

Abstract
The innate immune response plays a critical role in traumatic brain injury (TBI), contributing to ongoing pathogenesis and worsening long-term outcomes. Here we focus on neutrophils, one of the “first responders” to TBI. These leukocytes are recruited to the injured brain where they release a host of toxic molecules including free radicals, proteases, and pro-inflammatory cytokines, all of which promote secondary tissue damage. There is mounting evidence that the developing brain is more vulnerable to injury that the adult brain. This vulnerability to greater damage from TBI is, in part, attributed to relatively low antioxidant reserves coupled with an early robust immune response. The latter is reflected in enhanced sensitivity to cytokines and a prolonged recruitment of neutrophils into both cortical and subcortical regions. This review considers the contribution of neutrophils to early secondary pathogenesis in the injured developing brain and raises the distinct possibility that these leukocytes, which exhibit phenotypic plasticity, may also be poised to support wound healing. We provide a basic review of the development, life cycle, and granular contents of neutrophils and evaluate their potential as therapeutic targets for early neuroprotection and functional recovery after injury at early age. While neutrophils have been broadly studied in neurotrauma, we are only beginning to appreciate their diverse roles in the developing brain and the extent to which their acute manipulation may result in enduring neurological recovery when TBI is superimposed upon brain development.
Keywords: Granules, Neutropenia, Neutrophil Elastase, Oxidative Stress, Polarization, Recruitment
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability in children, with approximately 640,000 TBI-related visits to the emergency room in children under 14 in the United States in 2013 (Faul et al., 2010; Taylor et al., 2017). Infants and young children (aged 0-4) are at greatest risk of sustaining a TBI (Corrigan et al., 2013; Faul and Coronado, 2015; Faul et al., 2010), and compared to other age groups, young children show poorer functional recovery as measured by IQ and related cognitive performance tests, which may persist for years after injury (Anderson et al., 2000; Anderson et al., 2005; Catroppa et al., 2008). As processes such as myelination, synaptogenesis and pruning continue years beyond birth in humans (Koizumi, 2004), there is substantial risk that an early age injury will disrupt these developmental processes, and result in long-term behavioral consequences including anxiety, attention deficit hyperactivity disorder, psychosocial problems and increased risk-taking behavior (Corrigan et al., 2013).
This review will consider neutrophils, the most abundant leukocytes, which are recruited to the injured young brain, as mediators of early secondary pathogenesis and determinants of long-term neurological recovery. Infiltrated neutrophils are a common denominator of the human brain after a TBI (Gahm et al., 2002; Hausmann et al., 1999; Rhind et al., 2010) and rodent models of TBI, designed to interrogate their contribution to early secondary tissue damage (Clark et al., 1994; Claus et al., 2010). The latter have provided an opportunity to not only address the contribution of neutrophils to early secondary pathogenesis but the neurological consequences at adulthood after exposure of the developing brain to these leukocytes.
The Basics of Neutrophils
In order to understand the potential role(s) of neutrophils in early-age TBI, we first provide an overview of their general biology including their development, lifecycle, and granular contents.
Development and lifecycle of neutrophils
Neutrophils, basophils, and eosinophils are collectively known as granulocytes. Like monocytes, neutrophils descend from granulocyte-monocyte progenitor cells in the bone marrow. During the roughly 14 day process (in humans) of granulopoiesis, neutrophils mature, proliferate, differentiate, and develop their characteristic granules (Bainton et al., 1971; Mortaz et al., 2018).
The systemic response of neutrophils to brain injury is centered in the liver, which produces a host of cytokines in response to inflammation in the central nervous system (CNS), which drive the maturation and granulation of neutrophils in bone marrow (Anthony et al., 2012; Campbell et al., 2008; Furze and Rankin, 2008). Chief among the many cytokines capable of regulating granulopoiesis is granulocyte-colony stimulating factor (G-CSF), which is regulated by the interleukin (IL)-23/IL-17 axis (Aggarwal et al., 2003; Li et al., 2017; Schwarzenberger et al., 2000), and is elevated acutely after TBI in both clinical and preclinical studies (Banks et al., 2016). The factors that regulate neutrophil death are similar to those that regulate granulopoiesis, notably G-CSF (Colotta et al., 1992). In fact, there is a homeostatic feedback loop linking neutrophil birth and death via the IL-23/IL-17/G-CSF axis; phagocytosis of apoptotic neutrophils by macrophages and dendritic cells leads to upregulation of granulopoiesis via IL-17 and G-CSF (Stark et al., 2005).
Under basal conditions, neutrophils live only a few hours after being released, dying by apoptosis and being removed by macrophages in the liver and spleen (Kolaczkowska and Kubes, 2013; Stark et al., 2005), However, the lifespan and death of neutrophils can be altered by the cellular environment. Inflammatory conditions such as congestive heart failure and hypoxia increase neutrophil lifespan (Hannah et al., 1995; Tracchi et al., 2009), and under circumstances of extensive tissue damage, like TBI, clearance of circulating neutrophils by the liver or spleen is delayed (Kolaczkowska and Kubes, 2013).
Fully mature neutrophils are released from the bone marrow and transmigrate across the epithelium and into the blood stream. Under normal conditions, there are 5-6 times as many mature neutrophils in the bone marrow as in circulation (Cartwright et al., 1964; Cowland and Borregaard, 2016; Tak et al., 2013). The chemokine receptor (CXCR) type 4, a G-protein coupled receptor expressed at low levels on the cell surface of mature neutrophils (Martin et al., 2003), binds with its ligand stromal derived factor (SDF)-1 to retain neutrophils in the bone marrow (Eash et al., 2009; Summers et al., 2010). Downregulation of CXCR4 and SDF-1 by G-CSF allows mobilization of neutrophils from bone marrow (Kim et al., 2006; Semerad et al., 2005).
Neutrophil recruitment and migration
Mature neutrophils move through the body via the circulatory system, resulting in a distribution that can be subdivided into two subpopulations: circulating neutrophils, which flow freely in the vasculature, and marginated neutrophils, which travel more slowly, as they transit through organs (Summers et al., 2010). Both circulating and nearby marginated neutrophils are acutely recruited to the site of injury (de Oliveira et al., 2016). Notably, there is recent preclinical evidence that when stimulated by stroke or aseptic meningitis, the majority of responding neutrophils in the brain arise from the marrow of the skull rather than that of long bones such as the femur or tibia, and that some of these neutrophils pass directly from the bone marrow to the cerebrospinal fluid of the meningeal compartment, ostensibly bypassing the blood-brain barrier (Herisson et al., 2018). In the case of TBI, the number of circulating neutrophils does not seem to be a rate-limiting factor for neutrophil recruitment, as treatment with G-CSF prior to controlled cortical impact (CCI) causes dramatic increases in circulating neutrophils with no effect on neutrophils recruited to the injured cortex (Whalen et al., 2000).
Neutrophils are one of the first myeloid-derived cells to infiltrate the injured brain, peaking at approximately 24 hours post injury (See review; Jassam et al., 2017; Kochanek et al., 2017). Circulating neutrophils are recruited to the acutely injured brain through a series of steps along the endothelium that involve tethering and rolling and their subsequent migration into the parenchyma (Yilmaz and Granger, 2010). The first step of this process involves attachment of neutrophils to endothelial cells (Abbassi et al., 1993; Schmidt et al., 2011). Neutrophils tether to and roll along the endothelial cell layer through the action of a class of cell adhesion molecules, of which the P-selectin glycoprotein ligand is the primary ligand (Eriksson et al., 2001). These adhesion molecules are known as P, E and L-selectins and are each expressed in different cells with unique actions and kinetics (Kansas, 1996). L-selectin is expressed on the surface of neutrophils and other leukocytes, while endothelial cells present P- and E-selectin on their luminal-facing surface (Kansas, 1996). To enter the injured brain, rolling neutrophils must overcome the shear force of blood flow to slow down to a crawl and then fully arrest, adhering firmly to the endothelial layer (Yilmaz and Granger, 2010). This crawling and subsequent arrest is achieved through the action of integrins, transmembrane receptors which connect the extracellular matrix to the cytoskeleton of the cell and promote cell-to-cell binding (Kourtzelis et al., 2017). Endothelial intercellular cell adhesion molecule −1 and 2 facilitate the extravasation of neutrophils from blood vessels through the barrier via their strong transmembrane binding action (Gorina et al., 2014).
