3F). Consistent with this, qPCR on sorted microglia from both Apoe and Trem2 KO retinas showed diminished Itgax expression (Figure 3G). Lpl was reduced in Trem2 KOs and lysosomal gene, Cd68, was significantly decreased in microglia from both KOs, but other DAM-related genes were not significantly altered (Figure 3G). Homeostatic gene, Tmem119, was selectively increased in the absence of Trem2 (Figure 3G). By HCR, we confirmed that the proportion of Itgax-expressing microglia was reduced in both KOs (Figures 3H and 3J), but the percent of microglia expressing DAM genes (Apoe, Lyz2, Clec7a, Igf1, Spp1) was not dramatically reduced in either KO (Figures 3H and 3J). Consistent with qPCR (Figure 3G), Tmem119 levels and Tmem119+microglia were increased specifically in Trem2 KOs (Figures 3I and 3J). Thus, loss of Apoe slightly reduces the proportion of Itgax-expressing microglia but has little impact on other aspects of DAM gene expression,whileTrem2 contributes to the downregulation of Tmem119 and upregulation of select genes, including Lpl. Additional pathways, therefore, drive the full gene signature in postnatal retinal microglia.
CD11cHI and DAM-Related Gene Expression in Retinal Microglia Are Linked to CSF1R Independence
We next aimed to deplete postnatal retinal microglia by disrupting the CSF1R pathway. We first used a genetic approach to conditionally delete Csf1r from CX3CR1-expressing microglia, induced by tamoxifen treatment (Cx3cr1creERT2; Csf1rfl/fl, RosatdTomato). With this approach, we previously achieved a 70% reduction in microglial density in the embryonic retina (Anderson et al., 2019). Here, we administered tamoxifen to experimental and control littermates at P4 and P6 (and P8 in controls) (Figure 4A). We observed substantial persistence of retinal microglia in experimental animals with targeted deletion of CSF1R, despite efficient recombination (>95% tdTomato+) (Figure 4B). To corroborate our results, we administered CSF1R inhibitor, PLX3397 (PLX; Elmore et al., 2014) daily from P4 to P6 to CX3CR1-GFP pups (Figure 4C). Similarly, at P7, we found persistence of retinal microglia (Figure 4D), despite a clear loss of microglia in the brain (Figure S3A). We further validated the efficacy of PLX in eliminating retinal microglia by successful depletion in the adult retina (P140) (Figures S3B and S3C). PLX-treated P7 retinas had roughly 60% of WT density by flow cytometry, revealing that approximately one-third of microglia were susceptible to inhibition of CSF1R signaling at this age (Figures 4E and 4F). Persistence of postnatal retina microglia was not due to reduced CSF1R expression since Csf1r mRNA expression was maintained at P7 (Figure 1H), and flow cytometry confirmed that CSF1R protein was present on the cell surface in a similar proportion as adult microglia (Figure S3D).
Figure 4. CD11cHI Retinal Microglia Are More Resistant to Loss or Inhibition of CSF1R Signaling.
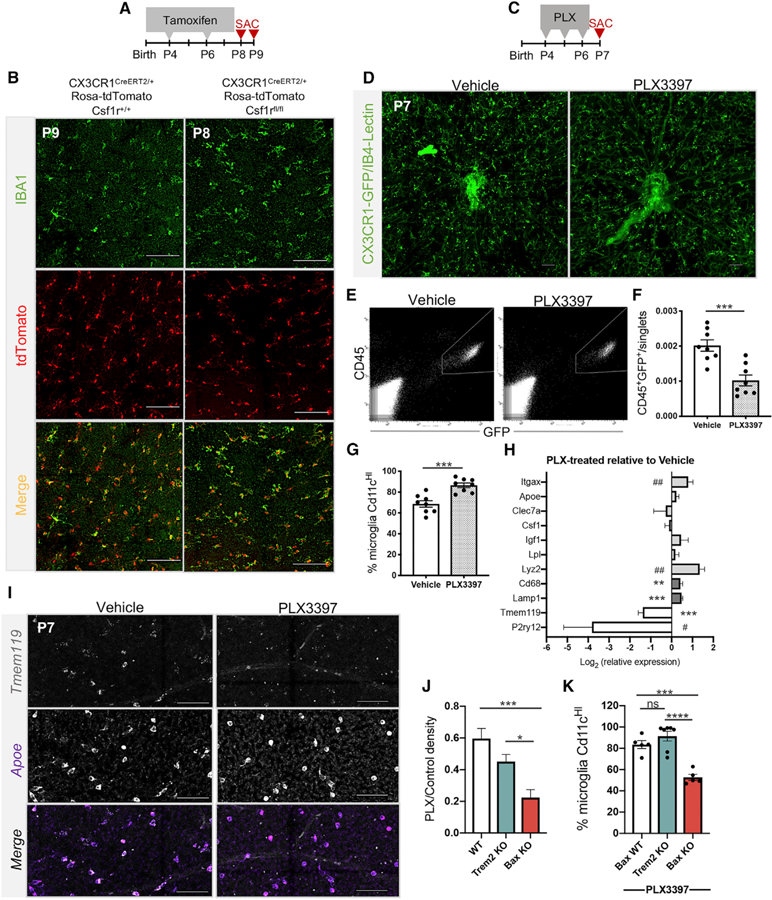
(A) Tamoxifen dosing regimen.
(B) Confocal images of retinal wholemounts of control at P9 (tamoxifen at P4, P6, and P8) (left) and homozygous floxed at P8 (tamoxifen at P4 and P6) (right). Scale bars, 100 µm. Experiment repeated twice, n = 4 animals each.
(C) PLX3397 (PLX) dosing regimen.
