

Abstract
A remaining challenge within microbial ecology is to understand the determinants of richness and diversity observed in environmental microbial communities. In a range of systems, including activated sludge bioreactors, the microbial residence time (MRT) has been previously shown to shape the microbial community composition. However, the physiological and ecological mechanisms driving this influence have remained unclear. Here, this relationship is explored by analyzing an activated sludge system fed with municipal wastewater. Using a model designed in this study based on Monod-growth kinetics, longer MRTs were shown to increase the range of growth parameters that enable persistence, resulting in increased richness and diversity in the modeled community. In laboratory experiments, six sequencing batch reactors treating domestic wastewater were operated in parallel at MRTs between 1 and 15 days. The communities were characterized using both 16S ribosomal RNA and non-target messenger RNA sequencing (metatranscriptomic analysis), and model-predicted monotonic increases in richness were confirmed in both profiles. Accordingly, taxonomic Shannon diversity also increased with MRT. In contrast, the diversity in enzyme class annotations resulting from the metatranscriptomic analysis displayed a non-monotonic trend over the MRT gradient. Disproportionately high abundances of transcripts encoding for rarer enzymes occur at longer MRTs and lead to the disconnect between taxonomic and functional diversity profiles.
Subject terms: Microbial ecology, Next-generation sequencing
Introduction
Environmental microbial communities often house a rich and diverse set of species and expressed enzymes [1, 2]. A remaining challenge within microbial ecology is to understand the mechanisms driving the differences in metabolic and taxonomic diversity between communities [3]. Of the influencing mechanisms, the microbial residence time (MRT; the average amount of time a microorganism resides in a system) has been postulated to be a key parameter influencing microbial diversity [4, 5]. Recent investigations in engineered systems showed that, as MRT increases, the diversity and richness of the community increases as well. However, specific studies exploring the relationship between MRT and community composition have shown opposing or more confounded trends [6, 7] (albeit with differing experimental setups and analysis methods), suggesting that the relationship between MRT of a system and community composition is complex. The influence of MRT is also relevant in natural [8] and host-associated systems [9], suggesting that more clearly identifying the influence of this parameter on community composition in engineered environments may provide insights that are also relevant to other systems.
In addition to the influence on taxonomic diversity and composition, communities also express more functions at longer MRT [4]. In wastewater treatment, functions related to substrate transformation have been demonstrated to emerge at longer MRT, e.g., nitrification and the biotransformation of trace organics [10, 11]. In a survey of 10 wastewater-treatment plants, functional richness was positively associated with taxonomic richness, and both parameters were in turn positively associated with plant performance in terms of trace organic contaminant removal [12]. By contrast, additional studies have noted that expressed functional richness and diversity may not be directly related to taxonomic parameters [13–15]. In streams [16], forests [17, 18], and host-associated communities [19], the monitored functional signals were independent of the parameters controlling the taxonomic profiles. Both the taxonomic and functional profiles must be monitored to understand further the linkage between community structure and function and to characterize more accurately the influence of an external variable (such as MRT) on the community [20].
In this study, the influence of the MRT on the observed taxonomic composition and functional profile of microbial communities cultivated in six parallel lab-scale sequencing batch reactors (SBR) treating domestic wastewater was explored experimentally and described using a Monod-model. Wastewater bioreactors provide a controllable experimental system [21] with available established computational models [22–24] that have provided previous insights into microbial ecological concepts including novel niches and community assembly [13]. Experimentally, the microbial communities were monitored using 16S-ribosomal RNA (rRNA) and messenger RNA (mRNA) metatranscriptomic non-target sequencing. Recently, 16S-rRNA sequencing has become an established method for analyzing bacterial communities [25, 26] in biotechnological applications, with detailed sample preparation and data-processing pipelines available [27]. To complement the taxonomic survey, mRNA sequencing (RNAseq) was performed to determine the functional profiles of the communities, for which Enzyme Commission (EC) numbers were used as a proxy for an expressed function. Numerically, Monod growth kinetics were employed in a simplistic MRT–diversity model to provide a concrete mechanistic basis for the connection between the MRT and the community composition within the SBR. This model uniquely investigates the underlying available range of growth parameters that result in persistence within the community. Organisms must survive substrate-rich and substrate-poor conditions, suggesting the importance of selecting growth parameters that in combination describe the ability of the organism to capture resources and to withstand starvation. Therefore, individual distinctive combinations of the maximum growth rate (μmax) and endogenous decay rate (be) are modeled and considered to be bounded in an ecological range of permissible values. Critically, allowing be, a parameter that has been previously shown to be species-specific [28], to vary between community members allows for the coexistence of multiple μmax values within a given community. This novel approach permits observing how the MRT, the independent factor in the model, influences the available set of μmax and be values. This simplified view of community composition leads to a better conceptual understanding of the influence of the MRT on the richness and diversity of microbial communities in the studied system.
Materials and methods
Activated sludge reactor configuration
Briefly, six automated sequencing batch reactors (6 × 12 L) treating local municipal wastewater after primary clarification were operated in parallel at MRTs of 1, 3, 5, 7, 10, or 15 days (d) as detailed previously [29] and summarized in Supplemental Table 1. Forty-eight days (time-point 1; TP1) and 187 days (time-point 2, TP2) after start-up, activated sludge samples were collected for DNA (at the start of the previously described biotransformation experiment [29]) and RNA (5 h after the start of the experiment) extraction.
Sample collection
To collect samples, culture (two 20-mL samples for TP1 and a 20 and 40-mL sample for TP2 for DNA and RNA analysis, respectively) was withdrawn and centrifuged at 3345 × g for 10 min at 4 °C. The supernatant was then discarded, and the pellets were stored at −80 °C until further processing.
DNA/RNA isolation
The total RNA and genomic DNA (gDNA) isolation protocol consisted of a phenol:chloroform:isoamylalcohol extraction followed by either a DNA PowerCleanup PRO Kit (Qiagen, Venlo, Netherlands) or MoBIO RNA Pro Clean-Up Kit (MoBio, Carlsbad, CA, USA) and purification with a TURBO DNase step (ThermoFisher Scientific, Waltham, MA, USA) as detailed in the Supplemental Materials and modified from Johnson et al. 2015. The RNA pellet was re-suspended with diethyl pyrocarbonate (DEPC)-treated RNase-free water to a total volume of 50 μL. DNA samples were quantified on a Qubit (Invitrogen, Waltham, MA, USA) analyzer following the manufacturer’s instructions, whereas RNA samples were quantified on a Nanodrop (Invitrogen) and quality-checked on a Bioanalyzer 2000 (RNA 6000 kit; Agilent Technologies, Santa Clara, CA, USA).
16S library preparation and sequencing
In preparation for the 16S sequencing, the total RNA was reverse transcribed into complementary DNA (cDNA) using the Superscript III Kit (Invitrogen) with random hexamer primers following the manufacturer’s instructions. The gDNA was used directly after the purification described above.
The 16S-rRNA or 16S-rDNA amplicon library preparation followed a standard procedure for the Illumina MiSeq platform (Illumina, San Diego, CA) that is detailed in Supplemental Materials 1. Two sets of 16S-rRNA primers (Integrated DNA Technologies, Inc., Skokie, IL, USA) were used in this analysis to amplify the sample cDNA and gDNA to account for the potential for the bias of a single primer-set [30]. The details of primers B1 and B2 are provided in Supplemental Table 2. The samples were sequenced using the PE 300 method on a MiSeq platform (Illumina) at the Genomics Diversity Centre at ETH Zurich, Switzerland. The raw data is publically available at EMBL-EBI under the study number PRJEB22087. The read count per sample and associated rarefaction curves are presented in Supplemental Figs. 1 and 2, respectively.