Once neutrophils have arrived at the site of injury and adhere to the endothelial cell layer, they traverse the endothelium in a process termed transmigration, which can occur by either paracellular or transcellular routes (Wittchen, 2009; Zen and Parkos, 2003). In paracellular transmigration, cells penetrate the barrier by traversing adjacent endothelial cells through tight junctions (Muller, 2015). In contrast, transcellular migration involves a neutrophil passing through the endothelial cell proper, emerging on the parenchymal side after full engulfment into the cytoplasmic compartment (Hyun and Hong, 2017). It is generally thought that most transmigration occurs via the paracellular route; however, the transcellular route may be preferred when endothelial cells express high levels of intercellular adhesion molecule-1 (Yang et al., 2005). In vitro studies that model the inflamed blood-brain barrier have demonstrated a neutrophilic preference for transcellular routes, with cells passing fully through the endothelial cell proper rather than the paracellular route (Gorina et al., 2014; von Wedel-Parlow et al., 2011).
Neutrophilic granules
Neutrophils influence their surroundings primarily through their granular contents, which are released either extracellularly or into phagosomes (Papayannopoulos, 2018). The immune response from TBI creates an environment where neutrophils readily degranulate, their granule contents acting in a variety of potentially harmful and helpful ways (Hager et al., 2010).
There are three general classifications of granules (Lacy, 2006): primary, which include myeloperoxidase, neutrophil elastase, and cathepsin G; secondary, which include lactoferrin; and tertiary granules, which include matrix metalloproteinases (MMPs). The primary functions of myeloperoxidase and lactoferrin are antimicrobial; the former generates cytotoxic hypochlorous acid, whereas the latter sequesters iron and degrades nucleic acid. MMP-9, a member of the family of matrix metalloproteinases, may facilitate paracellular migration of neutrophils as zonulae occludins-1 (a key component of the tight junctional complexes) is a known substrate of this protease (Turner and Sharp, 2016). Each type of granule also contains many other molecular contents, and the content of each granule seems to be determined by when in granulopoiesis it formed (Cowland and Borregaard, 2016). Classification of neutrophil granules (primary, secondary, tertiary) stems mainly from rodent models, but the nature of human neutrophil granules may be somewhat different, with more heterogeneity and overlap between granule contents (Bainton et al., 1971; Cowland and Borregaard, 2016; Kjeldsen et al., 1993).
Polarization of neutrophils and their phenotypic diversity
Emerging evidence supports the heterogeneity of neutrophils. Early studies suggested an N1 (pro-inflammatory)/N2 (anti-inflammatory) phenotypic polarization (Fridlender et al., 2009) modeled after the M1/M2 polarization identified in macrophages (Xu et al., 2017). Recently though, the concept of a binary M1/M2 classification has been replaced by the idea that a broader spectrum of macrophage phenotypic diversity exists (Morganti et al., 2016; Ransohoff, 2016). Similarly, neutrophils may not be so easily classified into two binary categories (Deniset and Kubes, 2016). One alternative scheme categorizes neutrophils by degrees of activation; namely, 1) naïve (circulating neutrophils), 2) mildly activated (wound- or tumor associated), 3) activated (acute infection) and 4) highly activated (cytotoxic; Houghton, 2010). Although neutrophil polarization has not been studied in great detail nor has the phenotypic spectrum been fully delineated, these differentially activated subsets of neutrophils have been reported in both humans and in animal models in different types of disease and infection (Silvestre-Roig et al., 2016; Tsuda et al., 2004).
Specific to inflammation in the brain, there is some evidence that neutrophils may be more likely to assume an anti-inflammatory phenotype, crucial to the resolution of inflammation and neuroprotection (Cuartero et al., 2013). For example, in a model of intracerebral hemorrhage, the anti-inflammatory cytokine IL-27 alters the phenotype of maturing neutrophils, decreasing their production of cytotoxic granular contents and increasing production of the beneficial iron-scavenging protein, lactoferrin (Zhao et al., 2017). No studies to date have considered the possibility that neutrophils may exhibit phenotypic diversity in the traumatically injured brain. However, it is clear that investigation of neutrophil polarization is crucial to understanding how these leukocytes behave in an evolving lesion where they may exhibit temporally distinct roles.
Pathobiology of the Injured Developing Brain
Long-term neurological deficits, reported in brain-injured children, are likewise paralleled in preclinical models. In response to CCI, rodents at either post-natal day (PND) 17 or 21, ages roughly equivalent to the toddler-aged child (Semple et al., 2016), exhibit a variety of abnormal behavioral phenotypes at adulthood, findings that speak to the long-term consequences of early age injury. These include deficits in spatial learning and memory (Adelson et al., 2013; Ajao et al., 2012; Kamper et al., 2013; Pop et al., 2013) and sensorimotor function (Kamper et al., 2013; Pullela et al., 2006). Hyperactivity is evident as early as adolescence, persisting into adulthood (Pullela et al., 2006). In contrast, deficits in sociosexual function are not detected at adolescence but rather present at adulthood (Semple et al., 2014).
Preclinical models of TBI at an early age have elucidated the pathobiology of secondary injury. In rodents exposed to a CCI at PND 21, neuronal cell loss is evident within the first several days of injury and coincides with activated microglia/macrophages in both the ipsi- and contralateral cortices, the ipsilateral hippocampus and the thalamus (Igarashi et al., 2007; Tong et al., 2002). Ongoing pathogenesis may persist for months after injury at either PND 17 or 21, culminating in reduced cortical thickness and numbers of neurons in the hippocampus as well as atrophy of the hippocampus and the corpus callosum (Ajao et al., 2012; Kamper et al., 2013; Tsuru-Aoyagi et al., 2009).
The brain at PND 21 is more vulnerable to damage as a result of TBI compared to that of the adult. This is due, in part, to inadequate antioxidant reserves (Tsuru-Aoyagi et al., 2009) coupled with prolonged recruitment of neutrophils that exceeds what is seen in adult injured brain (Claus et al., 2010; Potts et al., 2006). The injured brain is exposed to a high degree of oxidative stress resulting from reactive oxygen and nitrogen species, released by dying and damaged cells, infiltrating leukocytes, and free iron liberated by degradation of heme subsequent to hemorrhage (Potts et al., 2006). The young brain lacks adequate antioxidant reserves necessary to combat TBI-related oxidative damage (Bayir et al., 2002; Tsuru-Aoyagi et al., 2009) and the capacity to fully engage antioxidant pathways. For example, the antioxidant glutathione peroxidase is produced in the injured brains of rodents at PND 21 at levels similar to that seen in adults, but is not upregulated in response to injury as it is in adults (Fan et al., 2003). Overexpression of this enzyme in transgenic rodents results in improved long-term behavioral outcomes after TBI at PND 21, demonstrating the importance of this antioxidant in reducing pathogenesis (Tsuru-Aoyagi et al., 2009).
There is a growing interest in neutrophils in TBI, implicating their involvement in secondary injury (Dickens et al., 2017; Makinde et al., 2017; Roth et al., 2014; Russo et al., 2018; Zhao et al., 2017). Their recruitment to the injured brain (Biagas et al., 1992; Clark et al., 1994; Hartl et al., 1997; Schoettle et al., 1990; Soares et al., 1995) coincides with disruption of the blood-brain-barrier (Anthony et al., 1998; Anthony et al., 1997; Kaczorowski et al., 1995; Soares et al., 1995) and the release of free radicals, proteases, and pro-inflammatory cytokines, all of which promote tissue damage (Jickling et al., 2015; Kawabata et al., 2002; Keeling et al., 2000; Lee and Downey, 2001; Owen and Campbell, 1999).
The injured developing brain exhibits a unique inflammatory signature, characterized by prolonged recruitment of neutrophils into the damaged cortex. In a rodent model of CCI, neutrophil recruitment into the injured adult brain occurs over a period of about 3 days. In contrast, TBI at PND 21 results in recruitment of neutrophils that persists up to 14 days post injury (DPI) (Figure 1; Claus et al., 2010). These findings raise questions about the mechanism(s) underlying these differences in recruitment timecourse. An interesting series of studies, focused on cortical delivery of cytokines, have shown a dramatic age-dependency in neutrophil recruitment, cementing the idea of a developmental “window of susceptibility” in the response of neutrophils to neuroinflammation. In line with this age-dependent response of neutrophils to TBI (Claus et al., 2010), intraparenchymal delivery of IL-1β into the naive rodent brain at PND 21 results in far more recruitment of neutrophils and disruption of the blood-brain barrier compared to the adult rodent brain exposed to similar conditions, highlighting the enhanced sensitivity in the young brain to cytokines (Figure 2; Anthony et al., 1997). A recent study examining the response to intraparenchymal administration of IL-1β at PND 7, 14, 21, and 60 (young adult) has revealed an exquisite sensitivity to IL-1β at early developmental stages (Sa-Pereira et al., 2018). These results show elevated numbers of neutrophils in the brain with almost a 5-fold increase at PND 14 compared to both PND 7 and 21, with minimal to no detection at PND 60, indicating an age-dependent response by neutrophils specific to development. Lastly, intraparenchymal administration of the downstream cytokine-induced neutrophil chemoattractant 1 (CINC-1, also known as GRO in rat; homologue to human IL-8) produces a more rapid recruitment of neutrophils in rodents at PND 21 compared to adult (Anthony et al., 1998). These collective findings suggest that the time-course for neutrophil recruitment in response to neuroinflammation is directly impacted by age at time of injury.
