

Abstract
KGF-1 plays an important role in the wound healing process. Loss of the KGF-1 gene in diabetic mice attenuated the process of wound contraction, suggesting that KGF-1 contributes to wound contraction. However, the mechanism remains unclear. To investigate the role of KGF-1 in diabetic wound contraction, we established a keratinocyte–fibroblast co-culture system. Concentrations of transforming growth factor β1 (TGF-β1) in conditioned supernatant treated with KGF-1 (KGF-1 group), tk;4KGF-1-neutralizing antibody (anti-KGF-1 group), TGF-β1 (TGF-β1tk;1 group), KGF-1 and TGF-β1-neutralizing antibody (KGF-1 + anti-TGF-β1 group) were tested by ELISA. Conditioned medium was added to fibroblast-populated collagen lattice (FPCL) to investigate the effect of KGF-1 on fibroblastqj contraction. TGF-β1, Col-I, p-Smad2, p-Smad3, and α-smooth muscle actin (α-SMA) were examined by Western blotting. A diabetic rat wound model was utilized to evaluate wound morphology, histology, immunohistochemistry, and protein expression in wound tissue after treatment with KGF-1. ELISA assays revealed that the concentration of TGF-β1 in the conditioned supernatant in the KGF-1 group was significantly higher. The contractile capacity of FPCL stimulated by conditioned medium derived from the KGF-1 group was significantly elevated; however, the contractile activity of FPCL induced by KGF-1 was attenuated by TGF-β1–neutralizing antibody. The Western blot results suggest that KGF-1 is able to stimulate TGF-β1 activation with increased Col-I, p-Smad2, p-Smad3, and α-SMA expression. Diabetic wounds treated with KGF-1 had a higher degree of contraction with significantly higher expression of TGF-β1, Col-I, p-Smad2, p-Smad3, and α-SMA. Our findings demonstrate that KGF-1 promotes fibroblast contraction and accelerates wound contraction via the TGF-β1/Smad signaling pathway in a double-paracrine manner.
Keywords: wound healing, transforming growth factor β (TGF-β), cell migration, cell proliferation, cell–cell interaction, diabetic wound, epithelial–mesenchymal interaction, wound contraction
Introduction
Epithelial–mesenchymal interaction plays a critical role in the regulation of wound healing, mainly via growth factors and cytokines and through complicated signaling pathways (1). Previous studies have demonstrated that, after wound formation, skin fibroblasts (mesenchymal cells) can secrete growth factors such as keratinocyte growth factor 1 (KGF-1) and granulocyte macrophage colony-stimulating factor (GM-CSF),3 promoting keratinocyte (epithelial cell) proliferation, migration, and differentiation (2–5). On the other hand, epidermal cells also regulate mesenchymal cells through cytokines and growth factors. For example, keratinocytes act on fibroblasts by secreting IL-1 and activating protein 1 (AP-1) during wound healing (6). Epithelial–mesenchymal interaction affects the proliferation, migration, secretion, and differentiation of skin epidermal cells (such as keratinocytes) and mesenchymal cells (such as vascular endothelial cells and fibroblasts), thus influencing wound contraction, angiogenesis, and re-epithelialization.
KGF-1, also known as fibroblast growth factor 7, is secreted by various mesenchymal cells (predominately fibroblasts) and exerts its effects specifically via the KGF-1 receptor FGFR2-IIIb (7). Studies have shown that KGF-1 effectively promotes wound healing by facilitating re-epithelialization (8) and vascularization (9). Interestingly, our previous study found that loss of the KGF-1 gene attenuated the process of wound contraction in diabetic mice (10), and these results indicated that KGF-1 plays a critical role in wound contraction. However, the underlying mechanism of KGF-1 affecting wound contraction is currently not explicitly understood. It is known that myofibroblasts are responsible for contracting dermal tissue in wound healing (11). There are two major factors that affect the differentiation of fibroblasts into myofibroblasts: mechanical tension (12) and active transforming growth factor β1 (TGF-β1) (13). TGF-β1 is involved in various processes of wound healing, including extracellular matrix (ECM) synthesis, α-smooth muscle actin (α-SMA) expression, and fibroblast behavior (14, 15). When stimulated by active TGF-β1, fibroblasts can differentiate into myofibroblasts, which characteristically express α-SMA and develop contractile ability (16). Simultaneously, ECM is synthesized to accelerate the wound healing. Hsieh et al. (17) demonstrated that TGF-β1 has a synergistic effect with Col-I ECM gel stimulated by fibroblasts during wound contraction. The TGF-β1/Smad signaling pathway plays a crucial role in fibroblast contraction (18), which is initiated by binding of TGF-β1 with its receptors (TβRII and TβRI). The activated receptor complex then phosphorylates Smad proteins that propagate the signal (19, 20). KGF-1 is not able to directly act on fibroblasts, as there is no KGF-1 receptor on fibroblasts. However, keratinocytes have been demonstrated to secrete TGF-β1 (21). Hence, we assumed that KGF-1 has an indirect effect on fibroblasts by inducing TGF-β1 expression in keratinocytes, thus stimulating fibroblasts contraction. Therefore, in this study, we sought to prove our hypothesis that KGF-1 accelerates wound contraction by promoting TGF-β1–induced fibroblast contraction via the TGF-β1/Smad signaling pathway.
Results
KGF-1 stimulates active TGF-β1 secretion from HaCaT keratinocytes
TGF-β1 is regarded to be the main factor influencing fibroblast contraction (22). To determine whether KGF-1 can enhance TGF-β1 expression, we used a KGF-1–neutralizing antibody and a TGF-β1–neutralizing antibody to block KGF-1 or TGF-β1 protein in conditioned medium of keratinocyte–fibroblast co-cultures (Fig. 1, a and b). The concentration of TGF-β1 in the conditioned medium of each group in the HaCaT keratinocyte–fibroblast co-cultures at 48 h was analyzed by ELISA. The TGF-β1 protein concentration was significantly higher with addition of KGF-1 or TGF-β1 compared with the control group, anti-KGF-1 group, or KGF-1 + anti-TGF-β1 group. The concentration of TGF-β1 in the KGF-1 group was ∼1.5-fold higher than in the control group. On the other hand, the concentration of TGF-β1 in the anti-KGF-1 group was found to be significantly lower than in the control group (Fig. 1c). TGF-β1–neutralizing antibodies are capable of abolishing the elevation in TGF-β1 induced by KGF-1. Furthermore, RT-PCR and Western blot results showed that KGF-1 treatment induced HaCaT keratinocytes to secrete TGF-β1 in the absence of fibroblasts, whereas KGF-1 treatment had no effect on fibroblasts in the absence of keratinocytes (Fig. 1, d–g). These results indicate that KGF-1 is able to induce active TGF-β1 expression in keratinocytes.
