

Abstract
Neurons in the central nervous system (CNS) regenerate poorly compared to their counterparts in the peripheral nervous system. We previously showed that, in peripheral sensory neurons, nuclear HDAC5 inhibits the expression of regenerative associated genes. After nerve injury, HDAC5 is exported to the cytoplasm to promote axon regeneration. Here we investigated the role of HDAC5 in retinal ganglion cells (RGC), a CNS neuron which fails to survive and regenerate axons after injury. In contrast to PNS neurons, we found that HDAC5 is mostly cytoplasmic in naïve RGCs and its localization is not affected by optic nerve injury, suggesting that HDAC5 does not directly suppress regenerative associated genes in these cells. Manipulation of the PKCμ pathway, the canonical pathway that regulates HDAC5 localization in PNS neurons by phosphorylating serine 259 and 498, and other pathways that regulate nuclear/cytoplasmic transport did not affect HDAC5 cytoplasmic localization in RGC. Also, an HDAC5 mutant whose serine 259 and 488 were replaced by alanine (HDAC5AA) to prevent phosphorylation and nuclear export showed a predominantly cytoplasmic localization, suggesting that HDAC5 resides mostly in the cytoplasm in RGCs. Interestingly, expression of HDAC5AA, but not HDAC5 wild type, in RGCs in vivo promoted optic nerve regeneration and RGC survival. Mechanistically, we found that HDAC5AA stimulated the survival and regeneration of RGCs by activating the mTOR pathway. Consistently, the combination of HDAC5AA expression and the stimulation of the immune system by zymosan injection had an additive effect in promoting robust axon regeneration. These results reveal the potential of manipulating HDAC5 phosphorylation state to activate the mTOR pathway, offering a new therapeutic target to design drugs that promote axon regeneration in the optic nerve.
Keywords: Retinal ganglion cells, optic nerve regeneration, mTOR, HDAC5, zymosan
Introduction
Injuries to the optic nerve caused by ischemic optic neuropathy or glaucoma are followed by retinal ganglion cell (RGC) death and axon degeneration, leading to permanent vision loss (Quigley, 2016). The poor capacity of RGCs to survive and regenerate after injury was first described by Ramon y Cajal (SR, 1913). Over the last few decades, some success has been achieved in promoting axon regeneration and survival of RGCs. For example, inducing an inflammatory response in the retina by injecting zymosan in the eye promotes optic nerve regeneration (Yin et al., 2003). An even larger effect can be achieved when the PI3K/AKT/mTOR pathway is activated by deleting Phosphatase and Tensin homolog (PTEN) or Tuberous Sclerosis Complex 1 (TSC1) (Park et al., 2008; de Lima et al., 2012). However, the number of RGCs responding to these treatments is small, less than 10% of the total RGCs in the retina (Duan et al., 2015; Berry et al., 2018). In addition, overactivation of the mTOR pathway by PTEN deletion causes undesired secondary effects such as an increase in RGC size (Duan et al., 2015) and even cancer (Xie et al., 2015). Therefore, more research is still needed to promote robust optic nerve regeneration without secondary effects.
In contrast to RGCs, peripheral nervous system (PNS) neurons such as dorsal root ganglion (DRG) neurons are able to regenerate their axon following injury, leading to functional recovery. This is in part due to their ability to activate an intrinsic pro-regenerative response (Mahar and Cavalli, 2018). We previously demonstrated a dual role for the class II histone deacetylase 5 (HDAC5) in DRG axon regeneration (Cho and Cavalli, 2012; Cho et al., 2013). The function and localization of HDAC5 is regulated by Protein kinase C subtype μ (PKCμ) and, in some contexts, by Calmodulin Kinase (CamK) (McKinsey et al., 2000). PKCμ is phosphorylated by other PKCs, and achieves full activation when serine 916 is phosphorylated, which is a reliable activation marker (Iglesias and Rozengurt, 1998). Activated PKCμ regulates HDAC5 by phosphorylating serine 259 and 498 inducing HDAC5 interaction with 14-3-3 and HDAC5 nuclear export (Vega et al., 2004). On the other hand, phosphorylation of HDAC5 at serine 280 by protein kinase A blocks this interaction and induces nuclear import (Ha et al., 2010). After sciatic nerve injury, a calcium wave induces PKCμ activation in the DRG soma, leading to HDAC5 nuclear export, increased histone acetylation and expression of regenerative associated genes (Cho et al., 2013). In the cytoplasm, HDAC5 also plays an important role for promoting axon growth by regulating the acetylation levels of microtubules (Cho and Cavalli, 2012).
In RGC, class I HDACs have been shown to restrict RGC survival and regeneration after optic nerve injury. A broad histone deacetylases inhibitor, trichostatin A, induces RGC survival after optic nerve crush and expression of the histone acetyltransferase p300 promotes axon regeneration in the optic nerve (Gaub et al., 2011). The dual deletion of HDAC1 and HDAC2 (Lebrun-Julien and Suter, 2015) and the deletion or inhibition of HDAC3 (Schmitt et al., 2014; Schmitt et al., 2018) reduces RGC loss after acute optic nerve injury. These studies suggest that histone deacetylation mediated in part by HDAC3 nuclear accumulation after injury plays a central role in gene silencing and RGCs death after optic nerve injury (Pelzel et al., 2010; Gaub et al., 2011). HDAC3 is known to bind to HDAC5 (Fischle et al., 2002) and HDAC3 re-localizes to the cytoplasm in sensory neurons after sciatic nerve injury (Cho et al., 2013). These studies prompted us to examine the function of HDAC5 in RGCs and manipulate HDAC5 to promote RGC survival and axon regeneration after optic nerve injury.
Material and Methods
Mice lines
All animal experiments were performed according to the Washington University animal study guidelines. Animals used in this study include adult and early postnatal Sprague Dawley rats, adult C57Bl/6 mice, adult TSC2 floxed mice (floxed allele; RRID:MGI:3712786) (Hernandez et al., 2007), and adult Raptor floxed mice (floxed allele, MGI:3829755) (Polak et al., 2008).
Adeno Associated vectors and virus
Adenoassociated vector plasmid pscAAV-GFP (Gray and Zolotukhin, 2011) used to generate AAV2-GFP virus was obtained from Addgene (Plasmid #32396). For HDAC5 vector plasmids, GFP from the pscAAV-GFP vector was substituted by the FLAG-HDAC5WT, FLAG-HDAC5AA, or FLAG-HDAC5DAD. The CMV promoter was used in all AAV vectors - based manipulation in this study. AAV2 viral particles were generated by the Viral Core of Washington University. AAV2-Cre was purchased from the viral core of University of North Carolina. Viral titer ranged between 5 × 10^12 vg/mL to 1 × 10^13 vg/mL.
Retinal Ganglion Cell purification (adult and early postnatal) and culture
Sprague-Dawley early post–natal pups (P2–P3) rats were anesthetized by hypothermia and euthanized by decapitation with surgical scissors. Briefly, retinas were dissociated using 16.5 units/mL papain (Worthington) for 30 minutes. Next, RGCs were purified by immunopanning using antibodies against Anti-Rat Macrophage Polyclonal Antibody (Accurate Chemical) and Thy1 (T11D7 from ATCC), as previously described (Trakhtenberg et al., 2017). RGCs were either lysed with Cytoplasm Extraction buffer (CER) from the nuclear/cytoplasmic fractionation kit (see below) containing protease inhibitors (Roche) and phosphatase inhibitors (Roche). In other experiments, purified RGCs were incubated in an Eppendorf containing 1ml of media with DMSO, 100μM of KN-62 (Tocris) and CID755673 (Tocris), or 5 μM I3A (Fisher). After 8 hours incubation at 37°C in rotation, cells were centrifuged and lysed in 80 μL of Cell Lysis buffer (CST) plus protease and phosphatase inhibitors. For some experiments, RGCs were plated on 35 mm tissue culture plates (Falcon) pre coated with poly-D-lysine (PDL; 10 mg/mL; Sigma) and laminin (1 mg/mL; Life Technologies). RGCs were cultured in defined growth medium consisting of Neurobasal media (Life Technologies) supplemented with sodium pyruvate, N-acetyl cysteine, insulin, BDNF, CNTF, and GS21 supplement medium. In some experiments, forskolin (5μM; Sigma) or leptomycin (20ng/mL; Sigma) was added to the medium, and after 24 hours cells were washed three times with PBS and lysed in 100 μL of Cell Lysis buffer (CST), using a cell scraper (Falcon). For immunostaining, RGCs were plated glass bottom dishes (MatTek) also coated with poly-D-lysine and laminin. After 24 hours, cells were fixed for 30 minutes in 4% paraformaldehyde in the same plate followed by three PBS washes.
Adult Skeletal Muscle cell purification
Adult Skeletal Muscle Myofibers were purified as previously described (Pasut et al., 2013). Briefly, Extensor Digitorum Longus muscle of adult mice was dissected and incubated in 1.5 mg/mL collagenase (C6885; Sigma) in DMEM until the muscle starts to loosen up. Then digested muscle is transferred to a new dish where tissue is flushed with warm medium until myofibers are released from the muscle. Using a pipette, myofibers are transferred to a 1.5 mL tube and centrifuged at low speed. The supernatant was discarded and nuclear/cytoplasmic fractionation was performed.