Figure 1.
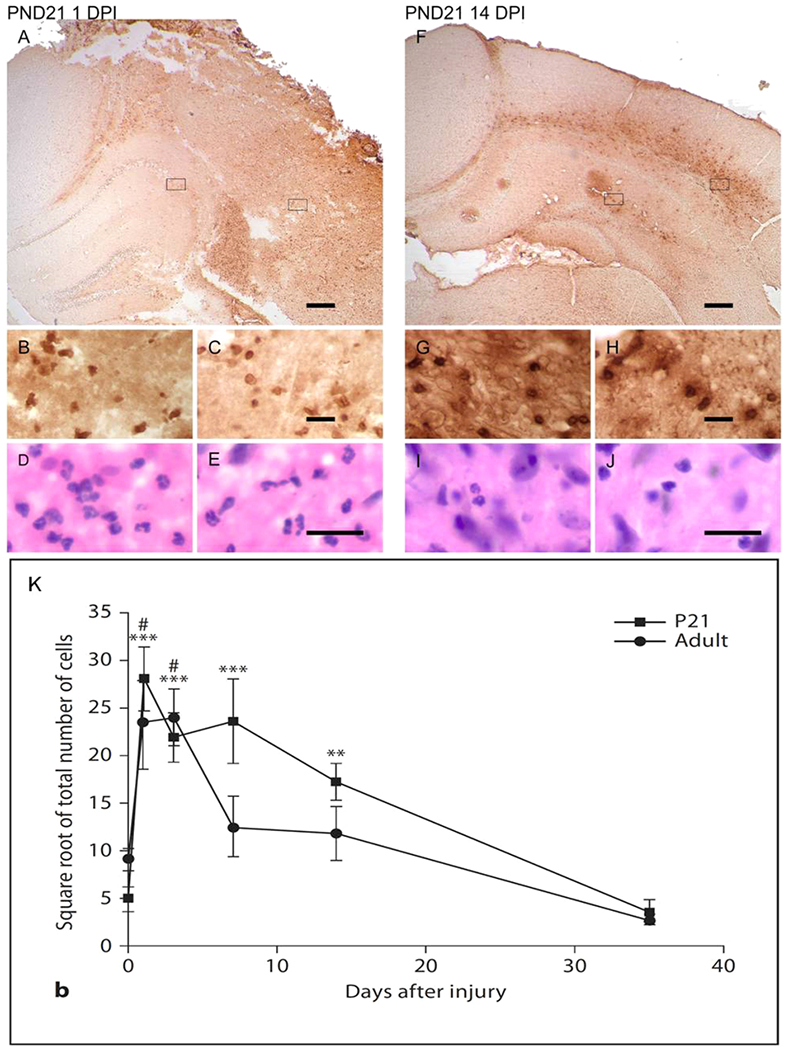
Recruitment of neutrophils into the injured brain at PND 21. GR-1+ neutrophils are immunolocalized in the cortex and hippocampus at 1 (A-E) and 14 days (F-J) post injury. Boxes in A, F are represented at higher magnification in the cortex (B,G) and hippocampus (C,H). Adjacent sections stained with hematoxylin and eosin show cells with lobulated nuclei in the cortex (D,I) and hippocampus (E,J). Scale bars = 200 μm (A,F) and 20 μm (C,E,H,J). (K) Quantification of neutrophils in sections of the cortex after injury at either PND 21 or at adulthood. GR-1+ neutrophils are significantly higher in the injured young brains within the first 2 weeks after injury relative to shams. In contrast, GR-1 is significantly elevated at only 1 and 3 days after injury to the adult brain. * * p < 0.01, * * * p < 0.001, one-way ANOVA followed by Newman-Keuls multiple comparison test (sham vs. PND 21); # p < 0.05, one-way ANOVA followed by Newman-Keuls multiple comparison test (sham vs. adult). Figures modified from Claus et al., 2010, with permission from S. Karger AG Publishers, Basel, Switzerland.
Figure 2.
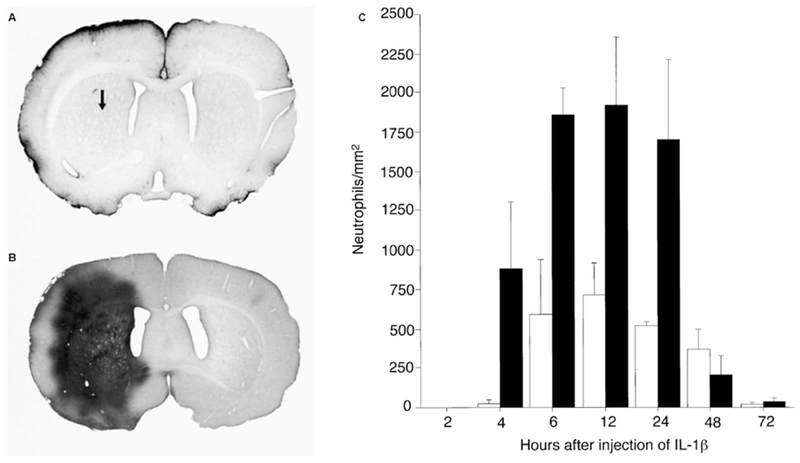
Breakdown of the blood-brain barrier to the vascular tracer horseradish peroxidase and recruitment of neutrophils after intracortical administration of interleukin-1β. There is more limited leakage to horseradish peroxidase at 4 hours after administration to the adult brain (3 months old) compared to the brain at PND 21 (A-B) (Arrow in A shows the position of the injection site for all sections). Dark staining indicates regions of abnormal vascular permeability to horseradish peroxidase. Intracortical administration of interleukin-1β likewise resulted in a more robust recruitment of neutrophils into the young compared to the adult brain (C). The number of neutrophils was quantified in a region of striatum immediately adjacent to the site of injection. Solid black bars represent mean number of neutrophils present in the PND21 brain, open bars represent mean number of neutrophils present in the adult brain. Values are the mean ± SD. Figure modified from Anthony et al., 1997, with permission from Oxford Press.
Neutrophils as Modifiers of Pathogenesis
A variety of approaches have been used to assess the pathogenicity of neutrophils in the injured brain. These include pharmacologic and immunologic depletion strategies, and the use of genetically modified animals to reduce the number of circulating neutrophils, target receptors that facilitate their transmigration, or address the consequences of selective deletion of their granular content. Surprisingly, very few of these studies in the injured adult brain have employed quantitative methods to address changes in recruitment and only one study has been conducted in the injured developing brain (Table 1). Importantly, some of these strategies may lack specificity to neutrophils, confounding interpretation of the results.
Table 1.
Summary of Studies Targeting Neutrophil Recruitment after TBI
Summary of strategies that have altered recruitment of neutrophils in pre-clinical rodent models of TBI at adulthood and at postnatal day (PND)21. This table considers only those studies that have modified neutrophil recruitment via either immunological depletion of these leukocytes or pharmacologic methods, or by use of genetically modified animals. Acute, subacute and long-term neurological consequences are summarized in accordance with each study.