Figure 1.

TGF-β1 concentration was examined by ELISA assay. Keratinocyte and fibroblast co-cultures were treated with PBS (Control), rhKGF-1 (KGF-1), KGF-1–neutralizing antibody (Anti-KGF-1), rhTGF-β1 (TGF-β1) and KGF-1 + TGF-β1–neutralizing antibody (KGF-1 + Anti-TGF-β1). a, the KGF-1–neutralizing antibody effectively blocked KGF-1 in conditioned medium. b, the TGF-β1–neutralizing antibody functionally blocked TGF-β1 in conditioned medium. c, the concentration of TGF-β1 in conditioned medium from each co-culture group. d, HaCaT cells and HDFs were treated with KGF-1. a.u., arbitrary unit. e–g, TGF-β1 mRNA expression, TGF-β1 protein level (e and f), and TGF-β1 concentration in conditioned medium (g) from each group were measured. Data are shown as mean ± S.D. *, p < 0.05; **, p < 0.01.
KGF-1 promotes TGF-β1–induced contraction of FPCL
To investigate the effect of KGF-1 on wound contraction in vitro, an FPCL assay was applied. In this study, FPCLs were cultured in the conditioned medium of co-cultures supplemented with either 50 ng/ml recombinant human KGF-1 (rhKGF-1, KGF-1 group), 0.5 μg/ml KGF-1–neutralizing antibody (anti-KGF-1 group), 5 ng/ml recombinant human TGF-β1 (rhTGF-β1, TGF-β1 group), 50 ng/ml rhKGF-1 + 10 μg/ml TGF-β1–neutralizing antibody (KGF-1 + anti-TGF-β1 group), or PBS (control group). Each FPCL was imaged at 0, 12, and 24 h, and contraction rates were measured using ImageJ. We found that FPCLs cultured in co-cultured conditioned medium pretreated with KGF-1 or TGF-β1 displayed a significantly greater contraction rate than that of the anti-KGF-1, KGF-1 + anti-TGF-β1, or control groups at 12 and 24 h (Fig. 2, a and b). Moreover, the elevated contractibility of FPCLs caused by KGF-1 was inhibited by the TGF-β1–neutralizing antibody, as demonstrated by less FPCL contraction in the KGF-1 + anti-TGF-β1 group relative to the KGF-1 group. The anti-KGF-1 group showed a lower contraction rate than the control group; however, there was no significant difference between the two groups. Afterward, FPCLs were cultured in medium conditioned only by HaCaT keratinocytes preincubated with either rhKGF-1 (kCM:KGF-1 group), TGF-β1–neutralizing antibody (kCM:anti-TGF-β1 group), rhKGF-1 + TGF-β1–neutralizing antibody (kCM:KGF-1:anti-TGF-β1 group), or PBS (kCM group). An additional control with FPCLs treated with rhKGF-1 (KGF-1 group) was also included. FPCLs in the kCM:KGF-1 group showed the strongest contractility (Fig. 2, c and d). However, the TGF-β1–neutralizing antibody severely impaired FPCL contractility; even the FPCL contraction rate increased by KGF-1 was attenuated dramatically when the TGF-β1–neutralizing antibody was added. Taken together, these results indicate that KGF-1 promotes FPCL contraction via TGF-β1.
Figure 2.

FPCLs were cultured with conditioned medium collected from each group. a, images of FPCLs at 0, 12, and 24 h. Scale bars = 3 mm. FPCLs were incubated with conditioned medium of co-cultures supplemented with rhKGF-1 (KGF-1), KGF-1–neutralizing antibody (anti-KGF-1), rhTGF-β1 (TGF-β1), or rhKGF-1 + TGF-β1–neutralizing antibody (KGF-1 + anti-TGF-β1). b, the relative contraction rates of FPCLs of each group. c, images of FPCLs at 0, 12, and 24 h. Scale bars = 5 mm. FPCLs were incubated with conditioned media of HaCaT keratinocytes (kCM) pretreated with rhKGF-1 (kCM:KGF-1), TGF-β1–neutralizing antibody (kCM:anti-TGF-β1), rhKGF-1 and TGF-β1–neutralizing antibody (kCM:KGF-1:anti-TGF-β1) and compared with FPCLs treated with rhKGF-1 (KGF-1). d, the relative contraction rates of FPCLs of each group. Data are shown as mean ± S.D. *, p < 0.05; **, p < 0.01.
KGF-1 induces fibroblast differentiation through the TGF-β1/Smad signaling pathway
Studies have shown that the TGF-β1/Smad signaling pathway can induce differentiation of fibroblasts into myofibroblasts (23). Myofibroblasts promote wound contraction by expressing α-SMA and secreting Col-I. Western blotting was used to determine changes in the levels of proteins associated with the TGF-β1/Smad signaling pathway and wound contraction. We therefore examined expression of the level of Col-I, the phosphorylation level of Smad2 (p-Smad2), the total level of Smad2 (Smad2), the phosphorylation level of Smad3 (p-Smad3), the total level of Smad3 (Smad3), and α-SMA in fibroblasts and TGF-β1 in HaCaT keratinocytes in a co-culture system after treatment with rhKGF-1 (KGF-1 group), KGF-1–neutralizing antibody (anti-KGF-1 group), rhKGF-1 + TGF-β1–neutralizing antibody (KGF-1 + anti-TGF-β1 group), and PBS (control group) for 48 h. We found that TGF-β1 and downstream p-Smad2 as well as p-Smad3 expression levels in the KGF-1 group were significantly higher than in the anti-KGF-1, KGF-1 + anti-TGF-β1, or control groups (Fig. 3, a–c). Similar changes in expression of α-SMA and Col-I that related to wound contraction and healing were found in each group. In addition, expression of TGF-β1, Col-I, p-Smad2, p-Smad3, and α-SMA was significantly inhibited by the TGF-β1–neutralizing antibody relative to the KGF-1 group. These findings suggest that KGF-1 induces fibroblast differentiation via the TGF-β1/Smad signaling pathway.
