

Abstract
The innate immune system plays an essential role in initial recognition of pathogen infection by producing inflammatory cytokines and type I interferons. cGAS is a cytoplasmic sensor for DNA derived from DNA viruses. cGAS binding with DNA induces the production of cGAMP, a second messenger that associates with STING in endoplasmic reticulum (ER). STING changes its cellular distribution from ER to perinuclear Golgi, where it activates the protein kinase TBK1 that catalyzes the phosphorylation of IRF3. Here we found that STING trafficking is regulated by myotubularin-related protein (MTMR) 3 and MTMR4, members of protein tyrosine phosphatases that dephosphorylate 3′ position in phosphatidylinositol (PtdIns) and generate PtdIns5P from PtdIns3,5P2 and PtdIns from PtdIns3P. We established MTMR3 and MTMR4 double knockout (DKO) RAW264.7 macrophage cells and found that they exhibited increased type I interferon production after interferon-stimulatory DNA (ISD) stimulation and herpes simplex virus 1 infection concomitant with enhanced IRF3 phosphorylation. In DKO cells, STING rapidly trafficked from ER to Golgi after ISD stimulation. Notably, DKO cells exhibited enlarged cytosolic puncta positive for PtdIns3P and STING was aberrantly accumulated in this puncta. Taken together, these results suggest that MTMR3 and MTMR4 regulate the production of PtdIns3P, which plays a critical role in suppressing DNA-mediated innate immune responses via modulating STING trafficking.
Keywords: phosphatidylinositol, phosphatidylinositol phosphatase, innate immunity, DNA viruses, interferon regulatory factor, IRF, DNA sensor, MTM, MTMR, PtdIns3P, STING, type I IFNs
Introduction
Host immune response is a defense system to eliminate invading pathogens, including bacteria, fungi, and viruses, and is largely classified into innate and acquired immune responses. Innate immune response is the first line of the host defense system that is triggered upon sensing molecules specific in pathogens, which are called pathogen-associated molecular patterns (PAMPs),4 with pattern-recognition receptors that are expressed in various types of host cells. Among them, dendritic cells and macrophages are the major innate immune cells that produce large amounts of pro-inflammatory cytokines and type I interferons (IFNs) and function as antigen-presenting cells that potentiate acquired immunity. Releasing cytokines and type I IFNs led to production of anti-bacterial or anti-viral proteins to prevent pathogen infections. So far, numerous pattern-recognition receptors have been identified, including toll-like receptors (TLRs), Retinoic acid-inducible gene I (RIG-I)-like receptor (RLRs), and intracellular sensor for DNA such as cGMP-AMP synthase (cGAS). TLRs localize to the cell surface or intracellular compartments such as the ER, endosome, lysosome, or endolysosome, and they recognize their PAMPs such as lipid, lipoprotein, protein, and nucleic acid. RLRs recognize cytosolic single-strand RNA and dsRNA derived from RNA virus, and cGAS recognizes cytosolic dsDNA derived for DNA virus. Each receptor activation by PAMP association leads to activating distinct signaling pathways which results in appropriate immune responses specific to given pathogens (1, 2).
Various DNA viruses such as human papillomavirus, herpes simplex virus (HSV), adenovirus, and hepatitis B virus release their own genomic DNA to the cytoplasm of host cells upon infection. Released dsDNA binds to a cytoplasmic DNA sensor cGAS (3). cGAS is an enzyme that synthesizes cGAMP from ATP and GTP following binding to viral and host DNA. cGAMP is a cyclic dinucleotide in that 3′-5′ and 2′-5′ ribose on ATP and GTP are connected via phosphodiester linkage, and it acts as the second messenger for adaptor protein STING (4). STING traffics from the ER to perinuclear region including the Golgi apparatus, then forms cytoplasmic punctate structures along with the protein kinase TBK1. TBK1 phosphorylates the transcription factor IRF3 and phosphorylated IRF3 translocates into nucleus and regulates transcription of type I IFN genes (5, 6). Therefore, translocation of STING to Golgi is a hallmark for activation of STING. Various molecules that regulate STING trafficking have been identified. STING trafficking from ER to Golgi apparatus is regulated via the translocon-associated protein (TRAP) complex TRAPβ (SSR2) that is recruited by inactive rhomboid protein 2 (iRhom2) and this complex reaches to Sec-5–containing perinuclear microsome or cytoplasmic punctate structures to assemble with TBK1 and the IKK complex (5). RAB2B-GARIL5 (Golgi-associated RAB2B interactor-like 5) complex is reported as a positive regulator of STING in Golgi apparatus by regulating phosphorylation of IRF3 by TBK1 (7). Moreover, the function of STING is regulated by posttranslational mechanism such as E3 ubiquitin ligases, TRIM32 and TRIM56, that conjugate Lys-63–linked polyubiquitination on STING and promote the recruitment of TBK1 (8–10).
Phosphatidylinositol (PtdIns) consists of an inositol ring and acid chains linked by phosphate. Inositol ring in PtdIns contains three phosphorylation sites (3′, 4′, and 5′) and are phosphorylated or dephosphorylated by different combinations. PtdIns are divided into eight types, and each PtdIns has a distinct function in terms of signal transduction, organelle trafficking, and cytoskeletal regulation (11). These PtdIns are phosphorylated and dephosphorylated at the specific sites in inositol ring by various types of phosphoinositide kinases and phosphatases. Some of these proteins such as PIKfyve, PI5P4K, and PI3K participate in innate immune responses (12).
The myotubularins (MTMs) are members of the phosphatase superfamily that consists of 1 MTM and 13 MTM-related (MTMR) members which potentially dephosphorylate 3′ site in inositol lipid (13). MTMR3 and MTMR4 are closely related members containing FYVE domain, PH-G domain, and the catalytic active site of phosphatase consensus site; catalytic Cys-X5-Arg (CX5R) motif. Biochemical analysis showed that both MTMR3 and MTMR4 mediate generation of PtdIns from PtdIns3P and PtdIns5P from PtdIns3,5P2 (14, 15). MTMR3 and MTMR4 are reported to contribute to the regulation of immune responses. MTMR3 is a risk factor for inflammatory bowel diseases wherein MTMR3 level is increased and PtdIns3P production is reduced (16). Reduction of PtdIns3P results in increased caspase-1–dependent cytokines production and inflammation by inhibiting autophagy formation (16, 17), and MTMR4-dependent PtdIns3P production is required for Salmonella-mediated vacuole formation (18).
