

Abstract
BACKGROUND
Waldenström macroglobulinemia (WM) is preceded by asymptomatic WM (AWM), for which the risk of progression to overt disease is not well defined.
METHODS
We studied 439 patients with AWM, who were diagnosed and observed at Dana-Farber Cancer Institute between 1992 and 2014.
RESULTS
During the 23-year study period, with a median follow-up of 7.8 years, 317 patients progressed to symptomatic WM (72%). Immunoglobulin M 4,500 mg/dL or greater, bone marrow lymphoplasmacytic infiltration 70% or greater, β2-microglobulin 4.0 mg/dL or greater, and albumin 3.5 g/dL or less were all identified as independent predictors of disease progression. To assess progression risk in patients with AWM, we trained and cross-validated a proportional hazards model using bone marrow infiltration, immunoglobulin M, albumin, and beta-2 microglobulin values as continuous measures. The model divided the cohort into three distinct risk groups: a high-risk group with a median time to progression (TTP) of 1.8 years, an intermediate-risk group with a median TTP of 4.8 years, and a low-risk group with a median TTP of 9.3 years. We validated this model in two external cohorts, demonstrating robustness and generalizability. For clinical applicability, we made the model available as a Web page application (www.awmrisk.com). By combining two cohorts, we were powered to identify wild type MYD88 as an independent predictor of progression (hazard ratio, 2.7).
CONCLUSION
This classification system is positioned to inform patient monitoring and care and, for the first time to our knowledge, to identify patients with high-risk AWM who may need closer follow-up or benefit from early intervention.
INTRODUCTION
Waldenström macroglobulinemia (WM) is a low-grade non-Hodgkin lymphoplasmacytic lymphoma of the bone marrow (BM), characterized by production of monoclonal immunoglobulin M (IgM) protein.1,2 WM is a rare malignancy with an incidence of 3.4 per million among the male population and 1.7 per million among the female population in the United States, and an incidence of 7.3 and 4.2 per million for males and females, respectively, in Europe.3-6
The phenotype of these clonal lymphoplasmacytic cells suggests that they are derived from IgM memory B cells that have undergone somatic hypermutation, but not isotype switching.7-9 Approximately 90% to 95% of patients with WM carry the MYD88 L265P mutation, whereas 40% carry mutations in CXCR4.10-12
WM is preceded by an early precursor stage named IgM monoclonal gammopathy of undetermined significance (IgM MGUS) and a later stage known as smoldering WM (SWM).13,14 Both stages are asymptomatic, although SWM exhibits an increased risk of progression, which warrants closer follow-up and monitoring.15 According to previous reports, 1.5% of patients with IgM MGUS and 12% of patients with SWM progress to WM per year, and the rate decreases with the years of follow-up.10,11,13,14 Currently, treatment initiation is recommended in the presence of symptoms, including symptomatic lymphadenopathy or splenomegaly, constitutional symptoms, anemia with a hemoglobin level of 10 g/dL or less, platelet count less than 100 × 109/L, hyperviscosity syndrome, symptomatic peripheral neuropathy, and symptomatic cryoglobulinemia.16,17
Nonetheless, it has been challenging to distinguish the asymptomatic patients who will progress from those who will not. A revised stratification is needed, yet the rarity of the disease and the ensuing scarcity of data represent a practical challenge. We assembled the largest cohort of patients with AWM (including IgM MGUS and SWM) to date with the aim of identifying risk factors for disease progression and generating an evidence-based risk stratification system to help clinicians improve the management of patients with this rare malignancy and identify those who need closer follow-up.
METHODS
Primary Cohort
After institutional review board approval, we identified all patients with WM who had been diagnosed and observed at Dana-Farber Cancer Institute from November 1992 to December 2014 (Data Supplement). The cutoff date for follow-up was January 2018. Only patients with AWM at the time of diagnosis were included in this cohort to identify risk factors for disease progression. We defined patients with AWM as those who had morphologic findings of lymphoplasmacytic lymphoma in the BM and monoclonal IgM protein, encompassing both IgM MGUS and SWM stages.2 Exclusion criteria are provided in the Data Supplement. Clinical data were analyzed after reviewing medical records and death certificates. Survival status, cause of death, and disease progression were identified at the end of follow-up from the National Death Index and the patients’ medical records.