Figure 3.

KGF-1 increased expression of Col-I and α-SMA via the TGF-β1/Smad signaling pathway in vitro. a, expression of Col-I, p-Smad2, Smad2, p-Smad3, Smad3, and α-SMA in fibroblasts and TGF-β1 in HaCaT was examined after HaCaT keratinocyte and fibroblast co-cultures were treated with rhKGF-1 (KGF-1), PBS (Control), KGF-1–neutralizing antibody (Anti-KGF-1), and KGF-1 + TGF-β1–neutralizing antibody (KGF-1 + Anti-TGF-β1), respectively, for 48 h. b, results expressed as a percentage of the control. Each protein level is standardized to that of GAPDH as the internal control. c, protein expression ratio of p-Smad2/Smad2 and p-Smad3/Smad3 (d) in each group. Data are shown as mean ± S.D. *, p < 0.05; **, p < 0.01.
KGF-1 accelerates wound contraction and healing in diabetic rats
Previous studies have shown that KGF-1 knockout diabetic mice have an attenuated process of wound contraction (10); therefore, we investigated the effects of KGF-1 on wound contraction in diabetic rats. We found that, after rhKGF-1 (50 ng/ml) treatment, both wound contraction and wound healing rates increased remarkably compared with the untreated wound (PBS) at each time point from day 0 to day 12 post-injury (Fig. 4, a–c). In addition, H&E staining of wound tissue sections showed that wound contraction was significantly greater in the KGF-1 group relative to the control group on day 12 post-injury (Fig. 5a). The wound contraction and wound healing rates in the KGF-1 group were significantly higher than that of the control group (Fig. 5b). The newly formed epithelial tissue in wounds treated with KGF-1 was observed to migrate further to the wound center (Fig. 5c). Taken together, the findings reveal that KGF-1 significantly promotes contraction and healing of diabetic wounds.
Figure 4.

KGF-1 promoted wound contraction and wound healing in diabetic rats. Full-thickness wounds were excised on each diabetic rat's dorsal side and treated separately with KGF-1 (50 ng/ml) and PBS for 12 consecutive days. a, representative images of wounds of the KGF-1 and control group from day 0 to day 12 after wound injury. b and c, graphical representation of wound contraction (b) and wound healing (c) rates from day 0 to day 12 post-injury in the KGF-1 and control groups. Scale bars = 1 cm. Data are shown as mean ± S.D. *, p < 0.05.
Figure 5.

H&E staining revealed the effects of KGF-1 on diabetic wound tissue on day 12 post-injury. a, photomicrograph of the wound histology of the control and KGF-1 groups. Black arrows, contraction; E, re-epithelialization; W, the length of the wound edge (epithelial gap distance); D, unwounded dermis; G.T, granulation tissue. b, wound contraction in both KGF-1–treated and untreated (Control) groups is depicted as a percentage of the original total wound and represents the contribution of contraction to wound closure. c, epithelial gap distance in both KGF-1–treated and control wounds. Data are shown as mean ± S.D. **, p < 0.01.
KGF-1 increased the expression levels of TGF-β1, Col-I, p-Smad2, p-Smad3, and α-SMA in diabetic wounds
To investigate how KGF-1 promotes wound contraction in vivo, both immunohistochemical staining and Western blotting were used to determine the expression of TGF-β1, Col-I, p-Smad2, p-Smad3, and α-SMA in KGF-1–treated and untreated diabetic rat wounds on day 12 post-injury. An increased presence of myofibroblasts is associated with increased wound contraction. Using TGF-β1, Col-I, and α-SMA antibodies as markers for myofibroblasts, we observed remarkably higher expression of TGF-β1, Col-I, and α-SMA in diabetic wounds treated with KGF-1 relative to the control group (Fig. 6, a and b). These findings were further supported by the Western blot results of TGF-β1, Col-I, and α-SMA (Fig. 6, c and d). We also found that p-Smad2 and p-Smad3 were significantly higher expressed in wounds treated with KGF-1 relative to the untreated (PBS) control group (Fig. 6, c–f). Taken together, these findings demonstrate that KGF-1 induces fibroblast differentiation and wound contraction via the TGF-β1/Smad signaling pathway.
Figure 6.

KGF-1 increased the expression of TGF-β1, Col-I, p-Smad2, p-Smad3, and α-SMA in diabetic wound tissues. a, representative ×20 images of immunohistochemical localization (brown) of TGF-β1, Col-I, and α-SMA expression in granulation tissue of diabetic wounds treated with KGF-1 (50 ng/ml) or PBS on day 12 post-wounding. Nuclei were counterstained with hematoxylin (blue). Arrows represent positive cells. Scale bars = 200 μm. b, percentages of positive cells in the total cell count from each staining per high-power field were calculated in the KGF-1–treated and untreated group. c, expression of TGF-β1, Col-I, p-Smad2, Smad2, p-Smad3, Smad3, and α-SMA in wound tissues on day 12 post-wounding were determined by Western blotting in the KGF-1 and control group. d, percentages of protein level in the KGF-1 group compared with the control group. e and f, protein expression ratio of p-Smad2/Smad2 (e) and p-Smad3/Smad3 (f) in each group. Data are shown as mean ± S.D. **, p < 0.01.