(D) Confocal images of retinal wholemounts at P7 from vehicle- (left) and PLX-treated (right) animals. Scale bar, 100 µm.
(E) Representative flow cytometry plot and gate for collecting GFP+CD45+ microglia in vehicle- (left) and PLX-treated (right) animals.
(F) Percent microglia (CD45+GFP+) of total single cells by flow cytometry. (±SEM; n = 8 each). Two-tailed unpaired t test t(14) = 4.438, ***p = 0.0006.
(G) Percent CD11cHIof total CD45+GFP+ microglia by flow cytometry. (±SEM; n = 8 each) Two-tailed unpaired t test t(14) = 4.662, ***p = 0.0004.
(H) Log-transformed relative gene expression by qPCR (±SEM) in sorted microglia from PLX-treated retinas relative to vehicle controls. WT (n≥8 except n = 5 Cd68, Lamp1, Lyz2, P2ry12); PLX (n = 9 except n = 8 for Itgax, n = 5 Cd68, Lamp1, Lyz2, P2ry12). Two-tailed unpaired t test *p ≤ 0.05, **p ɛ≤ 0.01, ***p ≤ 0.001. #, Welch’s correction.
(I) HCR of vehicle- and PLX-treated retinas. Scale bars, 50µm.
(J) Ratio (±SEM) of PLX-treated over genotyped-matched control density (CD45+Cd11b+/singlets). WT (n = 17); Trem2 KO (n = 6); Bax KO (n = 5). One-way BrownForsythe ANOVA F(2,24.29) = 11.76, p = 0.0003 and Games-Howell’s multiple comparisons test. *p ≤ 0.05, ***p ≤ 0.001.
(K) Percent (±SEM) of CD11cHImicroglia in total CD45+CD11b=population after PLX by flow cytometry. WT littermates (n = 5), Trem2 KO (n = 7); Bax KO (n = 5). One-way ANOVA F(2,14) = 24.55, p < 0.0001 and Tukey’s multiple comparisons test ***p = 0.0005, ****p < 0.0001.
See also Figures S3 and S4.
Although microglia density (CD45+GFP+) was reduced by PLX treatment, the proportion of CD11cHI microglia was increased to nearly 90% (Figure 4G), suggesting enrichment for this population. To test whether the resilient cells more highly expressed DAM-related genes, we performed qPCR on sorted microglia in vehicle- and PLX-treated animals. PLX-treated microglia had increased expression of Itgax, Lyz2, Cd68, and Lamp1 and reduced expression of Tmem119 and P2ry12 relative to vehicle controls (Figure 4H). HCR on vehicle- and PLX-treated P7 retinas found a reduced proportion of microglia expressing Tmem119 following PLX treatment compared with controls (Figure 4I). Thus, microglia resistant to loss of CSF1R signaling are preferentially CD11cHI, are enriched for phagocytic genes, and show reduced Tmem119 expression.
To test whether high CD11c and phagocytic gene expression are linked to independence from CSF1R signaling, we administered PLX daily from P4 to P6 (Figure 4C) to Bax KO and Trem2 KO pups and quantified the density of microglia at P7 with flow cytometry. Trem2 KO retinas treated with PLX had slightly reduced microglia density (45%) compared with WT PLX treated retinas (59%) but did not reach significance (Figure 4J), in line with the modest reduction in the proportion of CD11cHI microglia (Figure 3F). In contrast, P7 Bax KO retinas, which had a drastic reduction in CD11cHI and DAM-related and phagocytosis genes (Figures 3A–3E), had significantly reduced microglial density (22%) compared with both WT and Trem2 KO following PLX treatment (Figures 4J and S4E). To validate our flow cytometry data, we confirmed that all GFP+ microglia were CD11b+ and nearly all CD11b+ cells were GFP+, regardless of treatment (Figures S4A–S4D). Over 90% of remaining microglia in Trem2 KOs were CD11cHI (Figure 4K), which is similar to the enrichment we observed in WT after PLX (Figure 4G). Notably, the proportion of CD11cHI microglia in Bax KO retina was increased to 47% after PLX treatment (Figure 4K), up from 25% without treatment (Figure 3E). Altogether, these data suggest the majority of postnatal microglia do not require CSF1R signaling for survival, and this is linked to exposure to apoptotic neurons and the expression of high levels of CD11c and select DAM-related genes.
DISCUSSION
While microglia have emerging roles in developing and adult retina (Anderson et al., 2019; O’Koren et al., 2019; Silverman and Wong, 2018), we have limited understanding of microglial phenotype and function in this discrete CNS region. We find that retinal microglia are a heterogeneous population with distinctive transcriptional states across development, and that a proportion with peak density postnatally resemble microglia subsets previously identified in aging, disease, and in developing white matter. These findings suggest parallels in the mechanisms governing the molecular properties of microglia across multiple contexts including disease and provides a powerful platform for investigating the pathways involved.
There is growing appreciation that microglia share related remodeling functions in development and disease (Anderson and Vetter, 2019; Hammond et al., 2018), thus the overlap of postnatal retinal microglia with disease-related transcriptional profiles has interesting implications. Recent single-cell analysis of developing brain microglia has confirmed the presence of a distinct subpopulation in developing axon tracts and in proliferative neurogenic regions that is enriched for phagocytic genes and DAM-related genes (Hammond et al., 2019; Li et al., 2019). While retinal microglia are likely distinct from these other subpopulations, the overlap in gene expression is intriguing, especially since the retina is devoid of oligodendrocytes, myelination, and disease pathology.