Rodent Surgeries
All surgical procedures were performed under isofluorane anesthesia and approved by Washington University in St. Louis, School of Medicine Animal Studies Committee. Intravitreal injection was performed as previously described with minor modifications (Steketee and Goldberg, 2012). One drop of atropine solution (1%; Akorn) was applied topically to dilate the pupils. A fine tip of pulled glass micropipette was attached to a Hamilton syringe using a needle compression fitting (55750-01; Hamilton), and loaded with 1.25 μL of adeno-associated virus (AAV2) or 594 fluorescently labeled cholera toxin B (Life Technologies). The tip was then inserted through the sclera with a 45° angle and the solution was slowly released in the vitreal space.
Optic nerve crushed was performed as previously described with minor modifications (Ban et al., 2017). Investigator performing the surgery was blinded to the treatments. One hour before surgery, Buprenorphine (1.0 mg/kg) was injected subcutaneously. Animals were then anesthetized with isofluorane and 5% Sodium Chloride eye drops were applied preoperatively to protect the cornea during surgery. Left optic nerve was exposed intraorbitally and crushed with sterile jeweler’s forceps for 3 seconds around 1 mm behind the optic disc. To preserve the retinal blood supply, care was taken not to damage the underlying ophthalmic artery. The endpoint of this surgery was between 1 and 28 days.
In some experiments, eye tissues were harvested for biochemical purposes. After sacrificing the animal, the eye together with the proximal part of the optic nerve was dissected out. Retina was extracted by cutting out the sclera and the cornea. The proximal part to the injury site was also resected. Tissue was then transferred to a 1.5 mL tube containing 80 μL of either Cell Lysis Buffer (Cell Signaling Technology) or CER buffer plus protease inhibitor and phosphatase inhibitors (Roche), and homogenized using disposable pestles and a pestle motor (Kontes).
In another set of experiments, rapamycin was prepared according to Chen et al. (Chen et al., 2009). Briefly, Rapamycin (LC Laboratories) was resuspended in ethanol at 50 mg/mL and then diluted in 5% Tween-80 (Sigma) and 5% PEG-400 (Sigma) to a working solution of 1 mg/mL. Mice received 6 mg/kg every other day by intraperitoneal injection. Vehicle solution without rapamycin was injected in control animals.
Nuclear/Cytoplasmic fractionation
Nuclear and cytoplasmic fractionation was performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). Briefly, homogenized purified cells or dissected retinas in CER buffer were vortexed for 10 seconds and centrifuged at 12,000 g at 4°C for 30 minutes. Supernatant was collected and stored at −80°C while the pellet was washed once with PBS and then resuspended in Nuclear Extraction buffer or RIPA buffer to extract nuclear proteins.
Immunoprecipitation
Immunoprecipitation was performed using anti-flag antibody attached to protein G beads. Nuclear and cytoplasmic fractions obtained from one retina were diluted three times with water/0.02% tween and PBS/0.02% tween respectively. Next, ten μL of anti-flag beads were resuspended in each of these samples, and incubated overnight at 4°C in a rotary shaker. Beads were separated using a magnet and washed three times with PBS/0.02% tween, transferred to a new tube, and boiled in 1× Laemmli Buffer for 5 minutes. Beads were discarded and the supernatant was loaded in an electrophoresis followed by a western blot.
Electrophoresis and western blot
For Western blot, 5× SDS loading buffer, prepared according to Cold Spring Harbor protocols, was added to samples. These samples were heated at 95°C for 5 minutes and after cooling down on ice were run in on 8-16% NuPAGE SDS-PAGE gradient gel (Life Technologies) in MOPS SDS Running Buffer (Life Technologies), transferred to 0.2 μm nitrocellulose membranes (Amersham). Next, these membranes were blocked in 3% BSA, 0.1% Tween-20 at pH 7.6 for 1 hour, incubated at 4°C overnight in blocking buffer with 1:1000 primary antibodies, monoclonal anti-HDAC5 antibody (H4538; Sigma), monoclonal anti-phosphorylated (Ser 498) HDAC5 (sc101692; Santa Cruz), rabbit anti-HDAC3 antibody (sc11417; Santa Cruz), rabbit anti-Histone3 antibody (9715S; CST), rabbit anti-GAPDH antibody GAPDH (sc25778, Santa Cruz), rabbit anti-Phospho-PKCμ Ser916 (2051S; CST), rabbit anti-PKCμ (2052S; CST), rabbit anti-phospho-c-Jun antibody (9261s; CST), goat anti-Brn3a (sc-31984; Santa Cruz), and rabbit anti-β3 tubulin antibody (PRB-435P, Covance). Membranes were then washed in TBST 3 times, incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG (1:5000; Invitrogen) for 1 hour at room temperature, washed in TBST three times, and membrane was developed with SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific). The membranes were imaged using ChemiDoc System (Bio Rad) and analyzed with ImageLab software
Perfusion and cryostat sectioning
Mice were sacrificed by CO2 asphyxiation and then perfused with 15 mL of PBS followed by 15 mL of paraformaldehyde 4%. Eyes and optic nerve were dissected out and an incision was made in the cornea. These tissues were then fixed for 3 h in 4% paraformaldehyde, washed 3 times in PBS, and incubated in 30% sucrose in PBS at 4°C overnight. Optic nerves were separated from the retina using spring scissors, embedded in tissue tek and directly sectioned at 11 μm in a Cryostat. Eyes were embedded in tissue-tek and frozen at −80°C, and then sectioned at 11 μm thickness. Cryosections were always mounted on Superfrost Plus micro slides (Fisherbrand).
Immunohistochemistry and analysis
Cryosections or fixed cells were blocked with 20% goat serum and 0.2% Triton X-100 in PBS for 1 hour. Next, these sections were incubated overnight at 4°C in the same buffer containing primary antibodies goat anti-Brn3a (sc-31984; Santa Cruz) or monoclonal (sc-8429; Santa Cruz) antibody, rabbit anti-pS6 (Ser240/244) antibody (5364S; CST), rabbit anti-β3 tubulin antibody (PRB-435P, Covance). Then, samples were washed 3 times in PBS, incubated 2 hours at 4°C with DAPI (1:3,000, Life Technologies) and Alexa fluorophore-conjugated secondary antibodies (1:500, Life Technologies), washed three times in PBS and mounted in ProLong Gold (P36934, Life Technologies) with a glass coverslip. Last, samples were imaged using a Nikon TE-2000E fluorescence microscope and analyzed in FIJI (NIH). For survival analysis, investigator performing the quantification was blinded to the treatments. Number of Brn3a+ cells per section were counted and normalized to the total distance of the RGC layer in that section. Three to four sections per retina were counted. For optic nerve regeneration analysis, investigator performing the quantification was blinded to the treatments. Lines at 250, 500, 750 and 1000 μm were drawn using FIJI, and the number of CTXB+ segments contacting the lines were counted. Three to four sections were averaged to obtain the average number of regenerating axons/mm. The number of regenerating axons was estimated using the formula previously described (Park et al., 2008) with minor modifications: 2π × optic nerve radius × [average axons/μm]/11.
RNA-seq
To isolate the RNA for sequencing, retinas were dissected from four female C57BI/6J per condition, and homogenized in 300 μl lysis buffer using disposable pestles attached to a motor. RNA was purified using the PureLink RNA Mini kit with on column DNase treatment and submitted to Washington University Genome Technology Access Center for quality control, library preparation, and sequencing. Briefly, RNA was quality controlled and quantified using both a Nanodrop 2000 and an Agilent Bioanalyzer; all samples passed quality control (RIN > 8, A260/268 > 2). Next, rRNA was depleted using a Ribo-Zero rRNA removal kit, cDNA libraries were generated using a Clontech SMARTer kit, and libraries were sequenced on an Iliumina HiSeq3000 using 1×50 lanes. Quality control was then performed on fastq files using FastQC and reads aligned to mm10 using STAR-align. Next, aligned reads were counted using HTseq-count and differential expression determined using DESeq2. Genes with fewer than an average of 5 counts per million were removed from the analysis. RNA-seq FastQ files were deposited at the NCBI GEO database (http://www.ncbi.nlm.nih.gov). Accession number GSE120257.
Experimental Design and Statistical Analysis
In the experiments where Western blot statistical analysis was performed, at least three female C57BL/6J mice or female Sprague Dawley rats were used per condition. Bands were quantified using Image Lab software and One-way ANOVA analysis was performed using Statistica 7 software.
For optic regeneration assay experiments, four to seven C57BL/6J female mice were used per condition in each experiment. However, in the experiments performed in TSC2floxed mice the number of animals was reduced to three and four per condition and mice were used regardless of their sex. The reason was that littermates were preferred in this experiment to reduce variability and therefore the litter size of this particular mice line was a limiting factor. In experiments comparing different HDAC5 mutants, four experiments performed on different days were combined. In each of these experiments, an AAV-GFP control was always included. One-way ANOVA with Bonferroni post hoc analysis was performed using Statistica 7 software, similar to previously reported (Parket al., 2008). In the in vivo RGC survival assay and in the quantifications of the percentage of pS6+ RGCs, five C57BL/6J female mice were used per condition. One-way ANOVA with Tukey’s post hoc test analysis was performed using Statistica 7 software similarly to previously reported (Li et al., 2016).