Discussion
Wound healing is a process in which various skin cells are spatiotemporally coordinated with well-balanced properties (24). In the process of tissue repair, a series of cells, mainly skin epidermal cells (such as keratinocytes) and mesenchymal cells (such as vascular endothelial cells and fibroblasts), undergo proliferation, migration, secretion, differentiation, and other biological activities, contributing to wound contraction, wound vascularization, and wound re-epithelialization (25). The epidermis and dermis transmit signals to each other through an epithelial–mesenchymal interaction mechanism in which the network of growth factors and cytokines plays a significant role in regulating biological behaviors of epidermal cells and mesenchymal cells (26). Previous studies have shown that, after skin injury, fibroblasts can secrete growth factors such as KGF-1 and GM-CSF, acting on keratinocyte proliferation and differentiation (2–5), which confirms the stimulating effect of mesenchymal cells on epidermal cells. Epidermal cells also regulate mesenchymal cells by secreting cytokines and growth factors. In the keratinocyte–fibroblast co-culture system, keratinocytes released IL-1 and AP-1, which activated underlying fibroblasts. Interestingly, this was not the end of the story. KGF-1 was then secreted by fibroblasts upon IL-1 and AP-1 stimulation, which, in turn, acted on keratinocytes (6). Simultaneously with the AP-1 impact, GM-CSF, pleiotrophin (PTN) and stromal cell–derived factor 1 (SDF-1) were released by fibroblasts and promoted keratinocyte proliferation and differentiation (27). In short, IL-1 and AP-1 were secreted by keratinocytes and acted on fibroblasts in a paracrine manner. Upon IL-1 and AP-1 stimulation, a series of paracrine factors were then released by fibroblasts and, in turn, targeted keratinocytes. That feedback loop in epithelial–mesenchymal interaction is called double-paracrine signaling; it is involved in wound repair and plays significant roles.
KGF-1 is a paracrine growth factor whose expression increases 160-fold after skin injury (28), specifically and positively regulating the proliferation and migration of keratinocytes (7) and promoting wound epithelialization. Our previous study suggested that KGF-1 is involved in wound contraction (10). However, KGF-1 cannot directly stimulate fibroblast contraction because there is no KGF-1 receptor (FGFR2-IIIb) on fibroblasts. We assumed that KGF-1 has an indirect effect on fibroblast contraction. Wound contraction, as a key step in the wound healing process, determines the speed of wound repair. Fibroblasts, with the largest cell population in the skin, are activated after skin injury; they migrate and cover almost the entire wound. Some fibroblasts are differentiated into myofibroblasts, which express α-SMA and have contractile function, contributing to wound contraction and accelerated wound healing (16). TGF-β1 is regarded as the central regulator of differentiation of fibroblasts into myofibroblasts, which express α-SMA and ultimately lead to wound contraction (29–31). In addition, TGF-β1 can also synergize the contraction of fibroblasts on collagen (mainly Col-I), promoting contraction (15). TGF-β1 binds to transmembrane receptors and transmits signals to Smad proteins. Smads are signal transduction molecules in the cytoplasm that can transfer the TGF-β1 signal directly from the cell membrane into the nucleus (32). Therefore, the TGF-β1/Smad pathway participates in wound contraction and plays a vital role. Previous studies have shown that TGF-β1 can be secreted by keratinocytes (21). Therefore, although KGF-1 cannot act directly on fibroblasts, it can act indirectly on fibroblasts by affecting the expression of TGF-β1.
To prove our hypothesis, we chose an in vitro model of a keratinocyte–fibroblast monolayer co-culture system, as it is a more accurate representation of the anatomical relationship between keratinocytes and fibroblasts for research of cytokine interaction (33). ELISAs revealed that the concentration of TGF-β1 in conditioned supernatant from the co-culture system treated with exogenous rhKGF-1 was significantly higher than that of the untreated control group and that the KGF-1– or TGF-β1–neutralizing antibody can abolish this effect. FPCLs were established to study fibroblasts contraction (34). We observed that the contractile capacity of FPCLs stimulated with conditioned medium derived from the co-cultured group treated with exogenous rhKGF-1 was significantly enhanced compared with the control group and the group treated with KGF-1–neutralizing antibody. The contractile activity of FPCLs induced by KGF-1 was attenuated by the TGF-β1–neutralizing antibody. These results indicate that KGF-1 may enhance fibroblast-mediated collagen gel contraction via TGF-β1. Our Western blot results suggest that KGF-1 is able to stimulate TGF-β1 activation based on increased expression of Col-I, p-Smad2, p-Smad3, and α-SMA. Consistent with the results of in vitro experiments, we found that diabetic wounds treated with KGF-1 had a higher degree of contraction and a faster rate of healing, with significantly higher expression of TGF-β1, Col-I, p-Smad2, p-Smad3, and α-SMA compared with untreated wounds. Our results illustrate that keratinocytes release active TGF-β1 in a KGF-1–dependent manner, which, in turn, enhances fibroblast contraction (Fig. 7).
Figure 7.
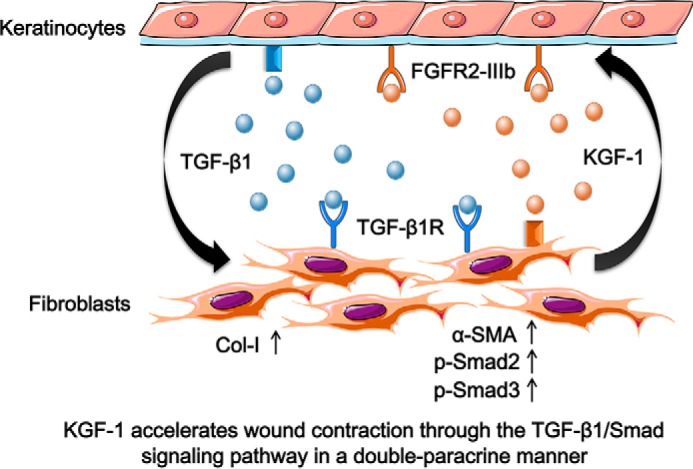
Schematic of the proposed molecular mechanism of KGF-1–induced fibroblast contraction through the TGF-β1/Smad pathway. KGF-1 bound to FGFR2-IIIb on keratinocytes, stimulating active TGF-β1 secretion, which activated the TGF-β1/Smad signaling cascade that enhanced the expression of Col-I and α-SMA in fibroblasts, promoting fibroblast contraction in wound healing.
