

Abstract
Fibroblast growth factor-23 (FGF-23) interacts with a binary receptor complex composed of α-Klotho (α-KL) and FGF receptors (FGFRs) to regulate phosphate and vitamin D metabolism in the kidney. Excess FGF-23 production, which causes hypophosphatemia, is genetically inherited or occurs with chronic kidney disease. Among other symptoms, hypophosphatemia causes vitamin D deficiency and the bone-softening disorder rickets. Current therapeutics that target the receptor complex have limited utility clinically. Using a computationally driven, structure-based, ensemble docking and virtual high-throughput screening approach, we identified four novel compounds predicted to selectively inhibit FGF-23-induced activation of the FGFR/α-KL complex. Additional modeling and functional analysis found that Zinc13407541 bound to FGF-23 and disrupted its interaction with the FGFR1/α-KL complex; experiments in a heterologous cell expression system showed that Zinc13407541 selectivity inhibited α-KL-dependent FGF-23 signaling. Zinc13407541 also inhibited FGF-23 signaling in isolated renal tubules ex vivo and partially reversed the hypophosphatemic effects of excess FGF-23 in a mouse model. These chemical probes provide a platform to develop lead compounds to treat disorders caused by excess FGF-23.
INTRODUCTION
Fibroblast growth factor-23 (FGF-23) is a member of the subfamily of hormonal FGF ligands that includes FGF-19 and FGF-21 (1). FGF-23 is secreted by osteoblasts and osteocytes in bone and circulating FGF-23 participates in several endocrine signaling networks by targeting the FGF receptor (FGFR) and the type I membrane β-glycosidase α-Klotho (α-KL) that constitute the FGF-23 receptor (2–7). Specifically, the N-terminal FGF homology domain of FGF-23 interacts with binding domains in FGFRs, and its unique 71-residue C terminus binds to α-KL, an obligate co-receptor for FGF-23 (7–10). α-KL forms a binary complex with FGFR1c, FGFR3c, or FGFR4, but not with FGFR2, to form functional FGF-23 receptor complexes in a limited number to tissues (11). This eliminates the requirement for heparin binding for receptor activation that characterizes paracrine FGFs. α-KL is mainly expressed in the kidney, parathyroid gland, and choroid plexus, thereby imparting selective FGF-23 activation of these tissues (12–16).
Physiologically, FGF-23 inhibits phosphate reabsorption and suppresses 1,25(OH)2D production in the proximal tubule of the kidney (2, 10, 17–20). However, FGF-23 also targets the distal renal tubule to stimulate renal sodium and calcium retention (21–23). Primary increases in the abundance of circulating FGF-23 concentration cause hereditarily acquired hypophosphatemic disorders, including X-linked hypophosphatemia (XLH), autosomal dominant and autosomal recessive hypophosphatemia, and tumor-induced osteomalacia (18, 24–27). FGF-23 is also purported to suppress parathyroid hormone (PTH) secretion from the parathyroid gland that expresses FGFR/α-KL complexes (15). In contrast, reductions in circulating FGF-23 concentration causes familial tumoral calcinosis and leads to early postnatal mortality due to hyperphosphatemia and excessive1,25(OH)2D production (4, 28–33).
Secondary increases in FGF-23 concentration contribute to the pathogenesis of mineral metabolism and cardiovascular disorders in chronic kidney disease (CKD) (34). Increased circulating concentrations of FGF-23 play an initial adaptive role in maintaining phosphate balance in CKD (35, 36) but become maladaptive with more advanced renal failure and are a strong independent risk factor for both renal failure progression and cardiovascular mortality (37–39).
Currently available treatments for hereditary hypophosphatemic disorders typically include calcitriol and phosphate supplements that are marginally effective and associated with side effects (40), including further stimulation of FGF-23 production (41, 42). Similarly, efforts to suppress secondary elevations of FGF-23 in CKD with phosphate binders and calcimimetics have been used with limited success (43). Inhibition of FGF-23 activation of FGFR/α-KL receptor complexes will likely be a clinically important treatment for hereditary and acquired hypophosphatemic disorders and may prevent cardiovascular complications associated with CKD. A recombinant human immunoglobulin G1 (IgG1) monoclonal antibody (mAb) that binds to FGF-23 and blocks the biological activity of FGF-23 has been shown to be effective in treating hypophosphatemia in both animal models and humans with X-linked hypophosphatemic rickets (44). The long duration of action and the requirement for systemic administration of FGF-23- blocking antibodies potentially limit their use to treat disorders of FGF-23 excess. There is an important unmet medical need to develop small-molecule antagonists of FGF-23/FGFR/α-KL signaling to treat disorders of FGF-23 excess (45–47).
FGFR tyrosine kinase inhibitors, which have been shown to block both the production and end-organ effects of FGF-23, lack selectivity for FGF-23/FGFR/α-KL signaling, and their generalized ability to inhibit FGFRs in multiple tissues would have undesirable effects (48, 49). A small molecule, SSR128129E, which binds to the extracellular part of FGFR, was reported to act as an FGFR antagonist (50), but at present, there are no small molecules that specifically modulate FGF-23 activation of FGFR/α-KL complexes. The discovery of such molecules would not only provide research tools to elucidate FGF-23 biological actions but also advance the discovery of new treatments based on this novel bone/kidney endocrine network.
Here, a computationally driven drug discovery approach using homology modeling, molecular dynamics (MD) simulations, and docking was used to rationally identify small molecules that modulate FGF-23 activation in the presence of FGFR and α-KL. MD simulations generated an ensemble of structures of the N-terminal domain of FGF-23, which were used to perform in silico virtual screening to identify candidate molecules that dose-dependently and selectively inhibited FGF-23 activation of the FGFR/α-KL complex both in cell culture and in animal models.
RESULTS
Identification of trial compounds
To identify compounds that bind to FGF-23, we performed structural modeling of FGF-23 and generated homology models for MD simulations. The bioactive region of FGF-23 is from residues Ala28 through Ile251 and contains N-terminal and C-terminal (FGF-23CT) domains (51). The crystal structure of FGF-23 from Ser29 through Asn170, corresponding to the N-terminal domain, has been solved (52). FGF-23CT spans Ser180 through Ile251, demarcated by an RXXR179 furin-like cleavage motif involved in FGF-23 metabolism (53). Residues Ser180 to Thr200 in the C terminus are critical for binding α-KL (51, 52), and a 21-residue peptide derived from the FGF-23CT inhibits the activity of FGF-23 (51). Analysis of FGF-23 with the DISOPRED Web server (54, 55) indicates that the C-terminal domain is disordered (Fig. 1A), consistent with the failure to crystallize the C-terminal domain (52). Hence, structural modeling was confined to the N-terminal domain. Homology models were generated with different initial configurations of the backbone, and short, constrained MD simulations were performed to refine the structural models. The refined homology models used for the in silico virtual screen show some diversity in backbone structure and side-chain conformational variability, indicating that protein flexibility is accounted for to a limited extent in this analysis (Fig. 1B). We used the refined models for in silico virtual screening to identify compounds predicted to bind to the N-terminal domain, screening both the NCI Diversity Set 2 and ZINC databases. We screened the ZINC database with a Tanimoto similarity cutoff of 0.8 (56, 57). The Tanimoto cutoff was used to search a subset of chemical diversity space that is uniquely different from the NCI Diversity Set 2. This resulted in a custom database of 84,589 compounds for computational screening. We selected chemical compounds for experimental validation based on these docking results and tested the best-scoring compounds in the experimental signaling assay below.
Fig. 1. FGF-23 homology modeling and in silico virtual screen.

(A) Intrinsic disorder profile of FGF-23 as predicted by DISOPRED3. (B) Side-chain variability of Arg140 captured in the refined homology models.