The environmental factors driving the expression of these shared DAM-related genes have not been fully elucidated in any context. Here, we provide evidence that apoptotic developmental cell death (Braunger et al., 2014; Péquignot et al., 2003) is necessary for the DAM-related signature in postnatal retinal microglia. This may occur in other contexts, since microglia indeveloping white matter tracts phagocytose dying oligodendrocytes in the corpus callosum and cerebellum (Li et al., 2019), and the introduction of apoptotic cells into adult cortex increases Apoe and Clec7a expression in microglia (Krasemann et al., 2017). In addition, microglia in the adult cerebellum are exposed to cell death, are phagocytic, and express DAM-related genes (Ayata et al., 2018). Thus, cell death and phagocytosis may be regulators of microglial gene expression in these diverse settings. Other developmental cues, such as synaptic remodeling or vascularization, may also play a role in the retina since a subset of cells expressing DAM-related genes remain in the Bax KO. Although a direct effect of Bax KO in microglia cannot be ruled out, Bak1 is redundant with Baxin non-neuronal cells (Degenhardt et al., 2002; Lindsten et al., 2000; Wei et al., 2001), and we find Bax is not required for microglial loss following CSF1R inhibition. Neuronal apoptosis could have other effects, since in zebrafish brain it is a signal for entry of microglial precursors (Casano et al., 2016; Wu et al., 2018; Xu et al., 2016), and in quail microglial retina, entry is coincident with peak neuronal apoptosis (Martín-Estebané et al., 2017). Interestingly, however, Bax is not required for microglial density or migration in the mouse brain (Eyo and Dailey, 2013).
At present, the signaling pathways driving a DAM-related signature in both development and disease are incompletely understood. In disease, TREM2 (Keren-Shaul et al., 2017) and ApoE (Krasemann et al., 2017) are important for the expression of select genes. In developing white matter, neither pathway seems to be required (Li et al., 2019). Similar to disease, we find that Trem2 mediates Itgax and Lpl upregulation but promotes the downregulation of Tmem119. Aside from CD11c (Itgax) regulation, loss of Apoe did not phenocopy the loss of Trem2, but TREM2 is a receptor for several lipids including exposed phosphatidylserine (Takahashi et al., 2005). Importantly, microglia can utilize a diverse array of other pathways to recognize and engulf dying cells, including MFG-E8 and TAM receptors, such as MerTK (Arandjelovic and Ravichandran, 2015). However, it remains unclear whether the modest change in DAM-related gene expression with loss of Trem2 is due to reduced phagocytosis of apoptotic cells or other mechanisms, such as synaptic pruning (Filipello et al., 2018). Importantly, we find significant heterogeneity within microglia expressing DAM-related genes. We identify low and high expressers of CD11c and find variable levels of Clec7a, Spp1, and Igf1, which may delineate subpopulations ofDAM-related cells with diverse phenotypes. Future work will define other molecular pathways regulating the DAM-related gene signature in the retina and determine potential differences with disease contexts.
Of significance, we find this signature is linked to CSF1R independence. CSF1R is a receptor tyrosine kinase important for microglial development and survival (Chitu et al., 2016) and the ligands, CSF1 and IL-34, are supplied by microglia and neurons respectively (Nandi et al., 2012). Loss of CSF1R signaling results in a near complete depletion of microglia, both in development and adulthood (Waisman et al., 2015). Here, we found that the majority of microglia persist after conditional KO or inhibition of CSF1R in the postnatal retina (see also Kuse et al., 2018). It is unlikely that CSF1R inhibition is altering microglial proliferation since few microglia in the postnatal retina are proliferating (Santos et al., 2008), consistent with low postnatal expression of proliferation genes in our sequencing data. In adult, peripheral monocytes expressing low CSF1R populate the retina after ocular injury, engraft permanently, and are resistant to CSF1R loss (Paschalis et al., 2018). In brain, at perinatal stages there is infiltration of monocytes from fetal liver, but these cells are rapidly eliminated and do not contribute to the adult microglia population (Askew and Gomez-Nicola, 2018). However, in postnatal retinal microglia, we find no evidence for gene expression characteristic of peripheral cells, have excluded CCR2+ cells from our RNA sequencing, and observe high expression of Csf1 and CSF1R. Notably, we find microglia that persist after CSF1R inhibition are CD11cHIand more highly express lysosomal genes, suggesting that more phagocytic cells may be less dependent upon CSF1R for survival. Concurrently, we observed a pronounced reduction in expression of homeostatic genes, Tmem119 and P2ry12, suggesting that more ‘‘homeostatic’’ microglia may be more dependent. Consistent with this, Bax KO microglia with reduced-related expression and increased homeostatic gene expression were more susceptible to CSF1R inhibition. It is unclear whether subpopulations with varying dependence on CSF1R exist. It is intriguing to speculate that microglia expressing similar genes in other contexts may be resistant to CSF1R loss, especially since many studies do not achieve 100% depletion (Dagher et al., 2015). Future experiments will determine the mechanisms behind CSF1R independence and whether microglia shift their dependence to another factor for survival.
Our data provide clear evidence for heterogeneity and dynamic regulation of microglia developmentally. These findings broaden our understanding of microglial transcriptional states across CNS regions and demonstrate parallels in development, aging, and disease, which has implications for our understanding of microglial phenotype and function in diverse contexts.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Monica Vetter (monica.vetter@neuro.utah.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal husbandry and procedures
All animals were treated within the guidelines of the University of Utah Institutional Animal Care and Use Committee (IACUC) and all experiments were IACUC approved. Mice were housed in an AAALAC accredited animal facility with 12h light/12h dark cycles and ad libitum access to food and water. Both sexes were used for all experiments. Information on the ages of mice used for each experiment can be found in the figures/text. Timed matings were used to determine embryonic stages. The morning a plug was detected was considered embryonic day 0.5. 10mg/ml tamoxifen (Sigma-Aldrich, T5648), dissolved in corn oil (Sigma-Aldrich, C8267) was administered to pups by intraperitoneal injection on P4, P6, and sometimes P8 at 0.25mg/g body weight. PLX3397 (AdooQ BioScience, A15520) was dissolved in corn oil and 10% DMSO and administered to postnatal pups by intraperitoneal injection on P4, P5, and P6 at 0.25mg/g body weight. Adult administration of PLX3397 was given at 290mg/kg BW once daily for 4 days by oral gavage. Mice were euthanized by isoflurane asphyxiation followed by cervical dislocation.