For RNAseq, principal component analysis was then performed to detect outliers. Due to this, one of the GFP samples was removed. A cutoff of a p-adjusted value of 0.1 was used to identify differentially expressed genes. Downstream analysis of biological and signaling pathways was performed using MetaCore using a p-value of 0.05 to determine enriched pathways.
Results
HDAC5 is largely cytoplasmic in naïve and injured RGCs
We have previously shown that the localization of HDAC5 is critical for its effects in axon regeneration in DRG neurons (Cho et al., 2013). Therefore, we first determined the localization of HDAC5 in naïve RGCs and in RGCs after optic nerve injury. Acutely purified RGCs by immunopanning represent a pure population, with more than >98% of the cells positive for the RGC marker Brn3a (Figure 1A), as previously reported (Barres et al., 1988). Nuclear and cytoplasmic fractionation of these acutely purified RGCs followed by western blot analysis revealed that HDAC5 is largely found in the cytoplasm, whereas HDAC3 is equally distributed in both fractions (Figure 1B).
Figure 1. HDAC5 is predominantly localized in the cytoplasm of RGCs and does not change its location after injury.
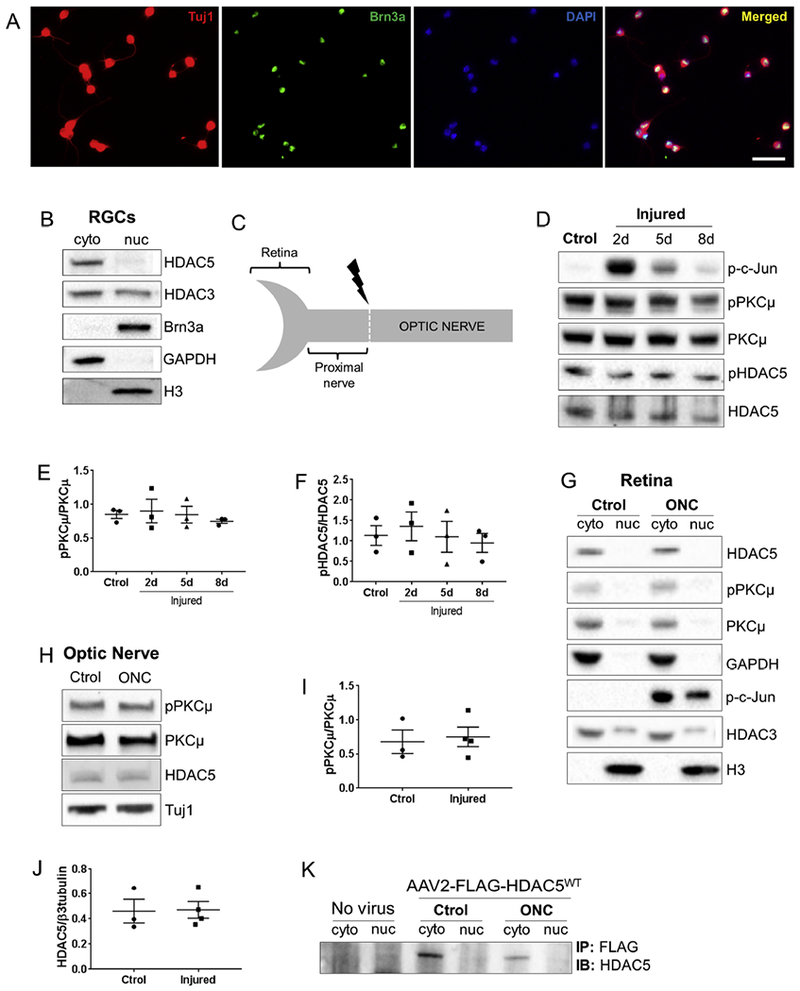
(A) Rat RGC cultures purified by immunopanning and stained with p3-tubulin (cytoplasm, red), Brn3a (nuclei, green), DAPI (nuclei, blue), shows that the majority of cells are triple positive, demonstrating the high purity of the culture. (B) Immunoblot for nuclear (nuc) and cytoplasmic (cyto) fractions of early postnatal rat purified RGCs shows that HDAC5 is predominantly in the cytoplasm. (C) Diagram showing the optic nerve regions dissected for following immunoblot analyses. (D) Immunoblot of mouse retina extracts isolated at 2, 5, and 8 days after optic nerve injury. (E) Quantification of (D) shows that there is no difference in the phosphorylation levels of PKCμ (serine 916) after optic nerve injury. (F) Quantification of (D) showing that there is no difference in the phosphorylation levels of HDAC5 (serine 498) after optic nerve injury. (G) Immunoblot of nuclear (nuc) and cytoplasmic (cyto) fractions of mouse uninjured retinas and retinas 2 days after optic nerve injury. (H) Immunoblot of the region proximal to the injury site 2 days after optic nerve injury in rats. (I) Quantification of (H) showing that there is no difference in the phosphorylation levels of PKCμ (serine 916) after optic nerve injury. (J) Quantification of (H) showing that there is no difference in the accumulation of HDAC5. (K) FLAG immunoprecipitation followed by immunoblot for HDAC5 in retinas infected with AAV2 virus encoding FLAG-HDAC5WT, uninjured and 2 days after optic nerve injury. Plotted values in (E), (F), (J) and (I) were quantified and expressed as mean ± SEM. Scale bar = 50μm. Statistical analysis is a One-way ANOVA. N at least3 in B, D, G, and L.
Since, it has been reported that injury induces a widespread silencing of genes in RGCs mediated in part by HDAC3 nuclear import (Pelzel et al., 2010), we asked whether HDAC5 also translocates to the nucleus after injury. We performed an optic nerve crush injury (ONC) and isolated retinas at various days after injury (Figure 1C). We observed that PKCμ activation is not increased (One-way ANOVA F(3, 8)=0.32, p=0.80) and HDAC5 phosphorylation at serine 498 does not increase (One-way ANOVA F(5, 6)=0.37, p=0.85) at 2, 5 or 8 days post injury (Figure 1D, E and F), suggesting that HDAC5 is not regulated by PKCμ phosphorylation in the retina after axon injury. Consistent with this result, nuclear fractionation showed that HDAC5 remains largely cytoplasmic in both control and injured retinas (Figure 1G). In contrast, HDAC3 was detected in both the nuclear and cytoplasmic fractions, but we did not observe HDAC3 nuclear accumulation after injury (Figure 1G). Additionally, and in contrast to what we have reported in sciatic nerve (Cho et al., 2013), we did not observe an accumulation of HDAC5 (One-way ANOVA F(1, 5)=0.009, p=0.92) or an activation of PKCμ (One-way ANOVA F(1, 5)=0.09, p=0.76) in the proximal site of the injured optic nerve (Figure 1H and 1I). Finally, we overexpressed FLAG-tagged HDAC5WT in RGCs in vivo using adeno-associated virus (AAV2) intravitreal injection, and immunoprecipitated FLAG-HDAC5WT from control and injured retinas. We found that FLAG-HDAC5WT is also localized in the cytoplasm and that injury does not change HDAC5 localization (Figure 1K). These results indicate that HDAC5 is largely cytoplasmic in RGC and that optic nerve injury does not change its localization.
HDAC5 subcellular localization in RGCs is not regulated by canonical signaling pathways
Next, we determined whether HDAC5 phosphorylation state has an effect on its cellular localization in RGCs. Since both PKCμ and CamK phosphorylate HDAC5 at serines 259 and 498 to promote its nuclear export in myocytes (McKinsey et al., 2000; Vega et al., 2004), we asked whether inhibiting PKCμ and CamK by incubating RGCs with their respective inhibitors CID755673 and KN62 prevent HDAC5 nuclear export. We found that these drugs clearly reduced the phosphorylation levels of HDAC5 at Serine 498 (Figure 2A), but the cytoplasmic localization of HDAC5 did not change (Figure 2B). To further support that HDAC5 phosphorylation at serines 259 and 498 does not induce HDAC5 nuclear export in RGCs, we overexpressed HDAC5WT and two other constructs of HDAC5 that mimic the phosphorylation states of HDAC5 in RGCs (Figure 2C). In one construct, serine 259 and 498 were substituted with alanine (HDAC5AA) to prevent phosphorylation. This mutation blocks interaction with the 14-3-3 protein (Vega et al., 2004), and prevents nuclear export in myocytes. In another, construct serine 259 and 498 were substituted with aspartic acid to mimic phosphorylation and serine 280 substituted with alanine to prevent phosphorylation by PKA (HDAC5DAD). HDAC5DAD interact with 14-3-3 to be transported to the cytoplasm in myocytes (Ha et al., 2010). All three constructs, expressed at similar levels in RGCs in vivo using AAV2 intravitreal injection, were predominantly cytoplasmic (Figure 2D). We also expressed these HDAC5 constructs in cultured RGCs and confirmed that both HDAC5DAD and HDAC5AA were predominantly in the cytoplasm (Figure 2E). These results suggest that the canonical pathways regulating HDAC5 localization (Vega et al., 2004) are not involved in HDAC5 subcellular localization in RGC.
