

Abstract
The NLR family pyrin domain-containing 3 (NLRP3) inflammasome is a multimeric protein complex that mediates maturation of the cytokines IL-1β and IL-18 as well as release of the proinflammatory protein high-mobility group box 1 (HMGB1) and contributes to several inflammatory diseases, including sepsis, gout, and type 2 diabetes. In this context, the well-studied active complement fragment C5a and its receptor C5aR1 or C5aR2 orchestrate the inflammatory responses in many diseases. Although a C5a-C5aR interaction in NLRP3-associated diseases has been suggested, little is known about the details of C5a–C5aR cross-talk with the NLRP3 inflammasome in macrophages. In this study, using mice and murine macrophages and cytokines, immunoblotting, siRNA, and quantitative real-time PCR assays, we demonstrate that C5aR2 deficiency restricts activation of the NLRP3 inflammasome and release of HMGB1 both in vitro and in vivo. Mechanistically, we found that C5aR2 promotes NLRP3 activation by amplifying dsRNA-dependent PKR expression, which is an important NLRP3-activating factor. We also observed that elevation of PKR expression because of the C5a–C5aR2 interaction depends on the mitogen-activated protein kinase/extracellular signal-regulated kinase kinase pathway and type I IFN signaling. In conclusion, these findings reveal that C5aR2 contributes to NLRP3 inflammasome activation and HMGB1 release from macrophages.
Keywords: immunology, complement system, IL-1, inflammasome, inflammation
Introduction
Inflammasomes are large multimeric protein complexes composed of a cytosolic sensor (e.g. NLRP3), an adaptor protein (apoptosis-associated speck-like protein containing a CARD (ASC)), and an effector, procaspase-1, which play a vital role in host defense against pathogens and inflammation (1, 2). Among these inflammasomes, the NLRP3 inflammasome has received much attention. A wide variety of pathogen-associated molecular patterns, such as nigericin (Nig),4 and damage-associated molecular patterns, including ATP and monosodium urate (MSU) crystals, can activate the NLRP3 inflammasome and lead to maturation of IL-1β and IL-18 (3). In addition, accumulated evidence shows that the NLRP3 inflammasome also mediates the release of high-mobility group box 1 (HMGB1), a late mediator of lethal sepsis, and contributes to the pathogenesis of septic shock (4–6). It has been well-documented that the NLRP3 inflammasome is involved in many inflammatory diseases and disorders, including sepsis, gout, type 2 diabetes, colitis, atherosclerosis, and arthritis (7–10). Consequently, proper regulation of the NLRP3 inflammasome is critical for the maintenance of immune homeostasis.
The active complement peptide C5a has been recognized as a powerful proinflammatory mediator, interacting with its receptors (C5aR1 and C5aR2) after onset of sepsis (11, 12). There are numerous studies suggesting that interaction of C5a with its receptors is linked to many inflammatory diseases, such as sepsis, septic cardiomyopathy, and gout (13–16). All of these studies indicate that blocking C5a or absence of the C5a receptor could limit inflammatory responses, decrease IL-1β, or suppress neutrophil recruitment and provide a protective role in such diseases (13–17). In addition, C5a–C5a receptor interaction in NLRP3-associated diseases has been suggested. However, the cell types used in these studies were cardiomyocytes (14, 15) rather than immune cells, and the underlying mechanism of C5a in NLRP3 inflammasome activation needs to be explored further. Recent studies have indicated that C5a–C5aR1 regulates lipopolysaccharide (LPS)–induced NLRP3 inflammasome activation in monocytes (18); therefore, we asked whether C5a–C5aR2 has similar effects on the modulation of NLRP3 inflammasome activation in macrophages and the associated disease models.
In this study, we demonstrated that C5aR2 deficiency dampens activation of the NLRP3 inflammasome and the release of HMGB1 in vitro and in vivo. Mechanistically, C5aR2 promotes NLRP3 activation by amplifying PKR expression, which is a well-studied NLRP3 activating factor. Moreover, C5a–C5aR2–promoting PKR expression depends on the MEK/ERK pathway and type I IFN signaling. Thus, our study reveals that C5aR2 contributes to NLRP3 inflammasome activation and HMGB1 release in macrophages.
Results
C5aR2 deficiency inhibits NLRP3 inflammasome activation and the release of HMGB1
To determine the role of C5aR2 in NLRP3 inflammasome activation, LPS-primed mouse peritoneal macrophages from C5aR2+/+ and C5aR2−/− mice were treated with NLRP3 agonists: ATP, nigericin, and MSU. We found that C5aR2 deficiency significantly reduced IL-1β production but not TNFα production (Fig. 1A). In addition, cleavage of caspase-1 and IL-1β was dampened by C5aR2 deficiency, whereas expression of NLRP3 was not affected by C5aR2 (Fig. 1B), and NLRP3 is required for IL-1β maturation in this process (Fig. S1, A–D). Previous research has suggested that C5aR2 is required for the release of HMGB1 (13). We therefore assessed the abundance and found that NLRP3-induced HMGB1 was also decreased in the absence of C5aR2 (Fig. 1C). Early studies indicated that NLRP3 is critical for IL-1β processing via noncanonical inflammasome activation (8), so we examined the role of C5aR2 in noncanonical inflammasome activation and obtained similar results as for canonical inflammasome activation (Fig. 1, D–F).
Figure 1.

C5aR2 deficiency inhibits activation of the NLRP3 inflammasome and the release of HMGB1. A–C, after priming with LPS (100 ng/ml) for 3 h, peritoneal macrophages collected from C5aR2+/+ and C5aR2−/− mice were treated with NLRP3 agonists: ATP (5 mm for 1 h), nigericin (10 μm for 1 h), and MSU (200 μg/ml for 6 h). Supernatants (Sup) were analyzed by ELISA for IL-1β and TNFα (A) and HMGB1 release (C). B, after stimulation with canonical NLRP3 agonists as in A, release of HMGB1 in supernatants, cleavage of caspase-1 and IL-1β, and expression of pro-caspase-1, pro-IL-1β, and NLRP3 in cell extracts were assayed by Western blotting. Ctrl, control. D–E, peritoneal macrophages collected from C5aR2+/+ and C5aR2−/− mice were treated with noncanonical NLRP3 agonists: LPS transfection (2 μg/ml of LPS transfected with 0.25% of FuGENE HD) and CTB+LPS (2 μg/ml of LPS combined with 5 μg/ml of CTB). Supernatants were analyzed by ELISA for HMGB1 (D) and IL-1β and TNFα release (E). F, after stimulation with noncanonical NLRP3 agonists as in D, release of HMGB1 in supernatants, cleavage of caspase-1 and IL-1β, and expression of pro-caspase-1, pro-IL-1β, and NLRP3 in cell extracts were measured by Western blotting. The results represent the mean ± S.D. of three independent experiments performed in triplicate (A and C–E). Two-tailed Student's t test was used (A and C–E). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. For Western blot analysis, data are representative of three independent experiments (B and F).