| Adult Brain | ||||||
|---|---|---|---|---|---|---|
| Reference | TBI Model | Approach | Treatment Strategy | Neutrophil Numbers | Acute Findings | Subacute & Chronic Findings |
| Uhl et al. 1994 | WD | Vinblastine Sulfate | Route: IP (5 d prior to TBI) | Absolute cell count in blood (prior to & 24 hrs PI) | ↓ CBF. No change-edema or lesion size (24 hrs PI) | Not Studied |
| Whalen et al. 1999 | CCI | Anti-RP-3 mAb to neutrophils | Route: IV (1 hr prior to TBI & for experimental period) | Absolute cell count in blood and histology: RP-3+ cells (4 hrs PI) | ↓ Neutrophil recruitment. No change- BBB permeability (4 hrs PI) | Not Studied |
| Semple et al. 2010 | WD | CXCR2 KO | Not Applicable | Histology: NIMP-R14+ cells (12 hrs & 7d PI) | ↑ Neutrophil chemokine production (12 & 24 hrs PI) | ↑ tissue damage. No change- BBB permeability or neurologic recovery (7 d PI) |
| Kenne et al. 2012 | CCI | Anti-GR-1 mAb RB6-8C5 to neutrophils | Route: IP (12 hrs prior & PI) | Histology: GR-1+ cells (24 hrs PI) | ↓ Edema & cell death (24 hrs PI) ↓edema (48 hrs PI) |
↓ lesion volume & microglial activation (7 d PI) ↓ lesion volume (14 d PI) |
| Roth et al. 2014 | CHI | Purinergic Receptor (P2) Antagonist Or Glutathione (GSH) | Route: Topical to skull (30 mins prior to TBI) | Histology: LysMGFP/+ cells (1, 3, & 6 hrs PI) | P2: ↓ Neutrophil recruitment (3 & 6 hrs PI) ↑ Meningeal cell death (12 hrs PI) GSH: ↓ Parenchymal cell death, microglial activation & breakdown of glial limitans (>1 hr PI) ↓ Neutrophil recruitment (3 & 6 hrs PI) |
Not Studied |
| Makinde et al. 2017 | CCI | Liposomal clodronate Or CX3CR1 KO | Route: IV (24 hrs prior to TBI) | Flow Cytometry: (24 hrs PI) | Clodronate and KO: ↓ Neutrophil recruitment & monocyte infiltration (24 hrs PI) | ↓ edema, ↑ motor function & working memory (assessed only in clodronate group; 1 mo post injury) |
| Developing Brain (Postnatal day 21) | ||||||
| Semple et al. 2015b | CCI | Neutrophil elastase (NE) Inhibitor Or NE KO | Route: IP (2, 6, & 12 hrs PI) | Histology: GR-1+ cells-drug study only (24 hrs PI) | NE KO: ↓ edema, cell death & oxidative stress (24 hrs PI) NE inhibitor: ↓ edema, cell death & oxidative stress (24 hrs PI) |
NE KO: ↑ spatial memory, ↓ hyperactivity; no change in anxiety or motor deficits (2 mos PI) NE inhibitor: No change in neurologic recovery (2 mos PI) |
Abbreviations/Explanations: BBB (blood-brain barrier), CBF (cerebral blood flow), CCI, (controlled cortical impact), CHI (Closed head injury), CXCR2 (Cytokine receptor 2, mediates neutrophil recruitment), CX3CR1 (C-X3-C motif chemokine receptor 1, impairs recruitment of monocyte-derived macrophages), d (day), hr (hour), IP (intraperitoneal), IV (intravenous), KO (knockout), LysMGFP/+ (neutrophils are brightly labeled in this construct), mAb (monoclonal antibody), mo (month), PI (post injury) TBI (traumatic brain injury), WD (weight drop)
Strategies to induce neutropenia
One of the earliest studies induced neutropenia (an abnormally low neutrophil cell count) via a relatively non-specific approach; namely, intraperitoneal administration of the antimitotic vinblastine sulfate prior to a weight drop injury in adult rodents (Uhl et al., 1994). The resulting neutropenia, confirmed by approximately a 90% depletion in circulating neutrophils, was associated with a reduction in cerebral blood flow in the absence of any changes in edema, as measured by brain water content, or lesion size at one day post injury. These findings suggest possible interactions between neutrophils and improved blood flow-findings that have been more recently linked to the protein cathepsin G, a serine protease that is stored in neutrophilic primary granules (Faraday et al., 2013). While this was one of the earliest studies to attempt to understand the role of neutrophils in the injured brain through their systemic depletion, the interpretation of these early findings are confounded by a number of factors, namely the lack of specificity of vinblastine and its potential toxicity and unanticipated findings including reduced cerebral blood flow in the sham group and the absence of any evidence of neutrophils in the injured brain.
Alternative, immunologic-based approaches have been used to induce neutropenia prior to TBI (Kenne et al., 2012; Whalen et al., 1999). Continuous intraventricular infusion of the RP-3 monoclonal antibody generated against neutrophils (Sekiya et al., 1989) before and after CCI induced neutropenia and resulted in an almost complete lack of neutrophil accumulation in the injured cortex but no change in blood-brain barrier permeability at 4 hours post injury, indicating that barrier leakage at early time-points is not mediated by neutrophils (Whalen et al., 1999). Systemic administration of an anti-GR-1 monoclonal antibody (RB6-8C5) 12 hours prior to and 12 hours following CCI to the adult brain resulted in decreased GR-1+ labeled neutrophils in the injured cortex and a reduction in the apoptotic marker cleaved-caspase 3, brain edema, and microglia/macrophage activation within the first week post injury that corresponded to an attenuation in lesion volume at 7 and 14 DPI (Kenne et al., 2012). This suggests an adverse role of neutrophils in early secondary pathogenesis. However, this interpretation is confounded by the use of the RB6-8C5 monoclonal antibody which reacts strongly with mouse Ly6G, a marker of neutrophils, but also weakly with mouse Ly6C, which is found on subsets of monocytes and lymphocytes (Daley et al., 2008). Thus, it remains unclear if these findings are solely due to neutropenia.
Purinergic signaling
Two-photon laser scanning microscopy, coupled with a mild closed head compression injury (CHI), has offered unique insights into the early dynamic roles of microglia and neutrophils in the meningeal and parenchymal compartments (Roth et al., 2014). The very acute signatures of this injury speak to the vulnerability of the meningeal compartment, where cell death precedes that in the parenchyma. Within 30 minutes post injury there is vascular leakage in the meninges, the emergence of reactive oxygen species, and the subsequent breach of the glial limitans. Microglia respond to the compromised glial limitans by forming a transient adjacent phagocytic cell layer. This early pathogenesis coincides with the recruitment of neutrophils within the meningeal compartment (and not the parenchyma) where they show marked motility and close interactions with dead cells. Subsequent experiments implicated purinergic (P2) receptors, which respond to ATP released by dying cells, in recruitment of these neutrophils. Antagonism of P2X7, a receptor involved in recruitment of these leukocytes in a model of sterile inflammation in the liver (McDonald et al., 2010), resulted in a near absence of LysM gfp/+ neutrophils in the meningeal space. Importantly, this resulted in increased cell death in the meningeal space with no impact on cell death in the parenchyma, suggesting that neutrophils may play a beneficial role in this acute scenario after a mild CHI.
Targeting the CXCR2 receptor
Neutrophil migration is regulated by chemokine signaling through CXCR2, which is elevated after TBI and has been associated with the progression of secondary damage (Kobayashi, 2008; Morganti-Kossmann et al., 2018). Performing CHI on wildtype and transgenic rodents deficient in CXCR2 showed that neutrophil recruitment in the injured hemisphere was greatly reduced in CXCR2 null rodents compared to wildtype at both 12 hours and 7 DPI (Semple et al., 2010). CXCR2 null rodents also had reduced tissue damage, neuronal loss, and cell death at 7 DPI, suggesting that neutrophil recruitment contributes to secondary tissue damage after TBI. It is noteworthy, however, that CXCR2 deficient rodents may demonstrate a compensatory increase in CXC neutrophil chemokines and G-CSF at 12 and 24 hours post injury, but no changes in other chemokines, indicating that the acute cytokine response may not be mediated primarily by neutrophils in CHI.
Another recent study showed that in two models of peripheral inflammatory response, CXCR2 deficient rodents displayed a transient exaggerated acute inflammatory response, including increased pro-inflammatory markers, reduced inflammatory resolution markers, and increased macrophage accumulation (Dyer et al., 2017). The marked contrast between the response of CXCR2 null animals to peripheral inflammation versus CHI highlights the complexity of the role neutrophils play in the immune response to injury. Though neutrophils may not directly mediate acute cytokine response, in certain circumstances, their recruitment may be vital to the early inflammatory response through their interaction with other immune cells, such as macrophages.