Validation Cohorts
Two different AWM cohorts were used for external validation. The first cohort was diagnosed and observed at Mayo Clinic, Rochester, MN, between 1996 and 2013. The second cohort was diagnosed and observed at the Department of Clinical Therapeutics, National and Kapodistrian University of Athens, Greece, between 1995 and 2014. Baseline characteristics of these cohorts are listed in the Data Supplement.
End Points
The primary end point of the study was progression to symptomatic WM that required treatment. Symptomatic WM was defined according to the criteria for treatment initiation by consensus panel recommendations from the Second International Workshop on Waldenström Macroglobulinemia.16-18
Risk Factors
Possible risk factors for disease progression were identified a priori and extracted from medical records. The presence of MYD88 L265P mutation was tested by allele-specific polymerase chain reaction on BM samples.11 All data were collected at the time of diagnosis.
Statistical Analysis
The primary end point with respect to progression to symptomatic WM was calculated in terms of the cumulative probability of progression. Time to progression (TTP) was calculated from the date of diagnosis to the date of the first assessment showing evidence of symptomatic disease and requiring treatment. Data for patients who died before disease progression, did not have disease progression during the study period or were lost to follow-up before progression were censored. For survival analysis, the Kaplan-Meier method was used to estimate the cumulative incidence of progression, and differences between the curves were tested by log-rank test. Disease-specific survival was calculated where patients whose death was deemed unrelated to WM or its complications were censored. A multivariable Cox proportional hazards model was used to identify risk factors for disease progression among patients with AWM.
RESULTS
Patient Baseline Characteristics
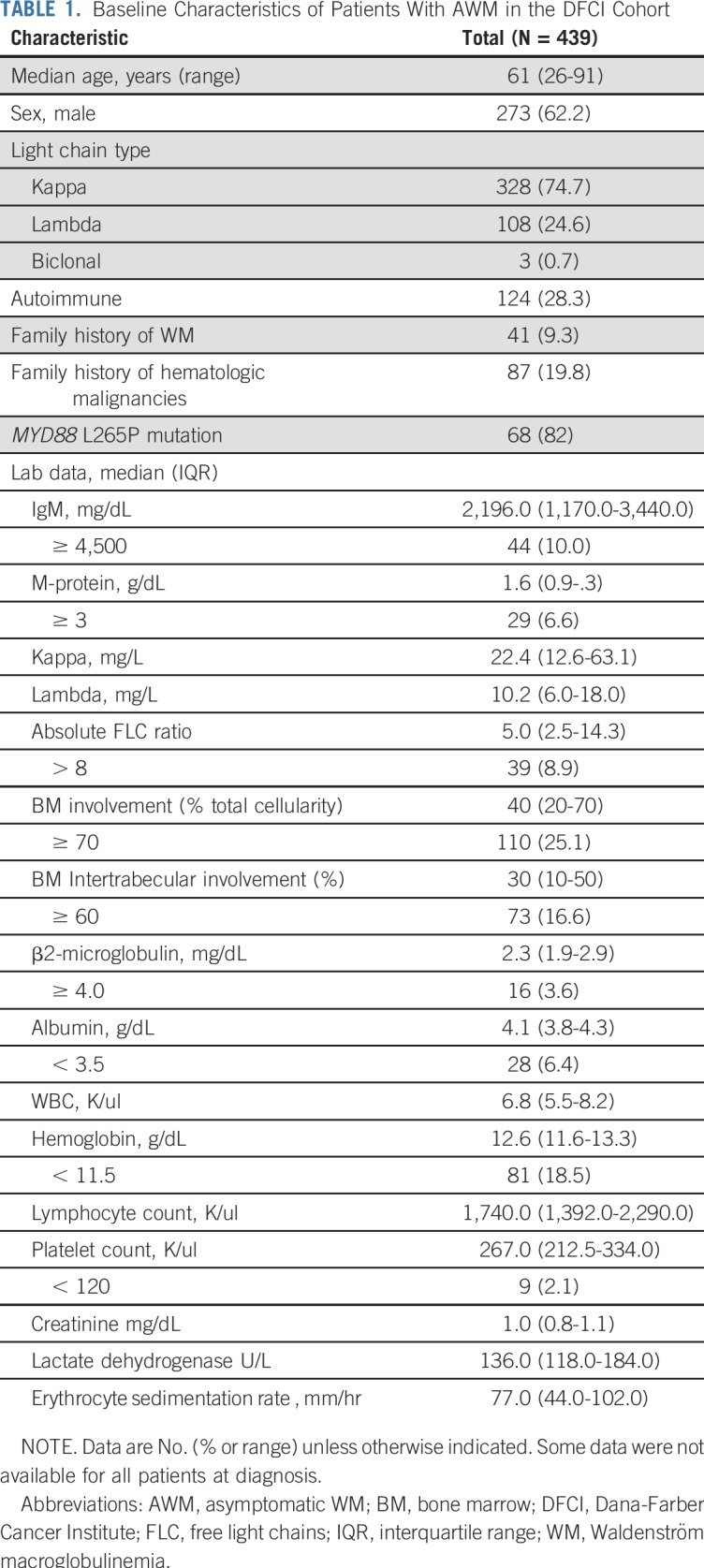
We identified 439 patients who were diagnosed with AWM at Dana-Farber Cancer Institute; 273 (62.2%) were men and 166 (37.8%) were women. The median age at WM diagnosis was 61 (range, 26 to 91) years. Forty-one patients (9.3%) had a family history of WM. All baseline characteristics are listed in Table 1.