Mouse strains
The B6.CX3CR1-gfp/+ mice (Jung et al., 2000) were a gift from Richard Lang with permission from Steffen Jung. B6.Cx3cr1creERT2 mice (Yona et al., 2013) were a gift from Steffen Jung. C57B6/2J, B6.Csf1rfloxed(021212), Trem2 KO (JAX 027197), Apoe KO (JAX 002052) and Bax KO (JAX 002994) mice were purchased from the Jackson Laboratory. B6.RosaTdtomato mice (Madisen et al., 2010) were a gift from Mario Capecchi. More details in the table.
METHOD DETAILS
Tissue Processing
Following euthanasia, retinas were dissected in ice cold 0.1M PBS.
Whole heads were fixed in 4% PFA for 45 minutes to an hour. Heads were washed 3 times for 15 minutes in PBS and underwent 12–16 hour consecutive treatments with 15% and 30% sucrose in PBS at 4°C. Heads were then embedded in OCT compound (Tissue-Tek), stored at 80°C, and sectioned at 16mm thickness. For retinal whole mounts, eyes were removed from the head and retinas were carefully dissected from the rest of the eye (cornea, lens, RPE, hyaloid vasculature, vitreous, ciliary body) in ice cold PBS. Whole neural retinas were washed in PBS for 10–20 minutes and then fixed in 4% PFA for 15–30 minutes at room temperature with rocking. For RNAase-free dissections for qHCR, FACs, and whole retina RT-qPCR, retinas were carefully dissected in RNase free conditions using ice cold, sterile RNase-free PBS, removing all non-neural eye tissue (ciliary body, pigmented epithelium, vitreous including hyaloid vasculature at embryonic stages).
Immunohistochemistry
Frozen sections were placed in ice cold PBS for 10 minutes, blocked for 1 hour at room temperature (0.2% triton-X, 10% BSA, 10 % normal donkey serum in 0.01M PBS), then incubated in primary antibody overnight at 4°C in (0.2% triton-X, 5% BSA in 0.01M PBS). The following day, sections were washed 3X with PBS and incubated in secondary antibodies (5% BSA in PBS) for 2 hours at room temperature, washed, and mounted with Vectashield mounting medium with DAPI (H-1200, Vector Laboratories). For whole mount immunostaining, retinas were incubated in primary antibody at 4C for 3 days. Antibody details in table.
In situ hybridization chain reaction (HCR)
Wholemount retinas were fixed overnight in 4% PFA in 4C. Retinas were washed and dehydrated in methanol at 25%, 50 %, 2X 100% for 15 minutes each, stored in 100% methanol overnight at 4°C and 20°C long term. In situ hybridization was performed as published (Choi et al., 2018) using v3.0 reagents from Molecular Instruments (https://www.molecularinstruments.com). Briefly, samples were rehydrated using (75% Methanol/25% PBST, 50% Methanol/50% PBST, 25%, Methanol/75% PBST, 2X 100 % PBST), treated with Proteinase K, and post fixed 20’ at room temperature (RT) in 4% PFA, and washed 3x with PBST. Pre-hybridization was performed in 30% probe Hybridization buffer for 30 minutes at 37°C, and retinas were placed in hybridization buffer at 37C overnight. Retinas were washed, placed in amplification buffer for 30 minutes at room temperature. Separately, hairpins used for amplification were denatured at 95C for 90 s and cooled to RT for 30 minutes. Retinas were placed in amplification buffer with hairpins at room temperature in the dark overnight. Retinas were washed, DAPI stained, and mounted on slides. 10 probes per gene recognizing all transcript variants for each of Cx3cr1, Itgax, Lyz2, Apoe, Igf1, Spp1,Clec7a and Tmem119 were generated by Molecular Technologies/Instruments. For Cx3cr1-GFP knockin mice, the fluorescent signal represents detection of Cx3cr1 mRNA by HCR and GFP fluorescence which persists through the procedure.
Confocal Microscopy
Confocal images were acquired on an inverted Nikon A1R Confocal Microscope. Images were acquired at 20X objective with a 3X digital zoom. Multi-points were stitched with a 10% overlap. Images of retinal whole mounts were 144 multi-point images (on average) for whole retinal images and 18 multi-points for HCR analysis. Stacks through the Z plane were at 0.8µm steps of about 30mm thickness at 0.2µm pixel resolution. Wholemount retina images represent max projections of inner retina (inner plexiform layer through nerve fiber layer) from the central retina (optic nerve to mid-periphery) of roughly 0.7 mm2. Image acquisition settings were consistent across ages and genotypes.
Sorted microglia RT-qPCR
Retinal microglia were sorted directly into RLT buffer (QIAGEN). Samples were frozen at 80C until extraction. RNA was extracted using an RNeasy Micro Kit (QIAGEN, 74004). First strand cDNA synthesis was performed using the SuperScript III First Strand cDNA synthesis kit (Invitrogen,11752). Quantitative real-time PCR was performed using SYBR Select Master mix (Applied Biosciences, 4472908) and run on the 7900 HT Fast Real-Time PCR system with QuantStudio 12K Flex software (Applied Biosystems) at the University of Utah Genomics Core. Oligonucleotides were synthesized by the DNA/Peptide Facility, part of the Health Sciences Center Cores at the University of Utah. Relative quantification was determined by DDCt using the QuantStudio Software. Genes of interest were normalized to Beta Actin. All qPCR graphs are log2 of relative expression compared to control with error bars representing the SEM of the delta Ct values. Primers were designed using Primer-BLAST software and when possible, primers spanned large introns to avoid genomic amplification.