Monocyte/neutrophil interactions
Monocytes infiltrate the injured adult brain through the C-C-chemokine receptor 2 (CCR2) and have been shown to adversely influence neuronal survival and neurological recovery (Hsieh et al., 2014). Recent studies have revealed that monocytes also modulate the recruitment of neutrophils into the injured adult brain. Systemic depletion of monocytes prior to injury, using liposome-entrapped clodronate, resulted in a reduction in neutrophils in the injured brain at 1 DPI, in the absence of any change in circulating neutrophils (Makinde et al., 2017). Clodronate-treated animals also showed reduced vasogenic edema and improved motor coordination and working memory, further supporting their adverse role in the injured brain. In rodents, monocytes are subdivided into classical monocytes (CD115+, Ly6Chi, CD62+, CCR2hi) and nonclassical monocytes (CD115+, Ly6Cb, CD62−, CCR2lo), which also have high levels of the CX3CR1 fractalkine receptor, involved in leukocyte adhesion and migration (Geissmann et al., 2003; Ziegler-Heitbrock et al., 2010). Immunological depletion of classical monocytes with an anti-CCR2 antibody that recognizes Ly6Chi monocytes (Shi and Pamer, 2011) did not impact the recruitment of neutrophils. Conversely, a transgenic rodent model lacking CX3CR1 with reduced nonclassical monocytes showed a decrease in neutrophil recruitment after CCI. Taken together, these results suggest that neutrophil recruitment may be specifically regulated by nonclassical monocytes (Makinde et al., 2017).
MMP-9 and Neutrophils
Clinical studies have reported an elevation of MMP-9 in cerebrospinal fluid and blood of patients with TBI (Grossetete et al., 2009; Guilfoyle et al., 2015), and preclinical studies demonstrate its presence in the acutely injured brain. Preclinical models have offered insights into the source and contributions of this protease to secondary pathogenesis. Neutrophils represent one potential source of MMP-9; it is stored in tertiary granules and in studies of the ischemic rodent brain, elevation of MMP-9 is attributed to infiltrating neutrophils (Gidday et al., 2005; Justicia et al., 2003). MMP-9 is likewise upregulated in the adult rodent brain after traumatic injury where it is responsible for the degradation of myelin and disruption of the blood-brain barrier (Wang et al., 2000). Such findings are in line with known substrates of MMP-9; namely, myelin basic protein and the tight junction protein zonulae occludens-1 (Hannocks et al., 2017; Liu et al., 2012). However, to the best of our knowledge, no studies have provided definitive evidence that infiltrating neutrophils are a key source of this protease in the brain after a traumatic injury or if its targeted deletion alters the recruitment of neutrophils. Only one study has studied MMP-9 in the injured developing brain; similar to the adult brain, it is upregulated acutely post injury (Semple et al., 2015a). However, its blockade using a gelatinase inhibitor does not alter early cell death nor long-term neurological function. Such findings contrast that of the injured adult brain, where blockade with a gelatinase inhibitor resulted in a smaller cortical lesion volume, reduced dendritic degeneration and microglial and astrocyte activation, and improvements in motor function (Hadass et al., 2013). Collectively, these findings support the upregulation of MMP-9 in both the injured adult and developing brain, but the extent to which neutrophils utilize this protease for transmigration into the parenchyma remains unclear. Moreover, the pathogenicity of MMP-9 may differ based upon age at time of injury. It is noteworthy that the activity of MMP-9 is more prominent in the acutely injured brain at PND 21 compared to that of the adult brain (Semple et al., 2015a; Wang et al., 2000). Such a distinction may impart greater vulnerability in the young brain to TBI as a consequence of enhanced proteolytic activity.
NeutrophilElastase
Neutrophil elastase is one of the most destructive enzymes in inflammatory mediated pathogenesis (Lee and Downey, 2001; Owen and Campbell, 1999). When neutrophils become activated, it is rapidly released from primary granules into the adjacent extracellular space (Lee and Downey, 2001). If left unchecked neutrophil elastase may not only intensify the host inflammatory response but can degrade a variety of matrix and nonmatrix proteins (i.e. plasma proteins, pro-inflammatory mediators, adhesion receptors) due to its broad specificity.
The destructive nature of neutrophil elastase is revealed in early preclinical models of spinal cord injury and cerebral ischemia. Pharmacologic blockade of this protease stabilized the barrier, resulted in an improvement in neurologic recovery in the injured spinal cord (Taoka et al., 1998; Tonai et al., 2001), reduced vasogenic edema and infarct volume (Shimakura et al., 2000), and protected hippocampal neurons (Matayoshi et al., 2009) in models of brain ischemia. The damaging consequences of neutrophil elastase are further realized in studies employing both a neutrophil elastase inhibitor and knockout models, where infarct volume is reduced in neutrophil elastase deficient animals after transient brain ischemia (Stowe et al., 2009). Such cross validation of genetic and pharmacologic approaches confirms the causal involvement of neutrophils in neurovascular pathology.
Most recently, neutrophil elastase has been studied in a model of CCI to the developing brain at PND 21 using both pharmacologic and genetic strategies (Semple et al., 2015b). A number of key findings have supported its potent pathogenicity and long-term impact on behavioral recovery. Neutrophil elastase activity is elevated in the cortex within the first 24 hours post injury, a finding that is consistent with the peak of infiltration of neutrophils into the injured brain (Claus et al., 2010). Brain-injured neutrophil elastase knockout animals show a reduction in vasogenic edema in the cortex and significant neuroprotection to the hippocampus as evidenced by reduced numbers of TUNEL and caspase+ cells. Upregulation of heme oxygenase, an indicator of oxidative stress, is likewise reduced in these animals. Subsequent behavioral assessments at adulthood reveals a reduction in injury-induced hyperactivity and improvement in spatial learning in the knockout compared to wildtype controls. Similar findings of early neuroprotection are reported in brain-injured wildtype rodents, treated with a neutrophil elastase inhibitor within the first 12 hours post injury (Semple et al., 2015b). These collective findings highlight the damaging consequences of neutrophil elastase. As neutrophils are recruited to the injured developing rodent brain over several weeks post injury (Claus et al., 2010) there is risk that prolonged exposure to this protease furthers secondary damage and thus contributes to the long-term neurological deficits.
Closing Remarks
The developing rodent brain shows greater vulnerability to damage from a traumatic injury compared to that of the injured adult brain (See review; Potts et al., 2006). This is, in part, attributed to a “window of susceptibility”, characterized by a more robust response to chemokines and a prolonged recruitment of neutrophils, compared to that of the injured adult brain. Activated neutrophils, recruited to the injured brain, have the capacity to support secondary pathogenesis through the generation of reactive oxygen species (superoxide anion radicals, hydrogen peroxide, hypochlorous acid) (Snelgrove et al., 2018) and release of granular content, including neutrophil elastase and MMP-9, that collectively mediate cell death, disruption of the blood-brain barrier and degradation of the extracellular matrix. With a high content of fatty acids that is further compounded by low antioxidant reserves, the developing brain is poorly positioned to respond to these toxic products produced by activated neutrophils (See review; Bayir et al., 2006).
We are only beginning to understand the role of neutrophils in the injured adult brain. Studies involving both systemic depletion of neutrophils and genetic deficiency in CXCR2 argue for their involvement in secondary pathogenesis (Dyer et al., 2017; Semple et al., 2010; Uhl et al., 1994). The caveats to this interpretation include the lack of specificity in methods of depletion which could result in changes in other leukocyte populations (Daley et al., 2008) and the presence of CXCR2 on other myeloid lineage cell types including monocytes, macrophages, and mast cells (Olson and Ley, 2002).
Beyond the traditional viewpoint which considered neutrophils as terminally differentiated leukocytes that served antimicrobial functions, there is a growing body of evidence that speak to their ability to perform specialized functions. This has led to a much broader perspective on neutrophils, as transcriptionally active leukocytes that assume distinct phenotypes; there is evidence for tissue/organ specificity during homeostasis as well as subpopulations that reflect disease states including inflammation and metabolic dysregulation (Rosales, 2018). While much of this work has been done outside of the CNS, early studies in stroke models have identified subpopulations of neutrophils that are defined as either pro- or anti-inflammatory (Cuartero et al., 2013) and may be, in part, related to a “threshold” number of neutrophils with higher numbers more closely associated with an anti-inflammatory phenotype (Easton, 2013).
To the best of our knowledge, no studies have yet examined neutrophil subpopulations in the brain after a TBI. As neutrophils show protracted recruitment to the injured developing brain (Claus et al., 2010), it is possible that they will exhibit phenotypic changes, reflecting diverse environments that span acute injury to wound healing. Based upon studies of the neutrophil elastase knockout, it is clear that neutrophils are damaging to the acutely injured brain (Semple et al., 2015b). However, no studies to date have evaluated the properties of these leukocytes during wound healing after a TBI. Neutrophils have the potential to support wound healing in the injured brain. This is exemplified in studies using a model of ischemia to skeletal muscle (Massena et al., 2015). Neutrophils, recruited to ischemic muscle, displayed a unique phenotype; namely, CD49dhi CXCR4hi VEGFR1, where VEGFR1 is the receptor for vascular endothelial growth factor (VEGF)-A. This study revealed that neutrophil recruitment to VEGF-A was dependent on activation of both VEGFR1 on neutrophils and VEGFR2 on endothelial cells and that this subpopulation of neutrophils was essential for angiogenesis (Massena et al., 2015). Such findings provide an additional perspective on neutrophils beyond their involvement in the inflammatory cascade, where they could be critical players in facilitating angiogenesis in the injured brain.