We previously reported that PtdInst5P that is generated by the lipid kinase PIKfyve plays a crucial role in innate immune responses to RNA virus infection (19). RNA virus infection results in increment of cellular PtdIns5P level in a PIKfyve-dependent manner. PtdIns5P physically binds to IRF3, which facilitates TBK1-mediated phosphorylation and activation of IRF3. In this study, we addressed roles of other phosphatases and kinases that mediate production of PtdIns in innate immune responses. We provided evidence showing that PtdIns3P produced by MTMR3 and MTMR4 plays an important role in the negative regulation of innate immune responses to DNA via modulating STING trafficking.
Results
MTMR3/MTMR4 expression in various types of cells
We first assessed the expression pattern of MTM and MTMR family members in RAW264.7 macrophage cells, murine bone marrow–derived dendritic cells, J774 macrophage-like cells, murine bone marrow-derived macrophages (BMMs), and murine embryonic fibroblast (MEFs) cells by RT-PCR. MTM family genes were widely expressed in all types of cells as tested (Fig. 1, A and B). We previously showed that PIKfyve, which contains the FYVE domain that potentially binds to PtdIns, regulates innate immune signaling by generating PtdIns5P production (19). As MTMR3 and MTMR4 also have the FYVE domain, here we focused on MTMR3 and MTMR4, and investigated whether they are involved in the regulation of innate immune signaling.
Figure 1.
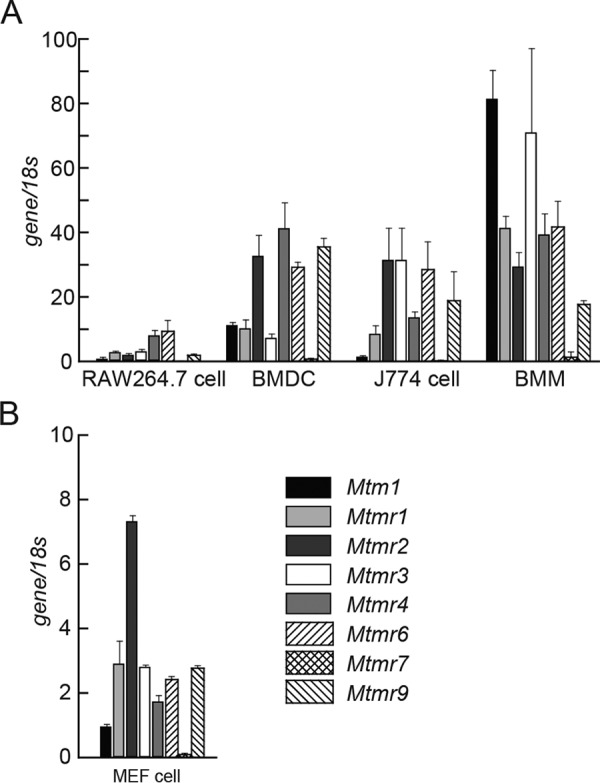
Expression of myotubularin family. A and B, relative expression of the genes encoding myotubularin family members in RAW264. 7, bone marrow–derived dendritic cells (BMDC), J774 cells, and BMMs (A) and MEF cells (B). Data are represented as mean ± SE.
Dispensable role of MTMR3 in cytokine production after TLR4, RLRs, DNA sensor stimulation
To understand the physiological function of MTMR3 and MTMR4 in innate immune responses, we generated MTMR3 knockout (KO) RAW264.7 cells by using CRISPR/Cas9 system. We designed gRNA in exon 3 regions that contained the PH Gram domain. MTMR3 KO cells showed 16-bp and 4-bp deletion in each allele (Fig. 2A). The expression of Mtmr3 was significantly decreased in MTMR3 KO cells as measured by RT-PCR (Fig. 2B). Our immunoprecipitation–Western blotting (IP-WB) analyses with anti-MTMR3 antibody demonstrated a loss of MTMR3 protein expression in MTMR3 KO cells (Fig. 2C). Then, we stimulated control and MTMR3 KO RAW264.7 cells with IFN-stimulatory DNA (ISD), poly(I:C) (RLRs ligand), and lipopolysaccharide (LPS) (TLR4 ligand) and measured Ifnb, Il6, and Cxcl10 mRNA by RT-PCR. Expression of these genes was comparable between control and MTMR3 KO cells (Fig. 2D). We also measured production of IL-6 and CXCL10 in the culture supernatant by ELISA, but these cytokines' production was also comparable between control and MTMR3 KO cells (Fig. 2E). These results suggest that MTMR3 is dispensable for cytokine expression induced by cGAS, RLRs, and TLR4 stimulation.
Figure 2.

Cytokines induction in MTMR3 KO cells. A, sequence of MTMR3 in KO cells generated by CRISPR/Cas9. Cells that have frame-shifted mutation were isolated and defined as MTMR3 KO cells. B, gene expression of Mtmr3 in control and MTMR3 KO cells. C, cell lysates of control and MTMR3 KO cells were subjected to IP-WB analysis with anti-MTMR3 antibody. D, control cells and MTMR3 KO cells were stimulated with ISD, poly(I:C), or LPS for 24 h, and expression of Ifnb, Il6, and Cxcl10 was measured by RT-PCR. E, cells were stimulated with ISD, poly(I:C), or LPS for 24 h, and IL-6 and CXCL10 levels in the culture supernatants were measured by ELISA. Data are represented as mean ± SE. *, p < 0.05 (Student's t-test).
Dispensable role of MTMR4 in cytokine production after TLR4, RLRs, DNA sensor stimulation
Next, we generated MTMR4 KO RAW264.7 cells by using the same strategy with MTMR3 KO cells. We designed the gRNA at the exon 3 and isolated MTMR4 KO cells that have 4-bp homozygous mutations (Fig. 3A). MTMR4 KO cells showed reduced Mtmr4 mRNA expression and a loss of MTMR4 protein expression (Fig. 3, B and C). Ifnb, Il6, and Cxcl10 mRNA expressions after ISD, poly(I:C), or LPS stimulation were comparable between control and MTMR4 KO cells (Fig. 3D). Moreover, IL-6 and CXCL10 production was also unimpaired in MTMR4 KO cells (Fig. 3E). These results suggest that MTMR4 is also dispensable for the regulation of innate immune responses.
Figure 3.