16S rRNA and rDNA sequencing data processing and analysis
The raw data was checked for quality using FastQC [31] v0.11.2. The reported nucleic sequence of the reads was then trimmed using PRINSEQ-lite [32] v0.20.4 to a length of 295 bp and merged using USEARCH [33] v8.1.1756 (with a minimum overlap of 15 bp, minimum merge-length of 100, and a maximum error of 5 bp). The primers were trimmed from the merged read using cutadapt [34] v1.5 with wildcards allowed, a full-length overlap, and an error rate of 0.01. The reads were then filtered using PRINSEQ-lite with an amplicon range of 431–506 and 252–254 for B1 and B2, respectively, a minimum quality mean of 15, and no ambiguous nucleotides allowed. USEARCH was employed to denoise the reads into exact sequence variants (ESVs; zero-level operational taxonomical units, ZOTUs using UNOISE3) and assign taxonomic origin (using usearch_global, 70% identity against the SILVA 16S database (release 128), followed by SINTAX with a 70% identity cutoff). The total number of raw and cleaned reads per sample for the B2 primer ranged from 59,311 to 191,897 with a median of 113,347 and 54,864–185,331 with a median of 104,537, respectively (the details for every sample are provided for primers B1 and B2 in Supplemental Tables 3 and 4, respectively). In total, 99.3% of these reads were binned into 10,644 ESVs, with 2918 ESVs displaying more than 10 reads in at least one sample. Primer B2 is considered in the main text because more positive and negative controls were analyzed for B2 than B1.
The resulting data was then analyzed in R v3.5.1 using phyloseq [35] v1.24.2 as detailed in Supplemental File 1. The bacterial 16S richness (°D; on rarefied data to remove potential sampling effort effects) and Shannon diversity index (ln(1D); on non-rarefied data) were calculated as n and , respectively, where n is the number of ESVs and pi is the abundance-weighted proportion of ESVi [36–38]. When relating metrics throughout this study, Spearman rank-correlation (denoted as r) analyses were employed to avoid imposing assumptions of linearity.
RNAseq library preparation and sequencing
The RNA samples were processed into libraries and sequenced following the Illumina TruSeq Single-End-Read 150 bp pipeline of the Genomics Facility at the University of Basel. In brief, the abundant ribosomal sequences in the samples were degraded to enhance the mRNA fraction using the Ribo-Zero Gold Epidemiology Kit (Illumina) to target eukaryotic, bacterial, and archaeal sequences. During testing, this Epidemiology Kit was found to outperform a sequential application of the Ribo-Zero Gold Bacterial and Eukaryotic Kits (Illumina) on the activated sludge samples (81.3 ± 5.2% versus 15.4 ± 1.8% of resulting reads of non-rRNA origin). The adapter addition, sample cleanup, and fragment selection were performed as outlined in the Illumina TruSeq protocol. The samples were then sequenced on a NextSeq 500 Platform (Illumina pipeline 2.4.11). The raw data are publically available at EMBL-EBI under the study number PRJEB22087. The quality of the RNA as extracted, RNA after depletion, and resulting fragments are provided in Supplemental Figs. 3–5.
RNAseq data processing, normalization, and analysis
The raw read files were trimmed of adapter sequences, index sequences, and low-quality reads using Trimmomatic [39] v0.33. The raw and trimmed reads were also checked for quality using FastQC [31] (Supplemental Figures 6–9). To remove contaminating rRNA reads in silico, the trimmed reads were compared against rRNA databases (Silva version 119 (Bacteria 16S and 23S, Archaea 16S and 23S, Eukaryota 18S and 28S) and RFAM (5S and 5.8S)) and filtered using SortMeRNA [40]. Sequences passing the quality control were annotated with the descriptors provided in the EC Number Uniprot database using DIAMOND [41] v0.2.1 with the blastx command and a minimum bitscore cutoff of 50 (all other parameters set to their default). Because we are primarily investigating EC annotation that can be shared across taxa and not specific genes from individual species, only the best annotation per read was recorded. The full Uniprot-TrEMBL database was created by downloading the database on March 6, 2018 (36.8 billion amino acids in 109 million sequences). The narrower Uniprot-EC database was created by searching for ec:* and downloading all matching hits on March 6, 2018 (5.8 billion amino acids in 14.9 million sequences). The script required to process the raw RNAseq files, generate the database, annotate the reads, and extract the taxonomic Uniprot identifiers is provided as Supplemental File 2. The resulting raw sequencing files contained 41.8–54.4 million reads, of which 72.5–87.7% remained in the dataset after quality and rRNA filtering. In total, 32.8–47.7 million reads per sample were submitted for annotation, resulting in 5.1–9.8 million reads being annotated per sample (Supplemental Table 5; Supplemental Figures 10 and 11).
The read counts were aggregated per EC number, and these EC numbers were used as a proxy of the functional profile in this study. When the Uniprot entry that provided the annotation of a read maintained multiple EC numbers, the read was assigned equally to each EC number (<5% of all annotations maintained multiple EC designations). The rarefaction curves showed that the richness of EC numbers saturated within the library’s sequencing depth (Supplemental Figure 12). For normalization, the count data was treated compositionally in that the abundance of a specific EC number was divided by the total number of reads identified to encode a protein. The total number of protein encoding reads was determined by first using 500k reads from each library to search against the full Uniprot-TrEMBL database and then multiplying the fraction annotated with the total number of reads submitted to the Uniprot-EC database (Supplemental Table 5; Supplemental Figure 13).
MRT–diversity model construction
MRT–diversity model approach, assumptions, and limitations
In the MRT–diversity model, Monod-type bacterial growth mathematics [42] were employed dynamically to approximate the linkage between the MRT and community composition in the experimental reactor (Fig. 1a). Monod-kinetics use the μmax, be, substrate affinity (Ks), and yield (Y) to describe the growth of an organism’s biomass (X) on a given substrate (S). The approach presented here utilizes these parameters in a novel manner by exploring the range of their combined values (an approximation of community diversity) that leads to persistence over a MRT gradient.
Fig. 1.

a Schematic diagram of the SBR used when running the model. All parameters are detailed in Table 1. b Iteratively solved 9 point persistence curves for the maximum growth rate (µmax) and endogenous decay (be) selection range at 1, 3, 5, 7, 10, and 15 days MRT. Constraints are placed on the range of maximal growth rates (μmax,eco; 0.2–9.8 d−1) and endogenous decay constants (be,eco; 0.02–0.2 d−1), defining the ecological space available. The model uses controlling growth parameters (µmax,constrain, be,constrain) of 5 and 0.11 d−1, respectively. c Example of the steady-state output for the volume, substrate concentration, and two biomasses for the 1 d MRT
In our approach, we apply a number of simplifications to typical considerations employed in other Monod-growth-based dynamic models [24] to determine the range of growth parameters leading to persistence. Specifically, the wastewater is considered a single substrate (e.g., no distinction of carbonaceous or nitrogenous compounds), growth limitations resulting from sources other than substrate availability are considered constant (e.g., mass transfer, toxic product formation, additional substrates), and competition is allowed only for this single substrate. When triggered, assigned flow rates and influent composition are also assumed to be temporally stable to remove variability resulting from other independent variables, and mixing within the reactors is considered perfect (except during the settle phase). Changes in steady-state growth depend only on the maximal gene expression and enzyme kinetics, and the availability of the enzyme pool is considered temporally stable thereby neglecting evolution. In turn, this stability is assumed to allow instantaneous adjustments of the growth rate to the change in the substrate concentrations (i.e., time lags have elapsed).