While most attention has been directed at understanding pathogenicity of activated neutrophils, there is a growing recognition of their potential benefits in the context of both early neuroprotection and tissue repair (See review; Liu et al., 2018; Snelgrove et al., 2018). From the perspective of neuroprotection, this is exemplified in recent studies of the meningeal compartment after a mild CHI to the adult brain where early purinergic-dependent recruitment of neutrophils results in reduced death of meningeal cells (Roth et al., 2014).
Such findings are in sharp contrast to a moderate CCI to the developing cortex, where neutrophil elastase, produced by infiltrating neutrophils, is a determinant of early neuronal death (Semple et al., 2015b). Neutrophils may behave differently in each of these scenarios, based on their known plasticity, where there is opportunity to change phenotypes and functions (Rosales, 2018); neutrophils may assume a more damaging phenotype with increasing injury severity, and/or when exposed to different environments (parenchyma of the brain versus meninges) and/or age at time of injury (young versus adult brain). These major unanswered questions may guide future investigations into heterogeneity of neutrophils in the context of injury (see Figure 3 for summary). Additionally, while data do support sex as a key biological variable in the young injured brain (Fraunberger et al., 2019; Spani et al., 2018), it has to be considered in the context of neutrophil function and warrants further investigation.
Figure 3.

Key emerging questions regarding neutrophil function.
There is now substantial evidence to support pleotrophic functions (Snelgrove et al., 2018); beyond their established ability to phagocytose and clear apopotic cells and debris, these leukocytes are immune-regulatory, spanning pro-inflammatory functions, facilitating resolution of inflammation via pro-resolving lipid mediators and production of anti-inflammatory cytokines, and supporting wound healing events including neovascularization. As noted by others (Snelgrove et al., 2018), it is time to shift the perspective of neutrophils as “indiscriminate killers” to a broader viewpoint whereby their function is defined by context, timing, and location. For the injured developing brain, neutrophil recruitment spans acute injury to wound healing. Thus, these leukocytes may change their functionality depending upon the local environment that is responding to injury while simultaneously evolving in accordance with ongoing maturational cues.
Abbreviations
- TBI
Traumatic brain injury
- PND
postnatal day
- CCI
controlled cortical impact
- CNS
central nervous system
- G-CSF
granulocyte-colony stimulation factor
- MMP
matrix metalloproteinase
- IL
interleukin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbassi O, Kishimoto TK, McIntire LV, Smith CW, 1993. Neutrophil adhesion to endothelial cells. Blood Cells 19, 245–260. [PubMed] [Google Scholar]
- Adelson PD, Fellows-Mayle W, Kochanek PM, Dixon CE, 2013. Morris water maze function and histologic characterization of two age-at-injury experimental models of controlled cortical impact in the immature rat. Childs Nerv Syst 29, 43–53. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL, 2003. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278, 1910–1914. [DOI] [PubMed] [Google Scholar]
- Ajao DO, Pop V, Kamper JE, Adami A, Rudobeck E, Huang L, Vlkolinsky R, Hartman RE, Ashwal S, Obenaus A, Badaut J, 2012. Traumatic brain injury in young rats leads to progressive behavioral deficits coincident with altered tissue properties in adulthood. J Neurotrauma 29, 2060–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson V, Catroppa C, Morse S, Haritou F, Rosenfeld J, 2000. Recovery of intellectual ability following traumatic brain injury in childhood: impact of injury severity and age at injury. Pediatr Neurosurg 32, 282–290. [DOI] [PubMed] [Google Scholar]
- Anderson V, Catroppa C, Morse S, Haritou F, Rosenfeld J, 2005. Functional plasticity or vulnerability after early brain injury? Pediatrics 116, 1374–1382. [DOI] [PubMed] [Google Scholar]
- Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K, 1998. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol 8, 923–926. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Bolton SJ, Fearn S, Perry VH, 1997. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain 120(3), 435–444. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Couch Y, Losey P, Evans MC, 2012. The systemic response to brain injury and disease. Brain Behav Immun 26, 534–540. [DOI] [PubMed] [Google Scholar]
- Bainton DF, Ullyot JL, Farquhar MG, 1971. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med 134, 907–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Dohi K, Hansen K, Thompson HJ, 2016. Assessing blood granulocyte colony-stimulating factor as a potential biomarker of acute traumatic brain injury in mice and humans. Brain Behav Immun 52, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM, 2002. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res 51, 571–578. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kochanek PM, Kagan VE, 2006. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci 28, 420–431. [DOI] [PubMed] [Google Scholar]
- Biagas KV, Uhl MW, Schiding JK, Nemoto EM, Kochanek PM, 1992. Assessment of posttraumatic polymorphonuclear leukocyte accumulation in rat brain using tissue myeloperoxidase assay and vinblastine treatment. J Neurotrauma 9, 363–371. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Anthony DC, Oakley F, Carlsen H, Elsharkawy AM, Blomhoff R, Mann DA, 2008. Hepatic nuclear factor kappa B regulates neutrophil recruitment to the injured brain. J Neuropathol Exp Neurol 67, 223–230. [DOI] [PubMed] [Google Scholar]
- Cartwright GE, Athens JW, Wintrobe MM, 1964. The kinetics of granulopoiesis in normal man. Blood 24, 780–803. [PubMed] [Google Scholar]
- Catroppa C, Anderson VA, Morse SA, Haritou F, Rosenfeld JV, 2008. Outcome and predictors of functional recovery 5 years following pediatric traumatic brain injury (TBI). J Pediatr Psychol 33, 707–718. [DOI] [PubMed] [Google Scholar]
- Clark RS, Schiding JK, Kaczorowski SL, Marion DW, Kochanek PM, 1994. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma 11, 499–506. [DOI] [PubMed] [Google Scholar]
- Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Manvelyan H, Whetstone W, Noble-Haeusslein LJ, 2010. Age is a determinant of leukocyte infiltration and loss of cortical volume after traumatic brain injury. Dev Neurosci 32, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A, 1992. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80, 2012–2020. [PubMed] [Google Scholar]
- Corrigan JD, Bogner J, Mellick D, Bushnik T, Dams-O’Connor K, Hammond FM, Hart T, Kolakowsky-Hayner S, 2013. Prior history of traumatic brain injury among persons in the Traumatic Brain Injury Model Systems National Database. Arch Phys Med Rehabil 94, 1940–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowland JB, Borregaard N, 2016. Granulopoiesis and granules of human neutrophils. Immunol Rev 273, 11–28. [DOI] [PubMed] [Google Scholar]
- Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, Corbi AL, Lizasoain I, Moro MA, 2013. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 44, 3498–3508. [DOI] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE, 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 83, 64–70. [DOI] [PubMed] [Google Scholar]
- de Oliveira S, Rosowski EE, Huttenlocher A, 2016. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 16, 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniset JF, Kubes P, 2016. Recent advances in understanding neutrophils. F1000Res 5, 2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, Megra B, Williams DW, Witwer KW, Gacias M, Tabatadze N, Cole RN, Casaccia P, Berman JW, Anthony DC, Haughey NJ, 2017. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer DP, Pallas K, Medina-Ruiz L, Schuette F, Wilson GJ, Graham GJ, 2017. CXCR2 deficient mice display macrophage-dependent exaggerated acute inflammatory responses. Sci Rep 7, 42681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eash KJ, Means JM, White DW, Link DC, 2009. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood 113, 4711–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AS, 2013. Neutrophils and stroke - can neutrophils mitigate disease in the central nervous system? Int Immunopharmacol 17, 1218–1225. [DOI] [PubMed] [Google Scholar]
- Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L, 2001. Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J Exp Med 194, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, Yamauchi T, Noble LJ, Ferriero DM, 2003. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma 20, 437–445. [DOI] [PubMed] [Google Scholar]
- Faraday N, Schunke K, Saleem S, Fu J, Wang B, Zhang J, Morrell C, Dore S, 2013. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS One 8, e71447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M, Coronado V, 2015. Epidemiology of traumatic brain injury. Handb Clin Neurol 127, 3–13. [DOI] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG, 2010. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. [Google Scholar]
- Fraunberger EA, Shutt TE, Esser MJ, 2019. Sex-dependent and chronic alterations in behavior and mitochondrial function in a rat model of pediatric mild traumatic brain injury. Brain Inj 33, 534–542. [DOI] [PubMed] [Google Scholar]