Cytokines induction in MTMR4 KO cells. A, sequence of Mtmr4 in KO cells generated by CRISPR/Cas9. Cells that have frame-shifted mutation were isolated and defined as MTMR4 KO cells. B, gene expression of Mtmr4 in control and MTMR4 KO cells. C, cell lysates of control and MTMR4 KO cells were subjected to IP-WB analysis with anti-MTMR4 antibody. D, control cells and MTMR4 KO cells were stimulated with ISD, poly(I:C), or LPS for 24 h, and expression of Ifnb, Il6, and Cxcl10 was measured by RT-PCR. E, cells were stimulated with ISD, poly(I:C), or LPS for 24 h, and IL-6 and CXCL10 levels in the culture supernatants were measured by ELISA. Data are represented as mean ± SE. *, p < 0.05 (Student's t-test).
Enhanced DNA-induced innate immune responses in MTMR3 and MTMR4 double KO and knockdown cells
MTMR3 and MTMR4 have similar secondary structure and show overlapped expression patterns in various cells as tested, suggesting that they may have complementary functions (Fig. 1). We therefore generated MTMR3 and MTMR4 double knockout (DKO) RAW264.7 cells using CRISPR/CAS9 system. Isolated cells having frameshift mutation in both alleles in both Mtmr3 and Mtmr4 genes were shown in Fig. 4A. Deficiency of these genes was confirmed by RT-PCR and IP-WB against anti-MTMR3 and anti-MTMR4 antibodies (Fig. 4, B and C).
Figure 4.

Enhanced innate immune response to DNA in MTMR3 and MTMR4 DKO cells. A, sequence of Mtmr3 and Mtmr4 in DKO cells generated by CRISPR/Cas9. Cells that have frame-shifted mutation in both genes were isolated and defined as MTMR3/4 DKO cells. B, gene expression of Mtmr3 and Mtmr4 in control and DKO cells. C, cell lysates of control and DKO cells were subjected to IP-WB analysis with the indicated antibodies. D, control and DKO cells were stimulated with ISD, poly(I:C), or LPS for 24 h, and expression of Ifnb, Il6, Cxcl10, and Il10 were measured by RT-PCR. E, cells were stimulated with ISD, poly(I:C), or LPS for 24 h, and the concentration of IL-6 and CXCL10 in the culture supernatant was measured by ELISA. F, Ifnb expression in control and DKO cells infected with HSV-1 was measured by RT-PCR. G, DKO cells were infected MTMR3 or MTMR4 expressing retro virus and expression of Ifnb (left), Mtmr3 (right upper) and Mtmr4 (right lower) were measured by RT-PCR. Data are represented as mean ± SE. *, p < 0.05 (Student's t-test).
We stimulated control and two different lines of MTMR3 and MTMR4 DKO (DKO1 and DKO2) cells with ISD, poly(I:C) and LPS and measured cytokine mRNA expression. Interestingly, expression of Ifnb, Il6, and Cxcl10 mRNA after ISD stimulation was significantly increased in both DKO1 and DKO2 cells compared with control cells, whereas Il10 mRNA expression was unaffected (Fig. 4D). On the other hand, Ifnb, Il6, and Cxcl10 mRNA expression after poly(I:C) and LPS stimulation was unimpaired. Consistent with these results, IL-6 and CXCL10 production after stimulation with ISD was also significantly increased in DKO1 and DKO2 cells whereas production of these cytokines after poly(I:C) or LPS was comparable among control, DKO1, and DKO2 cells (Fig. 4E). These results suggest that MTMR3 and MTMR4 negatively regulate DNA-mediated innate immune responses. To further analyze MTMR3 and MTMR4 function in DNA-sensing innate immune responses, control and DKO1 cells were infected with DNA virus HSV-1. Ifnb expression in DKO1 infected with HSV-1 was significantly higher than that in control cell (Fig. 4F). Then, we rescued MTMR3 or MTMR4 expression in DKO1 cells by retrovirus transfer system (Fig. 4G). Ifnb expression after ISD stimulation in DKO cells was higher than that in control cells, and this higher expression of Ifnb was significantly decreased by expression of MTMR3 or MTMR4. These results also suggest that MTMR3 and MTMR4 negatively regulate innate responses against DNA viruses.
A previous report (16) suggested that MTMR3 increases the activation of NLRP3 inflammasome, a protein complex that mediates caspase-1–dependent IL-1β release in response to various PAMPs or environmental stimuli via inducing autophagosome formation. Therefore, we knocked down MTMR3 and MTMR4 in primary macrophages and examined IFNβ and IL-1β induction. We electroporated siRNA for Mtmr3 and Mtmr4 into BMMs and verified knockdown efficacy by RT-PCR (Fig. 5A). Ifnb expression after ISD stimulation was increased in knockdown cells compared with control cells (Fig. 5B). IL-1β production by cells stimulated with LPS plus nigericin, a microbial toxin that triggers NLRP3 inflammasome, was significantly reduced by Mtmr3 and Mtmr4 knockdown, suggesting a crucial role of MTMR3 and MTMR4 in the NLRP3 inflammasome activation (Fig. 5C).
Figure 5.

Mtmr3 and Mtmr4 knockdown in BMMs enhances Ifnb expression after ISD stimulation. A, BMMs were electroporated with siRNA for control or Mtmr3/Mtmr4; then mRNA expression for Mtmr3 and Mtmr4 mRNA was measured by RT-PCR. B, Ifnb expression was measured by RT-PCR. C, BMMs primed with LPS were stimulated with nigericin for 3 h. The concentration of IL-1β in the culture supernatant was measured by ELISA. Data are represented as mean ± SE. *, p < 0.05 (Student's t-test).
MTMR3 and MTMR4 regulate production of PtdIns3P and suppress STING-mediated signaling
MTMR3 and MTMR4 are shown to dephosphorylate PtdIns3,5P2 and PtdIns3P and generate PtdIns5P and PtdIns, respectively (14, 15). It is therefore expected that MTMR3 and MTMR4 deficiencies result in increased PtdIns3P and reduced PtdIns5P production. To visualize cellular distributions of PtdIns3P and PtdIns5P, we expressed YFP-tagged PX domain in p40phox, an NADPH oxidase cytoplasmic component (YFP-PX p40phox) or YFP-tagged PH domain in docking protein 5 (DOK5) (YFP-PH DOK5), which binds to PtdIns3P or PtdIns5P, respectively, in RAW264.7 cells (20, 21). YFP-PX p40phox and YFP-PH DOK5 signals were observed as cytoplasmic puncta structures (Fig. 6A). To further understand cellular localization of YFP-tagged these markers preciously, cells were co-stained with a series of organelle makers EEA1 (early endosome), GM130 (Golgi), calnexin (ER), or LAMP1 (lysosome). YFP-PX p40phox localized mainly in the lysosome but partially with early endosome, whereas YFP-PH DOK5 localized mainly to unknown dotlike structures but partially localized in endosomal membrane (Fig. S1). We then expressed YFP-PX p40phox and YFP-PH DOK5 in control and MTMR3 and MTMR4 DKO cells. YFP-PX p40phox expression in DKO cells showed increased YFP-positive dotlike structures compared with control cells. By contrast, the cellular distribution of YFP-PH DOK5 signals was comparable between control and DKO cells (Fig. 6, A and B). These results suggest that DKO cells exhibited accumulation of PtdIns3P.