FGF-23 activation of FGFR/α-KL signaling assay
First, we established that human embryonic kidney (HEK) 293T cells expressed all of the FGFR isoforms, including FGFR1, FGFR2, FGFR3, and FGFR4, but not transcripts encoding α-KL (fig. S1). These cells lacking endogenous α-KL have minimal responses to FGF-23 (see below). To identify compounds generated from the in silico virtual screening that antagonize FGF-23 actions, we established an in vitro screening assay to test the inhibitory effects of these chemical probes in HEK293T cells that were engineered to overexpress membrane-localized α-KL (11, 58, 59) and an ELK1-GAL [extracellular signal-regulated kinase (ERK)] reporter construct (Fig. 2A) (60). Because HEK293T cells express all of the endogenous FGFR isoforms, FGF-23 stimulated ERK reporter activity in cells transfected with α-KL, reflecting the effects of FGFR1/α-KL, FGFR3/α-KL, and FGFR4/α-KL complexes (20, 51, 58, 61–63). We validated this screening assay by demonstrating that exogenously added recombinant FGF-23 (rFGF-23) stimulated ERK reporter luciferase activity in a dose-dependent manner (Fig. 2B). The median effective concentration (EC50) of rFGF-23 was ~0.1 nM (10−10 M). The maximal effect of rFGF-23 stimulating ERK reporter activity was achieved at 1 nM (10−9 M). To further validate that this assay measures α-KL-dependent FGFR activation, we showed that heparan sulfate (HS) had no additive effect on rFGF-23 activation, whereas an FGF-23CT-blocking peptide (100 nM), which disrupts FGF-23 binding to the FGFR/α-KL binary complex (51), abolished rFGF-23-induced reporter activity (Fig. 2C). A pan-FGFR tyrosine kinase inhibitor PD173074 (10 μM) also blocked FGF-23-induced reporter activity (Fig. 2D).
Fig. 2. Activation of FGF-23 signaling in HEK293T cells that were transfected with human membrane α-KL.
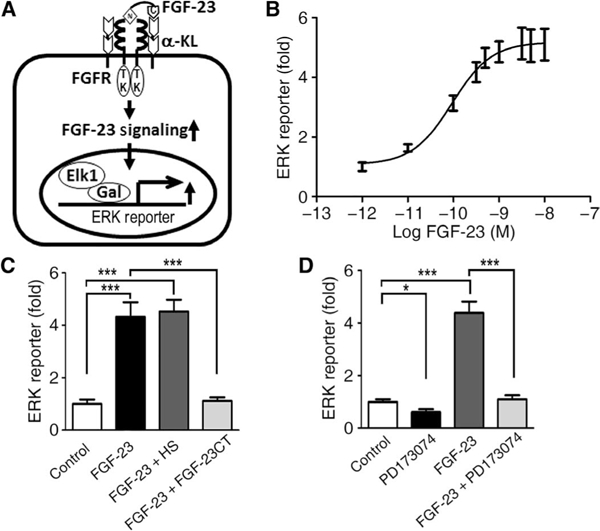
(A) Schematic of assay system. (B) Dose-response curve of FGF-23-mediated ERK reporter luciferase activity in α-KL-transfected HEK293T cells. (C and D) Inhibitory effects of the FGF-23 C-terminal peptides (FGF-23CT; C) or the pan-FGFR tyrosine kinase inhibitor (PD173074; D) on FGF-23-induced ERK reporter activity in transfected HEK293T cells. Data are means ± SD from three independent experiments. *P < 0.05, ***P < 0.001; one-way analysis of variance (ANOVA) with Newman-Keuls multiple comparison test.
Identification of chemical probes that antagonize FGF-23- mediated activation of FGFR/α-KL complex signaling in vitro
Using the above in vitro screening assay, we experimentally tested 16 high-scoring chemical probes identified from the in silico virtual screen at an initial concentration of 10 μM in the absence and presence of rFGF-23 (Fig. 3). Thirteen of the 16 compounds exhibited measurable effects inhibiting rFGF-23-stimulated ERK reporter activity (Fig. 3). Compounds NCI_61610, NCI_80313, and NCI_374204 at 10 μM concentration had no effect on FGF-23-induced signal transduction Compounds Zinc04769985, NCI_37553, NCI_102656, NCI_401490, Zinc00055523, NCI_293778, NCI_308835, NCI_84100, and NCI_93354 at 10 μM concentration exhibited partial (less than 50%) inhibition of FGF-23-induced ERK reporter activity, and compound Zinc01626100 achieved an intermediate (~60%) inhibitory effect on FGF-23-induced signal transduction. However, compounds Zinc13407541, NCI_116702, and NCI_97920 completely suppressed FGF-23-induced ERK reporter activity at a 10 μM concentration (Fig. 3). Only compounds NCI_97920 and NCI_116702 inhibited basal FGFR-α-KL activity in the absence of FGF-23 (Fig. 3).
Fig. 3. Effects of 16 chemical probes on ERK reporter activity in the absence or presence of rFGF-23.

ERK reporter activities in α-KL-transfected HEK293T cells cultured with rFGF-23 and the indicated compound. Data ar means ± SD from three independent experiments. ***P < 0.001; one-way ANOVA with Newman-Keuls multipl comparison test.
Dose-dependent response of chemical probes in antagonizing FGF-23 signaling
To explore the inhibitory effects of the four most potent chemical probes, we performed additional studies using doses ranging from 10−9 to 10−5 M. We observed that all four probes exhibited dose-dependent inhibition of FGF-23 signaling (Fig. 4). The estimated median inhibitory concentration (IC50) values for Zinc13407541, NCI_97920, NCI_116702, and Zinc01626100 were 0.45 ± 0.24 μM, 1.11 ± 0.22 μM, 1.14 ± 0.24 μM, and 4.57 ± 0.22 μM, respectively (Fig. 4, A to D). Next, we evaluated the potential draggability of each of the platforms using various drug-likeness filters (www.chemicalize.org) as previously described (64–70). In general, each of the platforms exhibited drug-likeness with ZINC13407541 and ZINC01626100 having the most drug-like features, including Lipinski’s rule of five, bioavailability, Ghose filter, lead likeness, Muegge filters, and Veber filter (table S1).
Fig. 4. Molecular structure and dose-response curves.

(A to D) Molecular structures and dose-response curves of Zinc13407541 (A), NCI_97920 (B), NCI_116702 (C), and Zinc01626100 (D) in α-KL-transfected HEK293T cells incubated with rFGF-23. Data are means ± SD from three independent experiments.
Multicenter ensemble docking to identify possible binding sites for FGF-23 antagonists
To better understand the structural basis for antagonist binding and to select compounds for additional testing, we performed additional docking using the two best candidate probes, Zinc13407541 and Zinc01626100. Because protein flexibility is an important factor in ligand binding (70–72), these docking calculations were refined by subjecting the protein to MD simulations with the backbone unrestrained, thus permitting a more complete exploration of structural fluctuations than in the original virtual screen.
Zinc13407541 and Zinc01626100 were docked to the potential binding centers identified in the multiple conformers of the protein (see Materials and Methods; Fig. 5). Briefly, the conformers were derived from clustering the MD trajectories, and the binding centers were identified by FTMap. The docking calculations were not sufficiently accurate to identify unambiguous high-precision three-dimensional structures of the two compounds in complex with the protein. Nevertheless, we gained insight by examining the highest-scoring poses. In most of the docking poses found within error of the highest-scoring position (Fig. 5), reflecting the orientation and conformation of the ligand and receptor when bound to each other, the ligands bind in areas predicted (from alignment with a crystal structure of FGF-2 bound to FGFR1) to be critical for the formation of the FGF-23/FGFR1 complex (8).
Fig. 5. Multicenter ensemble docking of Zinc13407541 and Zinc01626100.

(A) Best-scored poses of the docking of Zinc13407541. (B) Best-scored poses of the docking of Zinc1626100. The four binding epitopes in FGF-23 predicted by Yamazaki et al. (8) are colored cyan (epitope 1), gray (epitope 2), orange (epitope 3), and yellow (epitope 4), and the translucent green-shaded area identifies a potential distal binding pocket predicted by our modeling. Zinc13407541 and Zinc01626100 are predicted to bind to the four epitopes, and Zinc13407541 is also predicted to bind to the distal site.
Zinc13407541 poses mostly form multiple hydrogen bonds, with various side chains of Asn112, Gln131, Tyr132, Glu111, Leu138, and Arg140 (Fig. 6, A to H), consistent with the ability of lower concentrations of this compound to inhibit FGF-23 function (Fig. 4A). Arg140 is of particular note because it forms interactions with the ligand in the FGF-23 crystal structure (Fig. 6H) (52). Both Zinc13407541 and Zinc01626100 form hydrogen bonds with Ser155 in the previously predicted FGF-1 binding region (Fig. 6I) (8). Zinc01626100 also forms hydrogen-bonding interactions with the backbone oxygen of Tyr154 and the side chain of Thr44 (Fig. 6, J and K). On the basis of dose-response data (Fig. 4) and further modeling (Figs. 5 and 6), we selected Zinc13407541 for testing target engagement and functional response in vitro and in vivo.