Figure 2. The cytoplasmic localization of HDAC5 is independent of the phosphorylation states of key serines 259, 280, 498.
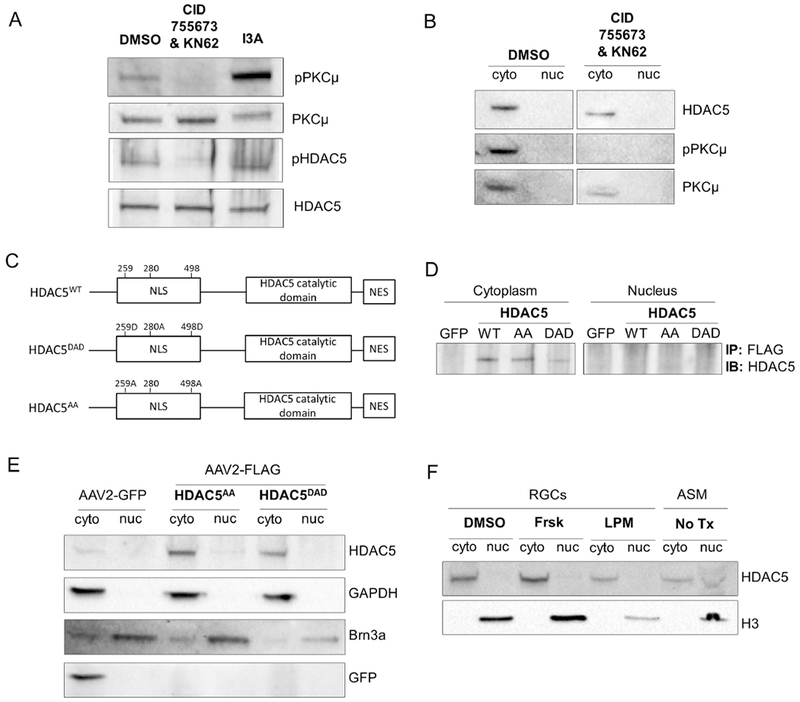
(A) Immunoblot of cultured rat RGCs treated for 8 hours with vehicle (DMSO), 100μM CID755673 + 100 μM KN62, or 1 μM I3A. (B) Immunoblot for nuclear (nuc) and cytoplasmic (cyto) fractions of cultured rat RGCs treated for 8 hours with vehicle (DMSO), or 100μM CID755673 + 100 μM KN62. (C) Diagram of the HDAC5 constructs, HDAC5WT , HDAC5AA, and HDAC5DAD, showing the serines that are substituted for alanine (A, phospho-dead) or aspartic acid (D, phospho-mimic), NLS, nuclear localization signal region, NES, nuclear export signal region. (D) FLAG immunoprecipitation followed by immunoblot for HDAC5 in retinas infected with AAV2 encoding GFP, FLAG-HDAC5WT FLAG-HDAC5AA, or FLAG-HDAC5DAD 15 days prior to sacrifice. (E) Immunoblot for nuclear (nuc) and cytoplasmic (cyto) fractions of cultured rat RGCs infected with AAV2-GFP, AAV2-FLAG-HDAC5AA, or AAV2-FLAG-HDAC5DAD 8 days prior to harvest. (F) Immunoblot for nuclear (nuc) and cytoplasmic (cyto) fractions of cultured rat RGCs treated for 24 hours with vehicle (DMSO), forskolin (frsk), or leptomycin (LPM). As a positive control for nuclear HDAC5, myofibers from adult skeletal muscle (ASM) were used. N=3 in B and C, and n=2 in D.
To further support that in RGCs, HDAC5 is insensitive to canonical signals that in other cell types regulate its localization, we treated RGCs with two drugs that have been previously demonstrated to promote HDAC5 nuclear localization, forskolin (Ha et al., 2010) and leptomycin (Harrison et al., 2004). Neither, forskolin, which activates PKA and phosphorylates HDAC5 serine 280 in cardiomyocyte (Ha et al., 2010), nor leptomycin, which inhibits nuclear export of proteins, were able to induce HDAC5 nuclear localization in RGCs (Figure 2F). As a control, we used purified adult skeletal muscle cells (ASM), a cell type in which HDAC5 nuclear localization is known to inhibit genes for muscle growth (Liu et al., 2009), and in which we clearly observed HDAC5 localization in the both cytoplasmic and nuclear fraction (Figure 2F). These results further support the notion that HDAC5 is largely cytoplasmic in RGC.
HDAC5AA activates mTOR and increases optic nerve regeneration and survival of injured RGCs
Because mutation of HDAC5 phosphorylation was shown to affect interactions with other proteins in addition to regulate subcellular localization (Greco et al., 2011), we tested if overexpressing HDAC5WT and HDAC5 mutants in RGCs in vivo has an effect on axon regeneration (Figure 3A). HDAC5WT and HDAC5DAD behaved similarly and were not significantly different than the control GFP virus, in which very limited axon growth past the injury site was observed (Figure 3B and 3C). However, HDAC5AA promoted optic nerve regeneration (One-way ANOVA F(8, 68)=5.03, p=0.00006 with Bonferroni’s post hoc test), doubling the number of regenerating axons at all distances past the injury site (Figure 3B and C). Therefore, although the localization of HDAC5AA and HDAC5DAD is indistinguishable using nuclear fractionation, their effects in optic nerve regeneration are very different.
Figure 3. Overexpression of HDAC5AA but not GFP, HDAC5WT, or HDAC5DAD promotes optic nerve regeneration and RGC survival.
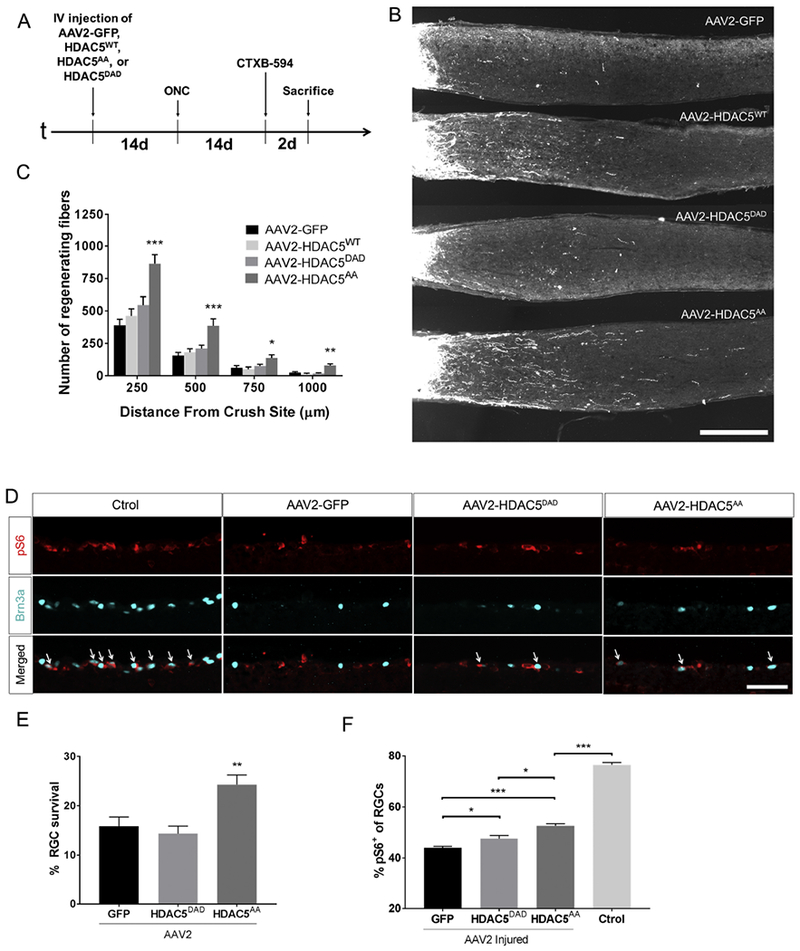
(A) Timeline showing experimental procedures, including intravitreal injection of AAV2-GFP, AAV2-FLAG-HDAC5WT, AAV2-FLAG-HDAC5AA, or AAV2-FLAG-HDAC5DAD, optic nerve crush (ONC), intravitreal injection of Alexa 594 fluorescently labeled cholera toxin b (CTXB-594), and sacrifice of mice. (B) Representative optic nerve sections showing that there are more regenerating axons in retinas infected with AAV2-FLAG-HDAC5AA than in the other conditions. (C) Quantification of (B) showing significant differences in optic nerve transducted with AAV2-FLAG-HDAC5AA compared to other conditions at all distances analyzed. Plotted values in (C) were quantified and expressed as mean ± SEM. One-way ANOVA with Bonferroni post hoc analysis, * p<0.05, ** p<0.01, *** p<0.001. Scale bar = 250 μm. N=20 for AAV2-GFP, n=4 for AAV2-FLAG-HDAC5WT, n=9 for AAV2-FLAG-HDAC5DAD and n=14 for AAV2-FLAG-HDAC5AA. (D) Retinal sections from mice that were injected with AAV2-GFP, AAV2-FLAG-HDAC5AA, or AAV2-FLAG-HDAC5DAD 15 days prior optic nerve injury and sacrificed 15 days post injury, were stained with Brn3a (nucleus, green, a marker for RGCs) and pS6 (cytoplasm, red, a marker of mTOR activation). The non-injured non-injected contralateral retina was included as Control. Arrows indicate colocalization of both markers. (E) Quantification of the percentage of Brn3a positive cells in injured retinas compared to the non-injured contralateral retina. (F) Quantification of the percentage of pS6+/Brn3a+ cells. Plotted values in (B) and (C) were quantified and expressed as mean ± SEM. Statistical Analysis was One-way ANOVA with Tukey’s post hoc test. B and D, Scale bar = 500 μm. N=5