TABLE 1.
Baseline Characteristics of Patients With AWM in the DFCI Cohort
Disease Progression
During the 23-year study period and a median follow-up of 7.8 years, 317 patients (72.2%) experienced disease progression. The median TTP from AWM diagnosis to symptomatic WM was 3.9 years (95% CI, 3.2 to 4.6 years). The probability of disease progression within 2 years was 30.8% (95% CI, 26.7% to 35.3%), as shown in Figure 1A.
FIG 1.

Cumulative probability of progression among patients. The Kaplan-Meier method was used for estimation of cumulative incidence of progression (A) among patients with asymptomatic Waldenström macroglobulinemia and stratified by (B) immunoglobulin M (IgM) levels, (C) bone marrow (BM) involvement, (D) β2-microglobulin, and (E) albumin.
To ensure there was no bias in TTP estimates because of changes in patient management during the study period of 23 years, we divided our cohort into two groups on the basis of the date of diagnosis (patients diagnosed between 1992 and 2003 and patients diagnosed between 2004 and 2014) and tested for a difference in TTP. No difference was observed between these two groups (log-rank test, P = .1; Data Supplement).
The 2-year progression rate was 63.6% (95% CI, 49.7% to 77.4%) for patients with IgM 4,500 mg/dL or greater and 25.7% (95% CI, 21.5% to 30.6%) for those with lower levels. The risk of progression was 45.5 and 15.1 events per 100 person-years, respectively (log-rank test, P < .001; Fig 1B).
The 2-year progression rate for patients with BM lymphoplasmacytic infiltration of 70% or greater was 61.0% (95% CI, 52.0% to 70.1%) and 20.6% (95% CI, 16.6% to 25.4%) for those with lower infiltration levels. The risk of progression was 37.5 and 13.6 events per 100 person-years, respectively (P < .001; Fig 1C).
The 2-year risk of progression among patients with β2-microglobulin of 4.0 mg/dL or greater was 65.3% (95% CI, 42.2% to 87.1%), whereas it was 28.1% (95% CI, 22.6% to 34.6%) in those with lower levels (P < .001; Fig 1D). Furthermore, the 2-year progression rate in patients with albumin less than 3.5 g/dL was 60.7% (95% CI, 43.5% to 78.3%), but only 27.1% (95% CI, 21.8% to 33.3%) in those with higher albumin levels (P < .001; Fig 1E).
The reasons for treatment initiation in the 317 patients who progressed to symptomatic WM were documented. Two thirds (67%) developed anemia associated with increasing IgM levels and constitutional symptoms. Peripheral neuropathy with increasing IgM levels were reported in 19.8% of the patients, whereas 15% developed hyperviscosity symptoms, and only 10.4% had organomegaly (lymphadenopathy and/or splenomegaly).
Of note, BM lymphoplasmacytic infiltration of 10% or more was associated with a significant increase in the risk of progression to symptomatic WM (log-rank test, P < .001). We also identified a small subgroup of patients (n = 24) with BM lymphoplasmacytic infiltration less than 10% and IgM less than 2,000 mg/dL, for which cumulative progression risk was particularly low, with 5- and 10-year progression-free survival of 100.0% and 78.7% (95% CI, 46.2 to 92.8), respectively (Data Supplement).