Fig. 6. Predicted intermodular interactions between the N-terminal domain of FGF-23 and the antagonist compounds.

(A to H) FTMap-based predictions of Zinc13407541 interaction with Asn112 (A and B), Gin131 (C), Tyr132 (D), Glu111 (E), Ser155 (F), Leu138 (G), and Arg140 (H) of the N terminus of FGF-23. (I) FTMap-based prediction of hydrogen bond formations between Zinc13407541 and Zinc 01626100 with the side chain of Ser155. (J and K) FTMap-based prediction of Zinc01626100 interaction with the backbone oxygen of Tyr154 (J) and the side chain of Thr44 (K).
Effects of Zinc13407541 on FGF23/FGFR1-α-KL complex formation and FGF-23 thermal stability
To assess engagement between Zinc13407541 and FGF-23, we performed coimmunoprecipitation and protein thermal shift assays. We performed coimmunoprecipitation assays using HEK293T cell lysates cotransfected with V5-tagged FGF-23, full-length FGFR1, and membrane α-KL in the presence of Zinc13407541 or dimethyl sulfoxide (DMSO) control. We found that the V5 antibody coprecipitated α-KL, FGFR1, and FGF-23, indicating that these three factors form a trimeric complex. Addition of Zinc13407541 (10 μM) markedly disrupted FGF-23 and FGFR1/α-KL complex formation (Fig. 7, A to C), consistent with Zinc13407541 inhibiting FGF-23/FGFR1-α-KL complex interaction, though these studies do not exclude potential effects of Zinc13407541 to interfere with the immunoprecipitation protocol or decrease FGF- 23 stability. We also confirmed target engagement by protein thermal shift assays. Using fluorescent SYPRO Orange to monitor FGF-23 thermal unfolding, we found that Zinc13407541 (200 μM) stabilized 5 nM FGF-23, resulting in a 16.5°C shift in melt temperature (Tm) (Fig. 7B).
Fig. 7. Target engagement assays of the FGF-23 antagonist (Zinc13407541) and the FGF-23 protein.

(A) Immunoprecipitation (IP) and immunobiotted (IB) blots are representative of three experiments from cotransfected HEK293T ceil lysates. (B and C) Quantification of α-KL and FGFR1 abundance relative to the amount of FGF-23. (D) Protein thermal shift assay in a real-time polymerase chain reaction (PCR) reaction containing fluorescent SYPRO Orange and FGF-23 protein with Zinc13407541 or DMSO control. Data are means ± SD from three independent experiments (n = 3). **P < 0.01, ***P < 0.001;n.s., not significant; one-way ANOVA with Newman-Keuls multiple comparison test.
Specificity of Zinc13407541 to antagonize FGF-23
Testing the specificity and selectivity of Zinc13407541 requires examining FGFR activation by other FGFs in the presence of cofactors required for optimal ligand-dependent FGFR activation. We examined the ability of nine different FGF ligands from six FGF subfamilies to activate endogenous FGFRs in HEK293T cells overexpressing α-KL signaling or in cells lacking α-KL but treated with HS, the cofactor for paracrine FGF activation of FGFRs (Fig. 8). We found that FGF-23 in the presence of α-KL markedly stimulated ERK reporter activity, whereas, in the absence of α-KL, the response was minimal (Fig. 8A). In contrast, expression of α-KL did not impart FGFR activation to either FGF-19 or FGF-21, hormonal FGFs that require β-KL. In addition, HS, which is the cofactor for paracrine FGF ligands, did not support FGF-23 activation of FGFRs. Using FGF-2 as a prototypic paracrine FGF ligand, we demonstrated that HS augmented FGF-2 activation of FGFR in HEK293T cells (Fig. 8A).
Fig. 8. Effects of different doses of the FGF-23 antagonist Zinc13407541 on ERK luciferase reporter activities in the absence and presence of FGF ligands from different FGF subfamilies.

(A) Different FGF ligands mediated ERK reporter luciferase activities in the absence or presence of α-KL and/or HS in nontransfected or α-KL-transfected HEK293T cells. (B) Dose-dependent inhibition of α-KL-dependent FGFR-mediated ERK reporter activation by Zinc13407541 in α-KL-transfected HEK293T cells. (C) Dose-dependent inhibition of HS-dependent FGFR-mediated ERK reporter activation by Zinc13407541 in either nontransfected or FGFR1-transfected HEK293T cells. Data are means ± SD from three independent experiments (n = 3). *P < 0.01, ***P < 0.001;one-way ANOVA with Newman-Keuls multiple comparison test.
Zinc13407541 resulted in a dose-dependent inhibition of FGF-23 activation of the FGFR/a-KL receptor complex. A significant ~60 and 100% inhibition of FGF-23 stimulation of ERK reporter activity was observed at concentrations of 2.5 and 10 μM, respectively (Fig. 8B). We also tested the effects of Zinc13407541 to inhibit the action of FGF-1, FGF-2, FGF-4, FGF-5, FGF-9, and FGF-18 on HS-dependent FGFR signaling. At a concentration of 2.5 μM, Zinc13407541 did not inhibit FGFR activation of these FGF ligands; however, it did (by ~30%) at a concentration of 10 μM (Fig. 8C). These findings suggest that lower doses of Zinc13407541 selectively inhibit FGF-23-mediated FGFR/ α-KL signaling, whereas higher doses display partial and nonspecific inhibition of nonhormonal FGF ligand activation of FGFR/HS signaling.
Effects of Zinc13407541 on FGF-23 regulation of gene expression in primary renal tubule cell cultures
The kidney is a physiologically important target for FGF-23, where it regulates phosphate transport and vitamin D metabolism in the proximal tubule (6, 34). Therefore, we tested the effects of Zinc13407541 blocking FGF-23 effects on primary renal tubule cell cultures. We confirmed that primary tubule cells expressed Fgfr1/α-KL complexes and responded to exogenous FGF-23 exposure as assessed by increased tyrosine phosphorylation of Fgfr1 and subsequent ERK activation (Fig. 9A and fig. S2). Zinc13407541 had no effect on the expression of the Fgfr1/α-KL complex (Fig. 9A and fig. S2) but significantly attenuated FGF-23-mediated tyrosine phosphorylation of Fgfr1 and downstream ERK activation in proximal tubule cells (Fig. 9B and fig. S2). FGF-23 may also act through Fgfr3 and Fgfr4, as well as through Fgfr1, to mediate downstream ERK activation. Renal tubule cells express several Fgfr isoforms, and we have shown by mouse genetic approaches that FGFR1, FGFR3, and FGFR4 mediate the renal effects of FGF-23 in the kidney (61, 73). To examine the downstream (transcriptional) effects of FGF-23, we assessed the effects of rFGF-23 on cytochrome P450 family 27 subfamily B member 1 (Cyp27b1), cytochrome P450 family 24 subfamily A member 1 (Cyp24a1), type IIa Na+-dependent phosphate cotransporter (Npt2a), and thiazide-sensitive Na+-Cl− cotransporter (NCC) expression in isolated tubule cells. Consistent with known actions of FGF-23, rFGF-23 inhibited Cyp27b1 and Npt2a expression, and stimulated Cyp24a1 and NCC message expression in isolated renal tubules. Treatment with Zinc13407541 (10 μM) inhibited FGF-23-mediated induction of Cyp27b1, Cyp24a1, Npt2a, and NCC expression in primary tubule cell cultures (Fig. 9, C to F).
Fig. 9. The effects of the FGF-23 antagonist (Zinc13407541) on FGF23-induced signaling in primary tubule cell cultures.

(A and B) Western blot analysis of FGFR1/α-KL signaling (A) and the quantification of phosphorylated ERK abundance relative to that of total ERK (B) in cultured mouse primary tubule cells. (C to F) Quantitative real-time reverse transcription (RT)-PCR analysis of total Cyp27b1, Cyp24a1, Npt2a, and NCC transcripts in cultured mouse primary tubule cells. Data are means ± SD from three independent experiments (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001; one-way ANOVA with Newman-Keuls multiple comparison test.