To capture competition over both substrate-rich and substrate-poor phases, individuals within this model are allowed to be distinct in two growth parameters only: μmax and be. Both internal (consumption of stored substrate) and external (adverse environmental conditions, cell programmed death, and viral attack) decay are considered incorporated in the be parameter [43]; higher order ecological considerations dependent on the consideration of additional substrates, such as predation and growth on lysis products are excluded from the model. The maximum and minimum μmax and be values are bounded by ecological limits, and a constraining combination of growth parameters must be satisfied by the range. In summary, the main uncertainties in the model include the appropriateness of restricting the analysis to a single substrate, the placement of the constraining growth parameters, the uncertain assignment of the ecological constraints, and the exclusion of other contributors to diversity, such as population oscillation and the time to reach equilibrium [44].
Role of μmax and be in the MRT–diversity model
To conceptualize the interaction of the μmax, be, and MRT within the MRT–diversity model of a SBR, the solution for the minimum substrate concentration (S*min) that leads to persistence in a continuously stirred tank reactor [45, 46] (Supplemental Materials 2) provides a simplified analogy that can be written without including differential equations:
| 1 |
where all parameters were defined previously. Organisms with the lowest calculated S*min values will persist in the reactor because they will outcompete other community members for the sole resource. In previous models, a single surviving species would be selected because of the hypothesized inability of other organisms to exactly match the μmax, Ks, and be combination required for persistence in the reactor [45]. Notably, we relax this constraint and allow multiple organisms to grow on a single substrate. Modeling co-existing combinations of growth parameters explores whether we can predict richness and diversity values similar to the experimentally observed values over a MRT gradient.
In developing this Monod-kinetics model of multiple organisms for the investigated SBR (Fig. 1a), the combination of growth parameters that are allowed at a given MRT is simply given by the maximum and minimum μmax and be values that persist in the reactor (Fig. 1b). Varying be influences the range of μmax values leading to persistence in the reactor more than Ks (Supplemental Figure 14) because be represents an additional component other than resource capture, i.e., survival during low or no production. Ks was therefore held constant to reduce model complexity. The line of growth parameter combinations that results in persistence (and determined by the equations detailed below) is required to fall within a roughly set ecological range and to pass through constraining growth parameters (μmax,constrain and be,constrain; values that are initially assumed to remain unchanged between reactors, arbitrarily set to the center of the range, and explored further in Supplemental Figure 15). To establish the permissible ecological values of be, the extremes of previously reported observations (from ~0.02 [47, 48] to ~0.2 d−1 [49]) were used as approximate boundaries (be,eco; 0.02–0.2 d−1), and an average value (0.11 d−1) was selected as the be,contrain (Table 1). The μmax,eco boundaries (0.2–9.8 d−1) were set to exceed the range of values reported for a previous MRT gradient [50], and an average value of 5 d−1 was selected as the μmax,contrain (Table 1). The model was found to be rather insensitive to the selection of these constraining points (Supplemental Figure 15).
Table 1.
Model nomenclature and parameter values
| Symbol | Parameter | Units | Value | Source | Model sensitivity |
|---|---|---|---|---|---|
| μ max | Maximum growth rate | d−1 | – | – | Modeled parameter |
| μ max,eco,min | Lowest maximum growth rate allowed | d−1 | 0.2 | 50 | Sensitivea |
| μ max,eco,max | Highest maximum growth rate allowed | d−1 | 9.8 | 50 | Sensitivea |
| μ max,constrain | Constraining growth rate used for modeling | d−1 | 5 | Set to range center | Sensitivea |
| b e | Endogenous decay | d−1 | – | – | Modeled Parameter |
| b e,eco,min | Lowest endogenous decay rate allowed | d−1 | 0.02 | 47, 48 | Sensitivea |
| b e,eco,max | Highest endogenous decay rate allowed | d−1 | 0.2 | 49 | Sensitivea |
| b e,constrain | Constraining endogenous decay used for modeling | d−1 | 0.11 | Set to range center | Sensitivea |
| K s | Half-saturation constant | mg/L | 50 | Typical value | Insensitiveb |
| Y | Yield | mg X/mg S | 0.3 | Typical value | Insensitive |
| S in | Influent substrate concentration | mg/L | 250 | Typical value | Insensitive |
| S | Substrate concentration in the reactor | mg/L | – | – | Modeled parameter |
| X | Biomass concentration | mg/L | – | – | Modeled parameter |
| X settle | Settled biomass concentration | mg/L | – | – | Modeled parameter |
| V full | Full volume of reactor | L | 12 | Experimental setup | – |
| V t | Time variable volume of reactor | L | – | – | Modeled Parameter |
| V mixed drain | Mixed volume removed, dependent on MRT | L | Variablec | Experimental setup | – |
| V clarified drain | Clarified volume removed, 4 L-Vmixed drain | L | Variablec | Experimental setup | – |
| t cycle | Total cycle time | h | 4 | Experimental setup | – |
| t react | React time | h | 3 | Experimental setup | – |
| t settle | Settle time | h | 0.5 | Experimental setup | – |
| t mixed drain | Time to drain the mixed volume | h | 0.167 | Experimental setup | – |
| t clarified drain | Time to drain the clarified volume | h | 0.167 | Experimental setup | – |
| t fill | Fill time | h | 0.333 | Experimental setup | – |
| Q mixed drain | Mixed drain flowrate | L/h | Vmixed drain/tmixed drain | Experimental setup | – |
| Q clarified drain | Clarified drain flowrate | L/h | Vclarified drain/tclarified drain | Experimental setup | – |
| Q in | Influent flowrate | L/h | 12 | Experimental setup | – |
SBR differential equations
The combination of growth parameter values resulting in persistence (i.e., non-zero steady-state concentrations) across the ecological range were determined with the following system of differential equations that describes the flow, biomass, and substrate concentrations within the SBR (Fig. 1a):
| 2 |
| 3 |
| 4 |
where the flowrates are triggered during their respective cycles (and are zero otherwise); the i subscript indicates parameters and biomass for the ith combination of growth parameters (ranging from 1 to n) that were modeled simultaneously; and all other parameters are defined in Table 1 and further described in Supplemental File 3. To ensure flow balance across the SBR cycle (Fig. 1a), the Qclarified drain is calculated to offset the Qin and Qmixed drain (outflow of suspended biomass):
| 5 |
where all parameters are defined in Table 1. Notably, the MRT is determined as the full volume of the reactor divided by the total volume of suspended biomass removed (Qmixed drain*tmixed drain) per six cycles (one day).
An iterative approach was used to calculate the μmax values resulting in persistence for nine be values distributed across the ecological range. A full solution line was then fit to these nine points (Fig. 1b). This solution line was found to depend only on those parameters directly influencing μmax, be, and MRT and was insensitive to changes in other global parameters, such as the Sin, Y, and Ks. All differential equations mentioned in this study were analyzed using deSolve [51] v1.21, and all calculations were performed in R v3.5.1 (Supplemental File 3).