Figure 6.

MTMR3 and MTMR4 are required for PtdIns3P production and suppression of STING pathway. A, confocal images of PX p40phox (PtdIns3P) and PH DOK5 (PtdIns5P) localization in control and DKO cells. Scale bar = 10 μm. B, number of YFP positive puncta in control and DKO cells was counted. C and D, HEK293T cells were overexpressed with MTMR3 and MTMR4 expression plasmids along with an expression plasmid for STING, IPS-1, MyD88, or TRIF and a reporter plasmid driven by the IFNβ promoter. After 24 h, cells were lysed and the luciferase expression was measured. E, control and DKO cells were stimulated with ISD, poly(I:C), or LPS for the indicated time points. Cells lysates were subjected to WB with the indicated antibodies. Data are represented as mean ± SE. *, p < 0.05 (Student's t-test).
To understand the contribution of MTMR3 and MTMR4 to innate immune signaling, we performed a reporter assay for the IFNβ promoter. MTMR3 and MTMR4 expression plasmids were transfected together with a luciferase reporter plasmid driven by the IFNβ promoter into HEK293T cells. Overexpression of either or both MTMR3 and MTMR4 did not influence IFNβ promoter activity (Fig. 6, C and D). Then, we co-overexpressed MTMR3 and MTMR4 along with an adaptor protein; STING (an adaptor for DNA sensor), IPS-1 (an adapter for RLRs), MyD88 (an adapter for TLRs except for TLR3), or TRIF (an adapter for TLR3 and TLR4) into HEK293T cells. Expression of each adaptor protein alone significantly increased the promoter activity. However, STING-dependent promoter activation was significantly suppressed by co-expression with MTMR3 and MTMR4 whereas promoter activation induced by other adapters was unaffected by MTMR3 and MTMR4 co-overexpression (Fig. 6C). In addition, overexpression of either MTMR3 or MTMR4 significantly suppressed the IFNβ promoter activity driven by STING overexpression (Fig. 6D). These results suggest that MTMR3 and MTMR4 specifically suppress STING-mediated DNA sensing pathway. Next, we examined phosphorylation of IRF3 and p65 (NF-κB) in control and DKO cells, and found that IRF3 phosphorylation after ISD stimulation was markedly increased in both DKO1 and DKO2 cells compared with control cells, whereas IRF3 phosphorylation after poly(I:C) and LPS stimulation was unimpaired (Fig. 6E). In addition, ISD-induced p65 phosphorylation was not augmented in DKO cells (Fig. 6E). Thus, these results suggest that MTMR3 and MTMR4 repress DNA-induced IRF3 activation.
Rapid trafficking of STING from ER to Golgi in Mtmr3 and Mtmr4 DKO cells
It has been shown that STING localizes to ER in steady state and changes its localization to perinuclear Golgi after DNA stimulation (6). To visualize STING trafficking in DKO cells, we established control and DKO cells expressing FLAG-tagged STING using retrovirus transfer system, and co-stained them with anti-FLAG and anti-GM130 antibodies. Co-localization of STING with perinuclear Golgi occurred within 4 h after ISD stimulation in both control and DKO1 and DKO2 cells (Fig. 7A). Interestingly, STING localization to the perinuclear Golgi was found to occur within 1 h after ISD stimulation in DKO1 and DKO2 cells whereas control cells did not show STING trafficking to the perinuclear Golgi at this time point, suggesting that STING trafficking from ER to Golgi is more rapid in DKO cells than in control cells, and this rapid translocation may be responsible for increased responses to DNA in DKO cells. Then, we examined activation of STING by utilizing semi-native WB that detects STING dimerization (Fig. 7B). STING dimerization after ISD stimulation was more rapid and increased in MTMR3/4 DKO cells than control cells. These results suggest that accumulation of PtdIns3P by MTMR3 and MTMR4 deficiencies is responsible for STING-dependent responses.
Figure 7.

MTMR3 and MTMR4 regulate STING translocation. A, FLAG-tagged STING was transferred in control and MTMR3/4 DKO1 and DKO2 cells. Cells were stimulated with and without ISD at the indicated time points and were stained with anti-FLAG (green) and anti-GM130 (red) antibodies, a Golgi marker. B, FLAG-STING expressing control and DKO cells were stimulated with ISD and cell lysates were subjected to semi-native WB. C, PX p40phox plasmid was transfected in control and MTMR3/4 DKO1 expressing FLAG-STING. Cells were stimulated with or without ISD for 4 h and visualized. Scale bar = 10 μm.
To further address functional relationship between PtdIns3P and STING trafficking, we examined cellular localization of PX p40phox (PtdIns3P) and STING in control and DKO cells (Fig. 7C). In unstimulated condition, STING was not co-localized with PX p40phox positive puncta in both control and DKO1 cells. However, STING was partially incorporated into PX p40phox positive puncta in DKO cells after 4h stimulation with ISD, and this overlap was not observed in control cells. These results also supported by the staining with recombinant fluorescent-labeled GST-PX p40phox domain (Fig. S2). These results suggest that STING is accumulated in PtdIns3P positive puncta in DKO cells, which potentiate STING-dependent signaling pathways.
Reduced PtdIns3P level suppresses ISD-mediated innate immune response
To understand the roles of PtdIns3P in STING activation, we used wortmannin, a PI3K inhibitor that is reported to reduce the production of PtdIns3P in cells (22). We visualized cellular PtdIns3P by staining RAW264.7 cells with Alexa Fluor 647–labeled GST-PX p40phox domain. GST-PX p40phox domain signals showed dotlike structures (Fig. 8A), which were abrogated by wortmannin treatment (Fig. 8B), suggesting that wortmannin treatment reduced PtdIns3P levels. Notably, wortmannin treatment reduced ISD-stimulated Ifnb expression and IRF3 phosphorylation in RAW264.7 cells and BMMs (Fig. 8, C and D). These results suggest that PtdIns3P is required for ISD-induced IRF3 activation.
Figure 8.