It is well established that activating mTOR by deleting TSC1, TSC2 (Park et al., 2008; Abe et al., 2010) or PTEN (Park et al., 2008; Christie et al., 2010) promotes axon regeneration. In RGC, these manipulations not only promote optic nerve regeneration but also RGC survival, by blocking the injury-induced dephosphorylation of the S6 ribosomal protein (S6) after optic nerve crush (Park et al., 2008). Since it has recently shown that, in a hypothalamic cell line, cytoplasmic HDAC5 can indirectly regulate mTOR activation (Ma et al., 2015), we tested if HDAC5AA activates the mTOR pathway to promote RGC survival and regeneration. We observed a 10% increase in the number of surviving RGCs (labeled with the nuclear marker Brn3a) after injury in retinas overexpressing HDAC5AA compared to the other treatments (One-way ANOVA F(2, 12)= 13.38, p=0.00089 with Tukey’s post hoc test; p=0.004 vs GFP and p=0.001 vs HDAC5DAD but not difference (p=0.75) between HDAC5DAD and GFP) (Figure 3D and E). We also observed a higher percentage of cells labeled with phosphorylated S6 (pS6) in the surviving RGCs in retinas expressing HDAC5AA compared to retinas expressing GFP or HDAC5DAD (One-way ANOVA F(2, 9)=25.56, p=0.00019 with Tukey’s post hoc test; p=0.003 vs GFP and p=0.02 vs DAD) (Figure 3D and 3F and C). We also observed a slight but significant increase in the number of pS6 positive RGCs in retinas expressing HDAC5DAD compared to GFP (p=0.011). These results suggest that HDAC5 is able to regulate mTOR activity but that, HDAC5DAD is less efficient than HDAC5AA and is unable to promote survival or axon regeneration of RGCs.
HDAC5 regulates the expression of genes in the PI3K-mTOR pathway
To determine the underlying gene expression changes leading to the phenotypical differences in optic nerve regeneration between control GFP, HDAC5DAD, and HDAC5AA we performed an RNAseq analysis. Whole retina were collected four days after ONC in animals injected with AAV2 two weeks before ONC, and compared them with non-injected/non-injured retinas. Since ~70% of cells transduced by intravitreal injection of AAV2 are RGCs- (Harvey et al., 2002), most gene expression changes between conditions should reflect changes occurring in RGCs, or changes in other cells as a result of HDAC5AA and HDAC5DAD expression in RGCs. As expected, we observed that retinas expressing HDAC5AA and HDAC5DAD showed 20% more total HDAC5 compared to GFP expressing retina (Figure 4A). To differentiate between endogenous HDAC5 and overexpressed HDAC5, we focused our analysis in the HDAC5 region around amino acid 498, where the mutations of these two constructs reside. We were able to detect these mutations and compared their expression levels to endogenous HDAC5, confirming that both mutated versions were expressed at similar levels (Figure 4B). Next, we identified the genes that are differentially expressed in GFP, HDAC5DAD, and HDAC5AA compare to uninjured, uninjected controls and found that a large proportion of the genes differentially expressed are shared between the three conditions (Figure 4C), indicating common genes affected by injury and AAV injection. We also observed that HDAC5DAD and HDAC5AA differentially regulated a specific set of genes compared to GFP, indicating that these genes are regulated by HDAC5 expression. To determine which pathways and processes are regulated by these specific sets of genes, we performed gene enrichment analysis using MetaCore software (Figure 4E), which contains a curated knowledge database allowing to obtain functional pathways and processes regulated by defined set of genes. This analysis revealed that the set of genes that are differentially upregulated in retinas overexpressing HDAC5AA relate to “positive regulation of locomotion”, “morphogenesis”, and “cell migration and motility” , which are in line with the effects observed in axon growth (Figure 4D). Pathways regulated by these set of genes are also known regulators of axon growth and include the “GDNF pathway”; the “TGF-beta pathway”; the “NGF-trkA pathway”; and the “H-Ras pathway” (Figure 4D). Interestingly, the GDNF pathway (Wang et al., 2007), the TGF beta pathway (Cheng and Hao, 2017), and the NGF-TrkA pathway (Nakamura et al., 2011) directly or indirectly activate the mTOR pathway which has drastic effects on axon regeneration (Park et al., 2008). The set of genes differentially downregulated in retinas overexpressing HDAC5AA compared to HDAC5DAD and GFP did not reveal pathways clearly related with axon growth (Figure 4D). However, analysis of the genes downregulated in both HDAC5DAD and HDAC5AA (Figure 4E) revealed pathways related to “development neurogenesis”, “mTORCI pathway”, and “p53 suppression pathway”. The p53 pathway is known to inhibit mTOR (Feng et al., 2005), suggesting that both HDAC5 mutants impact the mTOR pathway but only HDAC5AA has effects in axon growth.
Figure 4. RNAseq analysis demonstrates that HDAC5AA overexpression regulates pathways related to mTOR.
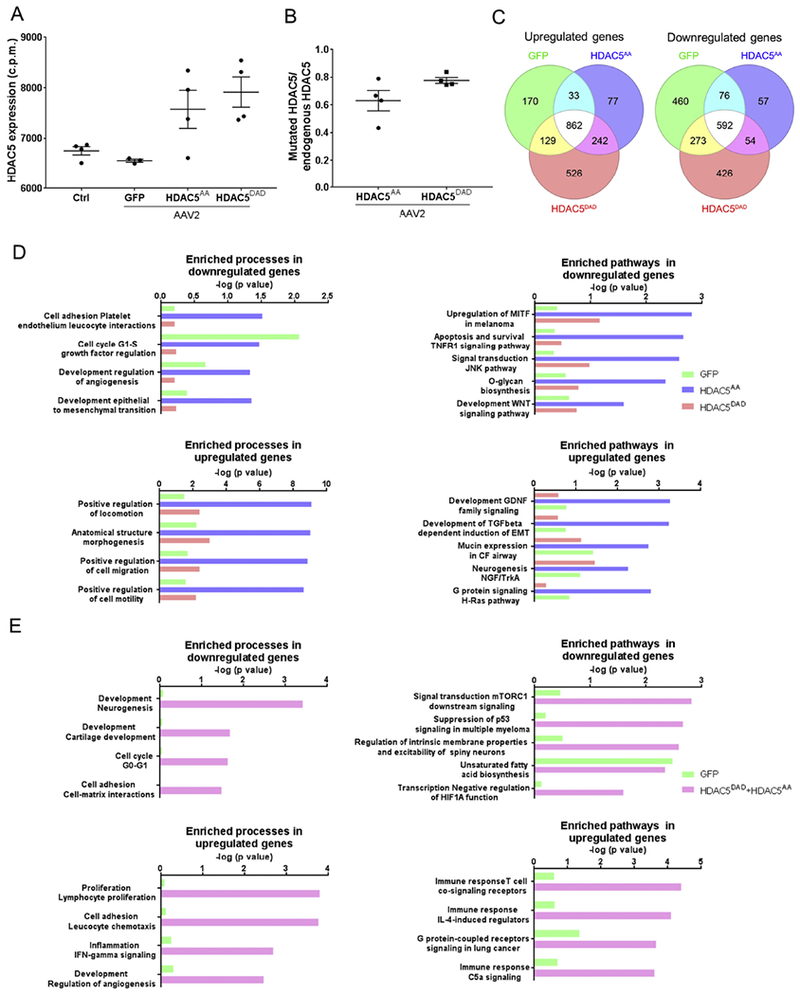
(A) Retinas were intravitreally injected with either AAV2-GFP, AAV2-FLAG-HDAC5AA or AAV2-FLAG-HDAC5DAD. 15 days later, optic nerve injury was performed. 4 days after optic nerve injury, retinas were processed for RNAseq. Control refers to non-injected, non-injured retina. N=4. HDAC5 mRNA expression levels in counts per million (c.p.m) of HDAC5 in the different conditions. (B) Ratio between the number of reads of HDAC5 mutants at the DNA location of serine 498 and number of reads in the homologous region in the endogenous HDAC5. Plotted values in (A) and (B)were quantified and expressed as mean ± SEM. (C) Venn diagram indicating the number of differentially expressed genes in each condition compared to the non-injected, non-injured control retina. (D) Enriched processes and pathways in the set of genes that are differentially expressed in retinas overexpressing GFP, HDAC5AA or HDAC5DAD. (E) Enriched processes and pathways in the set of genes that are differentially expressed in retinas overexpressing GFP compared to both HDAC5AA and HDAC5DAD.