Fluorescence-Activated Cell Sorting (FACS) and Flow cytometry
Except for RNA sequencing, we pooled 2 retinas from an individual animal for each sample for Flow and FACs. Freshly dissected pure retinas were dissociated in PBS, 50 mM HEPES, 0.05 mg/ml DNase I (Sigma D4513), 0.025 mg/ml Liberase (Sigma 5401119001) for 35 min with intermediate trituration. Cells were passed through a 70 mm nylon cell strainer, washed with staining buffer (1X PBS, 2 % BSA, 0.1% sodium azide, 0.05% EDTA), and red blood cells were lysed (eBioscience 00-4333-57). Cell counts were determined using a cell counter (Invitrogen Countess) and Fc block (BD Biosciences 553142) was added at 2 mL per 106 cells. Antibodies were applied for 30 min on ice (details for each in table). Cells were washed, pelleted, and resuspended in 500 mL staining buffer. FACS was performed using a BD FACS Aria cell sorter at the University of Utah Flow Cytometry Core. Forward and side scatter were used to eliminate debris and the height and area of the forward scatter was used to discriminate singlets. For Flow Analysis, roughly 1 million singlet events (300,000 in rare cases) were recorded for flow analysis using FlowJo software (Flowjo, LLC, Ashland, Oregon). The proportion of CD11c populations were analyzed gating on live CD45+CD11b+or GFP+ cells. Unstained dissociated retina was used as a negative control to set gates.
RNA sequencing
GFP+, CD45+, CCR2− were sorted directly into RLT buffer (QIAGEN 79216) and stored at 20C. Three biological replicates of retinal microglia were collected for each age. 1–2 litters were pooled for each sample at e16.5 for roughly 7400, 6800, and 4310 cells each, 5–12 retinas were pooled for P7 for 13292, 5300, and 4000 cells each, and 7–14 retinas were pooled for P60 for 9600, 2600, and 2200 cells each. RNA from sorted cells was purified using an RNeasy Plus Micro kit (QIAGEN 74034). The University of Utah HighThroughput Genomics Core ran Agilent High Sensitivity RNA ScreenTape Assay, generated cDNA libraries using the NuGEN Ovation RNA-seq System v2 for ultra-low input samples, and prepared individual bar-coded RNA-seq libraries for each sample. An Illumina HiSeq 2000 was used to sequence each sample and roughly 20–30 million reads per sample were obtained. Whole retina samples (central retina for e16.5) were purified with the RNeasy Plus Mini kit (QIAGEN 74134) and assayed with Agilent High Sensitivity RNA ScreenTape. High quality RNA samples were transformed into sequencing libraries with TruSeq Stranded RNA Kit with Ribo-Zero Gold (Illumina RS-122-2301 and RS-122-2302) and applied to HiSeq v4 and a 50 cycle single-read sequence run was performed (Illumina FC-401-4002).
QUANTIFICATION AND STATISTICAL ANALYSIS
Image Analysis
For HCR images, roughly 0.7 mm2(3×6 tiles) was analyzed from the central retina just adjacent to the optic nerve head, to the mid periphery. Images were max projected and roughly 30mm thick, spanning the nerve fiber layer to the inner nuclear layer, focusing on the inner retina where the majority of microglia reside. Manual counts were performed using Nikon Elements software (Melville, NY)
Bioinformatics
QC analysis of Fastq files was performed using FastQC (v0.10.1, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) with support from the University of Utah Bioinformatics core. Fastq files were aligned to ensembl Mus musculus cDNA annotation (release 92) with salmon (v0.9.1, Patro et al., 2017). The alignments were compiled with tximport (v1.8.0), and analyzed with DESeq2 (v1.20.0) in R (v.3.5.1) (Love et al., 2014; Patro et al., 2017; Soneson et al., 2015). Genes were excluded from downstream analysis if they did not have at least 10 reads in at least one of the 14 retinal microglia or whole retinal datasets.
Principal component analysis was performed using the 500 most variable rlog-transformed genes, and the Spearman correlation was calculated based on all transformed genes. For clustering analysis, a list of 2872 differentially expressed and microglia-enriched genes was generated. We included genes that are differentially expressed (padj % 0.05) in sorted microglia between any two time points: e16.5, P7, or P60. We also excluded genes that were expressed R 20-fold higher in whole retina samples compared to sorted microglia at any age. This was done to exclude any genes that are differentially expressed in the microglia samples due to contamination of genes from other retinal cells. K-means clustering was performed on this gene list using the scaled average FPKM values from each time point, with the optimal number of clusters determined by an elbow plot of within group sum of squares. The hypergeometric distributions were generated with the enricher function from clusterProfiler (Yu et al., 2012) and gene set enrichment analyses were performed with GAGE (Luo et al., 2009). Brain microglia Fastq files were obtained (GEO: GSM2104052, (Matcovitch-Natan et al., 2016) and processed as described above. Three sub-clusters were generated within each of the six retinal clusters by Kmeans clustering.
All bar graphs showing FPKM represent the average FPKM values for each condition ± standard error of the mean (SEM). Statistical significance of differential expression was determined by DESeq2 using Benjamini-Hochberg correction (padj).