Previous studies (19–21) have demonstrated the importance of C5aR1 in inflammatory responses, so we compared the contribution of C5aR1 and C5aR2, individually and collectively, to the activation of NLRP3 inflammasomes. We applied siRNAs to silence C5aR1 in C5aR2+/+ and C5aR2−/− mouse macrophages, followed by stimulation with NLRP3 agonists. The knockdown efficiency of C5aR1 is more than 70% (Fig. S2, A and B). The results showed that knockdown of C5aR1 in WT cells barely affects NLRP3 inflammasome activation. Further, interference of C5aR1 in C5aR2−/− macrophages (a mimic of a double defect of C5aR1 and C5aR2) had a similar effect on the production of IL-1β compared with C5aR2−/− cells (Fig. S2C). This is in accordance with a previous study in which C5aR1 deficiency had no effect on NLRP3 inflammasome activation (18) and also suggests a possible dominant role of C5aR2 rather than C5aR1 in activation of NLRP3. To determine whether C5aR2 specifically regulates the NLRP3 inflammasome, we stimulated LPS-primed macrophages from C5aR2+/+ and C5aR2−/− mice with poly(dA:dT) (an AIM2 inflammasome activator), flagellin (an NLRC4 inflammasome activator), and nigericin. The abundance of IL-1β induced by AIM2 and NLRC4 agonists was slightly decreased by C5aR2 deficiency (Fig. S3A), whereas there was no difference in production of TNFα (Fig. S3B). Additionally, we checked C5aR2's role in TLR signaling (TLR4 stimulated by LPS, TLR2 stimulated by Pam3CSK4, and TLR3 stimulated by poly(I:C)) and found that TNFα or IL-6 was barely influenced by C5aR2 deficiency (Fig. S3, C and D). Taken together, these data suggested that C5aR2 mainly contributes to activation of the NLRP3 inflammasome and the release of HMGB1.
C5aR2 deficiency inhibits NLRP3 inflammasome activation in vivo
To further investigate the function of C5aR2 in NLRP3 inflammasome activation in vivo, C5aR2−/− and C5aR2+/+ mice were injected with MSU to induce NLRP3 inflammasome-dependent peritoneal inflammation. The IL-1β level and peritoneal exudate cell numbers in the peritoneal lavage fluids were measured. Compared with C5aR2+/+ mice, C5aR2−/− mice showed an obvious reduction in peritoneal inflammation (Fig. 2, A and B). Additionally, we used a LPS-induced sepsis model to examine the effect of C5aR2. In response to LPS challenge, the serum levels of IL-1β and HMGB1, but not TNFα, were greatly reduced in C5aR2−/− mice compared with C5aR2+/+ mice (Fig. 2C). C5aR2−/− mice also exhibited higher resistance to LPS-induced lethality than C5aR2+/+ mice (Fig. 2D), which was consistent with an earlier report that C5aR2−/− mice have improved survival compared with C5aR2+/+ mice after cecal ligation and puncture (CLP)–induced sepsis (13). Collectively, these data suggested that C5aR2 is needed for activation of the NLRP3 inflammasome in vitro and in vivo.
Figure 2.

C5aR2 deficiency restricts NLRP3 inflammasome activation in vivo. A and B, C5aR2+/+ and C5aR2−/− mice were injected i.p. with 1 mg of MSU, and the level of IL-1β (A) and peritoneal exudate cell numbers (B) in the peritoneal lavage fluids were measured. C, C5aR2+/+ and C5aR2−/− mice were injected i.p. with LPS (10 mg/kg body weight) for 12 h, and the serum levels of IL-1β (left panel), TNFα (center panel), and HMGB1 (right panel) were measured. D, the survival of C5aR2+/+ and C5aR2−/− mice subjected to 10 mg/kg LPS was monitored. Data points represent individual animals. Data were combined from three independent experiments with n = 15 mice/group (A–C) and shown as mean ± S.D. Two-tailed Student's t test was used (A–C). *, p < 0.05; ***, p < 0.001; ns, not significant. The survival data in D are representative of two independent experiments with n = 20 mice/group and presented as Kaplan–Meier plot by log-rank (Mantel–Cox) test.
C5aR2 deficiency reduces the expression of PKR in macrophages
We and others have suggested that PKR is required for NLRP3 inflammasome activation and HMGB1 release (22–25). Thus, we wanted to find out whether PKR is involved in C5aR2-mediated NLRP3 inflammasome activation and HMGB1 release. Accordingly, we examined the expression and the phosphorylation level of PKR in NLRP3 agonist–treated macrophages from C5aR2−/− and C5aR2+/+ mice. Surprisingly, expression and the level of PKR phosphorylation were diminished in the absence of C5aR2 (Fig. 3, A and B). This gave us a clue to further explore the relationship between C5aR2 and PKR. Then we examined the expression of PKR in macrophages treated with LPS, the C5aR2 natural ligand C5a, or both. We found that C5a alone (1 μg/ml) could promote the expression of PKR as much as LPS (Fig. 3C and D). Although combining with LPS, C5a promoted the expression of PKR and increased the phosphorylation level of PKR in a dose-dependent manner (Fig. 3C and D). Moreover, LPS or C5a or the both slightly promoted PKR expression in macrophages in the absence of C5aR2. (Fig. 3C and D). Thus, C5aR2 deficiency reduces the expression of PKR as well as phosphorylation of PKR in macrophages upon LPS or C5a stimulation.