Risk Factors of Progression
We selected the previously mentioned cutoffs on the basis of their association with 60% or greater probability of progression to symptomatic WM within 2 years of diagnosis (Data Supplement). In the univariable analysis, the risk of progression to WM was significantly higher for patients with IgM 4,500 mg/dL or greater (hazard ratio [HR], 3.04), BM involvement percentage 70% or greater (HR, 2.78), β2-microglobulin 4.0 mg/dL or greater (HR, 3.01), and albumin less than 3.5 g/dL (HR, 2.79). In the multivariable model, IgM 4,500 mg/dL or greater (HR, 4.65; 95% CI, 2.52 to 8.58; P < .001), BM involvement percentage 70% or greater (HR, 2.56; 95% CI, 1.69 to 3.87; P < .001), β2-microglobulin 4.0 mg/dL or greater (HR, 2.31; 95% CI, 1.19 to 4.49; P = .014), and albumin less than 3.5 g/dL (HR, 2.78; 95% CI, 1.52 to 5.09; P = .001) were independent predictors of disease progression. The multivariable regression analysis results are listed in Table 2.
TABLE 2.
Risk Factors for Disease Progression in Patients With AWM in DFCI Cohort

Risk Stratification by a Proportional Hazards Model
The four factors that were significant in the multivariable analysis (BM infiltration, serum IgM, β2-microglobulin, and albumin) were then included as continuous variables in a proportional hazards model to predict TTP. The model was able to separate patients whose risk scores were below the first quartile (low risk) from those whose risk scores were in the interquartile range (intermediate risk) and those whose risk scores were above the third quartile (high risk). Effectively, this model divided the cohort into three distinct groups: (1) a high-risk group with a median TTP of 1.8 years (95% CI, 1.02 to 2.2 years), (2) an intermediate-risk group with a median TTP of 4.8 years (95% CI, 2.2 to 6.2 years), and (3) a low-risk group with a median TTP of 9.3 years (95% CI, > 5.5 years; Fig 2A). The model also successfully identified three risk groups with clear curve separation and similar medians and CIs when we divided the cohort into two groups on the basis of the diagnosis date (Data Supplement).
FIG 2.

Cumulative probability of disease progression among patients with different risk scores according to the proportional hazards model and model performance in external validation cohorts. (A) The model was built using four variables: BM involvement, immunoglobulin M (IgM) levels, β2-microglobulin, and albumin. It divided the cohort into three risk groups, corresponding to low-, intermediate-, and high-risk AWM with a median time to progression (TTP) of 9.3, 4.8, and 1.8 years, respectively. Dashed and solid lines represent the results of training set and cross-validation, respectively. (B) Mayo Clinic, Rochester, MN, cohort: median TTP of 10.2, 5.7, and 2.4 years, for the low-, intermediate-, and high-risk groups, respectively. (C) National and Kapodistrian University, Athens, Greece, cohort: median TTP of not reached, 7.3, and 2.9 years, for the low-, intermediate-, and high-risk groups, respectively. BM, bone marrow; AWM, Asymptomatic Waldenström macroglobulinemia.
Because of competing risks of death in our cohort with a median age of 61 years, disease-specific survival was calculated, censoring patients whose death was deemed unrelated to WM or its complications. We observed that high-risk AWM patients had significantly lower disease-specific survival compared to intermediate and low-risk groups (log-rank test, P = .029; Data Supplement).
External Validation of Predictive Model
With a median follow-up of 9.4 years, the first cohort, from Mayo Clinic, Rochester, MN, comprised 48 patients with a median TTP of 4.6 years and a progression rate of 77%. The model successfully divided the cohort into three groups with a median TTP of 2.4 years (95% CI, 1.4 to 4.9 years), 5.7 years (95% CI, 3.4 to 9.3 years), and 10.2 years (95% CI, > 5.58 years) for the high-, intermediate- and low-risk groups, respectively (Fig 2B). Similarly, with a median follow-up of 7.2 years, the second cohort, from Greece, which comprised 47 patients with a 34% progression rate, the model successfully divided the patients into three groups, with a median TTP of 2.92 years (95% CI, > 1.3 years), 7.25 years (95% CI, > 5.6 years), and not reached (95% CI, > 5.35 years) for the high-, intermediate-, and low-risk groups, respectively (Fig 2C).
Web Page Development
For clinical applicability, we designed a Web page application that allows clinicians to input patient laboratory values for the designated variables and obtain a risk score that places the patient in one of three risk groups: low, intermediate, or high (Data Supplement).