Statistical Methods
Detailed statistical information can be found in the figure legends, including tests used, number of n, and precision measures. All qPCR, image, and flow data were analyzed using Prism 8 software (GraphPad, La Jolla, CA). All data were first tested for normality using the Shapiro-Wilk test. For all t tests, we applied the Welch’s correction if the two samples had unequal variances determined by an F-test. We ran the Brown-Forsythe one-way ANOVA with posthoc Games-Howell’s multiple comparison’s test if the standard deviations were significantly different by a Bartlett test. For all data is presented as the mean with error bars indicating the standard error of the mean, SEM. We used a 95% confidence interval and a p value of < 0.05 was set for rejecting the null hypothesis.
DATA AND SOFTWARE AVAILABILITY
The sequence data reported in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO). The accession number for this sequence data is GEO: GSE123757.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| goat GFP (1:2000) | Abcam | Cat# ab5450; RRID:AB_304897 |
| rabbit IBA1 (1:1000) | Wako | Cat# 019–19741; RRID:AB_839504 |
| 488 Donkey anti-goat (1:400) | Invitrogen | Cat# A11055; RRID:AB_2534102 |
| 488 Donkey anti-rabbit (1:400) | Invitrogen | Cat# A21206; RRID:AB_2535792 |
| BV421 CD45 | BD Bioscience | Cat# 563890; RRID:AB_2651151 |
| 488 CD11b | BD Bioscience | Cat# 557672; RRID:AB_396784 |
| PE CD11b | BD Bioscience | Cat# 553311; RRID:AB_394775 |
| APC CD11c | BD Pharmingen | Cat# 561119; RRID:AB_10562405 |
| APC CCR2 | R&D | Cat# FAB5538P; RRID:AB_10718414 |
| PE CD115 | BioLegend | Cat# 135505; RRID:AB_1937254 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| IB4-Lectin (1:400) | Sigma-Aldrich | Cat# L9381 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Corn oil | Sigma-Aldrich | Cat# C8267 |
| Pexidartinib (PLX3397) | AdooQ BioScience | Cat# A15520 |
| Liberase TM | Sigma-Aldrich | Cat# 5401119001 |
| Red blood cell lysis buffer | eBioscience | Cat# 00-4333-57 |
| Mouse Fc Block | BD Biosciences | Cat# 553142 |
| DNase I | Sigma-Aldrich | Cat# D4513 |
| Critical Commercial Assays | ||
| in situ hybridization chain reaction v3.0 (HCR) | Molecular Instruments | N/A |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| SuperScript III First Strand cDNA synthesis kit | Invitrogen | Cat# 11752 |
| SYBR Select Master mix | Applied Biosciences | 4472908 |
| Deposited Data | ||
| RNA-seq files | https://www.ncbi.nlm.nih.gov/geo | GEO: GSE123757 |
| Experimental Models: Organisms/Strains | ||
| B6.129P2(Cg)-Cx3cr1tm1Litt/J or CX3CR1-GFP | gift from Richard Lang | JAX 005582 |
| B6.129P2(C)-Cx3cr1tm2.1(cre/ERT2)Jung/J | gift from Steffen Jung | JAX 020940 |
| B6.Cg-Csf1rtm1.2Jwp/J | Jackson Labs | JAX 021212 |
| C57BL/6J-Trem2em2Adiuj/J | Jackson Labs | JAX 027197 |
| B6.129P2-Apoetm1Unc/J | Jackson Labs | JAX 002052 |
| B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | gift from Mario Capecchi | JAX 007914 |
| B6.129X1-Baxtm1Sjk/J | Jackson Labs | JAX 002994 |
| C57BL/6J | Jackson Labs | JAX 000664 |
| Oligonucleotides | ||
| Beta Actin Forward: TGAGAGGGAAATCGTGCGTG | University of Utah,DNA/Peptide Facility | N/A |
| Beta Actin Reverse: TCGTTGCCAATAGTGATGACCTG | University of Utah,DNA/Peptide Facility | N/A |
| Tmem119 Forward: CTTCACCCAGAGCTGGTTCCATA | University of Utah, DNA/Peptide Facility | N/A |
| Tmem119 Reverse: CGCGATGAGCATCACGTACT | University of Utah, DNA/Peptide Facility | N/A |
| P2ry12 Forward: AGTGCAAGAACACTCAAGGC | University of Utah, DNA/Peptide Facility | N/A |
| P2ry12 Reverse: GTGTTGACACCAGGCACATC | University of Utah, DNA/Peptide Facility | N/A |
| Itgax Forward: CAAGACAGGACATCGCTCCC | University of Utah, DNA/Peptide Facility | N/A |
| Itgax Reverse: CCTGGAAATCTCTGCAGGTGT | University of Utah, DNA/Peptide Facility | N/A |
| Apoe Forward: GGGACAGGGGGAGTCCTATAAT | University of Utah, DNA/Peptide Facility | N/A |
| Apoe Reverse: TTTGCCACTCGAGCTGATCT | University of Utah, DNA/Peptide Facility | N/A |
| Clec7a Forward: CTTCACCTTGGAGGCCCATT | University of Utah, DNA/Peptide Facility | N/A |
| Clec7a Reverse: AGGGAGCCACCTTCTCATCT | University of Utah, DNA/Peptide Facility | N/A |
| Lpl Forward: CGAGAGCGAGAACATTCCCT | University of Utah, DNA/Peptide Facility | N/A |
| Lpl Reverse: GTCTCTCCGGCTTTCACTCG | University of Utah, DNA/Peptide Facility | N/A |
| Igf1 Reverse: GGCTTGTTGAAGTAAAAGCCCC | University of Utah, DNA/Peptide Facility | N/A |
| Cd68 Forward: GGACACTTCGGGCCATGTTT | University of Utah, DNA/Peptide Facility | N/A |
| Cd68 Reverse: CTTACACAGTGGACTGGGGC | University of Utah, DNA/Peptide Facility | N/A |
| Csf1 Forward: AGAACAAGGCCTGTGTCCGAA | University of Utah, DNA/Peptide Facility | N/A |
| Csf1 Reverse: CTGCTAGGGGTGGCTTTAGG | University of Utah, DNA/Peptide Facility | N/A |
| Lamp1 Forward: GGTAACAACGGAACCTGCCT | University of Utah, DNA/Peptide Facility | N/A |
| Lamp1 Reverse: TGGCATTCATCCCAAACTGC | University of Utah, DNA/Peptide Facility | N/A |
| Lyz2 Forward: TGAACGTTGTGAGTTTGCCAG | University of Utah, DNA/Peptide Facility | N/A |
| Lyz2 Reverse: CAGCAGAGCACTGCAATTGAT | University of Utah, DNA/Peptide Facility | N/A |
| B3-Apoe ISH probe | Molecular Instruments | N/A |
| B2-Clec7a ISH probe | Molecular Instruments | N/A |
| B1-Cx3cr1 ISH probe | Molecular Technologies | N/A |
| B3-Cx3cr1 ISH probe | Molecular Instruments | N/A |
| B4-Igf1 ISH probe | Molecular Technologies | N/A |
| B1-Itgax ISH probe | Molecular Instruments | N/A |
| B2-Lyz2 ISH probe | Molecular Technologies | N/A |
| B1-Spp1 ISH probe | Molecular Technologies | N/A |
| B4-Tmem119 ISH probe | Molecular Technologies | N/A |
| B1-Alexa 488 amplifier | Molecular Technologies | N/A |