Suppression of PtdIns3P production by PI3K inhibition reduces ISD response. A, confocal images of RAW264.7 cells stained with GST-PX p40phox (PtdIns3P) or GST (control). B, confocal image of RAW264.7 cells stained with GST-PX p40phox pretreated with DMSO or wortmannin. Scale bar = 10 μm. C and D, RAW264.7 cells or BMMs pretreated with DMSO or wortmannin were stimulated with ISD for the indicated periods, and Ifnb expression was measured by RT-PCR (C) or IRF3 phosphorylation was evaluated by WB with and anti-total IRF3 or anti-phosphorylated IRF3 antibodies (D). Data are represented as mean ± SE. *, p < 0.05 (Student's t-test).
Discussion
In this study, we generated single and double KO of MTMR3 and MTMR4 in RAW264.7 cells by Cas/CRISPR system. The Ifnb, Il6, and Cxcl10 mRNA level and IL-6 and CXCL10 production in MTMR3 or MTMR4 single KO cells were comparable with control cells during stimulation with ISD, poly(I:C), and LPS (Figs. 2 and 3), demonstrating that either MTMR3 or MTMR4 is dispensable for cGAS-, RLR-, and TLR4-mediated signaling. MTMR3 and MTMR4 have similar secondary structures and MTM family genes including Mtmr3 and Mtmr4 are widely expressed in MEF, macrophages, and dendritic cells, suggesting the possibility that their function is redundant (Fig. 1). Then, we generated MTMR3 and MTMR4 DKO cells and found that these cells showed significantly increased Ifnb, Il6, and Cxcl10 expression and IL-6 and CXCL10 production following ISD stimulation. Consistent with these results, IRF3 phosphorylation after ISD stimulation was increased in DKO cells (Fig. 4). These results were consistent with the results on the IFNβ promoter assay in which STING-mediated IFNβ promoter activity was repressed by overexpression of MTMR3 and MTMR4 together. Thus, both MTMR3 and MTMR4 play important roles in the negative regulation of DNA-sensing innate immune signaling pathways (Fig. 6).
MTMR3 and MTMR4 have abilities to dephosphorylate PtdIns3P and PtdIns3,5P2 to generate PI and PtdIns5P, respectively. MTMR3 and MTMR4 deficiencies may thus induce PtdIns3P increment/PI decrease, and/or PtdIns3,5P2 increment/PtdIns5P decrease. Notably, DKO cells showed increased size and number of YFP-PX p40phox (binding to PtdIns3P) positive dotlike structure compared with control cells, suggesting accumulation of PtdIns3P in DKO cells. In contrast, signals in YFP-PH DOK5 (binding to PtdIns5P)–expressing DKO cells did not show remarkable differences with those in control cells, suggesting that production of PtdIns5P was not abrogated in DKO cells (Fig. 6, A and B). Previous reports (23) demonstrated that PtdIns5P production was decreased by inhibition of MTMR3 or MTMR4 activity, whereas PtdIns3P production was increased by MTMR4 knockdown, expression of inactive mutant for MTMR4 (MTMR4C407), or MTMR3 knockdown (16, 24). Our results supported that PtdIns3P production was increased by MTMR3 and MTMR4 deficiencies, but did not support PtdIns5P reduction. It is unclear why MTMR3 and MTMR4 deficiencies in RAW264.7 cells did not affect PtdIns5P production. It is thought that PtdIns5P production in RAW264.7 cells may be preferentially regulated by other enzymes such as PIKfyve because we previously demonstrated that PtdIns5P production in murine macrophages is decreased by PIKfyve knockdown (19). Thus, PIKfyve may be responsible for PtdIns5P production in MTMR3 and MTMR4 DKO cells.
STING localizes in the ER in a steady state and traffics to perinuclear Golgi apparatus after stimulation with ISD (5, 6). We hypothesized that accumulation of PtdIns3P by MTMR3 and MTMR4 deficiencies may change the STING localization. Our results showed that STING reached to Golgi after 4-h stimulation in control and DKO cells, but it translocated more rapidly to Golgi in DKO cells than in control cells (Fig. 7A). It was shown that TBK1 is recruited to STING-positive perinuclear puncta after DNA stimulation and becomes activated, suggesting that TBK1 translocation to Golgi and subsequent activation is coupled to IRF3 activation. Thus, rapid translocation of STING from ER to Golgi may contribute to enhanced innate immune responses in DKO cells.
It was reported that PtdIns3P is predominantly accumulated in early endosome, but it also widely distributed in other cellular membranes (25). Moreover, PtdIns3P is well-known to regulate autophagosome formation. Autophagy initiates its biogenesis through a pre-autophagosomal double membrane structure that is a subdomain of ER membrane positive for PtdIns3P and PtdIns3P-binding proteins (26). Consistently, it is reported that knockdown of MTMR3 increased autophageosome formation (17) whereas increased MTMR3 expression reduced autophagy (16). Autophagy is shown to have an anti-inflammatory role, because disruption of autophagy-related protein in mice results in increased inflammation and type I IFN production during innate immune signaling pathways (27, 28). Our results showing that PtdIns3P is increased in DKO cells suggest that the autophagy formation is accumulated in these cells, and inflammation is suppressed. However, MTMR3/4 DKO cells and knockdown cells rather exhibited enhanced type I IFN responses during STING activation, whereas MTMR3/4 knockdown in BMMs results in reduced IL-1β production during NLR3 inflammasome activation. This suggests the possibility that autophagosome formation is unimpaired in DKO cells irrespective of increased PtdIns3P, or a rapid translocation of STING to Golgi may negatively regulate PtdIns3P-dependent autophagosome formation via unknown pathways. The relationship between autophagy and MTMR3- and MTMR4-dependent STING regulation should be understood in the future.
Our results demonstrate that STING translocates from ER to Golgi at an earlier time point in DKO cells than in control cells. This suggests that accumulation of PtdIsn3P by MTMR3/4 deficiencies enhances translocation of STING. ER has a dynamic and complex membrane network that extends to other endosomal organelle. Recent studies suggested that autophagosome biogenesis is initiated at a contact site of ER to other organelles, such as mitochondria and plasma membrane (22, 29), suggesting that PtdIns3P is generated at a specific site in ER. Because STING localizes to ER in the steady state, a functional interaction between STING and PtdIns3P in ER may be responsible for the trafficking of STING to Golgi. Indeed, reduced PtdIns3P by wortmannin treatment reduced ISD-stimulated Ifnb expression and IRF3 phosphorylation. Thus, it is possible that PtdIns3P is required for ISD-mediated innate immune response. However, it is still unclear how PtdIns3P regulates STING translocation, but several GPCR and ion channels are reported to bind to PtdIns4,5P2 and regulate their functions (30). One possibility is that PtdIns3P may also directly bind to STING and induce the change of STING conformation, which induces STING dimerization and translocation of STING to Golgi. This was also supported by our observation that STING dimer formation is enhanced by MTMR3 and MTMR4 deficiencies (Fig. 7B). Association of PtdIns3P with STING is really suggested by our observation in which STING is aberrantly incorporated into PtdIns3p positive puncta in DKO cells (Fig. 7C and Fig. S2). Another possibility is that PtdIns3P provides a platform for association of STING and STING-regulatory proteins such as iRhom2, TRIM32, and TRIM56.