MRT–diversity model alpha diversity calculation
After determining the solution (Fig. 1b), the length of the line representing all combinations of μmax and be leading to survival within the reactor was then calculated:
| 6 |
where the Scaling Factor is set to 0.25 to represent a case when the μmax range contributes more to the length than the be (emphasizing the fact that be serves more to allow the coexistence of different μmax values rather than contribute to diversity directly; see Supplemental File 3). The growth parameter solution length is utilized as a proxy for the richness of a community; this length will most likely be an underestimate of true richness as a result of binning organisms (or ESVs when comparing to 16S data) that display the identical combination of growth parameters. The Shannon diversity index was determined by numerically solving differential equations for the steady-state biomasses (Xi(steady-state)) when considering the number of distinct combinations of growth parameters within the community to be the length of the range multiplied by a constant value (n = 50; discretionarily set to achieve an integer value representative of community size and a timely computation of the differential equations). The instantaneous substrate utilization rate (ktheo) was calculated as the maximum substrate utilization rate determined at the beginning of one cycle.
Results and discussion
Observed taxonomic richness and diversity increases with MRT
The ESV richness increases monotonically across the MRT gradient for the active community members, i.e., the 16S rRNA (Spearman rank correlation r = 0.98 and 0.89 for TP1 and TP2, respectively), but displays a lower correlation to a monotonic trend for the present community members, i.e., the 16S rDNA (r = 0.81 and 0.77 for TP1 and TP2, respectively) (Fig. 2a, b). Additionally, the abundance weighted diversity metric, the Shannon diversity index, shows a decelerating increase in the rRNA transcripts which levels off above 5.1. Overall, the observed increase in the Shannon diversity value between 3 and 10 d (i.e., a mean ± s.d. of 4.6 ± 0.3 to 5.3 ± 0.2, respectively) agrees with a previous study investigating lab-scale synthetic wastewater-treating membrane bioreactors (MBRs) [52]. Other studies utilizing synthetic wastewater indicated no substantial difference between the community diversity metrics at ~2 and 10 d MRT in a MBR system [6, 7], or a decrease in diversity from 3 to 8 d MRT in a SBR system [53]. These studies also employed other sequencing techniques, such as denaturing gel gradient electrophoresis [7] and terminal restriction length polymorphism analysis [53] that can affect the exact quantified values, but are not expected to affect the reported trends of stable or decreasing diversity metrics. By contrast, investigations of full-scale WWTPs reported a comparable increase in diversity metrics at longer MRT [4, 5, 54], suggesting that real wastewater is required to consistently display a direct MRT–diversity relationship as also observed here.
Fig. 2.

a, b The calculated diversity metrics for the rarified B2-primer amplified 16S rRNA (black) and rDNA (red) data. c, d The abundance data distributed into taxonomic orders; the top 10 of the sums across each time-point and source (cDNA or gDNA) were assigned a color, resulting in 15 orders being represented overall. A replicate analysis is presented for the alternate B1 primer in Supplemental Figure 16
Of the 15 highlighted orders (Fig. 2c, d), six (Burkholderiales, Rhodocyclales, Myxococcales, Sphingobacteriales, Rhodobacteriales, and Pseudomonadales) were previously demonstrated to be commonly shared by a wide variety of activated sludge [55, 56]. Across a set of 13 Danish WWTPs, genera of the Thiotrichales order were abundantly observed in only two WWTPs, highlighting the potential transient nature of this population in WWTPs [56]. At both time-points, the relative 16S rRNA transcript abundance of Burkholderiales decreases by nearly a factor of two with increasing MRT (from 41.3 ± 0.30% to 19.6 ± 0.23% and 37.5 ± 0.60% to 21.1 ± 0.15% of the community for TP1 and TP2, respectively) consistent with a previous study [4], whereas Rhodocyclales (from 21.2 ± 0.44% to 41.3 ± 0.20% and 12.3 ± 0.21% to 19.6 ± 0.59%) and Myxococcales (from 0.11 ± 0.30% to 12.2 ± 0.38% and 0.14 ± 0.01% to 5.7 ± 0.12%) show increasing abundances. Additionally, a low abundance subpopulation capable of oxidizing ammonia to nitrate, the Nitrosomonadales, established at longer MRT when nitrification was noted [28] and expected [57].
The relative distribution of the orders are maintained in both the TP2 16S rRNA and rDNA profiles (Spearman r for the top 50 orders of 0.90, 0.88, 0.80, 0.74, 0.72, and 0.60 for 1, 3, 5, 7, 10, 15 d MRT, respectively). However, the profile in TP1 was substantially more variable (r = 0.27, 0.33, 0.47, 0.47, 0.38, and 0.25, respectively). This divergence is attributed to the detection of unique orders (Caldilineales, Lactobacillales, Micrococcales) and to the over-abundance of members within the Thiotrichales order in the TP1 rDNA profile (Fig. 2c). This over-abundance suggests that the filamentous Thiotrichales in TP1 causes a negative selection event in those reactors in which the most dominant organism by biomass (rDNA) is not the most active or productive (rRNA) [58]. This more variable signal also results in the Nitrosomonadales order displaying a 10-fold to 100-fold lower rDNA than rRNA signal (Fig. 2c, d), obscuring the ability to detect the known MRT-dependent emergence of this organism and its causal relationship to nitrification [59]. Overall, the 16S results suggest that the expression of 16S rRNA is more reflective of activity than the detection of an organism (16S rDNA) in the activated sludge experiments, supporting previous findings in surveys of other aerobic systems [60].
MRT is a driver of modeled taxonomic richness and diversity
In the constructed MRT–diversity model, increasing the MRT expands the range of combinations of μmax and be values that lead to persistence (Fig. 1b; Supplemental Table 6). Strikingly, the increase in the range of growth parameters (Fig. 3a) strongly correlates with the 16S rRNA observed richness (Fig. 2a, b) with r-values of 0.98 and 0.89 for TP1 and TP2, respectively (Fig. 3a inset). Substantially reducing the community complexity into parameters that describe resource capture and represent survival during low production (μmax and be, respectively; components of the Competition-Stress-Ruderal continuum [61, 62]) recaptures the trend of increasing richness across the MRT range. Alternatively, when varying parameters describing resource capture alone (μmax and Ks; components of the r/k-specialist theory [63]), the resulting range of growth parameter combinations is lower because the influence of variations in both μmax and Ks diminishes as the substrate concentration approaches zero (Supplemental Figure 14). Therefore, employing a variable parameter that is independent of the substrate concentration (i.e., be) results in a higher range of growth parameters leading to persistence.
Fig. 3.