| B1-Alexa 546 amplifier | Molecular Technologies | N/A |
| B2-Alexa 546 amplifier | Molecular Technologies | N/A |
| B2-Alexa 647 amplifier | Molecular Technologies | N/A |
| B3-Alexa 647 amplifier | Molecular Technologies | N/A |
| B4-Alexa 546 amplifier | Molecular Technologies | N/A |
| Software and Algorithms | ||
| Nikon Elements | Nikon | N/A |
| Prism | Graph Pad | V8 |
| FastQC | Babraham Bioinformatics | V0.10.1 |
| Salmon | http://combine-lab.github.io | V0.9.1 |
| R | https://www.r-project.org | V3.5.1 |
| Tximport | Bioconductor | V1.8.0 |
| DESeq2 | Bioconductor | V1.20.0 |
| clusterProfiler | Bioconductor | V3.10.1 |
| GAGE | Bioconductor | V2.32.1 |
Highlights.
Retinal microglia have distinct transcriptional states across development
Microglia in postnatal retina highly express disease associated genes
Developmental apoptosis is a major driver of the disease-related profile
High CD11c and disease-related gene expression are linked to CSF1R independence
ACKNOWLEDGMENTS
We thank C. Gregg, O. Shcheglovitov, and R. Dorsky for helpful comments on the manuscript. The work was supported by NIH EY025082 (M.L.V.) and EY025967 (S.R.A.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.062.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anderson SR, and Vetter ML (2019). Developmental roles of microglia: A window into mechanisms of disease. Dev. Dyn 248, 98–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SR, Zhang J, Steele MR, Romero CO, Kautzman AG, Schafer DP, and Vetter ML (2019). Complement targets newborn retinal ganglion cells for phagocytic elimination by microglia. J. Neurosci 39, 2025–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arandjelovic S, and Ravichandran KS (2015). Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol 16, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew K, and Gomez-Nicola D (2018). A story of birth and death: Insights into the formation and dynamics of the microglial population. Brain Behav. Immun 69, 9–917. [DOI] [PubMed] [Google Scholar]
- Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh YE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, et al. (2018). Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci 21, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad.Sci. USA 113, E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, and Barres BA (2018). A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, and Barres BA (2017). Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 94, 759–773.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunger BM, Demmer C, and Tamm ER (2014). Programmed Cell Death During Retinal Development of the Mouse Eye (New York, NY: Springer NewYork; ). [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. (2014). Identification of a unique TGF-b-dependent molecular and functional signature in microglia. Nat. Neurosci 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, and Greter M (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol 17, 1397–1406. [DOI] [PubMed] [Google Scholar]
- Casano AM, Albert M, and Peri F (2016). Developmental Apoptosis Mediates Entry and Positioning of Microglia in the Zebrafish Brain. Cell Rep 16, 897–906. [DOI] [PubMed] [Google Scholar]
- Checchin D, Sennlaub F, Levavasseur E, Leduc M, and Chemtob S (2006). Potential role of microglia in retinal blood vessel formation. Invest. Ophthalmol. Vis. Sci 47, 3595–3602. [DOI] [PubMed] [Google Scholar]
- Chitu V, Gokhan Ş, Nandi S, Mehler MF, and Stanley ER (2016). Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci 39, 378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generationin situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, and Butovsky O (2017). Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL, and Green KN (2015). Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflammation 12, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Van Deren D, Peden E, Hockin M, Boulet A, Titen S, and Capecchi MR (2018). Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development 145, dev152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Sundararajan R, Lindsten T, Thompson C, and White E (2002). Bax and Bak independently promote cytochrome C release from mitochondria. J. Biol. Chem 277, 14127–14134. [DOI] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, and Green KN (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, and Dailey ME (2013). Microglia: key elements in neural development, plasticity, and pathology. J. Neuroimmune Pharmacol 8, 494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah MH, and Easter SS Jr. (2005). Cell birth and death in the mouse retinal ganglion cell layer. J. Comp. Neurol 489, 120–134. [DOI] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, et al. (2018). The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48, 979–991.e8. [DOI] [PubMed] [Google Scholar]