Figure 3.

C5aR2 deficiency suppresses the expression of PKR in macrophages. A and B, after priming with LPS (100 ng/ml) for 3 h, C5aR2+/+ and C5aR2−/− mouse peritoneal macrophages were treated with canonical (LPS+ATP, LPS+Nig, or LPS+MSU) or noncanonical (LPS transfection or CTB+LPS) NLRP3 agonists. Total RNA was extracted and subjected to Q-PCR (A), and total protein was extracted and subjected to Western blot analysis of PKR expression and the phosphorylation level of PKR (B), respectively. Ctrl, control. C and D, peritoneal macrophages from C5aR2+/+ and C5aR2−/− mice were treated with LPS (100 ng/ml) and/or increasing concentrations of recombinant C5a protein (0.1, 0.5, and 1 μg/ml) for 4 h, and then total RNA (C) and protein (D) were extracted and subjected to Q-PCR and Western blot analysis of PKR expression and the phosphorylation level of PKR, respectively. The results represent the mean ± S.D. of three independent experiments performed in triplicate (A and C). Two-tailed Student's t test was used (A and C). For Western blot analysis, data are representative of three independent experiments (B and D). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
PKR rescues NLRP3 inflammasome activation in the absence of C5aR2
We tested whether PKR could rescue NLRP3 inflammasome activation in the absence of C5aR2. We electroporated PKR or an empty vector to macrophages from C5aR2+/+ and C5aR2−/− mice and then monitored the expression of IL-1β and HMGB1 after stimulation with NLRP3 agonists. The transfection efficiency was quantified by real-time qPCR (Q-PCR) (Fig. 4A), and the ELISA results showed that PKR transfection could partially rescue IL-1β and HMGB1 release in the absence of C5aR2 (Fig. 4, B and C). These data demonstrate that PKR is pivotal for C5aR2-mediated NLRP3 inflammasome activation and HMGB1 release.
Figure 4.

PKR rescues NLRP3 inflammasome activation in the absence of C5aR2. A–C, peritoneal macrophages from C5aR2+/+ and C5aR2−/− mice were electroporated with a PKR expression vector or an empty vector. 24 h later, cells were subjected to NLRP3 canonical stimuli as indicated. The transfection efficiency was quantified by Q-PCR (A), and the expression of IL-1β and TNFα (B) and HMGB1 (C) was measured. The results represent the mean ± S.D. of three independent experiments performed in triplicate. Two-tailed Student's t test was used. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Ctrl, control.
C5a induces proinflammatory mediators via PKR in vivo
To further verify the significance of PKR in C5a-mediated inflammation in vivo, we injected LPS plus recombinant C5a to PKR+/+ and PKR−/− mice and collected whole blood. In PKR+/+ mice, the levels of serum IL-1β and HMGB1 from the LPS plus C5a–treated group were significantly higher than in the LPS alone–treated group, whereas in PKR−/− mice, there was no statistically significant difference between the two groups (Fig. 5, A and B), which further demonstrated that PKR regulates the production of C5a-induced proinflammatory mediators in vivo. These results indicate that the C5a-C5aR2 axis promotes NLRP3 activation and HMGB1 release by amplifying the expression of PKR.
Figure 5.

PKR is involved in the production of C5a-induced proinflammatory mediators in the blood. A and B, ELISA of serum IL-1β, TNFα (A), and HMGB1 (B) in PKR+/+ and PKR−/− mice after i.p. injection of 10 mg/kg LPS with or without C5a (2.5 μg per mouse) for 12 h. Data points represent individual animals. Data were combined from three independent experiments with n = 15 mice/group and are shown as mean ± S.D. Two-tailed Student's t test was used. **, p < 0.01; ***, p < 0.001; ns, not significant.
C5a promotes the expression of PKR through MEK/ERK signaling and the type I IFN pathway
To determine the potential signal pathway underlying the C5aR2-mediated promotion of PKR expression, we used only C5a to stimulate the macrophages, as it is the natural ligand for C5aR2 and has a similar promoting effect with LPS. We examined MEK/ERK, mitogen-activated protein kinase 14 (p38), and PI3K regulatory subunit 1/Akt, which have been reported to mediate C5a's function in innate immune cells (18, 26, 27). After treatment with a MEK1/2 inhibitor (U0126), a p38 inhibitor (SB203580), or a PI3K inhibitor (wortmannin), macrophages were challenged with C5a. Our results indicated that the MEK1/2 inhibitor greatly reduced the expression of PKR induced by C5a, whereas the other two had no significant effect (Fig. 6A). The specificity of the inhibitors was detected according to previous research (28–33) (Fig. S4, A–C). In addition, IL-1β secretion was decreased when U0126 or SB203580 was added to macrophages under LPS+Nig stimulation (Fig. 6B). We also noticed that SB203580 could impair TNFα expression, whereas U0126 had no effect (Fig. 6B), which is because SB203580 could suppress NF-κB activation (33–35). To further validate the above signaling pathways, we synthesized siRNAs targeting MEK1, p38, and PI3K before C5a treatment. Only siMEK1 could reduce the expression of PKR following C5a stimulation (Fig. S5, A–D). Accordingly, MEK/ERK signaling is likely involved in C5aR2-mediated NLRP3 inflammasome activation.
Figure 6.