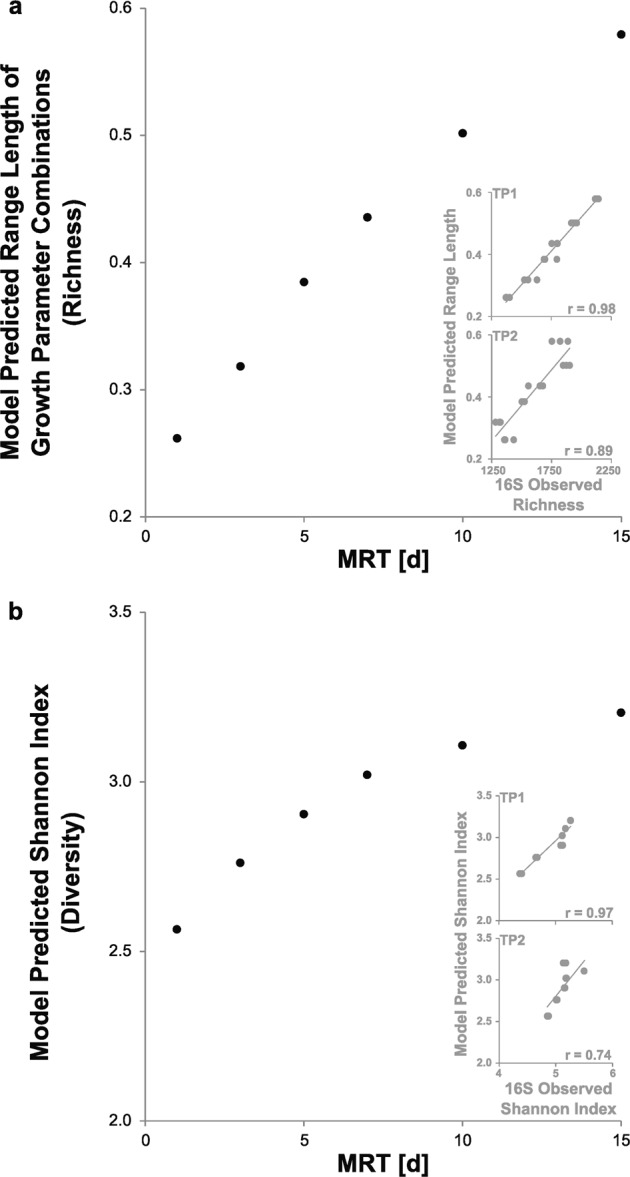
Alpha diversity results from the MRT–diversity model explaining the relationship between the community composition and the MRT. a The range of growth parameters, a proxy for the richness, with increasing MRT. The inset displays the linear relationship between the model predicted and the observed richness data. b The calculated Shannon diversity index. The number of species types modeled in the Shannon diversity index calculations is set to 50 times the length of the range of growth parameters. The inset displays the linear relationship between the model predicted and the observed Shannon Index data
Although a range of growth parameters will persist (Fig. 1b), the organisms they represent will be present at various abundances at steady-state (Xi in Fig. 1c). The Shannon diversity for the SBR shows a decelerating increase with MRT (Fig. 3b), matching the 16S rRNA observed data (r = 0.97 and 0.74 for TP1 and TP2, respectively (Fig. 3b inset)). An underlying assumption in this comparison is that the ratio of the biomass resulting from a given combination of growth parameters to the number of representative rRNA transcripts is constant; however, this ratio varies even at the gene copy per genome level [64]. Therefore, the general trend of the curve is informative of whether the MRT influences the diversity, but the magnitude of the shifts would be substantially affected by this rRNA-to-biomass ratio.
The substrate consumption rate of the entire activated sludge community is often monitored through respirogram bulk tests (i.e., biomass normalized maximal oxygen uptake rate (OUR) analyses) [65] and has been previously reported to slow with increasing MRT [50, 66], suggesting an adaptation of the community. Our model allows predicting the instantaneous substrate utilization rate (ktheo), and the previously published slowing trend is not observed from the initial default parameters (modeled ktheo of 5.0 and 5.8 d−1 at 1 and 15 d, respectively). This disconnect likely stems from underlying assumptions of our model, most notably fixing constraining central growth parameters (μmax,constrain and be,constrain). However, when using be,ecomax as be,constrain instead, the decreasing trend in the previously reported empirical values is successfully mirrored (ktheo of 4.8 and 4.1 d−1 at 1 and 15 d, respectively) while the diversity profiles are conserved. Therefore, ambiguity remains regarding the accurate placement of these controlling parameters, as well as the ecological range parameter values.
Overall, these observed and modeled results complement a previous study monitoring an activated sludge reactor for 313 d in a 30, 12, 3, 30 d MRT disturbance cycle, in which an increased diversity was noted for the higher MRT values [4, 67]. In that study, two mechanisms were proposed that contribute to the higher richness and diversity: decreases in unconsumed resources (analogous to decreases in the S*min in Eq. (1) or an increase in the length of the starvation phase in the SBR) and increases in niche space (represented by the range of growth parameters and assumed to represent the richness in the MRT–diversity model). Our model uses the MRT and growth parameters to represent these two mechanisms in separate equations and predicts that the richness increases as the availability of unconsumed resources decreases across the observed MRT range (Fig. 3). Notably, the MRT–richness profile can display a non-monotonic trend at higher MRT or by placing the constraining growth parameters (μmax,constrain or be,constrain) close to the ecological limits. By contrast, the availability of unconsumed resources will consistently decrease with increasing MRT. Observing a non-monotonic MRT–richness relationship would thus suggest that niche space contributes more to diversity. In future studies, the potential for this non-monotonic profile should be tested by establishing reactors exceeding the maximum MRT observed here (Fig. 2a, b). Additionally, uncovering a transition from a monotonic to non-monotonic MRT–richness relationship would assist in testing the underlying assumptions of the model and more accurately estimating the ecological ranges and constraining growth parameters.
Observed functional richness, but not diversity, increases with MRT
With increasing MRT, conceptually either an organism absent at lower MRT may occupy the additional growth parameter space or a shared microorganism across MRT expresses different functional enzymes. To test for shifts in the functional profile, the metatranscriptomes of the experimental communities were sequenced, annotated as EC numbers, and analyzed using alpha diversity indicators of the number of unique (richness) and evenness of the relative abundance (Shannon diversity index) of the EC sub-subclasses and numbers (Fig. 4). Similar to what was noted in a previous study, the 16S rRNA taxonomic and EC number richness of both time-points display a strong correlation (r = 0.98 and 0.97 for the TP1 and TP2 samples, respectively) [12] and a nearly monotonic increase with MRT in richness (Fig. 2a, b; Fig. 4). This relationship between the taxonomic and functional richness is not as consistently strong with the 16S rDNA (r = 0.81 and 0.94, respectively). These results again highlight that a measure more reflective of current activity within the cell (16S rRNA) links better with the overall functional profile (mRNA) than a survey of presence alone (16S rDNA). When considering the relative abundances of a given EC sub-subclass or number, the Shannon diversity displays a non-monotonic profile, contrasting the taxonomic profiles and the EC sub-subclass or number richness. This disconnect between taxonomic and functional diversity has been previously demonstrated in model wastewater reactors [13], suggesting that this result represents a true signal beyond simple limitations with the 16S rRNA measure (e.g., abundance not always correlating with growth rate, inter-species differences in copy numbers per cell [68], steep ecological gradients across the SBR cycle). However, the disconnect between the functional richness and Shannon diversity indicates that although the quantity of EC sub-subclasses or numbers increases across the gradient, specific categories increase in dominance at longer MRTs, offsetting the increased richness (Fig. 4).
Fig. 4.