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM, 2009. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furze RC, Rankin SM, 2008. Neutrophil mobilization and clearance in the bone marrow. Immunol 125, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahm C, Holmin S, Mathiesen T, 2002. Nitric oxide synthase expression after human brain contusion. Neurosurgery 50, 1319–1326. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR, 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS, 2005. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 289, H558–568. [DOI] [PubMed] [Google Scholar]
- Gorina R, Lyck R, Vestweber D, Engelhardt B, 2014. beta2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood-brain barrier. J Immunol 192, 324–337. [DOI] [PubMed] [Google Scholar]
- Grossetete M, Phelps J, Arko L, Yonas H, Rosenberg GA, 2009. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery 65, 702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilfoyle MR, Carpenter KL, Helmy A, Pickard JD, Menon DK, Hutchinson PJ, 2015. Matrix Metalloproteinase Expression in Contusional Traumatic Brain Injury: A Paired Microdialysis Study. J Neurotrauma 32, 1553–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadass O, Tomlinson BN, Gooyit M, Chen S, Purdy JJ, Walker JM, Zhang C, Giritharan AB, Purnell W, Robinson CR 2nd, Shin D, Schroeder VA, Suckow MA, Simonyi A, Sun GY, Mobashery S, Cui J, Chang M, Gu Z, 2013. Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS One 8, e76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager M, Cowland JB, Borregaard N, 2010. Neutrophil granules in health and disease. J Intern Med 268, 25–34. [DOI] [PubMed] [Google Scholar]
- Hannah S, Mecklenburgh K, Rahman I, Bellingan GJ, Greening A, Haslett C, Chilvers ER, 1995. Hypoxia prolongs neutrophil survival in vitro. FEBS Lett 372, 233–237. [DOI] [PubMed] [Google Scholar]
- Hannocks MJ, Zhang X, Gerwien H, Chashchina A, Burmeister M, Korpos E, Song J, Sorokin L, 2017. The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes. Matrix Biol. [DOI] [PubMed] [Google Scholar]
- Hartl R, Medary MB, Ruge M, Arfors KE, Ghajar J, 1997. Early white blood cell dynamics after traumatic brain injury: effects on the cerebral microcirculation. J Cereb Blood Flow Metab 17, 1210–1220. [DOI] [PubMed] [Google Scholar]
- Hausmann R, Kaiser A, Lang C, Bohnert M, Betz P, 1999. A quantitative immunohistochemical study on the time-dependent course of acute inflammatory cellular response to human brain injury. Int J Legal Med 112, 227–232. [DOI] [PubMed] [Google Scholar]
- Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, Wojtkiewicz GR, Masson GS, Vinegoni C, Kim J, Kim D-E, Weissleder R, Swirski FK, Moskowitz MA, Nahrendorf M, 2018. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci 21, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton AM, 2010. The paradox of tumor-associated neutrophils: fueling tumor growth with cytotoxic substances. Cell Cycle 9, 1732–1737. [DOI] [PubMed] [Google Scholar]
- Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J, Nakamura MC, 2014. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma 31, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun YM, Hong CW, 2017. Deep insight into neutrophil trafficking in various organs. J Leukoc Biol 102, 617–629. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Potts MB, Noble-Haeusslein LJ, 2007. Injury severity determines Purkinje cell loss and microglial activation in the cerebellum after cortical contusion injury. Exp Neurol 203, 258–268. [DOI] [PubMed] [Google Scholar]
- Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J, 2017. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 95, 1246–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR, 2015. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 35, 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM, 2003. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab 23, 1430–1440. [DOI] [PubMed] [Google Scholar]
- Kaczorowski SL, Schiding JK, Toth CA, Kochanek PM, 1995. Effect of soluble complement receptor-1 on neutrophil accumulation after traumatic brain injury in rats. J Cereb Blood Flow Metab 15, 860–864. [DOI] [PubMed] [Google Scholar]
- Kamper JE, Pop V, Fukuda AM, Ajao DO, Hartman RE, Badaut J, 2013. Juvenile traumatic brain injury evolves into a chronic brain disorder: behavioral and histological changes over 6months. Exp Neurol 250, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansas GS, 1996. Selectins and their ligands: current concepts and controversies. Blood 88, 3259–3287. [PubMed] [Google Scholar]
- Kawabata K, Hagio T, Matsuoka S, 2002. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol 451, 1–10. [DOI] [PubMed] [Google Scholar]
- Keeling K, Hicks RR, Mahesh J, Billings B, Kotwal G, 2000. Local neutrophil influx following lateral fluid-percussion brain injury in rats is associated with the accumulation of the third complement (C3) of the complement system. J Neuroimmunol 105, 20–30. [DOI] [PubMed] [Google Scholar]
- Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F, 2012. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, De La Luz Sierra M, Williams CK, Gulino AV, Tosato G, 2006. G-CSF down-regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood 108, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen L, Bainton DF, Sengelov H, Borregaard N, 1993. Structural and functional heterogeneity among peroxidase-negative granules in human neutrophils: identification of a distinct gelatinase-containing granule subset by combined immunocytochemistry and subcellula fractionation. Blood 82, 3183–3191. [PubMed] [Google Scholar]
- Kobayashi Y, 2008. The role of chemokines in neutrophil biology. Front Biosci 13, 2400–2407 [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Wallisch JS, Bayir H, Clark RSB, 2017. Pre-clinical models in pediatric traumatic brain injury-challenges and lessons learned. Childs Nerv Syst 33, 1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi H, 2004. The concept of ‘developing the brain’: a new natural science for learning and education. Brain Dev 26, 434–441. [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P, 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13, 159–175. [DOI] [PubMed] [Google Scholar]
- Kourtzelis I, Mitroulis I, von Renesse J, Hajishengallis G, Chavakis T, 2017. From leukocyte recruitment to resolution of inflammation: the cardinal role of integrins. J Leukoc Bio 102, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P, 2006. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol 2, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Downey G, 2001. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med 164, 896–904. [DOI] [PubMed] [Google Scholar]
- Li T, Zhang YM, Han D, Hua R, Guo BN, Hu SQ, Yan XL, Xu T, 2017. Involvement of IL-17 in Secondary Brain Injury After a Traumatic Brain Injury in Rats. Neuromolec Med 19, 541–554. [DOI] [PubMed] [Google Scholar]
- Liu J, Jin X, Liu KJ, Liu W, 2012. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci 32, 3044–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Li S, Dai SS, 2018. Neutrophils in traumatic brain injury (TBI): friend or foe? J Neuroinflammation 15, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinde HM, Cuda CM, Just TB, Perlman HR, Schwulst SJ, 2017. Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration afte Traumatic Brain Injury. J Immunol 199, 3583–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM, 2003. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19, 583–593. [DOI] [PubMed] [Google Scholar]
- Massena S, Christoffersson G, Vagesjo E, Seignez C, Gustafsson K, Binet F, Herrera Hidalgo C, Giraud A, Lomei J, Westrom S, Shibuya M, Claesson-Welsh L, Gerwins P, Welsh M, Kreuger J, Phillipson M, 2015. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 126, 2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi H, Hirata T, Yamashita S, Ishida K, Mizukami Y, Gondo T, Matsumoto M, Sakabe T, 2009. Neutrophil elastase inhibitor attenuates hippocampal neuronal damage after transient forebrain ischemia in rats. Brain Res 1259, 98–106. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM, 2018. The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol. [DOI] [PubMed] [Google Scholar]
- Morganti JM, Riparip LK, Rosi S, 2016. Call Off the Dog(ma): M1/M2 Polarization Is Concurrent following Traumatic Brain Injury. PLoS One 11, e0148001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortaz E, Alipoor SD, Adcock IM, Mumby S, Koenderman L, 2018. Update on Neutrophil Function in Severe Inflammation. Front Immunol 9, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, 2015. The regulation of transendothelial migration: new knowledge and new questions. Cardiovasc Res 107, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson TS, Ley K, 2002. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol 283, R7–28. [DOI] [PubMed] [Google Scholar]
- Owen C, Campbell E, 1999. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol 1999 February;65(2):137–50. 65, 137–150. [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V, 2018. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18, 134–147. [DOI] [PubMed] [Google Scholar]