C5a–C5aR2 promotes the expression of PKR by modulating the MEK/ERK pathway and the type I IFN pathway. A, WT mouse peritoneal macrophages were pretreated with U0126 (a pharmacologic MEK1/2 inhibitor), SB203580 (a selective p38 inhibitor), and wortmannin (a selective PI3K inhibitor) 1 h prior to C5a (1 μg/ml) stimulation (3 h), and the PKR mRNA level was assayed by Q-PCR. B, after pretreatment with inhibitors as in A, WT mouse macrophages were primed with LPS (100 ng/ml) with or without C5a (1 μg/ml) for 3 h, followed by stimulation with nigericin (10 μm) for 1 h, and then supernatants were analyzed by ELISA for IL-1β and TNFα. C, peritoneal macrophages from IFNαβR+/+ and IFNαβR−/− mice were treated with C5a (1 μg/ml) for 3 h, and PKR mRNA was detected via Q-PCR. D, peritoneal macrophages from IFNαβR+/+ and IFNαβR−/− mice were treated with LPS (100 ng/ml) with or without C5a (1 μg/ml) for 3 h, followed by stimulation with nigericin (10 μm) for 1 h, and then supernatants were analyzed by ELISA for IL-1β and TNFα. The results represent the mean ± S.D. of three independent experiments performed in triplicate. Two-tailed Student's t test was used. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Type I IFN signaling has been reported to regulate PKR expression (36), so we used type I IFN receptor knockout (IFNαβR−/−) mice to further the investigation, in which type I IFN signaling is blocked. We stimulated macrophages from IFNαβR+/+ and IFNαβR−/− mice with C5a protein. PKR expression was markedly decreased in IFNαβR−/− mice after C5a stimulation compared with IFNαβR+/+ mice (Fig. 6C), suggesting that IFN signaling is involved in C5a-mediated promotion of PKR expression. Moreover, C5a treatment could not increase the production of IL-1β and TNFα in IFNαβR−/− macrophages upon NLRP3 agonist exposure (Fig. 6D). Overall, these data suggest that C5a–C5aR2 promotes PKR expression via MEK/ERK signaling and the type I IFN pathway.
Discussion
The anaphylatoxin C5a is generated upon complement activation and contributes to the development of many NLRP3-associated inflammatory disorders, including sepsis (37), septic cardiomyopathy (15), and gout (38). However, little is known about C5a–C5a receptor cross-talk with the NLRP3 inflammasome in immune cells such as macrophages. In this study, we showed that, in the absence of C5aR2, macrophages produce less IL-1β and HMGB1 upon NLRP3 agonist treatment. Moreover, in the MSU-induced peritonitis model and LPS-induced sepsis model, C5aR2−/− mice exhibited obviously reduced inflammation. To further uncover the mechanism, we surprisingly found that C5aR2 promotes the expression of PKR, an NLRP3 inflammasome activator, which was identified by our previous research (22). Furthermore, PKR transfection rescued inhibition of IL-1β and HMGB1 secretions in the presence of NLRP3 agonists and C5aR2 deficiency. In addition, PKR-deficient mice show attenuated production of IL-1β and HMGB1 after LPS plus C5a injection intraperitoneally compared with WT mice. Last, we found that C5a elevated PKR expression via MEK/ERK signaling and type I IFN signaling. Thus, our study revealed a previously unknown role of C5aR2 in NLRP3 inflammasome activation in macrophages (Fig. 7).
Figure 7.
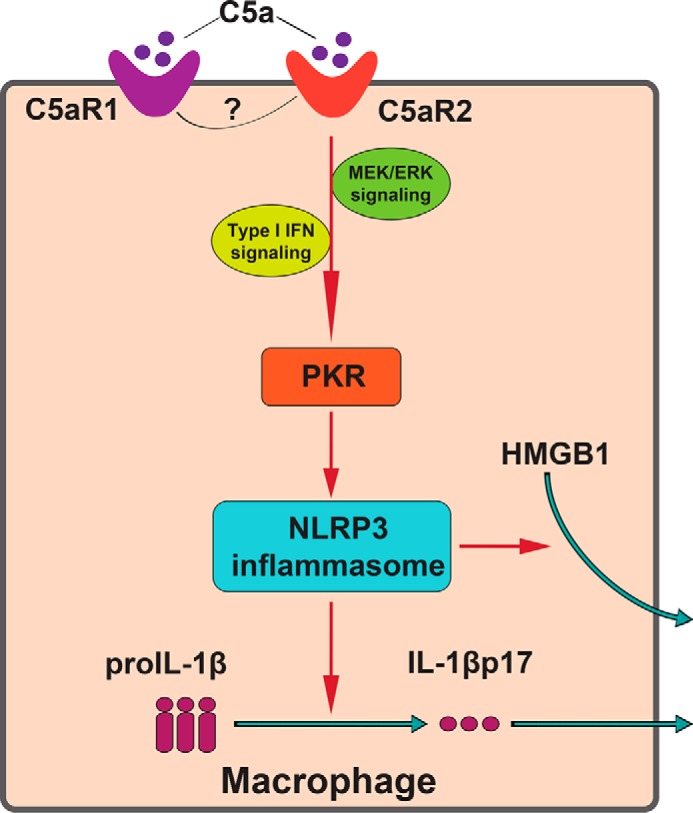
Model of C5aR2 modulation of NLRP3 inflammasome activation and HMGB1 release in macrophages. C5a–C5aR2 contributes to NLRP3 inflammasome activation and HMGB1 release by elevating the expression of PKR via the MEK/ERK and IFN pathways. The cross-talk between C5aR2 and C5aR1 remains elusive in macrophages during NLRP3 inflammasome activation.
C5a–C5a receptor interaction has attracted much attention in inflammatory disorders as studies reveal their proinflammatory roles (38, 39). An initial study found that, in a CLP-induced sepsis model, C5aR1- or C5aR2-deficient mice had improved survival compared with WT mice and that C5aR2 is required for HMGB1 release (13). Based on this study, our work showed similar results in an endotoxemia model and further identified that C5aR2 contributes to NLRP3 inflammasome activation and then HMGB1 release. In addition, our work utilized an MSU-induced peritonitis model to explore the role of C5aR2 in NLRP3 inflammasome activation in vivo. Our findings are consistent with earlier reports in which blocking the C5a receptor with an antagonist ameliorated inflammation (40, 41). In summary, our work and others all suggest that C5aR2 contributes to harmful consequences in NLRP3-related disorders. Although one study found that C5a could suppress the NLRP3 inflammasome in murine macrophages in vitro and hypothesized that C5aR2 may contribute to the suppressive effect (18), we did not observe this phenomenon in C5aR2-deficient mice. Notably, C5aR1 plays a negligible role in NLRP3 inflammasome activation in this study (Fig. S2, A–C), which is in agreement with previous research (18). Moreover, the absence of both C5aR1 and C5aR2 had a similar effect on NLRP3 activation as C5aR2 deficiency alone. These results indicate that C5aR2 might have a dominant role in regulation of the NLRP3 inflammasome. There has been some research concerning the cross-talk of these two receptors. In CLP-induced sepsis, C5aR1 and C5aR2 have been shown to play a synergistic role in activating the NLRP3 inflammasome in cardiocytes (14). However, in CD4+ T cells, C5aR2 seems to inhibit intracellular C5aR1 (42). Interestingly, it has been reported that C5aR2 can heterodimerize with C5aR1, mediating either pro- or anti-inflammatory responses (43). This may be attributed to the balance of C5aR1 versus C5aR2 activation or rely on the concentrations of C5a. A high local concentration of C5a induces heterodimerization of C5aR1 and C5aR2, which facilitates anti-inflammatory cytokine production to protect from excessive inflammation (39, 44). However, the underlying mechanisms of how this interplay happens and what it looks like in other immune cells remains to be explored in future studies.