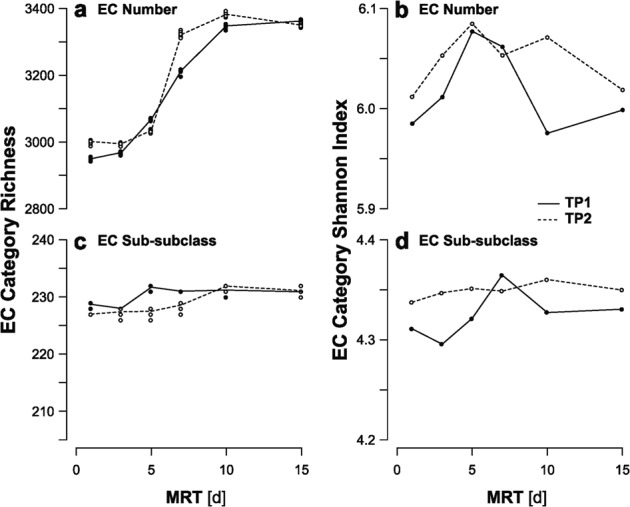
The richness (a, c) and Shannon diversity (b, d) values for the reaction-type annotated RNA data binned into Enzyme Commission (EC) number (a, b) and sub-subclass (c, d) for TP1 (solid) and TP2 (dashed) with a fractional abundance cutoff of 10−7. These diversity values were calculated for each reactor using the rtk v0.2.5.4 package in R v3.5.1 for (a, c) 10 bootstrap sub-selections of the annotations or (b, d) the full data. The lines trace the (a, c) bootstrap mean or (b, d) calculated value
The relative abundance shifts of “rare” enzyme classes drive the functional diversity profile
Several overrepresented EC sub-subclasses in terms of observed abundance (Fig. 5a), e.g., the 2.7.7 nucleotidyltransferase (containing 2.7.7.6 RNA-polymerase, RpoB) and 5.99.1 other-isomerase (containing 5.99.1.2 DNA topoisomerase) EC sub-subclass, decrease in their fractional share of the metatranscriptome as the MRT increases (Fig. 5b). Simultaneously, the fractional share of sub-subclasses that include oxidoreductases and nitrogen-processing-related enzymes that are linked to the emergence of nitrification over the MRT gradient (e.g., nitrogenous oxidoreductase with [1.7.2] and without [1.7.99] cytochrome) increase (Fig. 5a, b). The over-abundance of nitrogen metabolism-related gene transcripts has been previously noted in activated sludge even when nitrifiers are a minor fraction of the community [69]. Notably, these EC sub-subclasses and associated numbers that markedly increase in abundance over the MRT induced the non-monotonic functional diversity profile, indicating that substantially different abundances of mRNAs encoding for specific enzymes are likely required to achieve those growth parameters resulting in persistence. The discrepancy between the non-monotonic functional diversity profile of all EC numbers and the monotonic taxonomic diversity profile for a single-targeted transcript (e.g., 16S-rRNA) results from enzymes displaying additional properties [70], such as specific substrates affinities, product turnover rates [71], and protein-to-transcript ratios [72, 73] that affect their relative fractional abundances.
Fig. 5.

Summary of the sub-subclass Enzyme Commission (EC) numbers across the MRT gradient averaged between TP1 and TP2 samples that exceed 10,000 normalized reads in at least 1 reactor (n = 99). The heatmaps are organized hierarchically according to a Euclidean distance and Ward clustering of the scaled EC fraction across the MRT gradient. (a) Total relative abundance. (b) The 15/1 d MRT abundance log ratio. The within EC sub-subclass taxonomic (c) richness and (d) Shannon diversity metrics were calculated based on the Uniport identifiers of the organism of origin at the genus level that provided the annotation to the reads. (e) The fraction of the total reads that were annotated per EC sub-subclass originating from an eukaryotic sequence in the Uniprot database. Note: The red dashed boxes highlight the 2.7.7 and 1.7.99 EC numbers. The full sub-subclass and top 50 EC numbers heatmaps for both TP1 and TP2 samples are presented in Supplemental Figures 17 and 18
To compare the observed diversities within each EC sub-subclass (similar to the 16S-rRNA analysis), specific taxonomic richness and diversity values were calculated based on the putative genus-level organism-origin annotation that each mRNA read is assigned (Fig. 5c, d, respectively). Focusing on the aforementioned 2.7.7 and 1.7.99 sub-subclasses to highlight categories demonstrated to be common and rare, respectively (Fig. 5c), the diversity profile of the common EC sub-subclass 2.7.7 displays a positive relationship with that of the 16S rRNA (Fig. 2; r = 0.89 and 0.59 for TP1 and TP2 Shannon indices, respectively). In contrast, a divergent profile is seen for the taxonomic diversity of the rare 1.7.99 sub-subclass (r = −0.65 and −0.76 for TP1 and TP2, respectively), indicating that select organisms dominate the origin of the reads within this category at higher MRTs. Notably, reads from the nitrifying Nitrospira [74] dominate the 1.7.99 sub-subclass. The nitrification rate intensifies across the MRT gradient, suggesting that Nitrospira expressed the proper bulk-growth parameters to persist and thrive within the community. The greater share of the overall reads transcribed resulting from a single, nitrification-related organism contributed to the noted decrease in functional diversity of the overall community (Fig. 4).
When further binning reads into Domain-level taxonomic origin, a substantial fraction of annotations originating from eukaryotic organisms (Fig. 5e) were noted for certain EC sub-subclasses increasing in abundance over the MRT gradient (Fig. 5a). In activated sludge, increasing MRT over the studied range have been reported to promote a higher abundance of protozoa [75], organisms that are overlooked in bacterial-targeted taxonomic surveys of WWTPs. This signal in the mRNA data could confound the previous comparison between the taxonomic and functional diversity metrics. However, when reanalyzing the functional diversity metrics (Fig. 4) for the bacterial portion (eukaryotic-sequences removed), a similar profile is obtained (Supplemental Figure 19), supporting the detection of a true distinction between the taxonomic composition and the functional profile.
Conclusions
As demonstrated experimentally, increasing the MRT positively affects the taxonomic richness and diversity, as well as functional richness of the monitored activated sludge community. To conceptualize these findings, a naïve model was constructed that utilized Monod-kinetics in a novel manner by considering wastewater as a single substrate and the community as a collection of growth parameter combinations. Combinations of μmax and be values were selected to represent the two strategies of efficient resource capture and survival during low production, respectively. This MRT–diversity model predicted that the range of μmax and be values expands with increasing MRT for the studied system, suggesting a new, kinetic parameter-driven metric that correlates strikingly well with the observed taxonomic profile and the functional richness across the MRT gradient. For a new community member to occupy these opened growth parameter combinations and thereby increase the taxonomic richness, previously unobserved EC numbers are likely required because of the noted increases in functional richness. In contrast to the taxonomic abundance-weighted diversity, the functional diversity displayed a non-monotonic trend over the MRT range. Whereas more EC sub-subclasses and numbers are detected at higher MRTs, their fractional share of the overall activity of the community varies depending on the expressed function. For example, rare sub-subclasses related to nitrification substantially increase in dominance at longer MRTs in this system. Although the complexity of the relationship between EC numbers is not successfully captured, the simplification of the community into combinations of μmax and be values appears to be a useful approximation for predicting changes in taxonomic richness and diversity, as well as functional richness over a MRT gradient in this system. Because this study is the first to employ Monod-kinetics in this manner, future work should determine whether the approach and assumptions introduced here are valid when used to describe other systems, explore the concepts of the constraining growth–parameter combination and ecological boundary values, and subdivide influent resources into individual substrate types (e.g., nitrogen-containing compounds).
Supplementary information
Acknowledgements
Data produced and analyzed in this paper were generated in collaboration with the Genetic Diversity Centre (GDC), ETH Zurich, Switzerland and the Genomics Facility at the University of Basel, Switzerland. We thank the operators and the staff of the WWTP ARA Niederglatt for providing activated sludge. We acknowledge financial support from the European Research Council under the European Union’s Seventh Framework Program (ERC grant agreement no. 614768, PROduCTS). We also thank Dr. Paola Meynet for assistance in the preparation of the 16S control libraries.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Cresten Mansfeldt, Stefan Achermann
Supplementary information
The online version of this article (10.1038/s41396-019-0371-6) contains supplementary material, which is available to authorized users.