Therapeutic effects of the FGF-23 antagonist Zinc13407541 on a murine model of excess FGF-23
Finally, to evaluate the therapeutic potential of Zinc13407541, we examined its efficacy to block the biological effects of excess FGF-23 in 8-week-old wild-type and homozygous Dmp1 knockout mice. Intraperitoneal injection ofZinc13407541 (100 mg/kg) twice a day for 3 days elicited no effects on survival or other apparent toxicity in either group during the duration of the protocol. As previously reported (5, 74), we observed that the Dmp1 knockout mice exhibited hypophosphatemia and suppressed 1,25(OH)2D associated with increased serum abundance of FGF-23. Compared to those treated with vehicle [5% DMSO in phosphate-buffered saline (PBS) solution], Dmp1-null mice treated with Zinc13407541 had significantly increased serum phosphate, PTH, and 1,25(OH)2D concentrations after treatment (Fig. 10, A to C) but no change in the serum concentration of calcium (Fig. 10D). Unexpectedly, treatment with Zinc13407541 also significantly decreased serum abundance of FGF-23 by about 35% in Dmp1-null mice (Fig. 10E). We observed similar results with respect to changes in serum phosphate, calcium, PTH, and 1,25(OH)2D concentrations in Zinc13407541-treated wild-type mice (fig. S3, A to D), but the serum concentration of FGF-23 in these mice was unchanged by Zinc13407541 (fig. S3E).
Fig. 10. The improvement of hypophosphatemia, PTH imbalance, and 1,25(OH)2D metabolism.

Serum biochemical analyses of phosphate (Pi) (A), PTH (B), 1,25(OH)2D (C), calcium (Ca2+) (D), and FGF-23 (E) in the homozygous Dmp1 knockout mice after 3 days of FGF-23 antagonist (Zinc13407541) injection (n = 4). Data are means ± SD from four mice for each treatment (n = 4). *P < 0.05, ***P < 0.001; two-way ANOVA with Bonferroni post hoc test.
DISCUSSION
Using homology modeling, MD simulation, and virtual high-throughput screening to guide experiments, we identified novel small molecules that interact with the FGF-23 ligand to block its signaling, presumably by activation of the FGFR1/α-KL receptor complex. By targeting the FGF-23 ligand, these molecules differ from other small molecules that inhibit FGFR activation by binding to the extracellular domain or the internal tyrosine kinase domain of the receptor (48–50, 75, 76). The success of the structure-based computational approach is notable, given that no a priori mutagenesis or other experimental data guided the docking calculations. This validates the utility of the MD simulations to overcome limitations of interpreting docking results using a static protein target (77). Specifically, our computational strategy generated homology models with different initial configurations of the backbone that were refined by constrained MD simulations. Using these refined structures for in silico virtual screens, we identified 16 compounds predicted to bind to FGF-23. Experimental testing of these 16 compounds identified 4 that strongly disrupted FGF-23 activity. These compounds had no previously known biological functions. Thus, we have shown that our virtual high-throughput screening approach can successfully identify novel chemical probes for specific targets, complementing other recent successes using the same approach (78).
Of the compounds identified, Zinc13407541 and Zinc01626100 appear to be the best potential candidates for further lead development and preclinical testing. These two compounds are structurally unrelated. Zinc13407541, or N-[[2-(2-phenylethenyl)cyclopenten-1-yl]methylidene]hydroxylamine, has a molecular formula of C14H15NO and a molecular weight of 213. Zinc01626100, or 6-phenyl-2,7-diazatricyclo[6.4.0.02’6]dodeca-1(12),8,10-trien-3-one, has a molecular formula of C16H14N2O and a molecular weight of 250. On the basis of estimated IC50 values of functional assays and differences in binding to primary and secondary pockets of FGF-23, the computational model predicts that compound Zinc13407541 has more than 10-fold higher affinity than compound Zinc01626100. Compound Zinc13407541 showed preferential and complete inhibition of FGF-23 action of FGFR/α-KL signaling in a heterologous cell model, although it exhibited partial inhibitory effects on several other FGF family members tested at higher concentration.
Our docking simulations and target engagement assays suggest that Zinc13407541 directly binds FGF-23 proteins and inhibits its interaction with the FGFR1-α-KL complex. This premise is supported by target engagement studies showing that Zinc13407541 binds to and stabilizes FGF-23, immunoprecipitation studies showing that Zinc13407541 disrupts FGF-23 pull-down of the FGFR/α-KL binary complex (11, 58), and functional studies showing that Zinc13407541 blocks FGF-23 activation of FGFR/α-KL-dependent signaling and gene expression. However, these studies do not assess the effects of Zinc13407541 to disrupt the physical interactions between FGF-23, α-KL, and FGFRs, and additional techniques, such as surface plasmon resonance spectroscopy, will be needed to confirm whether FGF-23 disrupts this ternary complex formation (51, 62). Further computational modeling and medicinal chemistry investigations are also needed to define structure activity/function relationships around these probes to demonstrate feasibility of extensive lead optimization and developing compounds with greater specificity. Because the computations identified residues that may be important to antagonist binding, mutagenesis and additional target engagement studies can confirm these binding sites (50, 75, 76). Finally, studies of Zinc13407541 on FGF-23 metabolism are needed to explore alternative mechanisms of action.
To our knowledge, these are the first small molecules to be identified that inhibit FGF-23 activation of FGFR/α-KL complexes through targeting the ligand to disrupt receptor interactions and activation. Both thermal shift assays and immunoprecipitation studies confirm the effects of Zinc13407541 to bind to FGF-23 and disrupt its interaction with the FGFR/α-KL complex, and studies in heterologous cell reporter models show dose-dependent effects of Zinc13407541 to inhibit FGF-23 activation of FGFR/α-KL signaling. Zinc13407541 provides greater selectivity than the currently available receptor tyrosine kinase inhibitors that are used to inhibit all of FGF-FGFR signaling (48, 49). Further, we show the biological relevance of these observations, by the additional findings that Zinc13407541 inhibits FGF-23 regulation of genes in the proximal tubule cells that FGF-23 is known to regulate in vivo, including enzyme regulating vitamin D metabolism (Cyp27b1 and Cyp24a1), and genes controlling sodium phosphate (Npt2a) and sodium chloride (NCC) cotransporters. Consistent with Zinc13407541 inhibition of FGF23-regulated Cyp27b1 and Cyp24a1 expressions in renal tubule cells, Zinc13407541 administration to Dmp1-null mice (a model of FGF-23 excess) also raised the serum concentration of 1,25(OH)2D and alleviated hypophosphatemia. These data indicate that Zinc13407541 may have clinical utility to block the effects of excess FGF-23 on the kidney.
We found that Zinc13407541 did not affect circulating FGF-23 concentrations in wild-type mice but increases serum phosphate, 1,25(OH)2D, and PTH, consistent with inhibition of FGFR/α-KL signaling in kidney and parathyroid gland (fig. S3). The mechanism of Zinc13407541 to reduce circulating FGF-23 amounts in Dmp1-null but not wild-type mice is not clear. This disparity could reflect an inhibition of FGFR/α-KL in bone that underlies increased FGF-23 in Dmp1-null mice (79, 80). Effects of variably decreased and increased circulating FGF-23 by FGFR inhibition have been observed and are attributed to opposing effects of the inhibition of FGF-23 production by bone and secondary increases in FGF-23 due to end-organ resistance from blocking FGFRs in the kidney (48, 49). We did not assess the effects of Zinc13407541 on FGF-23 stability, but if Zinc13407541 stimulated FGF-23 degradation, then its concentration should be reduced in both wild-type and Dmp1-null mice. Zinc13407541 was designed to bind to regions in the N terminus of FGF-23, remote from the furin RXR cleavage site in FGF-23 (19), and our thermal-based target engagement studies suggest that effects of Zinc13407541 stabilize FGF-23.
Similar to our findings, a recombinant human IgG1 mAb, KRN23, that binds to FGF-23 has been shown to correct hypophosphatemia and abnormalities in vitamin D metabolism in both mouse homolog models and humans with XLH (44). Zinc13407541 treatment also increased circulating concentration of PTH in Dmp1-null mice without altering serum calcium concentrations. This response is consistent with inhibition of FGF-23 effects to activate FGFR1/α-KL in the parathyroid gland and suppress PTH secretion (21), but differs from effects of the FGFR inhibitor or KRN23, which suppress serum PTH levels in the Hyp mouse model of FGF-23 excess (44, 48, 81). There are other notable differences between Zinc13407541 and KRN23 as potential therapeutics. KRN23 requires parenteral administration and has a long half-life that limits dose titrations. Because over-suppression of FGF-23 leads to tumoral calcinosis, use of KRN23 may have a narrow therapeutic window compared to an orally administered small-molecule FGF-23 antagonist. These small-molecule drug antagonists identified herein may provide an important alternative to the biological molecules that are currently under development to inhibit FGF-23 actions. Future studies comparing Zinc13407541 and KRN23 will be needed to establish the relative safety and efficacy of these different approaches for blocking FGF-23.