PKR, a dsRNA-dependent protein kinase, revealed by our previous work to be an NLRP3 activator (22), is a surprising player to be involved in the C5a–C5aR2 axis. We observed that C5a–C5aR2 could promote the expression of PKR, almost at the transcription level. This amplifying effect depends on type I IFN signaling, which is critical for PKR expression, as PKR is an interferon-stimulated gene (45). In addition, we found that a MEK1/2 inhibitor as well as siRNA targeting MEK1 (Fig. 6A and Fig. S5, A and D) could reduce PKR expression promoted by C5a. Nevertheless, the details of MEK/ERK signaling and type I IFN signaling in C5a-mediated promotion of PKR expression need further investigation.
Abnormal activation of the NLRP3 inflammasome is related to inflammatory diseases and disorders, explaining why great attention has been paid to regulation of the NLRP3 inflammasome in these diseases (10, 46–48). Our previous study found that PKR is involved in NLRP3 activation and identified ethyl pyruvate as an NLRP3 inflammasome inhibitor (22, 49). In this study, we identified C5aR2 as an important modulator of NLRP3 inflammasome activation, identifying a potential target to treat NLRP3-associated inflammatory disorders (41). The benefits of targeting C5aR2 to treat inflammatory disorders linked to NLRP3 relies on the tendency of C5aR2 to selectively regulate the activity of the NLRP3 inflammasome and have a minimal effect on other inflammatory responses (Fig. 2, A–C). However, it should be noted that C5aR2 expression and localization are possibly dynamic and cell-specific, undergoing continuous changes and being dependent on diverse milieus that make it difficult to be targeted with certain drugs or antibodies under pathological conditions (38, 50). C5a, the natural ligand of C5aR2, could also bind to C5aR1 with high affinity and trigger C5aR1-dependent signal transduction, which probably has unknown effects on C5aR2-mediated inflammatory responses (16, 26, 51). Nevertheless, the latest discovery of C5aR2's role in inflammasomes has further enhanced our understanding of the mechanism of NLRP3 activation and might help to advance the treatment of NLRP3-related diseases.
Experimental procedures
Mice
WT C57BL/6 mice (8–10 weeks old) were purchased from Hunan SJA Laboratory Animal Co. Ltd. (Changsha, China). C5aR2−/− mice in a C57BL/6 background were purchased from Model Animal Research Center (Nanjing, China). The PKR−/− mice were generous gifts from Dr. Kevin J. Tracey. The IFNαβR−/− mice were generous gifts from Dr. Jin Hou. All animals were kept under specific pathogen free conditions. Experimental groups were sex-matched and 8–12 weeks of age. For gene knockout mice, WT littermate mice were used as controls. Animals were maintained in the Central South University Animal Facility. Studies were conducted in accordance with the Institutional Animal Care and Use Committee of Central South University.
Reagents
ATP (tlrl-atpl), ultrapure LPS (tlrl-eklps), MSU (tlrl-msu), nigericin (tlrl-nig), Pam3CSK4 (tlrl-pms), poly(I:C) (tlrl-picw), flagellin (tlrl-stfla), and poly(dA:dT) (tlrl-patn) were obtained from Invivogen. FuGENE HD transfection reagent (E2311) was purchased from Promega. Cholera toxin B (CTB) was purchased from Sigma. Mouse IL-1β (88-7013-77) and TNFα (88-7324-88) ELISA kits were purchased from eBioscience. The mouse HMGB1 ELISA kit (326054329) was purchased from Shino-Test. The rabbit monoclonal anti-PKR (phospho-Thr446) antibody (E120) and anti-mouse HMGB1 (EPR3507) were obtained from Abcam. Anti-mouse IL-1β (AF-401-NA) and C5a protein (2150-C5-025) were from R&D Systems. Anti-mouse caspase-1 (14F468) and anti-mouse C5aR1 (sc-53797) were from Santa Cruz Biotechnology. Anti-mouse NLRP3 (AG-20B-0014) was purchased from Adipogen. Anti-mouse MEK1 (2352), anti-mouse ERK1/2 (4695), anti-mouse phospho-ERK1/2 (4377), anti-mouse p38 MAPK (9212), anti-mouse phospho-p38 MAPK (9216), anti-mouse MAPKAPK2 (12155), anti-mouse phospho-MAPKAPK2 (3007), anti-mouse PI3K (4249), anti-mouse Akt (2920s), anti-mouse phospho-Akt (4060s), anti-mouse c-Jun (9165), anti-mouse phospho-c-Jun (9261), anti-mouse JNK (9252), anti-mouse phospho-JNK (9251), and anti-mouse β-actin (8H10D10) were from Cell Signaling Technology. Wortmannin (S2758), U0126 (S1102), and SB203580 (S1076) were from Selleck Chemicals.
Generation of elicited peritoneal macrophages
Mice were injected intraperitoneally with 3 ml of thioglycollate (3%). After 72 h, mice were sacrificed, and the elicited cells were harvested by peritoneal lavage with 15 ml of RPMI 1640 medium and resuspended in complete medium (RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin). For subsequent experiments, macrophages were seeded into 24-well plates (for ELISA or real time qPCR) or 6-well plates (for Western blotting) with a concentration of 1 × 106/ml, followed by replacement of medium 2 h later to remove nonadherent cells.