The mTOR pathway is necessary and sufficient for HDAC5AA induced axon regeneration
To determine if the mTOR pathway is required for the increased axon regeneration that we observed in RGC expressing HDAC5AA, we used two different approaches to block mTOR activity in vivo: 1) injecting rapamycin, an mTOR inhibitor (Park et al., 2008) every other day (Figure 5A, B and C), and 2) deleting the regulatory-associated protein of mTOR (Raptor), which is a necessary binding partner to the mTOR kinase and is required for mTOR function (Hara et al., 2002), by expressing AAV-Cre in the RGC of Raptor floxed mice (Polak et al., 2008) (Figure 5D, E and F). Both strategies reduced the number of regenerating axons after optic nerve injury to the levels of the control (One-way ANOVA F(4, 16)=3.21, p=0.04 and F(4, 4)=6.05, p=0.05 respectively, both with Bonferroni’s post hoc test). To test if the decreased axon regeneration observed by inhibiting mTOR results from a decreased survival of RGCs, we measured the number of surviving RGCs. Rapamycin treatment led to a significant increase in the survival of RGCs (Fig 5C) (One-way ANOVA F(1,6)=43.57, p=0.0006), consistent with previous studies (Rodriguez-Muela et al., 2012; Li et al., 2018). In contrast, we observed no difference in RGC survival by deleting Raptor in RGC (Fig 5F) (One-way ANOVA F(1, 8)=0.4729, p=0.51). These results demonstrate that the increased axon regeneration promoted by HDAC5AA requires mTOR activity and is independent of RGC survival.
Figure 5. The mTOR pathway is required for HDAC5AA induced axon regeneration.
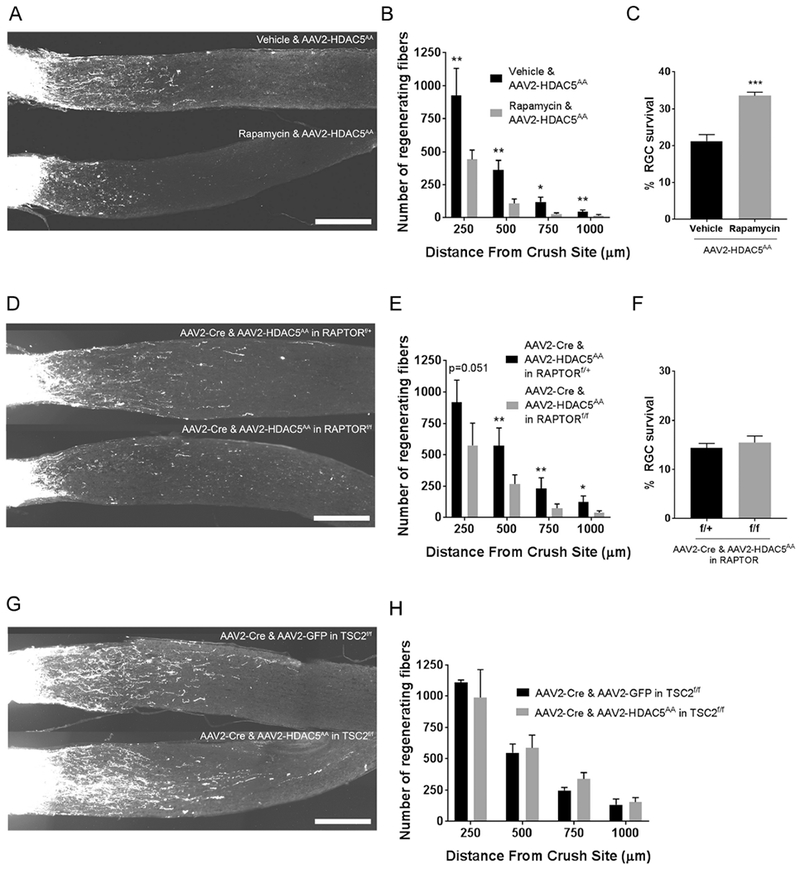
(A) Representative optic nerve sections showing axon regeneration in mice that were intravitreally injected with AAV2-FLAG-HDAC5AA 15 days prior injury, and then treated with rapamycin or vehicle until harvest (15 days post optic nerve injury). (B) Quantification of (A) showing that rapamycin injection significantly reduces the number of regenerating axons at all distances analyzed. (C) Quantification of the percentage of Brn3a positive cells in injured retinas of mice treated in (A) compared to the non-injured contralateral retina. (D) Representative optic nerve sections showing axon regeneration in RAPTORflox/flox or RAPTORflox/+ mice that were injected with AAV2-FLAG-HDAC5AA 15 days prior injury, and with AAV2-Cre one day after injection with AAV2-FLAG-HDAC5AA , and sacrificed 15 days post injury. (E) Quantification of (D) showing that RAPTOR deletion in RGCs significantly reduces the number of regenerating axons at all distances. (F) Quantification of the percentage of Brn3a positive cells in injured retinas of mice treated in (E) compared to the non-injured contralateral retina. (G) Optic nerve sections collected 15 days after injury from TSC2 flox/flox mice that were injected with either AAV2-FLAG-HDAC5AA, or AAV2-GFP 15 days prior injury, and one day after this injection with AAV2-Cre. (H) Quantification of (G) showing that when TSC2 is deleted (leading to mTOR activation), HDAC5AA overexpression is not significantly different than GFP overexpression. Plotted values in (B), (C), (E), (F) and (H) were quantified and expressed as mean ± SEM. Statistical analysis is a One-way ANOVA with Bonferroni’s post hoc test in (B), (E) and (H), and a One-way ANOVA with Tukey’s post hoc test in (C) and (F), * p<0.05, ** p<0.01, *** p<0.001. Scale bar= 250 μm. N=5 for vehicle and rapamycin injected mice in (B), n=3 for vehicle and n=5 for rapamycin injected mice in (C), n=5 for RAPTORflox/+ and n=6 for RAPTORflox/flox in E, n=5 in both conditions of (F), and n=4 and 3 for AAV2-FLAG-HDAC5AA and AAV2-GFP respectively in the TSC2flox/flox experiment.
Next, we expressed HDAC5AA in mice in which the mTOR pathway was genetically activated by deleting its negative regulator TSC2. Overexpressing HDAC5AA and Cre in TSC2 floxed mice (Hernandez et al., 2007) did not increase the number of regenerating axons (One-way ANOVA F(4, 2)=6.20, p=0.14, with Bonferroni’s post hoc test) compared to GFP and Cre controls (Figure 5E and F). Altogether, these results suggest that the increased axon regeneration observed in RGC expressing HDAC5AA is mediated by the mTOR pathway.
HDAC5AA synergizes with eye inflammation to stimulate axon regeneration
We next asked whether expression of HDAC5AA in combination with treatments that activate other regenerative pathways different than mTOR further increase axon regeneration. Inducing eye inflammation by injecting zymosan promotes axon regeneration (Leon et al., 2000), and this effect is partially independent of mTOR (Leibinger et al., 2012). We expressed HDAC5AA, and induced inflammation the same day that the optic nerve injury was performed by intravitreally injecting zymosan. We observed that HDAC5AA alone and zymosan treatment alone promotes axon regeneration (Figure 6A and B). When HDAC5AA was combined with zymosan treatment, an additive effect on axon regeneration was observed at all distances quantified (One-way ANOVA F(12, 45)=7.84, p=0.000001 with Bonferroni’s post hoc test). These results further support the notion that HDAC5AA promotes axon regeneration via the mTOR and that these effects can synergize with the pro-regenerative inflammatory responses.
Figure 6. HDACAA overexpression acts additively with an inflammatory stimulus to boost axon regeneration.