Although our findings suggest that Zinc13407541 targets the FGF-23 ligand, additional modifications will likely be needed to realize its therapeutic potential. At high concentrations, it also blocks other FGF ligands that lack the unique FGF-23 C terminus that interacts with α-KL. Other approaches to more selectively inhibit FGF-23 activity may be achieved by blocking interactions between the FGF-23 C terminus and α-KL (11, 51, 62). For example, an unstructured 26-residue sequence of the C-terminal domain was discovered to bind the FGFR1/ α-KL complex (51), leading to competitive inhibition of FGF-23 activation of the FGFR (82). Additional modeling may identify new chemical antagonists that target both the N terminus and the C terminus of FGF-23 that would likely impart greater efficacy and specificity in disrupting FGF-23/α-KL signaling. Regardless, Zinc13407541 defines a chemical platform for lead optimization that, in future studies, will include pharmacokinetics/pharmacodynamics testing, toxicity assessments, and chemical modifications necessary to explore the therapeutic potential of these compounds. Chemical compounds that antagonize FGF-23 activation of FGFRs provide a new tool to probe the functions of FGF-23 and set the stage for developing clinical drug candidates to treat disorders of FGF-23 excess.
In conclusion, we validate the utility of a computationally driven structure-based drug discovery approach using homology modeling, MD simulations, and docking to identify novel small molecules that inhibit FGF-23 activation in the presence of FGFR1 and α-KL. These chemical probes provide a new way to inhibit ligand-FGFR1-α-KL interactions and FGFR signaling inhibition without targeting the intrinsic tyrosine kinase activity and identify a chemical platform on which to develop lead compounds as potential treatments for disorders caused by FGF-23 excess. There are 22 mammalian FGFs and 4 alternative spliced FGFR genes (FGFR1 to FGFR4) that encode seven membrane-associated tyrosine kinase isoforms (FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, and FGFR4). An analogous approach might be used to disrupt other FGF ligand and FGFR interactions, thereby establishing a new paradigm for developing drugs that selectively disrupt FGFR activation by each of these FGF ligands.
MATERIALS AND METHODS
In silico virtual screening for the identification of trial compounds
Four computational models of FGF-23 were prepared using an FGF-23 crystal structure [Protein Data Bank ID (PDBID): 2p39 (52)], and three homology models were generated with the aid of the Max Planck Bioinformatics Toolkit (83). An HHPred sequence search (84) resulted in the selection of three crystal structures, two of FGF19 [PDBIDs: 1pwa (85) and 2p23 (52)] and one of FGF12 [PDBID: 1q1u (86)], to be used as templates to build the homology models. The structures for the three homology models were constructed with Modeller (87) by threading the FGF-23 sequence along the corresponding backbone geometry. The fourth model is based on the FGF-23 crystal structure [PDBID: 2P39 (52)]. The heparin analog and crystal-resolved solvent were removed from the crystal structure in preparation for short, constrained MD simulations.
All four models were prepared and simulated using CHARMM-GUI (88) and CHARMM (89). The proteins were protonated using the HBUILD facility of CHARMM (90), and each model was solvated in an octahedral unit cell (a = b = c = 85 Å). The structures were energy-minimized for 500 steps of steepest descent (91) and 500 steps of adopted basis Newton-Raphson (92) with the backbone and side chains restrained using 1.0 and 0.1 kcal mol−1 Å−2 harmonic potentials, respectively. Using the same restraints on the solute, the solvent was relaxed using MD with the NVE [a statistical thermodynamic ensemble where the number of particles (N), the volume (V), and the energy (E) of the system are held approximately constant through an iterative procedure] ensemble for 200 ps with a 1-fs time step. For production MD simulations, the same harmonic restraint was used to constrain the backbone, whereas the side chains were unrestrained. The SHAKE (93) algorithm was also used to constrain all bonds including hydrogen in the MD steps for solvent relaxation and the production simulations. The MD simulations were carried out using the NPT [number of particles (N), pressure (P), and temperature (T), which are held constant] ensemble at 1 atm and 298 K with a 2-fs time step. Eight independent seeds were carried out for each homology model; each seed was run with 2-fs time steps for 7 ns. Short MD simulations with the backbone constrained to the neighborhood of the initial starting structure have been shown to be adequate for the refinement of homology models (94).
The final configuration of each seed of each homology model was submitted to the FTMap Web server (95) to identify possible binding sites. FTMap has been shown to be adept at identifying druggable hotspots in proteins, particularly when used in conjunction with ensemble docking as in the present work (95–97). The consensus clusters generated by FTMap were culled to binding centers (at least 7.5 Å apart) using k-means clustering and HackaMol (98). The NCI Diversity Set 2 was used to carry out initial screens for each refined homology model. Subsequently, the ZINC database (99) was screened for molecules with a Tanimoto cutoff (100) of 0.8. This was done to ensure that the molecules screened from the ZINC database were chemically unique from those of the NCI Diversity Set 2. Open Babel (101) was used to build the starting configuration of each molecule from the respective SMILES representation (102). MGLTools (103) was used to generate the Protein Data Bank, Partial Charge and Atom Type for each candidate small molecule (ligand) and each FGF-23 configuration (receptor). The HackaMol interface to AutoDock Vina (104) was used for all screens. A 20 Å cubic screening box, centered at the clustered potential binding sites identified by FTMap, was used for each docking run. The AutoDock Vina exhaustiveness parameter was set to 24.
Chemicals and reagents
The most promising molecules from each virtual screen were tested experimentally. Four of the compounds (ZINC13407541, ZINC00055523, ZINC04769985, and ZINC01626100) were purchased from AKos Consulting and Solutions Deutschland GmbH. We obtained 12 chemicals [NCI_293778, NCI_37553, NCI_308835, NCI_84100_a, NCI_61610, NCI_80313, NCI_116702, NCI_97920, NCI_93354_a, NCI_374204 (ZINC01589294), NCI_401490 (ZINC01594155), and NCI_102656 (ZINC01674794)] from the Developmental Therapeutic Program of the National Cancer Institute. Recombinant human FGF-23 and FGF-2 were purchased from R&D Systems. Synthetic human C-terminal FGF-23 (residues 180 to 251) peptide (FGF-23CT), which binds to α-KL and blocks full-length FGF-23 binding to the FGFR/α-KL binary complex, was obtained from Phoenix Pharmaceuticals Inc. Recombinant human FGF-1, FGF-4, FGF-5, FGF-9, FGF-10, FGF-18, FGF-19, and FGF-21 were purchased from PeproTech. The laboratory of J.D.C. also synthesized compound ZINC13407541 at Tennessee Technological University for in vivo animal experiments. The nuclear magnetic resonance (NMR) spectra analysis showed that the purity of ZINC13407541 was more than 95%.
Cell culture, RT-PCR, and in vitro screening assays
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 1% penicillin and streptomycin (P/S). Total RNA was extracted by TRIzol Reagent (Thermo Fisher Scientific). Human kidney total RNA (AM7976, Thermo Fisher Scientific) was used as positive control. Regular RT-PCR was performed as previously described (105), using various sets of primers (table S2). For FGF-23-mediated activation of the FGFR1/α-KL complex, HEK293T cells were transiently transfected with either empty expression vector or full-length human α-KL along with the ERK luciferase reporter system (11) and Renilla luciferase-null as internal control plasmid. Transfections were performed by electroporation using Cell Line Nucleofector Kit R according to the manufacturer’s protocol (Amaxa Inc.). Thirty-six hours after transfection, the transfected cells were treated with the test compound in the presence or absence of 1 nM FGF-23. After 5 hours, the cells were lysed, and luciferase activities were measured using a Synergy H4 Hybrid Multi-Mode Microplate Reader and Promega Dual-Luciferase Reporter Assay System.