Stimulation of macrophages
For NLRP3 inflammasome activation, macrophages were primed with ultrapure LPS (100 ng/ml) for 3 h, followed by stimulation with ATP (5 mm, 1 h), nigericin (10 μm, 1 h), or MSU (200 μg/ml, 6 h). For noncanonical stimulation of the NLRP3 inflammasome, macrophages were treated with LPS (2 μg/ml) combined with CTB (5 μg/ml, CTB+LPS), or LPS (2 μg/ml) transfected with FuGENE HD (0.25% v/v) for 16 h for noncanonical NLRP3 inflammasome activation. To activate the AIM2 inflammasome, peritoneal macrophages were primed with ultrapure LPS (100 ng/ml) for 3 h, followed by transfection with poly(dA:dT) (1 μg/ml) for 16 h. To activate the NLRC4 inflammasome, peritoneal macrophages were primed with ultrapure LPS (100 ng/ml) for 3 h, followed by transfection with flagellin (2 μg/ml) for 1 h. For TLR signaling activation, macrophages were treated with ultrapure LPS (100 ng/ml), Pam3CSK4 (1 μg/ml), or poly(I:C) (1 μg/ml) for 3 h. After stimulation, cell culture supernatants were collected for ELISA of IL-β, TNFα, and IL-6. In some experiments, cells were stimulated simultaneously with LPS and a recombinant mouse C5a protein. Inhibitors of PI3K (wortmannin, 500 nm), MEK1/2 (U0126, 100 μm), and p38 (SB203580, 100 μm) were added 1 h before LPS priming.
MSU-induced mouse peritonitis
C5aR2+/+ and C5aR2−/− mice (8–10 weeks old and sex-matched) were injected intraperitoneally with 1 mg of MSU dissolved in 0.5 ml of sterile PBS. Mice were sacrificed 6 h later, and the peritoneal cavities were flushed with 5 ml of cold PBS. Peritoneal lavage fluids were collected, and cytokines were measured by ELISA after concentration using an Amicon Ultra 10 K filter (UFC900308) from Millipore. The pelleted cells were counted and analyzed for influx of neutrophils by cytometry. Trypan blue staining was used for exclusion of dead cells. The investigators were not blinded to allocation during experiments. The analysis was performed blindly by an independent researcher.
LPS-induced mouse endotoxemia
C5aR2+/+ and C5aR2−/− mice (8–10 weeks old and sex-matched) were injected intraperitoneally with LPS (10 mg/kg body weight) for 12 h, and the serum levels of IL-1β, TNFα, and HMGB1 were measured by ELISA. Survival of C5aR2+/+ and C5aR2−/− mice subjected to 10 mg/kg LPS were monitored for 4 days. The investigators were not blinded to allocation during experiments. The analysis was performed blindly by an independent researcher.
Transfection of the PKR expression vector to macrophages
We used a murine PKR expression vector described previously (22). For the rescue experiments, the recombinant PKR expression vector and the control vector were transiently transfected into peripheral macrophages of C5aR2+/+ and C5aR2−/− mice using an Amaxa P3 Primary Cell 4D-Nucleofector X kit (Lonza) according to the manufacturer's instructions.
ELISA assay for cytokines
The levels of IL-1β and TNFα collected from plasma of mice or from the culture medium in vitro were determined using quantitative ELISA kits (eBioscience) according to the manufacturer's instructions. The released HMGB1 in the serum or supernatant was determined using a commercial ELISA kit (Shino-Test).
Western blotting
Cell lysates were prepared by incubating treated macrophages with radioimmune precipitation assay buffer. Samples were fractionated by standard SDS-PAGE and transferred onto 0.2-μm PVDF membranes (Merck Millipore, ISEQ00010). Antibodies to mouse PKR, NLRP3, C5aR1, MEK1, p38, PI3K, ERK1/2, MAPKAPK2, Akt, c-Jun, JNK, caspase-1, IL-1β, and HMGB1 were used at 1:1000 dilution. Blots were normalized to β-actin expression (1:5000 dilution).
Design and transfection of siRNAs
Silencing the expression of mouse endogenous genes in primary peritoneal macrophages was achieved by a single gene-specific siRNA (Shanghai GenePharma Co. Ltd.). The sequences of siRNA used in this study were as follows: siC5aR1, 5′-GAU AAC AGC AGC UUU GAA A-3′; siNLRP3, 5′-GGU GAA AUG UAC UUA AAU C-3′; siMEK1, 5′-GGC CUG GUU AUG GCU AGA A-3′; sip38, 5′-GAC UGU GAG CUC AAG AUU C-3′; siPI3K, 5′-GCA GGA UCA AGU UGU CAA A-3′. The scrambled negative control siRNA was 5′-UUC UCC GAA CGU GUC ACG U-3′. Transfection of siRNAs was performed 24 h after plating using LipofectamineTM RNAiMAX transfection reagent (Invitrogen, 13778) according to the manufacturer's instructions.
Real-time qPCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, 74104) and reverse-transcribed using TransScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (TransGen Biotech, AT341), followed by real-time amplification with SYBR Green Master Mix (Roche, 29291200) on a 7500 real-time PCR instrument (Applied Biosystems). The 2−ΔΔCT method with normalization to β-actin and untreated controls was used for calculation of results. The primer sequences for mouse PKR were as follows: forward, 5′-ATG CAC GGA GTA GCC ATT ACG-3′; reverse, 5′-TGA CAA TCC ACC TTG TTT TCG T-3′. The primer sequences for mouse NLRP3 were as follows: forward, 5′-ATT ACC CGC CCG AGA AAG G-3′; reverse, 5′-TCG CAG CAA AGA TCC ACA CAG-3′. The primer sequences for mouse C5aR1 were as follows: forward, 5′-ATG GAC CCC ATA GAT AAC AGC A-3′; reverse, 5′-GAG TAG ATG ATA AGG GCT GCA AC-3′. The primer sequences for mouse MEK1 were as follows: forward, 5′-AAG GTG GGG GAA CTG AAG GAT-3′; reverse, 5′-CGG ATT GCG GGT TTG ATC TC-3′. The primer sequences for mouse p38 were as follows: forward, 5′-GGG ACA CCC CCT GCT TAT CT-3′; reverse, 5′-TCC CTG CTT TCA AAG GAC TGG-3′. The primer sequences for mouse PI3K were as follows: forward, 5′-ACA CCA CGG TTT GGA CTA TGG-3′; reverse, 5′-GGC TAC AGT AGT GGG CTT GG-3′. The primer sequences for mouse β-actin were as follows: forward, 5′-AGT GTG ACG TTG ACA TCC GT-3′; reverse, 5-GCA GCT CAG TAA CAG TCC GC-3′.