References
- 1.Curtis TP, Sloan WT, Scannell JW. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA. 2002;99:10494–9. doi: 10.1073/pnas.142680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Locey KJ, Lennon JT. Scaling laws predict global microbial diversity. Proc Natl Acad Sci USA. 2016;113:5970–5. doi: 10.1073/pnas.1521291113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antwis RE, Griffiths SM, Harrison XA, Aranega-Bou P, Arce A, Bettridge AS, et al. Fifty important research questions in microbial ecology. FEMS Microbiol Ecol. 2017;93:1–10. [DOI] [PubMed]
- 4.Vuono DC, Benecke J, Henkel J, Navidi WC, Cath TY, Munakata-Marr J, et al. Disturbance and temporal partitioning of the activated sludge metacommunity. ISME J. 2015;9:425–35. doi: 10.1038/ismej.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meerburg FA, Vlaeminck SE, Roume H, Seuntjens D, Pieper DH, Jauregui R, et al. High-rate activated sludge communities have a distinctly different structure compared to low-rate sludge communities, and are less sensitive towards environmental and operational variables. Water Res. 2016;100:137–45. doi: 10.1016/j.watres.2016.04.076. [DOI] [PubMed] [Google Scholar]
- 6.Bagchi S, Tellez BG, Rao HA, Lamendella R, Saikaly PE. Diversity and dynamics of dominant and rare bacterial taxa in replicate sequencing batch reactors operated under different solids retention time. Appl Microbiol Biotechnol. 2015;99:2361–70. doi: 10.1007/s00253-014-6134-4. [DOI] [PubMed] [Google Scholar]
- 7.Akarsubasi AT, Eyice O, Miskin I, Head IM, Curtis TP. Effect of sludge age on the bacterial diversity of bench scale sequencing batch reactors. Environ Sci Technol. 2009;43:2950–6. doi: 10.1021/es8026488. [DOI] [PubMed] [Google Scholar]
- 8.Fang H, Chen Y, Huang L, He G. Analysis of biofilm bacterial communities under different shear stresses using size-fractionated sediment. Sci Rep. 2017;7:1299. doi: 10.1038/s41598-017-01446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roager HM, Hansen LB, Bahl MI, Frandsen HL, Carvalho V, Gøbel RJ, et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat Microbiol. 2016;1:16093. doi: 10.1038/nmicrobiol.2016.93. [DOI] [PubMed] [Google Scholar]
- 10.Kreuzinger N, Clara M, Strenn B, Kroiss H. Relevance of the sludge retention time (SRT) as design criteria for wastewater treatment plants for the removal of endocrine disruptors and pharmaceuticals from wastewater. Water Sci Technol. 2004;50:149–56. doi: 10.2166/wst.2004.0322. [DOI] [PubMed] [Google Scholar]
- 11.Falås P, Andersen HR, Ledin A, la Cour Jansen J. Impact of solid retention time and nitrification capacity on the ability of activated sludge to remove pharmaceuticals. Environ Technol. 2012;33:865–72. doi: 10.1080/09593330.2011.601764. [DOI] [PubMed] [Google Scholar]
- 12.Johnson DR, Lee TK, Park J, Fenner K, Helbling DE. The functional and taxonomic richness of wastewater treatment plant microbial communities are associated with each other and with ambient nitrogen and carbon availability. Environ Microbiol. 2015;17:4851–60. doi: 10.1111/1462-2920.12429. [DOI] [PubMed] [Google Scholar]
- 13.Pholchan MK, Baptista JDC, Davenport RJ, Sloan WT, Curtis TP. Microbial community assembly, theory and rare functions. Front Microbiol. 2013;4:68. doi: 10.3389/fmicb.2013.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272–7. doi: 10.1126/science.aaf4507. [DOI] [PubMed] [Google Scholar]
- 15.Louca S, Jacques SM, Pires AP, Leal JS, Srivastava DS, Parfrey LW, et al. High taxonomic variability despite stable functional structure across microbial communities. Nat Ecol Evol. 2017;1:0015. doi: 10.1038/s41559-016-0015. [DOI] [PubMed] [Google Scholar]
- 16.Frossard A, Gerull L, Mutz M, Gessner MO. Disconnect of microbial structure and function: enzyme activities and bacterial communities in nascent stream corridors. ISME J. 2012;6:680–91. doi: 10.1038/ismej.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purahong W, Schloter M, Pecyna MJ, Kapturska D, Däumlich V, Mital S, et al. Uncoupling of microbial community structure and function in decomposing litter across beech forest ecosystems in CentralEurope. Sci Rep. 2014;4:7014. doi: 10.1038/srep07014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kivlin SN, Hawkes CV. Temporal and spatial variation of soil bacteria richness, composition, and function in a neotropical rainforest. PLoS One. 2016;11:e0159131. doi: 10.1371/journal.pone.0159131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci USA. 2011;108:14288–93. doi: 10.1073/pnas.1101591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boon E, Meehan CJ, Whidden C, Wong DHJ, Langille MG, Beiko RG. Interactions in the microbiome: communities of organisms and communities of genes. FEMS Microbiol Rev. 2014;38:90–118. doi: 10.1111/1574-6976.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daims H, Taylor MW, Wagner M. Wastewater treatment: a model system for microbial ecology. Trends Biotechnol. 2006;24:483–9. doi: 10.1016/j.tibtech.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Downing AL, Hopwood AP. Some observations on the kinetics of nitrifying activated-sludge plants. Schweiz Z Hydrol. 1964;26:271–88. [Google Scholar]
- 23.Lawrence AW, McCarty PL. Unified basis for biological treatment design and operation. J Sanit Eng Div. 1970;96:757–78. [Google Scholar]
- 24.Henze M., Gujer W., Mino T., van Loosedrecht M. Activated Sludge Models ASM1, ASM2, ASM2d and ASM3. Water Intelligence Online. 2015;5(0):9781780402369–9781780402369. doi: 10.2166/9781780402369. [DOI] [Google Scholar]
- 25.Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity. ISME J. 2012;6:183–94. doi: 10.1038/ismej.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinclair L, Osman OA, Bertilsson S, Eiler A. Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the illumina platform. PLoS One. 2015;10:e0116955. doi: 10.1371/journal.pone.0116955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, et al. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16:410–22. doi: 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- 28.van Bodegom P. Microbial maintenance: a critical review on its quantification. Microb Ecol. 2007;53:513–23. doi: 10.1007/s00248-006-9049-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achermann S, Falas P, Joss A, Mansfeldt C, Men Y, Vogler B, et al. Trends in micropollutant biotransformation along a solids retention time gradient. Environ Sci Technol. 2018;52:11601–11. doi: 10.1021/acs.est.7b06716. [DOI] [PubMed] [Google Scholar]
- 30.Guo F, Ju F, Cai L, Zhang T. Taxonomic precision of different hypervariable regions of 16S rRNA gene and annotation methods for functional bacterial groups in biological wastewater treatment. PLoS One. 2013;8:e76185. doi: 10.1371/journal.pone.0076185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 32.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 34.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 35.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill MO. Diversity and evenness: a unifying notation and its consequences. Ecology. 1973;54:427–32. doi: 10.2307/1934352. [DOI] [Google Scholar]
- 37.Chao A, Chiu CH, Jost L. Phylogenetic diversity measures based on Hill numbers. Philos Trans R Soc B—Biol Sci. 2010;365:3599–609. doi: 10.1098/rstb.2010.0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chao A, Gotelli NJ, Hsieh TC, Sander EL, Ma KH, Colwell RK, et al. Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species diversity studies. Ecol Monogr. 2014;84:45–67. doi: 10.1890/13-0133.1. [DOI] [Google Scholar]