Primary tubule cell cultures, Western blot, and quantitative real-time RT-PCR
The animal protocols and procedures have been approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (ID, 15–138.0). Wild-type C57BL/6 mice at 12 to 16 weeks of age were used in the current experiments. The isolation of tubular cells was performed as described by Weinman et al. (106) with some modifications. Briefly, kidney cortices were decapsulated and dissected to obtain 1-mm3 fragments. The fragments were then digested in 10 ml of Hanks’ balanced salt solution (Invitrogen Corp.) with 1% Worthington collagenase type II (200 U/ml) and 0.025% soybean trypsin inhibitor. This step was performed three times at 37°C for 15 min, and the suspension was filtered through filters with a mesh size of 100 μm after each digestion. The cell suspension was washed twice in PBS and centrifuged for 5 min at 200g. Cells were cultured in Matrigel-coated six-well plates with complete DMEM/F12 1:1 medium containing epidermal growth factor (10 ng/ml), 1% P/S, 1% L-glutamine, 15 mM Hepes, 0.11 mM hydrocortisone (40 ng/ml), and insulin/transferrin/selenium (Invitrogen; 5 mg/ml, 2.75 mg/ml, and 3.35 ng/ml, respectively) at 37°C under 5% CO2 in a humidified atmosphere. The culture medium was changed after 24 hours to eliminate nonadherent cells and residual cellular fragments. The tubule cells were grown in the growth medium for 4 days and treated with FGF-23 and the FGF-23 inhibitor for 4 hours. Western blotting and quantitative real-time RT-PCR were performed as previously described (79). Antibodies to FGFR1 (D8E4, #9740), phospho-ERK1/2 (Thr202/Tyr204; D13.14.4E, #4370), and ERK1/2 (#9102) were purchased from Cell Signaling Technologies. Antibody to phospho-FGFR1 (Tyr653, Tyr654; 44–1140G) was purchased from Thermo Fisher Scientific. Antibody to KL (KAL-KO604) was purchased from Cosmo Bio USA Inc. Antibody to β-actin (sc-47778) was obtained from Santa Cruz Biotechnology. Band intensity was quantified using ImageJ software (http://rsb.info.nih.gov/ij/).
Ensemble docking of experimentally verified hits on the N-terminal domain of FGF-23
To probe possible binding modes of the two lead compounds in more detail, we performed unconstrained simulations of the target. These additional MD simulations were carried out with the Amber suite of programs (107). The 2P39 crystal structure was used as the starting structure for these simulations (52). The system was solvated with an octahedral, periodic box consisting of 6810 TIP3P water molecules, and three Cl− atoms were added to maintain electrostatic neutrality of the system. Here, the crystallographic water molecules were retained. The protein topology file was built with the parm99SB (108) version of the Cornell force field (109). The system was energy-minimized via a two-step process. First, FGF-23 was held fixed with a force constant of 500 kcal mol−1 Å−1, whereas the system was minimized with 500 steps of steepest descent (91) performed followed by 500 steps of the conjugate gradient method (110). In the second minimization step, the restraint on FGF-23 was removed, and 1000 steps of steepest decent were performed followed by 1500 steps of conjugate gradient. The system was heated to 300 K while holding the protein fixed with a force constant of 10 kcal mol−1 Å−1 while running 1000 MD steps. Then, the restraints were released and 1000 MD steps were run. The SHAKE (93) algorithm was used to constrain all bonds involving hydrogen in the simulations as in the in silico virtual screen. A 200-ns MD run was completed from a randomly generated seed. A snapshot from the trajectory was written every 1000 MD steps. This yielded a total of 100,000 snapshots for further analysis.
These MD trajectories were analyzed with cluster analysis as implemented in the ptraj software in Amber12 (111). The cluster analysis resulted in the identification of 36 clusters. One representative structure from each cluster was used to create a small ensemble of structures for docking of the experimentally verified antagonist compounds. From this small ensemble, four structures were randomly selected for binding site identification and docking of the experimentally verified FGF-23 inhibitors with AutoDock Vina (104). FTMap was used to identify potential binding sites for the inhibitor compounds (112, 113). The experimentally identified chemical probes were then docked to each binding site identified by FTMap. To identify locations where the inhibitors bind FGF-23’s N-terminal domain, we used all the centers identified by FTMap to probe a larger portion of the protein surface along with a large search box size. Zinc13407541 and Zinc01626100 were docked to each potential binding center on each of the four randomly selected structures from the ensemble of structures created from the MD simulations. This is in contrast to other studies where only the top five consensus clusters are used for drug design (114). A 20 × 20 × 20 Å search box was used, and the exhaustiveness parameter was set to 25. The binding site centers were not combined in an effort to allow the docking simulations to search a larger part of the protein surface for stable binding locations.
Target engagement assays
We performed coimmunoprecipitation assays using HEK293T cell lysates cotransfected with V5-tagged FGF-23 (2.0 μg), full-length FGFR1 (2.0 μg), and membrane α-KL (2.0 μg). The cells were lysed with ice-cold 1× immunoprecipitation lysis buffer (Pierce Biotechnology) containing 1× protease inhibitors. Fifty microliters of anti-V5 magnetic beads (MBL International Corporation) was added into 400 μl of the lysate supernatant in the presence of Zinc13407541 (10 μM) or DMSO control. Equal amount of normal IgG magnetic beads (Sigma-Aldrich) served as a negative control. The mixtures were incubated with gentle agitation for 2 hours at 4°C. The mixtures were placed into the tube on the magnetic rack for a few seconds, and the magnetic beads were washed three times with cold lysis buffer and suspended in 30 μl of Laemmli sample buffer. Ten microliters of the supernatant sample per lane was loaded in 4 to 12% bistris gradient gels (Invitrogen) and carried out for electrophoresis and Western blot analysis. Antibody to α-KL (rat mAb KM2076) was purchased from TransGenic Inc. Antibody to FGFR1 (D8E4, #9740) was purchased from Cell Signaling Technologies. Antibody to FGF-23 (MAB2629) was obtained from R&D Systems Inc. Band intensity was quantified using ImageJ software (http://rsb.info.nih.gov/ij/).
Next, we conducted protein thermal shift assay using a real-time PCR instrument as previously described. Fifty microliters of solutions consisting of 5 nM FGF-23, 5×SYPRO Orange (Sigma-Aldrich), 10 mM Hepes-NaOH (pH 7.5), 150 mM NaCl, and either vehicle DMSO or Zinc13407541 (200 μM) was added to the wells of the 96-well iCycler iQ PCR plate. The plate was heated from 25° to 70°C with a heating rate of 0.5°C/30 s. The fluorescence intensity was measured with an excitation of 490 nm and an emission of 530 nm.
Animal experiments
All animal research was conducted according to the guidelines provided by the National Institutes of Health and the Institute of Laboratory Animal Resources, National Research Council. The University of Tennessee Health Science Center’s Animal Care and Use Committee approved all animal studies (protocol number, 15–138.0). All mice were maintained in our vivarium on a standard diet (7912; Harlan Teklad). To generate homozygous Dmp1 knockout mice, we crossed male heterozygous Dmp1 knockouts with female heterozygous Dmp1 knockouts to obtain homozygous Dmp1 knockouts as previously described (74). At 8 weeks of age, homozygous Dmp1 knockout mice were selected to collect blood for serum before treatments. Then, the mice were treated with intraperitoneal injection of Zinc13407541 (100 mg/kg) or vehicle control (5% DMSO in PBS solution) twice a day for 3 days. The serum samples were collected 4 hours after the last dose administration. Serum FGF23 concentration was measured using the FGF23 enzyme-linked immunosorbent assay (ELISA) kit (Kainos Laboratories). Serum phosphorus concentration was measured using a Phosphorus Liqui-UV kit (Stanbio Laboratory), and serum calcium levels were measured using a Calcium (CPC) Liquicolor kit (Stanbio Laboratory). Serum PTH concentration was measured using the Mouse Intact PTH ELISA kit (Immutopics). Serum 1,25(OH)2D concentration was measured using the 1,25-Dihydroxy Vitamin D EIA kit (Immunodiagnostic Systems) as previously described (79).
Statistical analysis
We evaluated differences between two groups, between multiple groups, and between two groups over time by unpaired t test, by one-way ANOVA, and by two-way ANOVA with interactions, respectively, as indicated in the figure legends. All values are expressed as means ± SD. All computations were performed using commercial biostatistics software (GraphPad Software Inc.).
Supplementary Material
Acknowledgments
Funding: This work was supported by grant R01-AR045955–15 and a supplement, R01-AR045955–15S1,to L.D.Q.from National Institute of Arthritis and Musculoskeletal and Skin Diseases Building Interdisciplinary Research Team Revision Award.