- Frade JM, and Barde Y-A (1998). Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 20, 35–41. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, and Glass CK (2014). Environment dr ives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeyer N, Hanft K-M, Akriditou M-A, Unger N, Park ES, Stanley ER, Staszewski O, Dimou L, and Prinz M (2017). Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol 134, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Robinton D, and Stevens B (2018). Microglia and the Brain: Complementary Partners in Development and Disease. Annu. Rev. Cell Dev.Biol 34, 523–544. [DOI] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Möller T, Orre M, Kamphuis W, et al. (2015). Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol. Commun 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Skola D, and Glass CK (2017). Transcriptional control of microglia phenotypes in health and disease. J. Clin. Invest 127, 3220–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Cui J, Li L, Hitchcock PF, and Li Y (2012). The role of microglia in the neurogenesis of zebrafish retina. Biochem. Biophys. Res. Commun 421, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling AI, Waugh M, Vessey KA, Phipps JA, Trogrlic L, Greferath U, Mills SA, Tan ZL, Ward MM, and Fletcher EL (2018). The Role of the Microglial Cx3cr1 Pathway in the Postnatal Maturation of Retinal Photoreceptors. J. Neurosci 38, 4708–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, and Littman DR (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol.Cell. Biol 20, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Kooijman L, Schetters S, Orre M, and Hol EM (2016). Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys.Acta 862, 1847–1860. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, HÖlscher C, et al. (2013). Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci 16, 273–280. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuse Y, Ohuchi K, Nakamura S, Hara H, and Shimazawa M (2018). Microglia increases the proliferation of retinal precursor cells during postnatal development. Mol. Vis 24, 536–545. [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, and Barres BA (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol 18, 225–242. [DOI] [PubMed] [Google Scholar]
- Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al. (2019). Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. (2000). The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Nair CE, Schlamp CL, Montgomery AD, Shestopalov VI, and Nickells RW (2016). Retinal glial responses to optic nerve crush are attenuated in Bax-deficient mice and modulated by purinergic signaling pathways. J. Neuroinflammation 13, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Estebané M, Navascués J, Sierra-Martín A, Martín-Guerrero SM, Cuadros MA, Carrasco M-C, and Marín-Teva JL (2017). Onset of microglial entry into developing quail retina coincides with increased expression of active caspase-3 and is mediated by extracellular ATP and UDP. PLoS ONE 12, e0182450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada Gonzalez F, Perrin P, et al. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science 353, aad8670. [DOI] [PubMed] [Google Scholar]
- Nandi S, Gokhan S, Dai X-M, Wei S, Enikolopov G, Lin H, Mehler MF, and Stanley ER (2012). The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev. Biol 367, 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Koren EG, Yu C, Klingeborn M, Wong AYW, Prigge CL, Mathew R, Kalnitsky J, Msallam RA, Silvin A, Kay JN, et al. (2019). Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 50, 723–737.e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschalis EI, Lei F, Zhou C, Kapoulea V, Dana R, Chodosh J, Vavvas DG, and Dohlman CH (2018). Permanent neuroglial remodeling of the retina following infiltration of CSF1R inhibition-resistant peripheral monocytes. Proc. Natl. Acad. Sci. USA 115, E11359–E11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pequignot MO, Provost AC, Salle S, Taupin P, Sainton KM, Marchant D, Martinou JC, Ameisen JC, Jais JP, and Abitbol M (2003). Major role of BAX in apoptosis during retinal development and in establishment of a functional postnatal retina. Dev. Dyn 228, 231–238. [DOI] [PubMed] [Google Scholar]
- Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, Mantingh-Otter IJ, Van Dam D, De Deyn PP, den Dunnen W, et al. (2017). Increased White Matter Inflammation in Aging- and Alzheimer’s Disease Brain. Front. Mol. Neurosci 10, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AM, Calvente R, Tassi M, Carrasco MC, Martín-Oliva D, MarínTeva JL, Navascues J, and Cuadros MA (2008). Embryonic and postnatal development of microglial cells in the mouse retina. J. Comp. Neurol 506, 224–239. [DOI] [PubMed] [Google Scholar]
- Silverman SM, and Wong WT (2018). Microglia in the Retina: Roles in Development, Maturity, and Disease. Annu. Rev. Vis. Sci 4, 45–77. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Rochford CDP, and Neumann H (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med 201, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waisman A, Ginhoux F, Greter M, and Bruttger J (2015). Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol 36, 625–636. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, and Korsmeyer SJ (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk A, Holtman IR, Krueger M, Yogev N, Bruttger J, Khorooshi R, Benmamar-Badel A, de Boer-Bergsma JJ, Martin NA, Karram K, et al. (2017). A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J 36, 3292–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Xue R, Hassan S, Nguyen TML, Wang T, Pan H, Xu J, Liu Q, Zhang W, and Wen Z (2018). ). Il34-Csf1r Pathway Regulates the Migration and Colonization of Microglial Precursors. Dev Cell 46, 552–563.e4. [DOI] [PubMed] [Google Scholar]
- Xu J, Wang T, Wu Y, Jin W, and Wen Z (2016). Microglia Colonization of Developing Zebrafish Midbrain Is Promoted by Apoptotic Neuron and Lysophosphatidylcholine. Dev. Cell 38, 214–222. [DOI] [PubMed] [Google Scholar]
- Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, Eggen BJL, Möller T, Tamm JA, Abdourahman A, et al. (2017). Immune hyperreactivity of Abplaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 55, 115–122. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data reported in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO). The accession number for this sequence data is GEO: GSE123757.