MYD88 Mutation Status Is an Independent Risk Factor of Progression
We combined available data on MYD88 mutation status from the Dana-Farber Cancer Institute cohort and the Greek cohort. In total, 106 patients from both cohorts had been genotyped; 89 patients (84%) carried MYD88 L265P and 17 (16%) were wild type (WT). The median TTP was 4.9 years (95% CI, 3.1 to 6.2 years) versus 1.8 years (95% CI, > 1.5 years) in patients with MYD88 L265P and MYD88 WT, respectively (log-rank test, P < .001; Fig 3). Wild type MYD88 was a significant independent risk factor for progression in multivariable analysis (HR, 2.7; 95% CI, 1.5 to 5; P < .001).
FIG 3.

MYD88 mutation status is an independent risk factor for progression to symptomatic Waldenström macroglobulinemia. The Kaplan-Meier method was used to compare progression-free survival between patients with MYD88 L265P and wild-type (WT) disease.
DISCUSSION
Because of its rarity, AWM has not been extensively studied, and only a few studies have been conducted in small cohorts.13,19,20 In fact, there are two sets of diagnostic criteria for AWM.2,13 The one proposed by Mayo Clinic requires 10% or greater BM lymphoplasmacytic cells for an SWM diagnosis and recommends a diagnosis of IgM MGUS for patients with less than 10% lymphoplasmacytic cells,13,15 whereas the recommendations of the Second International Workshop on Waldenström Macroglobulinemia panel defined AWM as the presence of any percentage of BM infiltration in the absence of WM symptoms, reserving the diagnosis of IgM MGUS for patients in whom there is no immunophenotypic evidence of WM.2,15-17 Until now, there has been no clear answer regarding which threshold is more appropriate. In our cohort, a BM lymphoplasmacytic infiltration of 10% or more was associated with a significantly increased risk of progression to WM. At the same time, we identified a small subgroup of patients with less than 10% infiltration and IgM less than 2,000 mg/dL, whose progression rate over time was similar to what is reported by studies on patients with IgM MGUS.14
However, it remains to be seen whether there is such a high-risk subgroup of patients with AWM for which progression is imminent enough to necessitate treatment. Progression risk in AWM has been previously studied by few groups. In 2003, Alexanian et al19 reported on a small cohort of 31 patients with AWM at MD Anderson Cancer Center. In that cohort, 19 of the 31 patients had BM infiltration less than 10%, and the median TTP was 6.9 years. Prognostic factors for early progression were hemoglobin less than 11.5 g/dL, β2-microglobulin 3.0 mg/L or greater, and IgM monoclonal protein greater than 3.0 g/dL. In 2005, Baldini et al20 reported results from a European cohort of 201 patients with SWM. With a median follow-up of only 5 years, approximately 22% of the patients progressed to overt disease. Independent risk factors of progression included increasing serum M-spike, decreasing hemoglobin, and male sex. In 2012, Kyle et al13 reported their results from a prospective cohort of 46 patients with SWM at Mayo Clinic, wherein approximately 71% of the patients had progressed to overt disease after a median follow-up of 15.4 years. In their study, independent risk factors for progression included BM lymphoplasmacytic infiltration percentage, serum M-spike, hemoglobin, and reduced serum IgA levels.
In this cohort of 439 patients with AWM, approximately 72% of the patients progressed to overt disease. In that respect, our cohort was more similar to that reported by Kyle et al.13 Independent risk factors of progression included BM lymphoplasmacytic infiltration 70% or greater, serum IgM 4,500 mg/dL or greater, β2-microglobulin 4 mg/dL or greater, and albumin less than 3.5 g/dL. These cutoffs were associated with a greater than 60% probability of disease progression within 2 years. Serum IgM was preferred as a biomarker over serum M-spike because of its ease of measurement and broad applicability. Hemoglobin was purposefully avoided as a predictor, given that its levels are already used as a threshold for treatment initiation in patients with AWM.