Footnotes
Competing interests: J.B. is a part owner of Minerva Discovery. The other authors declare that they have no competing interests.
Data and materials availability: Microgram quantities of Zinc13407541 are commercially available from AKos Consulting and Solutions Deutschland GmbH.
REFERENCES AND NOTES
- 1.Itoh N, Hormone-like (endocrine) Fgfs: Their evolutionary history and roles in development, metabolism, and disease. Cell Tissue Res. 342, 1–11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu S, Guo R, Simpson LG, Xiao Z-S, Burnham CE, Darryl Quarles L, Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J. Biol. Chem. 278, 37419–37426 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Quarles LD, Evidence for a bone-kidney axis regulating phosphate homeostasis. J. Clin. Investig. 112, 642–646 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Darryl Quarles L, Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol. Endocrinol. Metab. 291, E38–E49 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD, Pathogenic role of Fgf23 in Dmp1-null mice. Am. J. Physiol. Endocrinol. Metab. 295, E254–E261 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quarles LD, The bone and beyond: ‘Dem bones’ are made for more than walking. Nat. Med. 17, 428–430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pi M, Quarles LD, Multiligand specificity and wide tissue expression of GPRC6A reveals new endocrine networks. Endocrinology 153, 2062–2069 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H, Fujita T, Kuroki R, Yamashita T, Fukumoto S, Shimada T, Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J. Bone Miner. Res. 23, 1509–1518 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M, Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292–15297 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Yamashita T, Yoshioka M, Itoh N, Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 277, 494–498 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Urakawa I, Yamazaki Y, Shimada T, lijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T, Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774 (2006).17086194 [Google Scholar]
- 12.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y-I, Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Mitobe M, Yoshida T, Sugiura H, Shirota S, Tsuchiya K, Nihei H, Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp. Nephrol. 101, e67–e74 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW, Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 24, 3438–3450 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J, The parathyroid is a target organ for FGF23 in rats. J. Clin. Invest. 117, 4003–4008 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou L, Li Y, Zhou D, Tan RJ, Liu Y, Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 24, 771–785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki M, Uehara Y, Motomura-Matsuzaka K, Oki J, Koyama Y, Kimura M, Asada M, Komi-Kuramochi A, Oka S, Imamura T, βKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 22, 1006–1014 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T, Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. U.S.A. 98, 6500–6505 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ, Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 60, 2079–2086 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M, FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am. J. Physiol. Renal Physiol. 297, F282–F291 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, Pohl EE, Erben RG, FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med.6, 744–759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai B, David V, Martin A, Huang J, Li H, Jiao Y, Gu W, Quarles LD, A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLOS ONE 7, e44161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R, Shalhoub V, Mohammadi M, Pohl EE, Lanske B, Erben RG, FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 33, 229–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, Ono K, Kakitani M, Tomizuka K, Fujita T, Fukumoto S, Yamashita T, Vitamin D receptor- independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Physiol. Renal Physiol. 289, F1088–F1095 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Bai X, Miao D, Li J, Goltzman D, Karaplis AC, Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145, 5269–5279 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren Ö, Tenenhouse HS, Jüppner H, Jonsson KB, Transgenic mice expressing fibroblast growth factor 23 under the control of the a1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145, 3087–3094 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Fukumoto S, Yamashita T, Fibroblast growth factor-23 is the phosphaturic factor in tumor-induced osteomalacia and may be phosphatonin. Curr. Opin. Nephrol. Hypertens. 11, 385–389 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T, Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 113, 561–568 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benet-Pagès A, Orlik P, Strom TM, Lorenz-Depiereux B, An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet. 14, 385–390 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Larsson T, Davis SI, Garringer HJ, Mooney SD, Draman MS, Cullen MJ, White KE, Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differentially processed. Endocrinology 146, 3883–3891 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Sitara D, Razzaque MS, St-Arnaud R, Huang W, Taguchi T, Erben RG, Lanske B, Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am. J. Pathol. 169, 2161–2170 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Jüppner H, Lanske, Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 23, 421–432 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato K, Jeanneau C, Tarp MA, Benet-Pagès A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H, Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis: Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 281, 18370–18377 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Quarles LD, Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 8, 276–286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komaba H, Fukagawa M, FGF23-parathyroid interaction: Implications in chronic kidney disease. Kidney Int. 77, 292–298 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Craver L, Marco MP, Martínez I, Rue M, Borras M, Martín ML, Sarró F, Valdivielso JM, Fernández E, Mineral metabolism parameters throughout chronic kidney disease stages 1–5—Achievement of K/DOQI target ranges. Nephrol. Dial. Transplant. 22, 1171–1176 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M, Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 359, 584–592 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F; MMKD Study Group, Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The mild to moderate kidney disease (MMKD) study. J. Am. Soc. Nephrol. 18, 2600–2608 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Mirza MAI, Alsiö J, Hammarstedt A, Erben RG, Michaëlsson K, Tivesten Å, Marsell R, Orwoll E, Karlsson MK, Ljunggren Ö, Mellström D, Lind L, Ohlsson C, Larsson TE, Circulating fibroblast growth factor-23 is associated with fat mass and dyslipidemia in two independent cohorts of elderly individuals. Arterioscler. Thromb. Vasc. Biol. 31, 219–227 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ, Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J. Clin. Endocrinol. Metabol. 95, 1846–1850 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Georgoulas TI, Tournis S, Lyritis GP, Changes of fibroblast growth factor 23 (FGF23) levels following calcitriol treatment in a vitamin D deficient patient. J. Musculoskelet. Neuronal Interact. 14, 398–400 (2014). [PubMed] [Google Scholar]
- 42.Burnett-Bowie S-AM, Leder BZ, Henao MP, Baldwin CM, Hayden DL, Finkelstein JS, Randomized trial assessing the effects of ergocalciferol administration on circulating FGF23. Clin. J. Am. Soc. Nephrol. 7, 624–631 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wesseling-Perry K, Salusky I, Phosphate binders, vitamin D and calcimimetics in the management of chronic kidney disease-mineral bone disorders (CKD-MBD) in children. Pediatr. Nephrol. 28, 617–625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M, Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J. Clin. Invest. 124, 1587–1597 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stubbs JR, Idiculla A, Slusser J, Menard R, Quarles LD, Cholecalciferol supplementation alters calcitriol-responsive monocyte proteins and decreases inflammatory cytokines in ESRD. J. Am. Soc. Nephrol. 21, 353–361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wetmore JB, Quarles LD, Calcimimetics or vitamin D analogs for suppressing parathyroid hormone in end-stage renal disease: Time for a paradigm shift? Nat. Clin. Pract. Nephrol. 5, 24–33 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wetmore JB, Liu S, Krebill R, Menard R, Quarles LD, Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin. J. Am. Soc. Nephrol. 5, 110–116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wöhrle S, Henninger C, Bonny O, Thuery A, Beluch N, Hynes NE, Guagnano V, Sellers WR, Hofmann F, Kneissel M, Graus Porta D., Pharmacological inhibition of fibroblast growth factor (FGF) receptor signaling ameliorates FGF23-mediated hypophosphatemic rickets. J. Bone Miner. Res. 28, 899–911 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Wöhrle S, Bonny O, Beluch N, Gaulis S, Stamm C, Scheibler M, Müller M, Kinzel B, Thuery A, Brueggen J, Hynes NE, Sellers WR, Hofmann F, Graus-Porta D, FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J. Bone Miner. Res. 26, 2486–2497 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Herbert C, Schieborr U, Saxena K, Juraszek J, De Smet F, Alcouffe C, Bianciotto M, Saladino G, Sibrac D, Kudlinzki D, Sreeramulu S, Brown A, Rigon P, Herault JP, Lassalle G, Blundell TL, Rousseau F, Gils A, Schymkowitz J, Tompa P, Herbert JM, Carmeliet P, Gervasio FL, Schwalbe H, Bono F, Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell 23, 489–501 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M, Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. U.S.A. 107, 407–412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M, Molecular Insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 27, 3417–3428 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H., Koller A, Nizet V, White KE, Dixon JE, Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. U.S.A. 111, 5520–5525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT, The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Jones DT, Cozzetto D, DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Reynolds CH, Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J. Chem. Inf. Comput. Sci. 42, 1407–1414 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Bajusz D, Racz A, Héberger K, Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminf. 7, 20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M, Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 281, 6120–6123 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, Michigami T, Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J. Cell. Biochem. 