Although the regulatory role of translocation of STING by MTMR3/4 is still unclear, this study showed that MTMR3/4-dependent PtdIns metabolism is important in fine tuning innate immune responses to DNA. Self-DNA is aberrantly recognized by cGAS when it is released from the cytoplasm after host cells have died by necrosis or are damaged by stresses, drugs, aging, and infection. These results in unwanted activation of STING pathway appear to be highly associated with inflammatory and autoimmune diseases (31). Thus, MTMR3 and MTMR4 may be potential targets of drug design that can prevent these diseases. Also, it is important to analyze a mutation or SNP in MTMR3 and MTMR4 genes in these diseases.
Experimental procedures
Cells and reagents
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Nacalai Tesque) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies), and Plat-E cells were cultured in DMEM containing 100 μm 2-mercaptoethanol (Nacalai Tesque), 1 μg/ml puromycin (InvivoGen), and 10 μg/ml Blasticidin (InvivoGen) with 10% FBS at 37 °C in humidified 5% CO2. Murine bone marrow cells were treated with 10 ng/ml M-CSF (Proteo Tech) or 10 ng/ml GM-CSF (Proteo Tech), 100 μm 2-mercaptoethanol, and incubated for differentiation 6 to 8 days at 37 °C and 5% CO2 with Roswell Park Memorial Institute (RPMI) 1640 (Nacalai Tesque) containing 100 units/ml penicillin, 100 μg/ml streptomycin (Nacalai Tesque), and 10% FBS and used as BMM and BM-DC, respectively.
Generation of MTMR3 or MTMR4 KO cells
To establish single MTMR3 or MTMR4 KO RAW264.7 cells, guide sequences located in exon 3 of Mtmr3 and exon 3 of Mtmr4 were inserted into pX330-U6-Chimeric_BB-CBh-hSpCas9 (42230, Addgene) that expresses Cas9 and gRNA (Mtmr3: sense 5′-GCATAGCCTTGAGTGCATCC-3′; Mtmr4: sense 5′-TGAGGAGGGCCCCCCCAGCC-3′). Target DNA sequences for Cas9 were subcloned into px330 plasmid that expresses hCas9 and trans-activating CRISPR RNA (trcrRNA). gRNA is located next to PAM sequence (NGG) and 20-bp sequence which potentially shows low off-target effects that do not crossreact with other sites in genome. Genomic regions containing guide sequences were inserted into the pCAG EGxxFP plasmid (32), which is a reporter for genome editing. Then these plasmids were electroporated into RAW264.7 cells by NEON (Invitrogen) at 1680 V and 20 ms. GFP-positive cells were sorted by a BD FACS Aria (BD Bioscience). To obtain MTMR3 and MTMR4 DKO cells, stably expressing Cas/CRISPR RAW264.7 cells were generated. Synthesized trcrRNA and gRNA (or crRNA) were electroporated into the cells (target gRNA for MTMR3 exon 2: sense 5′-GTTCCTTTCCTTGAACTTCA-3′; MTMR4 exon 2: sense 5′-GGGTGAGGGAGTGGAATTCC-3′). One μl of 1.0 μg/ml crRNA and 3 μl of 0.5 μg/ml tracrRNA were mixed with 6 μl of cell suspension (1 × 107/ml). RNA and cell mixture were subjected to electroporation using NEON. Electroporated cells were titrated and suspended to 96-well plates. Cells were cultured for around 2 weeks until cellular density reached 70%. Then, cells were transferred to 24-well plates and DNA were isolated for sequence analysis.
Knockdown
Knockdown for targeting sequence is as follows: Mtmr3, ATGTTACTCGAAGATAAGGTA; Mtmr4, AAGTTGTTCTCTTAGTTATAA. Sense and antisense oligo RNA were synthesized with AA overhang (FASMAC). BMMs (4 × 105) were mixed with 3 μm siRNA and were electroplated with 1400 V, 20 ms, and 1 plus by NEON transfection system (Thermo Fisher Scientific).
Western blotting and immunoprecipitation
Cells were stimulated with the indicated ligand and were lysed with RIPA buffer (50 mm Tris HCl, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS). The supernatant obtained by centrifuging at 800 × g for 10 min was used as whole cell lysates. Whole cell lysates were mixed with 2 × SDS sample buffer (1 m Tris-HCl, 10% SDS, 20% glycerol, 0.01% bromphenol blue, 0.2 m DTT) and heat-treated at 95 °C for 5 min. Samples were subjected to SDS-PAGE and proteins were transferred to PVDF membrane (Bio-Rad). Transferred membranes were applied for blocking by TBST buffer (0.5 m Tris, 1.38 m NaCl, 0.027 m KCl, 0.05% Tween 20) containing 5% skim milk and were incubated with anti-pIRF3, IRF3, pp65, or p65 antibody (Cell Signaling Technology). For IP-WB, control, and KO cells cultured in 6-well plates were lysed with IP buffer (150 mm NaCl, 10 mm Tris-HCl, pH8.0, 10 mm EDTA, 2 mm EGTA, 0.2% Tween 20). Lysates were incubated with anti-MTMR3 (Cell Signaling Technology) or anti-MTMR4 antibody (Abgent) with Protein A agarose beads (Thermo Fisher Scientific) and were rotated for over night at 4 °C. Antibody-bounded Protein A beads were collected by spin down and were washed for three times with IP buffer. Sample buffer was applied to Protein A beads and samples were performed for WB against anti-MTMR3 antibody (Cell Signaling Technology) or anti-MTMR4 antibody (Proteintech Group). As an internal control, cell lysates were blotted with anti–actin β antibody (Santa Cruz Biotechnology). After incubation with HRP-conjugated secondary antibodies (Sigma) for 30 min, membranes were reacted with a luminescent reagent Western Lightning Plus-ECL (PerkinElmer), and proteins were detected by Image Quant LAS-4000 (GE Healthcare).