- 39.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 41.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 42.Monod J. The growth of bacterial cultures. Annu Rev Microbiol. 1949;3:371–94. doi: 10.1146/annurev.mi.03.100149.002103. [DOI] [Google Scholar]
- 43.Liu G, Wang J. Modeling effects of DO and SRT on activated sludge decay and production. Water Res. 2015;80:169–78. doi: 10.1016/j.watres.2015.04.042. [DOI] [PubMed] [Google Scholar]
- 44.Saikaly PE, Oerther DB. Bacterial competition in activated sludge: theoretical analysis of varying solids retention times on diversity. Microb Ecol. 2004;48:274–84. doi: 10.1007/s00248-003-1027-6. [DOI] [PubMed] [Google Scholar]
- 45.Hsu SB, Hubbell S, Waltman P. A mathematical theory for single-nutrient competition in continuous cultures of micro-organisms. SIAM J Appl Math. 1977;32:366–83. doi: 10.1137/0132030. [DOI] [Google Scholar]
- 46.Saikaly PE, Oerther DB. Diversity of dominant bacterial taxa in activated sludge promotes functional resistance following toxic shock loading. Microb Ecol. 2011;61:557–67. doi: 10.1007/s00248-010-9783-6. [DOI] [PubMed] [Google Scholar]
- 47.Habermacher J, Benetti AD, Derlon N, Morgenroth E. The effect of different aeration conditions in activated sludge–side-stream system on sludge production, sludge degradation rates, active biomass and extracellular polymeric substances. Water Res. 2015;85:46–56. doi: 10.1016/j.watres.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 48.Martínez-García CG, Fall C, Olguín MT. Activated sludge mass reduction and biodegradability of the endogenous residues by digestion under different aerobic to anaerobic conditions: comparison and modeling. Bioresour Technol. 2016;203:32–41. doi: 10.1016/j.biortech.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 49.Friedrich M, Takács I. A new interpretation of endogenous respiration profiles for the evaluation of the endogenous decay rate of heterotrophic biomass in activated sludge. Water Res. 2013;47:5639–46. doi: 10.1016/j.watres.2013.06.043. [DOI] [PubMed] [Google Scholar]
- 50.Friedrich M, Jimenez J, Pruden A, Miller JH, Metch J, Takács I. Rethinking growth and decay kinetics in activated sludge–towards a new adaptive kinetics approach. Water Sci Technol. 2017;75:501–6. doi: 10.2166/wst.2016.439. [DOI] [PubMed] [Google Scholar]
- 51.Soetaert KER, Petzoldt T, Setzer RW. (2010). Solving differential equations in R: package deSolve. J Stat Softw. 2010;33:1–25.
- 52.Duan L, Moreno-Andrade I, Huang CL, Xia S, Hermanowicz SW. Effects of short solids retention time on microbial community in a membrane bioreactor. Bioresour Technol. 2009;100:3489–96. doi: 10.1016/j.biortech.2009.02.056. [DOI] [PubMed] [Google Scholar]
- 53.Saikaly PE, Stroot PG, Oerther DB. Use of 16S rRNA gene terminal restriction fragment analysis to assess the impact of solids retention time on the bacterial diversity of activated sludge. Appl Environ Microbiol. 2005;71:5814–22. doi: 10.1128/AEM.71.10.5814-5822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzalez-Martinez A, Rodriguez-Sanchez A, Lotti T, Garcia-Ruiz MJ, Osorio F, Gonzalez-Lopez J, et al. Comparison of bacterial communities of conventional and A-stage activated sludge systems. Sci Rep. 2016;6:18786. doi: 10.1038/srep18786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang T, Shao MF, Ye L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2011;6:1137–47. doi: 10.1038/ismej.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saunders AM, Albertsen M, Vollertsen J, Nielsen PH. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 2016;10:11–20. doi: 10.1038/ismej.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollice A, Tandoi V, Lestingi C. Influence of aeration and sludge retention time on ammonium oxidation to nitrite and nitrate. Water Res. 2002;36:2541–6. doi: 10.1016/S0043-1354(01)00468-7. [DOI] [PubMed] [Google Scholar]
- 58.Jiang L. Negative selection effects suppress relationships between bacterial diversity and ecosystem functioning. Ecology. 2007;88:1075–85. doi: 10.1890/06-1556. [DOI] [PubMed] [Google Scholar]
- 59.Prosser JI, Head IM, Stein LY. The family Nitrosomonadaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompso F (eds.). The Prokaryotes, Berlin, Heidelberg: Springer; 2014. p. 901–18.
- 60.Vuono DC, Regnery J, Li D, Jones ZL, Holloway RW, Drewes JE. rRNA gene expression of abundant and rare activated-sludge microorganisms and growth rate induced micropollutant removal. Environ Sci Technol. 2016;50:6299–309. doi: 10.1021/acs.est.6b00247. [DOI] [PubMed] [Google Scholar]
- 61.Grime JP, Pierce S. The evolutionary strategies that shape ecosystems. John Wiley & Sons; West Sussex, UK, 2012.
- 62.Ho A, Di Lonardo DP, Bodelier PL. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol Ecol. 2017;93:1–14. [DOI] [PubMed]
- 63.Andrews JH, Harris RF. Advances in microbial ecology. Boston, MA: Springer; 1986. r-and K-selection and microbial ecology. In; pp. 99–147. [Google Scholar]
- 64.Větrovský T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One. 2013;8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chandran K, Smets BF. Estimating biomass yield coefficients for autotrophic ammonia and nitrite oxidation from batch respirograms. Water Res. 2001;35:3153–6. doi: 10.1016/S0043-1354(01)00022-7. [DOI] [PubMed] [Google Scholar]
- 66.Friedrich M, Takács I, Tränckner J. Physiological adaptation of growth kinetics in activated sludge. Water Res. 2015;85:22–30. doi: 10.1016/j.watres.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 67.Vuono DC, Munakata‐Marr J, Spear JR, Drewes JE. Disturbance opens recruitment sites for bacterial colonization in activated sludge. Environ Microbiol. 2016;18:87–99. doi: 10.1111/1462-2920.12824. [DOI] [PubMed] [Google Scholar]
- 68.Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 2013;7:2061–8. doi: 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu K, Zhang T. Metagenomic and metatranscriptomic analysis of microbial community structure and gene expression of activated sludge. PLoS One. 2012;7:e38183. doi: 10.1371/journal.pone.0038183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prosser JI. Dispersing misconceptions and identifying opportunities for the use of omics’ in soil microbial ecology. Nat Rev Microbiol. 2015;13:439–46. doi: 10.1038/nrmicro3468. [DOI] [PubMed] [Google Scholar]
- 71.Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS, et al. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry. 2011;50:4402–10. doi: 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- 72.de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol Biosyst. 2009;5:1512–26. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–32. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daims H, Lücker S, Wagner M. A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol. 2016;24:699–712. doi: 10.1016/j.tim.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salvado H. Effect of mean cellular retention time on ciliated protozoan populations in urban wastewater treatment plants based on a proposed model. Water Res. 1994;28:1315–21. doi: 10.1016/0043-1354(94)90296-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.