To predict progression to symptomatic WM, we proceeded to build a proportional hazards model using the four risk factors that were significant in the multivariable analysis. Given that these factors were continuous variables, we avoided dichotomizing them on the basis of artificial cutoffs and instead included them in the model as such. Because of that, our model was more flexible and comprehensive in stratifying patients with AWM. The model effectively divided the cohort into three risk groups—low, intermediate, and high-risk AWM—with a median TTP of 9.3, 4.8, and 1.8 years, respectively. The clear separation of risk group curves, as well as their corresponding TTP medians spanning a wide time interval, indicates that this model is representative of the whole spectrum of the asymptomatic disease state and can thus be used for comprehensive stratification of patients with AWM. Moreover, high-risk patients were shown to have significantly lower disease-specific survival, indicating a worse prognosis and suggesting that a potential intervention in this risk group might be warranted. To address generalizability concerns, we proceeded to validate our predictive model using two external cohorts from Mayo Clinic, Rochester, MN, and from the National and Kapodistrian University of Athens, Greece. The former was more enriched in high-disease-burden patients (77% progression rate), whereas the latter comprised more low-burden patients, with a progression rate of 34%. Our model had good discrimination properties in both cohorts, where it was able to identify three distinct risk groups with median times to progression that were similar to the ones in our cohort. These results serve to underline the robustness of our model, which was impervious to differences among cohorts spanning different centers and countries.
To simplify the application of the model, which we consider a key characteristic for clinical applicability, we made it available as an interactive Web application (www.awmrisk.com), where oncologists can enter an individual patient’s values and obtain information regarding their risk group and estimated risk of progression to symptomatic WM at any given timepoint.
As its most significant contribution, our model has managed to identify in all three cohorts a high-risk group of patients with AWM with an increased probability of progression within a short period of time from diagnosis. Such patients should be followed more closely or perhaps considered for early intervention in a clinical trial setting.
A previous study on patients with SWM (n = 66) showed that median TTP was 2.8 versus 1.9 years in patients with MYD88 L265P and MYD88 WT disease, respectively (P = .21).21 To investigate the role of MYD88 mutation status in disease progression in our study, we increased the number of patients with available MYD88 data by combining our cohort with the Greek cohort. In the combined data set (n = 106), patients with MYD88 WT disease had a significantly shorter TTP, and patients who carried MYD88 L265P mutation were 6.7 times less likely to progress to symptomatic WM, compared with patients with WT. Of note, all patients with WT disease in our data set progressed within 5 years from diagnosis. These results are in line with previous studies showing that MYD88 WT represents a more aggressive disease with a higher risk of transformation, resistance to therapy, and worse survival.22,23
Drawing on this large study on AWM, and with two external validation cohorts spanning the United States and Europe, we are uniquely positioned to address AWM diagnosis and stratification and lay the foundation for a broader consensus in the future. Our progression risk-based classification could help physicians improve the management of patients with AWM by distinguishing those who need closer follow-up or early intervention from those who do not and facilitate the design and implementation of clinical trials at the AWM stage, in the hopes of preventing disease progression with end-organ damage and improving patient outcomes.
ACKNOWLEDGMENT
We acknowledge the generous support of the Michele and Stephen Kirsch Fund for Waldenström Macroglobulinemia. We also acknowledge the generous support of Tone and Jørgen Heje family for Waldenstrom macroglobulinemia research. We are grateful to all the patients and their families for their contribution to this study.
Footnotes
Supported in part by Dana- Farber Cancer Institute to the Center for Prevention of Progression of Blood Cancers; National Institutes of Health Grants No. NIH R01 CA 205954 and F32 CA220859; Leukemia and Lymphoma Society; International Waldenström Macroglobulinemia Foundation; and Michele and Stephen Kirsch Fund for Waldenström Macroglobulinemia. I.M.G is a Scholar in Clinical Research of the Leukemia and Lymphoma Society.