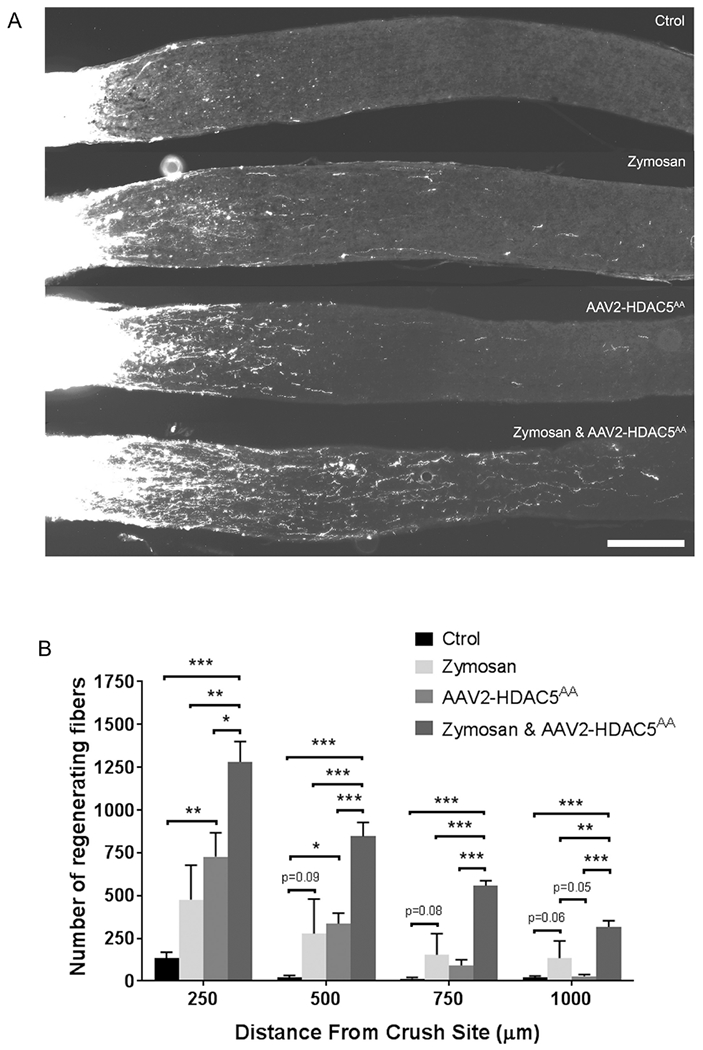
(A) Representative optic nerve sections showing axon regeneration in mice that were either untreated, or intravitreally injected with AAV2-FLAG-HDAC5AA 15 days prior injury, injected with zymosan the day of injury, or the combination of these two last treatments. (B) Quantification of (A) showing that the combination of HDAC5AA overexpression and zymosan injection significantly increases the number of regenerating axons at all distances analyzed when compared to the single treatments. Plotted values in (C) were quantified and expressed as mean ± SEM. Statistical analysis is a One-way ANOVA with Bonferroni’s post hoc test, * p<0.05, ** p<0.01, *** p<0.001. Scale bar = 250 μm. N=5 for control, HDAC5AA injected, and zymosan injected, and n=7 for the combination of zymosan injection and HDAC5AA overexpression.
Discussion
Our study reveals that HDAC5 is predominantly cytoplasmic in RGC and its localization is unaffected by injury or manipulation of canonical signaling pathways that regulate transport of class II HDACs between nucleus and cytoplasm. Phosphorylation at serines 259, 280, and 498 are critical for regulating the localization of HDAC5 in cell types such as muscle cells or peripheral nervous system neurons (McKinsey et al., 2000; Ha et al., 2010; Cho et al., 2013). However, our results in RGC showed a clear cytoplasmic localization of HDAC5 that is unaffected by PKCμ or PKA pathways. Additionally, blocking the CRM1 cytoplasmic transport using leptomycin (Harrison et al., 2004) does not induce HDAC5 nuclear accumulation, suggesting that there is no shuttle of HDAC5 between cytoplasm and nucleus and HDAC5 is for the most part localized in the cytoplasm. The difference between the capacity of HDAC5 to shuttle between cytoplasm and nucleus in PNS neurons and its persistent localization in the cytoplasm of RGCs might be explained by the interaction with different binding partners in each case. These results are also interesting in view of the known interaction between HDAC5 and HDAC3 (Fischle et al., 2002), and the fact that HDAC3 is transported to the nucleus of RGC after optic nerve injury, turning off many genes in injured RGC, thereby contributing to promote apoptosis (Pelzel et al., 2010). Our results suggest that HDAC5 is unlikely to modulate the localization of HDAC3 and does not follow HDAC3 to the nucleus. Either these two proteins do not interact in the cytoplasm of RGCs, or HDAC5 interaction with cytoskeletal proteins such as Ankyrin-repeat family proteins, tubulins (McKinsey et al., 2006; Greco et al., 2011; Cho and Cavalli, 2012), or other unidentified proteins sequester HDAC5 in the cytoplasm and prevent HDAC5 of following HDAC3 to the nucleus.
Indeed, beyond subcellular localization, HDAC5 phosphorylation is known to regulate HDAC5 binding with interacting partners (Greco et al., 2011). Our results indicate that HDAC5AA promotes optic nerve regeneration by activating the mTOR pathway in RGCs. There are several experiments in our study that demonstrate that HDAC5AA activates the mTOR pathway to promote optic nerve regeneration. First, axon regeneration induced by HDAC5AA overexpression is prevented by inhibiting the mTOR pathway using rapamycin or by deletion of Raptor, without reducing the number of surviving RGCs. Interestingly, similar to the control condition, we still observed a number of regenerating axons after blocking mTOR suggesting that blocking mTOR pathway does not indiscriminately block all regeneration. Indeed, a previous study demonstrated that although rapamycin reduces the number of regenerating axons at long distances from the injury site (1000 μm) in response to an inflammatory stimulus applied to the retina, it does not affect the number of regenerating axons at short distances (500μm) (Leibinger et al., 2012). Moreover, this study also showed that purified RGCs stimulated with CNTF grow axons at the same rate when the mTOR pathway is inhibited with rapamycin (Leibinger et al., 2012). Our study also reveals that applying an inflammatory stimulus by injecting intravitreally zymosan does have an additive effect with HDAC5AA. In contrast, deleting TSC2, which activates the mTOR pathway, does not have an additive effect, emphasizing the link between HDAC5AA overexpression and mTOR activation.
Whereas activation of mTOR by PTEN deletion completely reverts the decrease in pS6 staining produced by injury (Park et al., 2008), our data show that HDAC5AA reverts part of the pS6 decay after injury, suggesting that even with a modest activation of mTOR, a substantial axon regeneration can be achieved. However, a direct comparison between these studies is difficult because of the different antibodies used. Whereas we used an antibody that recognizes the phosphorylated serine 240/244, Park et al. used an antibody that recognizes phosphorylated serine 235/236 (Park et al., 2008). Both phosphorylation sites are phosphorylated by pS6 kinase, which is activated by mTOR. However, serine p240/244 is a more reliable marker of pS6 kinase activation since it is exclusively phosphorylated by pS6 kinase while p235/236 is also phosphorylated by other kinases (Roux et al., 2007). Of note, it has been previously shown that CNS neurons have a higher phosphorylation at serine 240/244 compared to serine 235/236 (Liu et al., 2014), which may explain the higher number of pS6 positive RGCs observed in our study compared to others studies.
It is also noteworthy that blocking mTOR activity in RGCs overexpressing HDAC5AA did not reduce their survival. Other studies reported that the increase in surviving RGCs mediated by PTEN deletion is prevented by rapamycin treatment (Park et al., 2008). We found that rapamycin treatment in HDAC5AA expressing mice increased RGC survival. The neuroprotective effect of rapamycin is suggested to occur through either an inhibition of retinal gliosis (Li et al., 2018) or an activation of autophagy (Rodriguez-Muela et al., 2012). Since, rapamycin treatment can increase or decrease survival depending on the context, it might be difficult to tease apart the two mechanisms in our experiment, especially considering that, the increase of RGC survival mediated by rapamycin (Rodriguez-Muela et al., 2012), and the increase mediated by HDAC5AA are relatively modest compared to the PTEN deletion (Park et al., 2008). Interestingly, blocking mTOR by Raptor deletion did not reduce the number of surviving RGCs, suggesting that the increase in survival mediated by HDAC5AA is caused by an mTOR independent mechanism, or that HDAC5AA overexpression does not increase survival in the Raptor floxed mouse background.
The exact mechanism by which HDAC5AA activates the mTOR pathway in RGC remains to be elucidated. There is evidence in a hypothalamic cell line that HDAC5 interacts and deacetylates Raptor, thereby activating mTOR (Ma et al., 2015). A similar mechanism might be taking place in RGCs. Both HDAC5DAD and HDAC5AA are able to activate mTOR pathway according to the pS6 staining in injured retinas and the RNAseq pathway analysis. However, these same results also showed that HDAC5AA is more effective in activating mTOR and probably for this reason is the only one that significantly promotes axon regeneration. It is possible that HDAC5AA has more affinity for Raptor compared to HDAC5DAD and therefore favors its deacetylation. Indeed, HDAC5 phosphorylation is known to regulate HDAC5 binding with interacting partners (Greco et al., 2011). Another possibility is that HDAC5AA activates the expression of pro-growth genes in the nucleus that leads to the activation of the mTOR pathway, since our RNAseq data reveals that both HDAC5 mutants can regulate mTOR by regulating the expression of genes in the mTORC1 pathway or in the p53 pathway (Feng et al., 2005). Similarly, deletion of class I histone deacetylases HDAC1 and HDAC2 in RGCs induce a downregulation of p53 and promotes RGC survival (Lebrun-Julien and Suter, 2015). These results suggest that activation of mTOR by HDAC5AA regulates directly or indirectly signaling pathways that establish a pro-growth program. However, our RNAseq analysis used whole retina and other cell types, such as reactive glia, might also contribute to the observed difference in gene expression. Future studies will be required to confirm these hypotheses.