- Pop V, Sorensen DW, Kamper JE, Ajao DO, Murphy MP, Head E, Hartman RE, Badaut J, 2013. Early brain injury alters the blood-brain barrier phenotype in parallel with beta-amyloid and cognitive changes in adulthood. J Cereb Blood Flow Metab 33, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ, 2006. Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx 3, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullela R, Raber J, Pfankuch T, Ferriero DM, Claus CP, Koh SE, Yamauchi T, Rola R, Fike JR, Noble-Haeusslein LJ, 2006. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev Neurosci 28, 396–409. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, 2016. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19, 987–991. [DOI] [PubMed] [Google Scholar]
- Rhind SG, Crnko NT, Baker AJ, Morrison LJ, Shek PN, Scarpelini S, Rizoli SB, 2010. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J Neuroinflammation 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C, 2018. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front Physiol 9, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB, 2014. Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, Latour LL, McGavern DB, 2018. Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat Immunol 19, 442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa-Pereira I, Roodselaar J, Couch Y, Consentino Kronka Sosthenes M, Evans MC, Anthony DC, Stolp HB, 2018. Hepatic acute phase response protects the brain from focal inflammation during postnatal window of susceptibility. Brain Behav Immun 69, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EP, Lee WL, Zemans RL, Yamashita C, Downey GP, 2011. On, around, and through: neutrophil-endothelial interactions in innate immunity. Physiology 26, 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoettle RJ, Kochanek PM, Magargee MJ, Uhl MW, Nemoto EM, 1990. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cereb,ral edema in rats. J Neurotrauma 7, 207–217. [DOI] [PubMed] [Google Scholar]
- Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, Bagby G, Nelson S, Kolls JK, 2000. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol 164, 4783–4789. [DOI] [PubMed] [Google Scholar]
- Sekiya S, Gotoh S, Yamashita T, Watanabe T, Saitoh S, Sendo F, 1989. Selective depletion of rat neutrophils by in vivo administration of a monoclonal antibody. J Leukoc Biol 46, 96–102. [DOI] [PubMed] [Google Scholar]
- Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, Levesque JP, Chappel J, Ross FP, Link DC, 2005. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood 106, 3020–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC, 2010. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis 40, 394–403. [DOI] [PubMed] [Google Scholar]
- Semple BD, Carlson J, Noble-Haeusslein LJ, 2016. Pediatric Rodent Models of Traumatic Brain Injury. Methods Mol Biol 1462, 325–343. [DOI] [PubMed] [Google Scholar]
- Semple BD, Noble-Haeusslein LJ, Gooyit M, Tercovich KG, Peng Z, Nguyen TT, Schroeder VA, Suckow MA, Chang M, Raber J, Trivedi A, 2015a. Early Gelatinase Activity Is Not a Determinant of Long-Term Recovery after Traumatic Brain Injury in the Immature Mouse. PLoS One 10, e0143386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Noble-Haeusslein LJ, Jun Kwon Y, Sam PN, Gibson AM, Grissom S, Brown S, Adahman Z, Hollingsworth CA, Kwakye A, Gimlin K, Wilde EA, Hanten G, Levin HS, Schenk AK, 2014. Sociosexual and communication deficits after traumatic injury to the developing murine brain. PLoS One 9, e103386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Trivedi A, Gimlin K, Noble-Haeusslein LJ, 2015b. Neutrophil elastase mediates acute pathogenesis and is a determinant of long-term behavioral recovery after traumatic injury to the immature brain. Neurobiol Dis 74, 263–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Pamer EG, 2011. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakura A, Kamanaka Y, Ikeda Y, Kondo K, Suzuki Y, Umemura K, 2000. Neutrophil elastase inhibition reduces cerebral ischemic damage in the middle cerebral artery occlusion. Brain Res 858, 55–60. [DOI] [PubMed] [Google Scholar]
- Silvestre-Roig C, Hidalgo A, Soehnlein O, 2016. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127, 2173–2181. [DOI] [PubMed] [Google Scholar]
- Snelgrove RJ, Patel DF, Patel T, Lloyd CM, 2018. The enigmatic role of the neutrophil in asthma: Friend, foe or indifferent? Clin Exp Allergy 48, 1275–1285. [DOI] [PubMed] [Google Scholar]
- Soares HD, Hicks RR, Smith D, McIntosh TK, 1995. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci 15, 8223–8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spani CB, Braun DJ, Van Eldik LJ, 2018. Sex-related responses after traumatic brain injury: Considerations for preclinical modeling. Front Neuroendocrinol 50, 52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K, 2005. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22, 285–294. [DOI] [PubMed] [Google Scholar]
- Stowe AM, Adair-Kirk TL, Gonzales ER, Perez RS, Shah AR, Park TS, Gidday JM, 2009. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis 35, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER, 2010. Neutrophil kinetics in health and disease. Trends Immunol 31, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L, 2013. What’s your age again? Determination of human neutrophil half-lives revisited. J Leukoc Biol 94, 595–601. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K, Murakami K, Johno M, Naruo M, 1998. Role of neutrophil elastase in compression-induced spinal cord injury in rats. Brain Research 799, 264–269. [DOI] [PubMed] [Google Scholar]
- Taylor CA, Bell JM, Breiding MJ, Xu L, 2017. Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths - United States, 2007 and 2013. MMWR Surveill Summ 66, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonai T, Shiba K, Taketani Y, Ohmoto Y, Murata K, Muraguchi M, Ohsaki H, Takeda E, Nishisho T, 2001. A neutrophil elastase inhibitor (ONO-5046) reduces neurologic damage after spinal cord injury in rats. J Neurochem 78, 1064–1072. [DOI] [PubMed] [Google Scholar]
- Tong W, Igarashi T, Ferriero D, Noble L, 2002. Traumatic brain injury in the immature mouse brain: characterization of regional vulnerability. Exp Neurol 176, 105–116. [DOI] [PubMed] [Google Scholar]
- Tracchi I, Ghigliotti G, Mura M, Garibaldi S, Spallarossa P, Barisione C, Boasi V, Brunelli M, Corsiglia L, Barsotti A, Brunelli C, 2009. Increased neutrophil lifespan in patients with congestive heart failure. Eur J Heart Fail 11, 378–385. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F, 2004. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 21, 215–226. [DOI] [PubMed] [Google Scholar]
- Tsuru-Aoyagi K, Potts MB, Trivedi A, Pfankuch T, Raber J, Wendland M, Claus CP, Koh S-E, Ferriero D, Noble-Haeusslein LJ, 2009. Glutathione peroxidase activity modulates recovery in the injured immature brain. Ann Neurol 65, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR, 2016. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci 10, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl MW, Biagas KV, Grundl PD, Barmada MA, Schiding JK, Nemoto EM, Kochanek PM, 1994. Effects of neutropenia on edema, histology, and cerebral blood flow after traumatic brain injury in rats. J Neurotrauma 11, 303–315. [DOI] [PubMed] [Google Scholar]
- von Wedel-Parlow M, Schrot S, Lemmen J, Treeratanapiboon L, Wegener J, Galla HJ, 2011. Neutrophils cross the BBB primarily on transcellular pathways: an in vitro study. Brain Res 1367, 62–76. [DOI] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH, 2000. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci 20, 7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Kochanek PM, Clark RS, Heineman S, Schiding JK, Franicola D, Memarzadeh F, Lo W, Marion DW, Dekosky ST, 1999. Neutrophils do not mediate blood-brain barrier permeability early after controlled cortical impact in rats. J Neurotrauma 16, 583–594. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Wisniewski SR, Clark RS, Mellick JA, Marion DW, Kochanek PM, 2000. Effect of neutropenia and granulocyte colony stimulating factor-induced neutrophilia on blood-brain barrier permeability and brain edema after traumatic brain injury in rats. Crit Care Med 28, 3710–3717. [DOI] [PubMed] [Google Scholar]
- Wittchen ES, 2009. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci (Landmark Ed) 14, 2522–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wang Z, Li J, Wu H, Peng Y, Fan L, Chen J, Gu C, Yan F, Wang L, Chen G, 2017. The Polarization States of Microglia in TBI: A New Paradigm for Pharmacological Intervention. Neur Plast 2017, 5405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW, 2005. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood 106, 584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN, 2010. Leukocyte recruitment and ischemic brain injury. Neuromolec Med 12, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K, Parkos CA, 2003. Leukocyte-epithelial interactions. Curr Opin Cell Biol 15, 557–564. [DOI] [PubMed] [Google Scholar]
- Zhao S, Yu Z, Liu Y, Bai Y, Jiang Y, van Leyen K, Yang YG, Lok JM, Whalen MJ, Lo EH, Wang X, 2017. CD47 deficiency improves neurological outcomes of traumatic brain injury in mice. Neurosci Lett 643, 125–130. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB, 2010. Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–80. [DOI] [PubMed] [Google Scholar]