In a study of cutaneous fibrosis, increased KGF-1 expression induced fibroblast activation through epithelial–mesenchymal double-paracrine signaling. KGF-1 induced keratinocytes to secrete oncostatin M, which, in turn, activated fibroblasts reacting with the increased expression of Col-Iα1, fibroblast activation protein, and enhanced migration. In this study, we discovered a new epithelial–mesenchymal interaction loop in wound healing. After skin injury, fibroblasts secreted KGF-1, stimulating keratinocytes to secrete TGF-β1 and promoting wound contraction through the TGF-β1/Smad signaling pathway. Therefore, KGF-1 has an effect on wound contraction through the double-paracrine signaling pathway.
In conclusion, we demonstrate that KGF-1 induces secretion of active TGF-β1 from keratinocytes and accelerates fibroblast contractibility as well as wound contraction through the TGF-β1/Smad signaling pathway in a double-paracrine manner. This study provides an important basis to better understand the mechanisms underlying wound contraction and healing and the interaction between epithelial and mesenchymal cells while also exploring novel ways to promote diabetic wound healing.
Experimental procedures
Cell isolation and culture
Human dermal fibroblasts (HDFs) were obtained from surgical resections of fresh foreskin tissue. All donors signed the informed consent form. The tissue was cleaned and cut into 1 × 0.5 cm strips. After being placed in Dispase II solution (8 mg/ml, Sigma) at 4 °C for 12 h, dermal tissues were spun off, shredded into pieces, and incubated in 0.25% trypsin solution at 37 °C for 15 min. Subsequently, DMEM (high-glucose, HyClone) supplemented with 10% FBS (Gibco) was used to terminate tissue digestion. The filtered fibroblasts were seeded at a density of l × l05/cm2 and cultured at 37 °C in a 5% CO2 humidified atmosphere with DMEM supplemented with 10% FBS, 2 mmol/liter l-glutamine, 100 units/ml penicillin, 100 g/ml streptomycin, and 50 g/ml ascorbic acid. The medium was changed every 3 days. Human epithelial cells, HaCaTs (35), were cultured in DMEM containing 10% fetal bovine serum, 100 units/ml penicillin, 100 g/ml streptomycin, and 2 mmol/liter l-glutamine. The medium was changed every day (36).
Co-culture experiments
A noncontact cell co-culture system was established using a 6-well plate Transwell (Corning). HaCaT cells (passage 40) and HDFs (passage 8) were plated in the upper and lower compartment, respectively, with density at a ratio of 1:6 (HaCaT:HDFs). Cells were co-cultured in DMEM containing 10% FBS at 37 °C in a 5% CO2 humidified incubator. The medium was subsequently replaced every 2 days. After HaCaT keratinocytes reached 80% confluence, each group was designated to receive either rhKGF-1 (50 ng/ml, PeproTech; KGF-1 group), KGF-1–neutralizing antibody (0.5 μg/ml, monoclonal, Abcam; anti-KGF-1 group), rhTGF-β1 (5 ng/ml, PeproTech; TGF-β1 group), rhKGF-1 (50 ng/ml) and TGF-β1–neutralizing antibody (10 μg/ml, monoclonal, Abcam; KGF-1 + anti-TGF-β1 group), or PBS (control group). The cells were co-cultured for 48 h. Subsequently, the conditioned medium from each group was analyzed by ELISA and FPCL contraction assays (37).
TGF-β1 ELISA assay
A human TGF-β1 ELISA kit (Elabscience) was used to detect the protein level of TGF-β1 in the conditioned medium of each co-culture group. Standard and activated samples (100 μl) were first added to each well and incubated at 37 °C for 90 min, washed with 100 μl of biotinylated antibody working solution, and then incubated again at 37 °C for 60 min. After washing with 100 μl of enzyme conjugate working solution and adding 90 μl of substrate solution, the mixture was incubated at 37 °C for 15 min. Finally, 50 μl of stop solution was added, and the optical density value was measured immediately at a wavelength of 450 nm.
TGF-β1 quantitative RT-PCR experiments
Total RNA was purified from cultured cells as recommended by the manufacturers using the E.Z.N.A. total RNA isolation kit (Omega Bio-Tek). Extracted RNA was then reverse-transcribed to complementary DNA using the Hiscript First-Strand cDNA Synthesis Kit (Vazyme Biotech Co., Ltd., Nanjing, China). Quantitative RT-PCR was performed with specific primers and ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd.) on an ABI 9700 sequence detection system (Applied Biosystems). The following primers were obtained from Tsingke Biological Technology: TGF-β1 forward, 5′-GGGACTATCCACCTGCAAGA-3′; TGF-β1 reverse, 5′-CCTCCTTGGCGTAGTAGTCG-3′; β-actin forward, 5′-CTGGCACCCAGCACAATG-3′; β-actin reverse, 5′-AGCGAGGCCAGGATGGA-3′.
Preparation of FPCLs and measurement of gel contraction
FPCLs were constructed as described previously using a modified method (38). Briefly, HDFs (passage 6) at the logarithmic growth phase were digested and prepared at a density of 5 × 105 cells/ml. Rat tail tendon type I collagen (5 mg/ml, Shengyou, Hangzhou, China), cellular suspension, and DMEM with 10% FBS were mixed rapidly at a volume ratio of 1:1:3 to give a final concentration of 1 mg/ml of type I collagen and a final cell concentration of 1 × 105 cells/ml. The mixture was added to a 24-well culture plate at a volume of 0.7 ml/well and incubated at 37 °C for 10 min. The resultant gel-like mixture formed the FPCL. Equivalent medium from each group was added into each FPCL. The diameter of each FPCL was imaged and measured at 0, 12, and 24 h. FPCL contraction was evaluated as the percentage of the resultant lattice area compared with the initial area before addition of conditioned medium. The relative contraction rates of FPCLs were calculated using the following formula: (1 − (current gel area/initial gel area)) × 100%.