Semi-native WB
STING dimerization was assayed under semi-native condition. FLAG-STING expressing cells were stimulated with ISD and were lysed with native sample buffer (1.5 mm Tris HCl, pH 6.8, 15% glycerol, 1% deoxycholate). Whole cell lysates were run on the standard SDS-PAGE and proteins were transferred to PVDF membrane (Bio-Rad), and blotted with anti-FLAG antibody.
RNA isolation and RT-PCR
Total RNA were isolated with TRIzol reagent (Invitrogen) and reverse transcribed with ReverTra Ace (Toyobo) according to the manufacturers' instructions. RT-PCR was performed with the following primers: Mtm1, sense 5′-ATCTTAGGAGGATCGCAACG-3′, reverse 5′-TACCGACAAGAGGCTGACTG-3′; Mtmr1, sense 5′-TCCATTTATGGGAGCAGTGA-3′, reverse 5′-TGCACCAATCTTCTCCACTC-3′; Mtmr2, sense 5′-AGCAGAAATGGAGGAACCAG-3′, reverse 5′-CGCTCCATGCTCTTGAAATA-3′; Mtmr3, sense 5′-TGGGAATGTATTCTGCTCCA-3′, reverse 5′-CAATGCTGGAGCTTGTAGGA-3′; Mtmr4, sense 5′-TGTGTCCTGGAAATGAAGA-3′, reverse 5′-AAACTGTTCAGGTGGCTTCC-3′; Mtmr6, sense 5′-TTCCGGTCCTATCCTACTGC-3′, reverse 5′-TGAAGCAAATGCTCATCCTC-3′; Mtmr7, sense 5′-CCAGTTTGGGAACTTCCTGT-3′, reverse 5′-GGCAGCTCGATTCTTCCACA-3′; Mtmr8 sense 5′-GGCTCTCTGGGTACCATCAT-3′, reverse 5′-GAGTGGACAATGCCTCAATG-3′. Primer for Ifnb, Il6, Il10, Cxcl10, and 18s were described previously (33, 34).
ELISA
RAW264.7 cells seeded in 96-well plates were stimulated with 2 μg/ml ISD, 1 μg/ml poly(I:C) (InvivoGen) or 100 ng/ml LPS (InvivoGen) for 24 h. Sense and antisense ISD sequences were synthesized (Fasmac) and annealed manually. ISD and poly(I:C) were incubated with Lipofectamine 2000 (Life Technologies) at a ratio of 1:1 in Opti-MEM (Life Technologies). The IL-6 and CXCL10 concentration in the culture supernatants was measured by a mouse Duoset (R&D Systems) according to the manufacturer's instructions.
Luciferase reporter assay
HEK293T cells were seeded on 24-well plates and were transfected with 100 ng of IFNβ reporter plasmids along with 500 ng of an expression plasmid for MTMR3, MTMR4, STING, IPS-1, MyD88, or TIRF. Ten ng of pRL-TK (Promega) was co-transfected as internal control. MTMR3 and MTMR4 were amplified from mouse cDNA and were subcloned into pFLAG-CMV2 (Sigma). Other plasmids were described previously (33). After 24-h transfection, luciferase activity was measured as described previously (33, 34).
Fluorescent microscope
Mouse STING gene was subcloned into pMRX-Puro vector. pMRX-STING-FLAG-Puro was transfected into plat-E cells and the supernatants were transferred to RAW264.7 cells. Then, cells were stained with anti-FLAG antibody (Sigma), anti-GM130 (BD Bioscience), and DAPI (Dojin). PX domain in p40phox and PH domain in DOK5 were amplified by PCR and subcloned into pYpetC3 vector. pYpetC3-PX p40phox and pYpetC3-PH DOK5 were electroporated into RAW264.7 cells using NEON. Cells were observed with fluorescence microscope LSM 700 (Zeiss) and the image was processed with ZEN software (Zeiss).
Staining with recombinant PX domain
PX P40phox domain was subcloned into pGEX-6p1 (GE Healthcare). GST-PX P40phox domain was purified from BL21 Escherichia coli (Takara) after induction with isopropyl β-d-thiogalactopyranoside (IPTG). GST-PX p40phox domain was labeled with Alexa Fluor 647 (Thermo Fisher). RAW264.7 cells were fixed with 4% paraformaldehyde and blocked with 10% FBS/PBS. Then, cells were permeated by flash frozen liquid nitrogen. Cells were stained with 20 ng/ml GST-PX p40phox domain and washed with PBS. To further stain with antibody, cells were fixed with 4% paraformaldehyde.
Statistics
All experiments were independently repeated at least three times. Statistical significance was determined by the Student's t test. A p value of less than 0.05 was considered significant.
Author contributions
D. D. P. P. and T. Kawasaki investigation; D. D. P. P. and T. Kawasaki writing-original draft; T. Kawasaki data curation; T. Kawasaki, M. M., T. S., T. D., D. O., and S. S. methodology; T. Kawasaki and T. Kawai writing-review and editing; T. Kawai conceptualization; T. Kawai formal analysis; T. Kawai supervision; T. Kawai funding acquisition; T. Kawai validation; T. Kawai project administration.
Supplementary Material
Acknowledgments
We thank K. Abe and C. Suzuki for secretarial assistance.
This study was supported by MEXT KAKENHI Grant-in-Aid for Scientific Research B 17H04066 and Young Scientist B 17K15598 and by Takeda Science Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
- PAMP
- pathogen-associated molecular pattern
- TLR
- toll-like receptors
- RLR
- RIG-I–like receptor
- HSV
- herpes simplex virus
- ER
- endoplasmic reticulum
- PtdIns
- phosphatidylinositol
- MTM
- myotubularin
- BMM
- bone marrow–derived macrophage
- MEF
- murine embryonic fibroblast
- gRNA
- guide RNA
- IP-WB
- immunoprecipitation–Western blotting
- ISD
- IFN-stimulatory DNA
- LPS
- lipopolysaccharide
- DKO
- double knockout.