Statistical analysis
All statistical analyses were performed with GraphPad Prism software. Statistical significance was determined with two-tailed unpaired Student's t test. Mouse survival data were plotted as Kaplan–Meier curves and compared by log-rank (Mantel–Cox) test. p < 0.05 was considered statistically significant, with increasing levels of confidence displayed as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
S. Y., D. W., L. H., Y. Z., and R. L. performed the experiments. Y. T. helped to conduct animal experiment and supply technical support. S. Y., D. W., K. Z., and B. L. analyzed data and wrote the manuscript. D. A. helped to revise the manuscript. K. Z. and B. L. conceived and supervised the whole project.
Supplementary Material
Acknowledgments
We thank Dr. Kevin J. Tracey for providing PKR−/− mice and Dr. Hou Jin for providing IFNαβR−/− mice. We thank Qianqian Xue for assistance with raising the animals.
This work was supported by National Key Scientific Project Grant 2015CB910700; National Natural Science Foundation of China Grants 81422027, 81400149, 81470345, and 81801967; and Innovation-driven Project of Central South University Grant 2018CX030 and 2019CX013. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
- Nig
- nigericin
- MSU
- monosodium urate
- LPS
- lipopolysaccharide
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- ERK
- extracellular signal-regulated kinase
- TNF
- tumor necrosis factor
- CLP
- cecal ligation and puncture
- Q-PCR
- real-time quantitative PCR
- qPCR
- quantitative PCR
- CTB
- cholera toxin B
- MAPK
- mitogen-activated protein kinase
- JNK
- c-Jun N-terminal kinase
- TLR
- Toll-like receptor.
References
- 1. Lamkanfi M., and Dixit V. M. (2014) Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 10.1016/j.cell.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 2. Broz P., and Dixit V. M. (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- 3. Gross O., Thomas C. J., Guarda G., and Tschopp J. (2011) The inflammasome: an integrated view. Immunol. Rev. 243, 136–151 10.1111/j.1600-065X.2011.01046.x [DOI] [PubMed] [Google Scholar]
- 4. Willingham S. B., Allen I. C., Bergstralh D. T., Brickey W. J., Huang M. T., Taxman D. J., Duncan J. A., and Ting J. P. (2009) NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 183, 2008–2015 10.4049/jimmunol.0900138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qin S., Wang H., Yuan R., Li H., Ochani M., Ochani K., Rosas-Ballina M., Czura C. J., Huston J. M., Miller E., Lin X., Sherry B., Kumar A., Larosa G., Newman W., et al. (2006) Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 203, 1637–1642 10.1084/jem.20052203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lamkanfi M., Sarkar A., Vande Walle L., Vitari A. C., Amer A. O., Wewers M. D., Tracey K. J., Kanneganti T. D., and Dixit V. M. (2010) Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185, 4385–4392 10.4049/jimmunol.1000803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinon F., Pétrilli V., Mayor A., Tardivel A., and Tschopp J. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 10.1038/nature04516 [DOI] [PubMed] [Google Scholar]
- 8. Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., Zhang J., Lee W. P., Roose-Girma M., and Dixit V. M. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 10.1038/nature10558 [DOI] [PubMed] [Google Scholar]
- 9. Lamkanfi M., and Dixit V. M. (2012) Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 10.1146/annurev-cellbio-101011-155745 [DOI] [PubMed] [Google Scholar]
- 10. Wen H., Ting J. P., and O'Neill L. A. (2012) A role for the NLRP3 inflammasome in metabolic diseases: did Warburg miss inflammation? Nat. Immunol. 13, 352–357 10.1038/ni.2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rittirsch D., Flierl M. A., and Ward P. A. (2008) Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8, 776–787 10.1038/nri2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Markiewski M. M., DeAngelis R. A., and Lambris J. D. (2008) Complexity of complement activation in sepsis. J. Cell. Mol. Med. 12, 2245–2254 10.1111/j.1582-4934.2008.00504.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rittirsch D., Flierl M. A., Nadeau B. A., Day D. E., Huber-Lang M., Mackay C. R., Zetoune F. S., Gerard N. P., Cianflone K., Köhl J., Gerard C., Sarma J. V., and Ward P. A. (2008) Functional roles for C5a receptors in sepsis. Nat. Med. 14, 551–557 10.1038/nm1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalbitz M., Fattahi F., Grailer J. J., Jajou L., Malan E. A., Zetoune F. S., Huber-Lang M., Russell M. W., and Ward P. A. (2016) Complement-induced activation of the cardiac NLRP3 inflammasome in sepsis. FASEB J. 30, 3997–4006 10.1096/fj.201600728R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fattahi F., Frydrych L. M., Bian G., Kalbitz M., Herron T. J., Malan E. A., Delano M. J., and Ward P. A. (2018) Role of complement C5a and histones in septic cardiomyopathy. Mol. Immunol. 102, 32–41 10.1016/j.molimm.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khameneh H. J., Ho A. W., Laudisi F., Derks H., Kandasamy M., Sivasankar B., Teng G. G., and Mortellaro A. (2017) C5a Regulates IL-1β production and leukocyte recruitment in a murine model of monosodium urate crystal-induced peritonitis. Front. Pharmacol. 8, 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Czermak B. J., Sarma V., Pierson C. L., Warner R. L., Huber-Lang M., Bless N. M., Schmal H., Friedl H. P., and Ward P. A. (1999) Protective effects of C5a blockade in sepsis. Nat. Med. 5, 788–792 10.1038/10512 [DOI] [PubMed] [Google Scholar]