AUTHOR CONTRIBUTIONS
Conception and design: Mark Bustoros, Romanos Sklavenitis-Pistofidis, Chia-Jen Liu, Carl Jannes Neuse, Catherine R. Marinac, David Liu, Lorenzo Trippa, Irene M. Ghobrial
Administrative support: Robert J. Soiffer, Irene M. Ghobrial
Provision of study materials or patients: Mark Bustoros, Romanos Sklavenitis-Pistofidis, Efstathios Kastritis, Steven P. Treon, Jorge J. Castillo, Stephen M. Ansell, Irene M. Ghobrial
Collection and assembly of data: Mark Bustoros, Romanos Sklavenitis-Pistofidis, Prashant Kapoor, Chia-Jen Liu, Efstathios Kastritis, Saurabh Zanwar, Jithma P. Abeykoon, Kalvis Hornburg, Carl Jannes Neuse, Maria Gavriatopoulou, Joseph M. Cappuccio, Henry Dumke, Kaitlen Reyes, Jorge J. Castillo, Meletios A. Dimopoulos, Irene M. Ghobrial
Data analysis and interpretation: Mark Bustoros, Romanos Sklavenitis-Pistofidis, Prashant Kapoor, Chia-Jen Liu, Geoffrey Fell, Jithma P. Abeykoon, Carl Jannes Neuse, David Liu, Jenny Soiffer, Cody Boehner, Henry Dumke, Robert J. Soiffer, Robert A. Kyle, Steven P. Treon, Jorge J. Castillo, Meletios A. Dimopoulos, Stephen M. Ansell, Lorenzo Trippa, Irene M. Ghobrial
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Progression Risk Stratification of Asymptomatic Waldenström Macroglobulinemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Mark Bustoros
Honoraria: Takeda, DAVAOncology
Consulting or Advisory Role: Takeda
Prashant Kapoor
Honoraria: Takeda
Consulting or Advisory Role: Sanofi (Inst)
Speakers' Bureau: Axis Medical Education
Research Funding: Amgen (Inst), Takeda (Inst), Sanofi (Inst)
Travel, Accommodations, Expenses: GlaxoSmithKline, Janssen, Sanofi, Takeda, Axis Pharma
Efstathios Kastritis
Honoraria: Amgen, Genesis Pharma, Janssen Oncology, Takeda
Consulting or Advisory Role: Amgen, Janssen Oncology, Takeda
Research Funding: Janssen Oncology (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: Janssen Oncology, Genesis Pharma
Jenny Soiffer
Leadership: Kiadis Pharma (I)
Consulting or Advisory Role: Merck (I), Gilead Sciences (I), Juno Therapeutics (I), Astellas Pharma (I)
Expert Testimony: Pfizer (I)
Maria Gavriatopoulou
Honoraria: Amgen, Janssen, Celgene, Takeda
Consulting or Advisory Role: Amgen, Karyopharm Therapeutics
Research Funding: Novartis
Travel, Accommodations, Expenses: Takeda, Genesis Pharma, Janssen
Robert J. Soiffer
Leadership: Kiadis Pharma
Consulting or Advisory Role: Juno Therapeutics, Gilead Sciences, Merck, Astellas Pharma
Expert Testimony: Pfizer
Travel, Accommodations, Expenses: Gilead Sciences, Astellas Pharma, Merck
Robert A. Kyle
Consulting or Advisory Role: Celgene, Bristol-Myers Squibb, Pharmacyclics, Pfizer
Steven P. Treon
Consulting or Advisory Role: Janssen, Pharmacyclics
Research Funding: Janssen, Pharmacyclics
Travel, Accommodations, Expenses: Janssen Oncology
Other Relationship: Janssen, Pharmacyclics
Jorge J. Castillo
Consulting or Advisory Role: Pharmacyclics, Janssen, Vical, Genentech
Research Funding: Pharmacyclics (Inst), AbbVie (Inst), Janssen (Inst), BeiGene (Inst), TG Therapeutics (Inst)
Meletios A. Dimopoulos
Honoraria: Amgen, Celgene, Takeda, Janssen-Cilag, Bristol-Myers Squibb
Consulting or Advisory Role: Amgen, Janssen-Cilag, Takeda, Celgene, Bristol-Myers Squibb
Stephen M. Ansell
Honoraria: WebMD, Research to Practice, Bristol-Myers Squibb (Inst), Seattle Genetics (Inst), Affimed Therapeutics (Inst), Regeneron (Inst), Pfizer (Inst), LAM Therapeutics (Inst), Trillium Therapeutics (Inst)
Irene M. Ghobrial
Honoraria: Celgene, Bristol-Myers Squibb, Millennium, Takeda, Amgen, Noxxon Pharma
Consulting or Advisory Role: Onyx, Bristol-Myers Squibb, Novartis, Amgen, Takeda, Noxxon Pharma, Celgene
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Novartis, Onyx, Millennium, Celgene, Takeda, Janssen Oncology
No other potential conflicts of interest were reported.