References
- 1. Kawai T., and Akira S. (2010) The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 11, 373–384 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 2. Wu J., and Chen Z. J. (2014) Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488 10.1146/annurev-immunol-032713-120156 [DOI] [PubMed] [Google Scholar]
- 3. Ma Z., and Damania B. (2016) The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19, 150–158 10.1016/j.chom.2016.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun L., Wu J., Du F., Chen X., and Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishikawa H., Ma Z., and Barber G. N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Q., Sun L., and Chen Z. J. (2016) Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 10.1038/ni.3558 [DOI] [PubMed] [Google Scholar]
- 7. Takahama M., Fukuda M., Ohbayashi N., Kozaki T., Misawa T., Okamoto T., Matsuura Y., Akira S., and Saitoh T. (2017) The RAB2B-GARIL5 complex promotes cytosolic DNA-induced innate immune responses. Cell Rep. 20, 2944–2954 10.1016/j.celrep.2017.08.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seo G. J., Kim C., Shin W. J., Sklan E. H., Eoh H., and Jung J. U. (2018) TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 9, 613 10.1038/s41467-018-02936-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J., Hu M. M., Wang Y. Y., and Shu H. B. (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655 10.1074/jbc.M112.362608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsuchida T., Zou J., Saitoh T., Kumar H., Abe T., Matsuura Y., Kawai T., and Akira S. (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 10.1016/j.immuni.2010.10.013 [DOI] [PubMed] [Google Scholar]
- 11. Wymann M. P., and Schneiter R. (2008) Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 9, 162–176 10.1038/nrm2335 [DOI] [PubMed] [Google Scholar]
- 12. Liu Y., and Bankaitis V. A. (2010) Phosphoinositide phosphatases in cell biology and disease. Prog. Lipid Res. 49, 201–217 10.1016/j.plipres.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson F. L., and Dixon J. E. (2006) Myotubularin phosphatases: Policing 3-phosphoinositides. Trends Cell Biol. 16, 403–412 10.1016/j.tcb.2006.06.001 [DOI] [PubMed] [Google Scholar]
- 14. Taylor G. S., Maehama T., and Dixon J. E. (2000) Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. U.S.A. 97, 8910–8915 10.1073/pnas.160255697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walker D. M., Urbé S., Dove S. K., Tenza D., Raposo G., and Clague M. J. (2001) Characterization of MTMR3, an inositol lipid 3-phosphatase with novel substrate specificity. Curr. Biol. 11, 1600–1605 10.1016/S0960-9822(01)00501-2 [DOI] [PubMed] [Google Scholar]
- 16. Lahiri A., Hedl M., and Abraham C. (2015) MTMR3 risk allele enhances innate receptor-induced signaling and cytokines by decreasing autophagy and increasing caspase-1 activation. Proc. Natl. Acad. Sci. U.S.A. 112, 10461–10466 10.1073/pnas.1501752112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taguchi-Atarashi N., Hamasaki M., Matsunaga K., Omori H., Ktistakis N. T., Yoshimori T., and Noda T. (2010) Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic 11, 468–478 10.1111/j.1600-0854.2010.01034.x [DOI] [PubMed] [Google Scholar]
- 18. Teo W. X., Kerr M. C., and Teasdale R. D. (2016) MTMR4 is required for the stability of the Salmonella-containing vacuole. Front. Cell. Infect. Microbiol. 6, 91 10.3389/fcimb.2016.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kawasaki T., Takemura N., Standley D. M., Akira S., and Kawai T. (2013) The second messenger phosphatidylinositol-5-phosphate facilitates antiviral innate immune signaling. Cell Host Microbe 14, 148–158 10.1016/j.chom.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 20. Guittard G., Mortier E., Tronchère H., Firaguay G., Gérard A., Zimmermann P., Payrastre B., and Nunès J. A. (2010) Evidence for a positive role of PtdIns5P in T-cell signal transduction pathways. FEBS Lett. 584, 2455–2460 10.1016/j.febslet.2010.04.051 [DOI] [PubMed] [Google Scholar]
- 21. Ueyama T., Tatsuno T., Kawasaki T., Tsujibe S., Shirai Y., Sumimoto H., Leto T. L., and Saito N. (2007) A regulated adaptor function of p40phox: Distinct p67phox membrane targeting by p40phox and by p47phox. Mol. Biol. Cell 18, 441–454 10.1091/mbc.e06-08-0731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nascimbeni A. C., Giordano F., Dupont N., Grasso D., Vaccaro M. I., Codogno P., and Morel E. (2017) ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 36, 2018–2033 10.15252/embj.201797006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oppelt A., Lobert V. H., Haglund K., Mackey A. M., Rameh L. E., Liestøl K., Schink K. O., Pedersen N. M., Wenzel E. M., Haugsten E. M., Brech A., Rusten T. E., Stenmark H., and Wesche J. (2013) Production of phosphatidylinositol 5-phosphate via PIKfyve and MTMR3 regulates cell migration. EMBO Rep. 14, 57–64 10.1038/embor.2012.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naughtin M. J., Sheffield D. A., Rahman P., Hughes W. E., Gurung R., Stow J. L., Nandurkar H. H., Dyson J. M., and Mitchell C. A. (2010) The myotubularin phosphatase MTMR4 regulates sorting from early endosomes. J. Cell Sci. 123, 3071–3083 10.1242/jcs.060103 [DOI] [PubMed] [Google Scholar]
- 25. Marat A. L., and Haucke V. (2016) Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 35, 561–579 10.15252/embj.201593564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., Griffiths G., and Ktistakis N. T. (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., and Akira S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- 28. Saitoh T., Fujita N., Hayashi T., Takahara K., Satoh T., Lee H., Matsunaga K., Kageyama S., Omori H., Noda T., Yamamoto N., Kawai T., Ishii K., Takeuchi O., Yoshimori T., and Akira S. (2009) Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U.S.A. 106, 20842–20846 10.1073/pnas.0911267106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y., Amano A., and Yoshimori T. (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 10.1038/nature11910 [DOI] [PubMed] [Google Scholar]
- 30. Hille B., Dickson E. J., Kruse M., Vivas O., and Suh B. C. (2015) Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 1851, 844–856 10.1016/j.bbalip.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahn J., and Barber G. N. (2014) Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol. 31, 121–126 10.1016/j.coi.2014.10.009 [DOI] [PubMed] [Google Scholar]
- 32. Mashiko D., Fujihara Y., Satouh Y., Miyata H., Isotani A., and Ikawa M. (2013) Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 3, 3355 10.1038/srep03355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murase M., Kawasaki T., Hakozaki R., Sueyoshi T., Putri D. D. P., Kitai Y., Sato S., Ikawa M., and Kawai T. (2018) Intravesicular acidification regulates lipopolysaccharide inflammation and tolerance through TLR4 trafficking. J. Immunol. 200, 2798–2808 10.4049/jimmunol.1701390 [DOI] [PubMed] [Google Scholar]
- 34. Sueyoshi T., Kawasaki T., Kitai Y., Ori D., Akira S., and Kawai T. (2018) Hu antigen R regulates antiviral innate immune responses through the stabilization of mRNA for Polo-like kinase 2. J. Immunol. 200, 3814–3824 10.4049/jimmunol.1701282 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.