Detection of TGF-β1, Col-I, P-Smad2, P-Smad3, and α-SMA proteins
For Western blotting, HDFs and HaCaT cells were isolated from the co-cultures and lysed with preconfigured RIPA buffer. The protein concentration of each group was determined using the BCA kit (Keygen Biotech, Nanjing, China). Samples (10 μg protein/lane) were resolved by 10% SDS-PAGE. The separated protein bands were then transferred to PVDF membranes (Millipore, Billerica, MA) and blocked with Tris-buffered saline containing 0.5% Tween in 5% nonfat milk powder for 1 h at room temperature. To detect the protein levels of TGF-β1, type I collagen (Col-I), phospho-Smad2 (p-Smad2), total Smad2 (Smad2), phospho-Smad3 (p-Smad3), total Smad3 (Smad3), and α-SMA, membranes were incubated with rabbit anti-TGF-β1 antibody (1:1000, Abcam), rabbit anti-Col-I antibody (1:1000, Abcam), rabbit anti-phospho-Smad2 antibody (1:500, Cell Signaling Technology), rabbit anti-Smad2 antibody (1:1000, Cell Signaling Technology), rabbit anti-phospho-Smad3 antibody (1:2000, Cell Signaling Technology), rabbit anti-Smad3 antibody (1:1000, Cell Signaling Technology), rabbit anti-α-SMA antibody (1: 2000, Abcam), and rabbit anti-GAPDH antibody (1:3000, Santa Cruz Biotechnology), respectively, for 1 h each and subsequently washed three times in Tris-buffered saline containing 0.5% Tween. Primary antibody was detected using horseradish peroxidase–conjugated secondary goat anti-rabbit antibody (1:5000, Beyotime, Shanghai, China) followed by chemiluminescence (ECL). Chemiluminescent signals were detected using an ECL Luminous Detector (MiniChemi 610, Sage Creation Science, Peking, China). The images of protein bands obtained from Western blotting were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD).
Establishing a diabetic rat wound model
Six male Sprague-Dawley rats (8 weeks old), supplied and housed by the Laboratory Animal Center of Xiangya Medical College of Central South University (Changsha, Hunan, China), were injected with streptozotocin (Sigma-Aldrich) in the abdominal cavity to induce diabetes, as described previously (39). After 3 weeks, the blood glucose levels of all animals were measured to be higher than 500 mg/dL, considered hyperglycemia. The rats were first anesthetized with an intraperitoneal injection of 1% sodium pentobarbital (6 ml/kg, Nembutal; Ovation Pharmaceuticals, Inc., Deerfield, IL). The dorsal hairs of the rats were then shaved, and the bare skin was disinfected with iodine solution. Eight square shaped wounds measuring 1 cm2 each were surgically inflicted on the back of each rat (four wounds on the left and four on the right). After covering with dressings, rats were then returned to their cages and raised separately. The experimental protocol was reviewed and approved by the Animal Care and Use Committee of Central South University, Changsha, Hunan, China.
Wound morphometric analysis
rhKGF-1 (PeproTech) was prepared with PBS at a concentration of 50 ng/ml. Wounds were divided into two groups: a control group (wound + PBS) and the KGF-1 group (wound + 50 ng/ml rhKGF-1). There were four repeating groups of wounds on each rat. Each wound in the KGF-1 group received 50 μl of 50 ng/ml rhKGF-1, whereas the control group received 50 μl of PBS. All solutions were applied to wounds with a fine brush. This process was done daily for a total of 12 days. To evaluate the rate of wound healing and contraction, a digital camera was used to record images of the wounds on the day of wounding (day 0) and on each day after wounding. The images were processed with Adobe Photoshop CS5.1 and measured using ImageJ. Wound contraction and healing rates were then quantified as follows. The percentages of wound healing and wound contraction were calculated with the following formulas, respectively: (1 − (current wound area/original wound area)) × 100% and (1 − (current wound area + epithelialized area)/original wound area) × 100% (10).
Histological analysis
On post-injury day 12, all rats were euthanized with an intraperitoneal injection of pentobarbital sodium salt. The wounded area and surrounding skin were collected for histological evaluation, immunohistochemical staining, and Western blotting. The excised tissues were fixed in 10% formalin for 48 h. After dehydration, the tissues were embedded in a paraffin wax block and sectioned into slices of 5-μm thickness. The sections were then deparaffinized in xylene, hydrated in a series of ethanol rinses (100%, 95%, 80%, and 75%), followed by washing with distilled water and staining with H&E. Wound contraction and epithelialization were measured by the distance between the wound edge and epithelialized length using Adobe Photoshop CS5.1.
Immunohistochemical staining
Biotin streptavidin HRP detection kits (ZSGB-BIO, Peking, China) were used for immunohistochemical staining of α-SMA, Col-I, and TGF-β1. Paraffin sections were deparaffinized, rehydrated, and retrieved using citric acid buffer (pH 6.0) at 95 °C for 30 min. After washing, sections were incubated in 3% H2O2 for 10 min to eliminate endogenous peroxidase activity and subsequently incubated in goat serum at room temperature for 30 min. Sections were then immersed in rabbit-anti-rat α-SMA (1:200, Abcam), Col-I (1:400, Abcam), and TGF-β1 (1:200, Abcam) antibodies at 4 °C overnight. After immersion, sections were then incubated with biotinylated goat-anti-rabbit secondary antibody (1:200) for 30 min at room temperature. Sections were then incubated in horseradish peroxidase protein, developed with diaminobenzidine, and counterstained with hematoxylin. The number of positive cells and the total number of cells were quantified in two sections of eight high-power fields using Image Pro. The percentages of each positive cell type based on the total number of cells were calculated.
Western blotting of wound tissues
A portion of the wound tissue was placed in liquid nitrogen to detect the tissue protein expression of TGF-β1, Col-I, p-Smad2, Smad2, p-Smad3, Smad3, and α-SMA. For Western blotting of wound tissues, 100 mmol/liter of PMSF (Keygen Biotech) was added to cold RIPA lysate (Keygen Biotech) with a volume ratio of 1:100. The RIPA buffer was mixed well and placed on ice for several minutes. The wound tissues were cut into sizes of 3 × 3 mm. Pre-cooled RIPA buffer (0.5 ml) was added to 100 mg of tissue. The mixtures were manually homogenized 15 times with a glass homogenizer until fully lysed. Each homogenized slurry was then transferred to a precooled centrifuge tube and centrifuged at 10,000 rpm at 4 °C for 10 min. The protein concentration of the resultant supernatant was determined using a BCA kit (Keygen Biotech). The Western blot assay was performed in the same manner as described for intracellular protein detection.