In conclusion, our study demonstrates that HDAC5 is largely localized in the cytoplasm of RGC and is unaffected by the manipulation of PKCμ, PKA, or even the inhibition nuclear export. It further demonstrates that blocking the phosphorylation of HDAC5 at serine 259 and 498 by substitution with alanine does also not induce nuclear localization, but partially prevents the decline in mTOR activity caused by injury (Park et al., 2008) and promotes optic nerve regeneration and RGC survival. Our results suggest a new therapeutic target for future studies that aim to promote axon regeneration through mTOR activation.
Highlights.
HDAC5 is localized primarily in the cytoplasm of RGCs
Phosphorylation of key serine residues does not regulate HDAC5 localization in RGCs
HDAC5AA overexpression promotes optic nerve regeneration and RGC survival
HDAC5AA induced regeneration is mTOR dependent
HDAC5AA synergizes with zymosan to promote robust axon regeneration
Acknowledgements
This work was supported in part by National Institutes of Health Grants EY029077, NS082446 and NS096034 (to V.C.), a Philip and Sima K. Needleman Doctoral Fellowship (to M.M) and a BioSURF fellowship (to A.J). We would like to thank the Hope Center Viral Vectors Core at Washington University School of Medicine for generating the AAV virus, the Genome Technology Access Center at Washington University for performing the RNA sequencing, Guoyan Zhao for help with HDAC5 expression analysis in the RNAseq data, Alan Sang for technical assistance and Amber Hackett for her constructive comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
The authors declare no conflict of interest.
References
- Abe N, Borson SH, Gambello MJ, Wang F, Cavalli V (2010) Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem 285:28034–28043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Siegfried CJ, Lin JB, Shui YB, Sein J, Pita-Thomas W, Sene A, Santeford A, Gordon M, Lamb R, Dong Z, Kelly SC, Cavalli V, Yoshino J, Apte RS (2017) GDF15 is elevated in mice following retinal ganglion cell death and in glaucoma patients. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL (1988) Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1:791–803. [DOI] [PubMed] [Google Scholar]
- Berry M, Ahmed Z, Logan A (2018) Return of function after CNS axon regeneration: Lessons from injury-responsive intrinsically photosensitive and alpha retinal ganglion cells. Prog Retin Eye Res. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu Y, Zheng P (2009) mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2:ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KY, Hao M (2017) Mammalian Target of Rapamycin (mTOR) Regulates Transforming Growth Factor-beta1 (TGF-beta1)-Induced Epithelial-Mesenchymal Transition via Decreased Pyruvate Kinase M2 (PKM2) Expression in Cervical Cancer Cells. Med Sci Monit 23:2017–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Cavalli V (2012) HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J 31:3063–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V (2013) Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155:894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW (2010) PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci 30:9306–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima S, Koriyama Y, Kurimoto T, Oliveira JT, Yin Y, Li Y, Gilbert HY, Fagiolini M, Martinez AM, Benowitz L (2012) Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc Natl Acad Sci U S A 109:9149–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Qiao M, Bei F, Kim IJ, He Z, Sanes JR (2015) Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron 85:1244–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 102:8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E (2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9:45–57. [DOI] [PubMed] [Google Scholar]
- Gaub P, Joshi Y, Wuttke A, Naumann U, Schnichels S, Heiduschka P, Di Giovanni S (2011) The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134:2134–2148. [DOI] [PubMed] [Google Scholar]
- Gray JT, Zolotukhin S (2011) Design and construction of functional AAV vectors. Methods Mol Biol 807:25–46. [DOI] [PubMed] [Google Scholar]
- Greco TM, Yu F, Guise AJ, Cristea IM (2011) Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol Cell Proteomics 10:M110 004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG (2010) PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 107:15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189. [DOI] [PubMed] [Google Scholar]
- Harrison BC, Roberts CR, Hood DB, Sweeney M, Gould JM, Bush EW, McKinsey TA (2004) The CRM1 nuclear export receptor controls pathological cardiac gene expression. Mol Cell Biol 24:10636–10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AR, Kamphuis W, Eggers R, Symons NA, Blits B, Niclou S, Boer GJ, Verhaagen J (2002) Intravitreal injection of adeno-associated viral vectors results in the transduction of different types of retinal neurons in neonatal and adult rats: a comparison with lentiviral vectors. Mol Cell Neurosci 21:141–157. [DOI] [PubMed] [Google Scholar]
- Hernandez O, Way S, McKenna J 3rd, Gambello MJ (2007) Generation of a conditional disruption of the Tsc2 gene. Genesis 45:101–106. [DOI] [PubMed] [Google Scholar]
- Iglesias T, Rozengurt E (1998) Protein kinase D activation by mutations within its pleckstrin homology domain. J Biol Chem 273:410–416. [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F, Suter U (2015) Combined HDAC1 and HDAC2 Depletion Promotes Retinal Ganglion Cell Survival After Injury Through Reduction of p53 Target Gene Expression. ASN Neuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibinger M, Andreadaki A, Fischer D (2012) Role of mTOR in neuroprotection and axon regeneration after inflammatory stimulation. Neurobiol Dis 46:314–324. [DOI] [PubMed] [Google Scholar]
- Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI (2000) Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci 20:4615–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang F, Zhang Q, Jin M, Lu Y, Chen S, Guo C, Zhang X (2018) Rapamycin mediates mTOR signaling in reactive astrocytes and reduces retinal ganglion cell loss. Exp Eye Res 176:10–19. [DOI] [PubMed] [Google Scholar]
- Li S, Yang C, Zhang L, Gao X, Wang X, Liu W, Wang Y, Jiang S, Wong YH, Zhang Y, Liu K (2016) Promoting axon regeneration in the adult CNS by modulation of the melanopsin/GPCR signaling. Proc Natl Acad Sci U S A 113:1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, Thom M (2014) Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol Commun 2:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Contreras M, Shen T, Randall WR, Schneider MF (2009) Alpha-adrenergic signalling activates protein kinase D and causes nuclear efflux of the transcriptional repressor HDAC5 in cultured adult mouse soleus skeletal muscle fibres. J Physiol 587:1101–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tang H, Yin Y, Yu R, Zhao J, Li Y, Mulholland MW, Zhang W (2015) HDAC5-mTORC1 Interaction in Differential Regulation of Ghrelin and Nucleobindin 2 (NUCB2)/Nesfatin-1. Mol Endocrinol 29:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar M, Cavalli V (2018) Intrinsic mechanisms of neuronal axon regeneration. Nat Rev Neurosci 19:323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Kuwahara K, Bezprozvannaya S, Olson EN (2006) Class II histone deacetylases confer signal responsiveness to the ankyrin-repeat proteins ANKRA2 and RFXANK. Mol Biol Cell 17:438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Tan F, Li Z, Thiele CJ (2011) NGF activation of TrkA induces vascular endothelial growth factor expression via induction of hypoxia-inducible factor-1alpha. Mol Cell Neurosci 46:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008) Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322:963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasut A, Jones AE, Rudnicki MA (2013) Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp:e50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelzel HR, Schlamp CL, Nickells RW (2010) Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci 11:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN (2008) Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8:399–410. [DOI] [PubMed] [Google Scholar]
- Quigley HA (2016) Understanding Glaucomatous Optic Neuropathy: The Synergy Between Clinical Observation and Investigation. Annu Rev Vis Sci 2:235–254. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Germain F, Marino G, Fitze PS, Boya P (2012) Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ 19:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J (2007) RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem 282:14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt HM, Schlamp CL, Nickells RW (2018) Targeting HDAC3 Activity with RGFP966 Protects Against Retinal Ganglion Cell Nuclear Atrophy and Apoptosis After Optic Nerve Injury. J Ocul Pharmacol Ther 34:260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt HM, Pelzel HR, Schlamp CL, Nickells RW (2014) Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol Neurodegener 9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SR C (1913) Degeneration and Regeneration of the Nervous System: Oxford University Press. [Google Scholar]
- Steketee MB, Goldberg JL (2012) Signaling endosomes and growth cone motility in axon regeneration. Int Rev Neurobiol 106:35–73. [DOI] [PubMed] [Google Scholar]
- Trakhtenberg EF, Pita-Thomas W, Fernandez SG, Patel KH, Venugopalan P, Shechter JM, Morkin MI, Galvao J, Liu X, Dombrowski SM, Goldberg JL (2017) Serotonin receptor 2C regulates neurite growth and is necessary for normal retinal processing of visual information. Dev Neurobiol 77:419–437. [DOI] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 24:8374–8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HJ, Cao JP, Yu JK, Gao DS (2007) Role of PI3-K/Akt pathway and its effect on glial cell line-derived neurotrophic factor in midbrain dopamine cells. Acta Pharmacol Sin 28:166–172. [DOI] [PubMed] [Google Scholar]
- Xie C, Lu H, Nomura A, Hanse EA, Forster CL, Parker JB, Linden MA, Karasch C, Hallstrom TC (2015) Co-deleting Pten with Rb in retinal progenitor cells in mice results in fully penetrant bilateral retinoblastomas. Mol Cancer 14:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI (2003) Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci 23:2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]