REFERENCES
- 1.Castillo JJ, Ghobrial IM, Treon SP. Biology, prognosis, and therapy of Waldenström Macroglobulinemia. Cancer Treat Res. 2015;165:177–195. doi: 10.1007/978-3-319-13150-4_7. [DOI] [PubMed] [Google Scholar]
- 2.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30:110–115. doi: 10.1053/sonc.2003.50082. [DOI] [PubMed] [Google Scholar]
- 3.Gertz MA. Waldenström macroglobulinemia: 2017 update on diagnosis, risk stratification, and management. Am J Hematol. 2017;92:209–217. doi: 10.1002/ajh.24557. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Chen Y, Li F, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: A large population-based study. Cancer. 2012;118:3793–3800. doi: 10.1002/cncr.26627. [DOI] [PubMed] [Google Scholar]
- 5. doi: 10.1093/annonc/mdt298. Kastritis E, Leblond V, Dimopoulos MA, et al: Waldenstrom’s macroglobulinaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 24:vi155-vi159, 2018. [DOI] [PubMed] [Google Scholar]
- 6.Kyle RA, Larson DR, McPhail ED, et al. Fifty-year incidence of Waldenström macroglobulinemia in Olmsted County, Minnesota, from 1961 through 2010: A population-based study with complete case capture and hematopathologic review. Mayo Clin Proc. 2018;93:739–746. doi: 10.1016/j.mayocp.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. doi: 10.1182/blood-2003-11-4024. Kriangkum J, Taylor BJ, Treon SP, et al: Clonotypic IgM V/D/J sequence analysis in Waldenstrom macroglobulinemia suggests an unusual B-cell origin and an expansion of polyclonal B cells in peripheral blood. Blood 104:2134-2142, 2004. [DOI] [PubMed] [Google Scholar]
- 8. doi: 10.1182/blood-2005-09-3613. Kriangkum J, Taylor BJ, Strachan E, et al: Impaired class switch recombination (CSR) in Waldenstrom macroglobulinemia (WM) despite apparently normal CSR machinery. Blood 107:2920-2927, 2006. [DOI] [PubMed] [Google Scholar]
- 9. doi: 10.3324/haematol.10755. Martín-Jimenez P, Garcia-Sanz R, Balanzategui A, et al: Molecular characterization of heavy chain immunoglobulin gene rearrangements in Waldenstrom's macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Haematologica 92:635-642, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–1646. doi: 10.1182/blood-2013-09-525808. [DOI] [PubMed] [Google Scholar]
- 11.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367:826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 12.Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121:2522–2528. doi: 10.1182/blood-2012-09-457101. [DOI] [PubMed] [Google Scholar]
- 13.Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: Long-term results. Blood. 2012;119:4462–4466. doi: 10.1182/blood-2011-10-384768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378:241–249. doi: 10.1056/NEJMoa1709974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk-Adapted Therapy (mSMART) guidelines 2016. JAMA Oncol. 2017;3:1257–1265. doi: 10.1001/jamaoncol.2016.5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30:116–120. doi: 10.1053/sonc.2003.50038. [DOI] [PubMed] [Google Scholar]
- 17.Dimopoulos MA, Kastritis E, Owen RG, et al. Treatment recommendations for patients with Waldenström macroglobulinemia (WM) and related disorders: IWWM-7 consensus. Blood. 2014;124:1404–1411. doi: 10.1182/blood-2014-03-565135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castillo JJ, Garcia-Sanz R, Hatjiharissi E, et al. Recommendations for the diagnosis and initial evaluation of patients with Waldenström macroglobulinaemia: A task force from the 8th International Workshop on Waldenström Macroglobulinaemia. Br J Haematol. 2016;175:77–86. doi: 10.1111/bjh.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexanian R, Weber D, Delasalle K, et al. Asymptomatic Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30:206–210. doi: 10.1053/sonc.2003.50051. [DOI] [PubMed] [Google Scholar]
- 20.Baldini L, Goldaniga M, Guffanti A, et al. Immunoglobulin M monoclonal gammopathies of undetermined significance and indolent Waldenstrom’s macroglobulinemia recognize the same determinants of evolution into symptomatic lymphoid disorders: Proposal for a common prognostic scoring system. J Clin Oncol. 2005;23:4662–4668. doi: 10.1200/JCO.2005.06.147. [DOI] [PubMed] [Google Scholar]
- 21.Abeykoon JP, Paludo J, King RL, et al. MYD88 mutation status does not impact overall survival in Waldenström macroglobulinemia. Am J Hematol. 2018;93:187–194. doi: 10.1002/ajh.24955. [DOI] [PubMed] [Google Scholar]
- 22.Treon SP, Cao Y, Xu L, et al. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123:2791–2796. doi: 10.1182/blood-2014-01-550905. [DOI] [PubMed] [Google Scholar]
- 23.Treon SP, Gustine J, Xu L, et al. MYD88 wild-type Waldenstrom macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018;180:374–380. doi: 10.1111/bjh.15049. [DOI] [PubMed] [Google Scholar]