Data analysis
Results were analyzed using unpaired, two-tailed Student's t tests for comparisons between two groups and one-way analysis of variance for multiple comparisons. The results are expressed as mean ± S.D. A p value of less than 0.05 was considered statistically significant.
Author contributions
Y. P., S. W., Q. T., S. L., and C. P. data curation; Y. P., S. W., Q. T., S. L., and C. P. formal analysis; Y. P., S. W., and Q. T. methodology; Y. P. writing-original draft; Y. P., S. W., Q. T., S. L., and C. P. writing-review and editing; S. W., S. L., and C. P. supervision; S. W., Q. T., and S. L. investigation; S. W., S. L., and C. P. project administration; C. P. conceptualization; C. P. resources; C. P. funding acquisition; C. P. validation.
This work was supported by National Natural Science Foundation of China Grant 81301636. The authors declare that they have no conflicts of interest with the contents of this article.
- GM-CSF
- granulocyte macrophage colony-stimulating factor
- TGF
- transforming growth factor
- ECM
- extracellular matrix
- α-SMA
- α-smooth muscle actin
- rh
- recombinant human
- HDF
- humanqj dermal fibroblast
- RIPA
- radioimmune precipitation assay
- Col-I
- type I collagen.
References
- 1. Ghahary A., and Ghaffari A. (2007) Role of keratinocyte-fibroblast cross-talk in development of hypertrophic scar. Wound Repair Regen. 15, S46–S53 10.1111/j.1524-475X.2007.00225.x [DOI] [PubMed] [Google Scholar]
- 2. Maas-Szabowski N., Szabowski A., Stark H. J., Andrecht S., Kolbus A., Schorpp-Kistner M., Angel P., and Fusenig N. E. (2001) Organotypic cocultures with genetically modified mouse fibroblasts as a tool to dissect molecular mechanisms regulating keratinocyte growth and differentiation. J. Invest. Dermatol. 116, 816–820 10.1046/j.1523-1747.2001.01349.x [DOI] [PubMed] [Google Scholar]
- 3. Maas-Szabowski N., Stark H. J., and Fusenig N. E. (2000) Keratinocyte growth regulation in defined organotypic cultures through IL-1-induced keratinocyte growth factor expression in resting fibroblasts. J. Invest. Dermatol. 114, 1075–1084 10.1046/j.1523-1747.2000.00987.x [DOI] [PubMed] [Google Scholar]
- 4. Szabowski A., Maas-Szabowski N., Andrecht S., Kolbus A., Schorpp-Kistner M., Fusenig N. E., and Angel P. (2000) c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell 103, 745–755 10.1016/S0092-8674(00)00178-1 [DOI] [PubMed] [Google Scholar]
- 5. Werner S., and Smola H. (2001) Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell Biol. 11, 143–146 10.1016/S0962-8924(01)01955-9 [DOI] [PubMed] [Google Scholar]
- 6. Maas-Szabowski N., Shimotoyodome A., and Fusenig N. E. (1999) Keratinocyte growth regulation in fibroblast cocultures via a double paracrine mechanism. J. Cell Sci. 112, 1843–1853 [DOI] [PubMed] [Google Scholar]
- 7. Finch P. W., and Rubin J. S. (2004) Keratinocyte growth factor/fibroblast growth factor 7, a homeostatic factor with therapeutic potential for epithelial protection and repair. Adv. Cancer Res. 91, 69–136 10.1016/S0065-230X(04)91003-2 [DOI] [PubMed] [Google Scholar]
- 8. Marti G., Ferguson M., Wang J., Byrnes C., Dieb R., Qaiser R., Bonde P., Duncan M. D., and Harmon J. W. (2004) Electroporative transfection with KGF-1 DNA improves wound healing in a diabetic mouse model. Gene. Ther. 11, 1780–1785 10.1038/sj.gt.3302383 [DOI] [PubMed] [Google Scholar]
- 9. Peng C., He Q., and Luo C. (2011) Lack of keratinocyte growth factor retards angiogenesis in cutaneous wounds. J. Int. Med. Res. 39, 416–423 10.1177/147323001103900209 [DOI] [PubMed] [Google Scholar]
- 10. Peng C., Chen B., Kao H. K., Murphy G., Orgill D. P., and Guo L. (2011) Lack of FGF-7 further delays cutaneous wound healing in diabetic mice. Plast. Reconstr. Surg. 128, 673e–684e 10.1097/PRS.0b013e318230c521 [DOI] [PubMed] [Google Scholar]
- 11. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest Dermatol. 127, 526–537 10.1038/sj.jid.5700613 [DOI] [PubMed] [Google Scholar]
- 12. Hinz B., Mastrangelo D., Iselin C. E., Chaponnier C., and Gabbiani G. (2001) Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 159, 1009–1020 10.1016/S0002-9440(10)61776-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Desmoulière A., Geinoz A., Gabbiani F., and Gabbiani G. (1993) Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 10.1083/jcb.122.1.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Massagué J. (2000) How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 1, 169–178 10.1038/35043051 [DOI] [PubMed] [Google Scholar]
- 15. Meckmongkol T. T., Harmon R., McKeown-Longo P., and Van De Water L. (2007) The fibronectin synergy site modulates TGF-β-dependent fibroblast contraction. Biochem. Biophys. Res. Commun. 360, 709–714 10.1016/j.bbrc.2007.06.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., and Brown R. A. (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- 17. Hsieh S. C., Wu C. C., Hsu S. L., Feng C. H., and Yen J. H. (2016) Gallic acid attenuates TGF-β1-stimulated collagen gel contraction via suppression of RhoA/Rho-kinase pathway in hypertrophic scar fibroblasts. Life Sci. 161, 19–26 10.1016/j.lfs.2016.07.011 [DOI] [PubMed] [Google Scholar]