111, 1210–1221 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Hodge C, Liao J, Stofega M, Guan K, Carter-Su C, Schwartz J, Growth hormone stimulates phosphorylation and activation of elk-1 and expression of c-fos, egr-1, and junB through activation of extracellular signal-regulated kinases 1 and 2. J. Biol. Chem. 273, 31327–31336 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Han X, Yang J, Li L, Huang J, King G, Quarles LD, Conditional deletion of Fgfr1 in the proximal and distal tubule identifies distinct roles in phosphate and calcium transport. PLOS ONE 11, e0147845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goetz R, Ohnishi M, Kir S, Kurosu H, Wang L, Pastor J, Ma J, Gai W, Kuro-o M, Razzaque MS, Mohammadi M, Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J. Biol. Chem. 287, 29134–29146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu A-L, Feng B, Chen MZ, Kolumam G, Zavala-Solorio J, Wyatt SK, Gandham VD, Carano RAD, Sonoda J, Antibody-mediated activation of FGFR1 induces FGF23 production and hypophosphatemia. PLOS ONE 8, e57322 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD, Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Walters WP, Murcko MA, Prediction of ‘drug-likeness’. Adv. Drug Deliv. Rev. 54, 255–271 (2002). [DOI] [PubMed] [Google Scholar]
- 66.Lipinski CA, Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 1, 337–341 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Ghose AK, Viswanadhan VN, Wendoloski JJ, A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1, 55–68 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Muegge I, Pharmacophore features of potential drugs. Chemistry 8, 1976–1981 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Carlson HA, McCammon JA, Accommodating protein flexibility in computational drug design. Mol. Pharmacol. 57, 213–218 (2000). [PubMed] [Google Scholar]
- 71.Lin J-H, Perryman AL, Schames JR, McCammon JA, Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 124, 5632–5633 (2002). [DOI] [PubMed] [Google Scholar]
- 72.McCammon JA, Target flexibility in molecular recognition. Biochim. Biophys. Acta 1754, 221–224 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Li H, Martin A, David V, Quarles LD, Compound deletion of Fgfr3 and Fgfr4 partially rescues the Hyp mouse phenotype. Am. J. Physiol. Endocrinol. Metab. 300, E508–E517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE, Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 38, 1310–1315 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Batley BL, Doherty AM, Hamby JM, Lu GH, Keller P, Dahring TK, Hwang O, Crickard K, Panek RL, Inhibition of FGF-1 receptor tyrosine kinase activity by PD 161570, a new protein-tyrosine kinase inhibitor. Life Sci. 62, 143–150 (1998). [DOI] [PubMed] [Google Scholar]
- 76.Thompson AM, Connolly CJC, Hamby JM, Boushelle S, Hartl BG, Amar AM, Kraker AJ, Driscoll DL, Steinkampf RW, Patmore SJ, Vincent PW, Roberts BJ, Elliott WL, Klohs W, Leopold WR, Showalter HD, Denny WA, 3-(3,5-Dimethoxyphenyl)-1,6-naphthyridine-2,7-diamines and related 2-urea derivatives are potent and selective inhibitors of the FGF receptor-1 tyrosine kinase. J. Med. Chem. 43, 4200–4211 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Ho J, Perez-Aguilar JM, Gao L, Saven JG, Matsunami H, Eckenhoff RG, Molecular recognition of ketamine by a subset of olfactory G protein-coupled receptors. Sci. Signaling 8, ra33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kapoor K, McGill N, Peterson CB, Meyers HV, Blackburn MN, Baudry J, Discovery of novel non-active site inhibitors of the prothrombinase enzyme complex. J. Chem. Inf. Model. 56, 535–547 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD, Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLOS ONE 9, e104154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith RC, O’Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, Cass TA, Saha J, Broderick C, Ma YL, Zeng QQ, Kharitonenkov A, Wilson JM, Guo Q, Sun H, Allen MR, Burr DB, Breyer MD, White KE, Circulating αKlotho influences phosphate handling by controlling FGF23 production. J. Clin. Invest. 122, 4710–4715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S, Shimada T, Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J. Bone Miner. Res. 24, 1879–1888 (2009). [DOI] [PubMed] [Google Scholar]
- 82.Mohammadi M, GOETZ R. (Google Patents, 2013).
- 83.Biegert A, Mayer C, Remmert M, Soding J, Lupas AN, The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 34, W335–W339 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Söding J, Biegert A, Lupas AN, The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harmer NJ, Pellegrini L, Chirgadze D, Fernandez-Recio J, Blundell TL, The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry 43, 629–640 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, Mohammadi M, Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 278, 34226–34236 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M, Pieper U, Sali A, Current Protocols in Bioinformatics (John Wiley & Sons Inc., 2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Brooks BR, Brooks CL III, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, f A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M, CHARMM: The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brunger AT, Karplus M, Polar hydrogen positions in proteins: Empirical energy placement and neutron diffraction comparison. Proteins 4, 148–156 (1988). [DOI] [PubMed] [Google Scholar]
- 91.Arfken G, in Mathematical Methods for Physicists (Academic Press, 1985), pp. 428–436. [Google Scholar]
- 92.Chu J-W, Trout BL, Brooks BR, A super-linear minimization scheme for the nudged elastic band method. J. Chem. Phys. 119, 12708–12717 (2003). [Google Scholar]
- 93.Ryckaert J-P, Ciccotti G, Berendsen HJC, Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977). [Google Scholar]
- 94.Raval A, Piana S, Eastwood MP, Dror RO, Shaw DE, Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins 80, 2071–2079 (2012). [DOI] [PubMed] [Google Scholar]
- 95.Brenke R, Kozakov D, Chuang G-Y, Beglov D, Hall D, Landon MR, Mattos C, Vajda S, Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 25, 621–627 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grant BJ, Lukman S, Hocker HJ, Sayyah J, Brown JH, McCammon JA, Gorfe AA, Novel allosteric sites on Ras for lead generation. PLOS ONE 6, e25711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miao Y, Nichols SE, McCammon JA, Mapping of allosteric druggable sites in activation-associated conformers of the M2 muscarinic receptor. Chem. Biol. Drug Des. 83, 237–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Riccardi D, Parks JM, Johs A, Smith JC, HackaMol: An object-oriented Modern Perl Library for molecular hacking on multiple scales. J. Chem. Inf. Model. 55, 721–726 (2015). [DOI] [PubMed] [Google Scholar]
- 99.Irwin JJ, Shoichet BK, ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.T. Tanimoto (McGraw-Hill, 1968).
- 101.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR, Open Babel: An open chemical toolbox. J. Cheminf. 3, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O’Boyle NM, Towards a universal SMILES representation—A standard method to generate canonical SMILES based on the InChI. J. Cheminf. 4, 22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanner MF, Python: A programming language for software integration and development. J. Mol. Graph. Model. 17, 57–61 (1999). [PubMed] [Google Scholar]
- 104.Trott O, Olson AJ, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xiao ZS, Thomas R, Hinson TK, Quarles LD, Genomic structure and isoform expression of the mouse, rat and human Cbfa1/Osf2 transcription factor. Gene 214, 187–197 (1998). [DOI] [PubMed] [Google Scholar]
- 106.Weinman EJ, Steplock D, Shenolikar S, Biswas R, Fibroblast growth factor-23- mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J. Biol. Chem. 286, 37216–37221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salomon-Ferrer R, Case DA, Walker RC, An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 198–210 (2013). [Google Scholar]
- 108.Hornak V, Abel E, Okur A, Strockbine B, Roitberg A, Simmerling C, Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA, A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 (1995). [Google Scholar]
- 110.Hestenes MR, Stiefel E, Methods of conjugate gradients for solving linear systems. J. Res. Natl. Bur. Stand. 49, 409–436 (1952). [Google Scholar]
- 111.Case D, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Gotz AW, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA, AMBER 12 (University of California, San Francisco, 2012). [Google Scholar]
- 112.Kozakov D, E Grove L, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D, Vajda S, The FTMap family of web servers for determining and characterizing ligand- binding hot spots of proteins. Nat. Protoc. 10, 733–755 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ngan CH, Bohnuud T, Mottarella SE, Beglov D, Villar EA, Hall DR, Kozakov D, Vajda S, FTMAP: Extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 40, W271–W275 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hall DR, Ngan CH, Zerbe BS, Kozakov D, Vajda S, Hot spot analysis for driving the development of hits into leads in fragment-based drug discovery. J. Chem. Inf. Model. 52, 199–209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.