- 18. Chin G. S., Liu W., Peled Z., Lee T. Y., Steinbrech D. S., Hsu M., and Longaker M. T. (2001) Differential expression of transforming growth factor-β receptors I and II and activation of Smad 3 in keloid fibroblasts. Plast. Reconstr. Surg. 108, 423–429 10.1097/00006534-200108000-00022 [DOI] [PubMed] [Google Scholar]
- 19. Pohlers D., Brenmoehl J., Löffler I., Müller C. K., Leipner C., Schultze-Mosgau S., Stallmach A., Kinne R. W., and Wolf G. (2009) TGF-β and fibrosis in different organs: molecular pathway imprints. Biochim. Biophys. Acta 1792, 746–756 10.1016/j.bbadis.2009.06.004 [DOI] [PubMed] [Google Scholar]
- 20. Weber C. E., Li N. Y., Wai P. Y., and Kuo P. C. (2012) Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound healing and tissue remodeling after injury. J. Burn Care Res. 33, 311–318 10.1097/BCR.0b013e318240541e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberts A. B. (1998) Molecular and cell biology of TGF-β. Miner. Electrolyte Metab. 24, 111–119 10.1159/000057358 [DOI] [PubMed] [Google Scholar]
- 22. Lichtman M. K., Otero-Vinas M., and Falanga V. (2016) Transforming growth factor β (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 24, 215–222 10.1111/wrr.12398 [DOI] [PubMed] [Google Scholar]
- 23. Verrecchia F., and Mauviel A. (2002) Control of connective tissue gene expression by TGFβ: role of Smad proteins in fibrosis. Curr. Rheumatol. Rep. 4, 143–149 10.1007/s11926-002-0010-4 [DOI] [PubMed] [Google Scholar]
- 24. Martin P. (1997) Wound healing: aiming for perfect skin regeneration. Science 276, 75–81 10.1126/science.276.5309.75 [DOI] [PubMed] [Google Scholar]
- 25. Kössi J., Elenius K., Niinikoski J., Peltonen J., and Laato M. (2001) Overview of wound healing. Ann. Chir. Gynaecol. 90, 15–18 [PubMed] [Google Scholar]
- 26. Barrientos S., Stojadinovic O., Golinko M. S., Brem H., and Tomic-Canic M. (2008) Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601 10.1111/j.1524-475X.2008.00410.x [DOI] [PubMed] [Google Scholar]
- 27. Florin L., Maas-Szabowski N., Werner S., Szabowski A., and Angel P. (2005) Increased keratinocyte proliferation by JUN-dependent expression of PTN and SDF-1 in fibroblasts. J. Cell Sci. 118, 1981–1989 10.1242/jcs.02303 [DOI] [PubMed] [Google Scholar]
- 28. Werner S., Peters K. G., Longaker M. T., Fuller-Pace F., Banda M. J., and Williams L. T. (1992) Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc. Natl. Acad. Sci. U.S.A. 89, 6896–6900 10.1073/pnas.89.15.6896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Serini G., Bochaton-Piallat M. L., Ropraz P., Geinoz A., Borsi L., Zardi L., and Gabbiani G. (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J. Cell Biol. 142, 873–881 10.1083/jcb.142.3.873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaughan M. B., Howard E. W., and Tomasek J. J. (2000) Transforming growth factor-β1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 257, 180–189 10.1006/excr.2000.4869 [DOI] [PubMed] [Google Scholar]
- 31. Shannon D. B., McKeown S. T., Lundy F. T., and Irwin C. R. (2006) Phenotypic differences between oral and skin fibroblasts in wound contraction and growth factor expression. Wound Repair Regen. 14, 172–178 10.1111/j.1743-6109.2006.00107.x [DOI] [PubMed] [Google Scholar]
- 32. Derynck R., Zhang Y., and Feng X. H. (1998) Smads: transcriptional activators of TGF-β responses. Cell 95, 737–740 10.1016/S0092-8674(00)81696-7 [DOI] [PubMed] [Google Scholar]
- 33. Rheinwald J. G., and Green H. (1975) Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6, 331–343 10.1016/S0092-8674(75)80001-8 [DOI] [PubMed] [Google Scholar]
- 34. Ehrlich H. P., and Rajaratnam J. B. (1990) Cell locomotion forces versus cell contraction forces for collagen lattice contraction: an in vitro model of wound contraction. Tissue Cell 22, 407–417 10.1016/0040-8166(90)90070-P [DOI] [PubMed] [Google Scholar]
- 35. Boukamp P., Petrussevska R. T., Breitkreutz D., Hornung J., Markham A., and Fusenig N. E. (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761–771 10.1083/jcb.106.3.761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shephard P., Martin G., Smola-Hess S., Brunner G., Krieg T., and Smola H. (2004) Myofibroblast differentiation is induced in keratinocyte-fibroblast co-cultures and is antagonistically regulated by endogenous transforming growth factor-β and interleukin-1. Am. J. Pathol. 164, 2055–2066 10.1016/S0002-9440(10)63764-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mukhopadhyay A., Tan E. K., Khoo Y. T., Chan S. Y., Lim I. J., and Phan T. T. (2005) Conditioned medium from keloid keratinocyte/keloid fibroblast coculture induces contraction of fibroblast-populated collagen lattices. Br. J. Dermatol. 152, 639–645 10.1111/j.1365-2133.2005.06545.x [DOI] [PubMed] [Google Scholar]
- 38. Fang Q., Schulte N. A., Kim H., Kobayashi T., Wang X., Miller-Larsson A., Wieslander E., Toews M. L., Liu X., and Rennard S. I. (2013) Effect of budesonide on fibroblast-mediated collagen gel contraction and degradation. J. Inflamm. Res. 6, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tan A. M., Samad O. A., Dib-Hajj S. D., and Waxman S. G. (2015) Virus-mediated knockdown of Nav1.3 in dorsal root ganglia of STZ-induced diabetic rats alleviates tactile allodynia. Mol. Med. 21, 544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]