- 18. Haggadone M. D., Grailer J. J., Fattahi F., Zetoune F. S., and Ward P. A. (2016) Bidirectional crosstalk between C5a receptors and the NLRP3 inflammasome in macrophages and monocytes. Mediators Inflamm. 2016, 1340156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu G. L., Chen J., Yang F., Li G. Q., Zheng L. X., and Wu Y. Z. (2014) C5a/C5aR pathway is essential for the pathogenesis of murine viral fulminant hepatitis by way of potentiating Fgl2/fibroleukin expression. Hepatology 60, 114–124 10.1002/hep.27114 [DOI] [PubMed] [Google Scholar]
- 20. Riedemann N. C., Guo R. F., and Ward P. A. (2003) A key role of C5a/C5aR activation for the development of sepsis. J. Leukocyte Biol. 74, 966–970 10.1189/jlb.0403137 [DOI] [PubMed] [Google Scholar]
- 21. Gerard N. P., and Gerard C. (1991) The chemotactic receptor for human C5a anaphylatoxin. Nature 349, 614–617 10.1038/349614a0 [DOI] [PubMed] [Google Scholar]
- 22. Lu B., Nakamura T., Inouye K., Li J., Tang Y., Lundbäck P., Valdes-Ferrer S. I., Olofsson P. S., Kalb T., Roth J., Zou Y., Erlandsson-Harris H., Yang H., Ting J. P., Wang H., et al. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 10.1038/nature11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hett E. C., Slater L. H., Mark K. G., Kawate T., Monks B. G., Stutz A., Latz E., and Hung D. T. (2013) Chemical genetics reveals a kinase-independent role for protein kinase R in pyroptosis. Nat. Chem. Biol. 9, 398–405 10.1038/nchembio.1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie M., Yu Y., Kang R., Zhu S., Yang L., Zeng L., Sun X., Yang M., Billiar T. R., Wang H., Cao L., Jiang J., and Tang D. (2016) PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7, 13280 10.1038/ncomms13280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boriushkin E., Wang J. J., Li J., Bhatta M., and Zhang S. X. (2016) p58(IPK) suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages. Sci. Rep. 6, 25013 10.1038/srep25013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bosmann M., Haggadone M. D., Zetoune F. S., Sarma J. V., and Ward P. A. (2013) The interaction between C5a and both C5aR and C5L2 receptors is required for production of G-CSF during acute inflammation. Eur. J. Immunol. 43, 1907–1913 10.1002/eji.201243075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarma J. V., and Ward P. A. (2012) New developments in C5a receptor signaling. Cell Health Cytoskeleton 4, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rouse J., Cohen P., Trigon S., Morange M., Alonso-Llamazares A., Zamanillo D., Hunt T., and Nebreda A. R. (1994) A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78, 1027–1037 10.1016/0092-8674(94)90277-1 [DOI] [PubMed] [Google Scholar]
- 29. Powis G., Bonjouklian R., Berggren M. M., Gallegos A., Abraham R., Ashendel C., Zalkow L., Matter W. F., Dodge J., and Grindey G. (1994) Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 54, 2419–2423 [PubMed] [Google Scholar]
- 30. Walker E. H., Pacold M. E., Perisic O., Stephens L., Hawkins P. T., Wymann M. P., and Williams R. L. (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6, 909–919 10.1016/S1097-2765(05)00089-4 [DOI] [PubMed] [Google Scholar]
- 31. Duan W., Chan J. H., Wong C. H., Leung B. P., and Wong W. S. (2004) Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J. Immunol. 172, 7053–7059 10.4049/jimmunol.172.11.7053 [DOI] [PubMed] [Google Scholar]
- 32. Hotokezaka H., Sakai E., Kanaoka K., Saito K., Matsuo K., Kitaura H., Yoshida N., and Nakayama K. (2002) U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. J. Biol. Chem. 277, 47366–47372 10.1074/jbc.M208284200 [DOI] [PubMed] [Google Scholar]
- 33. Cuenda A., Rouse J., Doza Y. N., Meier R., Cohen P., Gallagher T. F., Young P. R., and Lee J. C. (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233 10.1016/0014-5793(95)00357-F [DOI] [PubMed] [Google Scholar]
- 34. Kumar S., Jiang M. S., Adams J. L., and Lee J. C. (1999) Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 263, 825–831 10.1006/bbrc.1999.1454 [DOI] [PubMed] [Google Scholar]
- 35. Simon C., Goepfert H., and Boyd D. (1998) Inhibition of the p38 mitogen-activated protein kinase by SB 203580 blocks PMA-induced Mr 92,000 type IV collagenase secretion and in vitro invasion. Cancer Res. 58, 1135–1139 [PubMed] [Google Scholar]
- 36. García M. A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., and Esteban M. (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 70, 1032–1060 10.1128/MMBR.00027-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ward P. A. (2004) The dark side of C5A in sepsis. Nat. Rev. Immunol. 4, 133–142 10.1038/nri1269 [DOI] [PubMed] [Google Scholar]
- 38. Hajishengallis G., Reis E. S., Mastellos D. C., Ricklin D., and Lambris J. D. (2017) Novel mechanisms and functions of complement. Nat. Immunol. 18, 1288–1298 10.1038/ni.3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo R. F., and Ward P. A. (2005) Role of C5a in inflammatory responses. Annu. Rev. Immunol. 23, 821–852 10.1146/annurev.immunol.23.021704.115835 [DOI] [PubMed] [Google Scholar]
- 40. Strachan A. J., Woodruff T. M., Haaima G., Fairlie D. P., and Taylor S. M. (2000) A new small molecule C5a receptor antagonist inhibits the reverse-passive Arthus reaction and endotoxic shock in rats. J. Immunol. 164, 6560–6565 10.4049/jimmunol.164.12.6560 [DOI] [PubMed] [Google Scholar]
- 41. Monk P. N., Scola A. M., Madala P., and Fairlie D. P. (2007) Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 152, 429–448 10.1038/sj.bjp.0707332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arbore G., West E. E., Spolski R., Robertson A. A. B., Klos A., Rheinheimer C., Dutow P., Woodruff T. M., Yu Z. X., O'Neill L. A., Coll R. C., Sher A., Leonard W. J., Köhl J., Monk P., et al. (2016) T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 352, aad1210 10.1126/science.aad1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Croker D. E., Halai R., Kaeslin G., Wende E., Fehlhaber B., Klos A., Monk P. N., and Cooper M. A. (2014) C5a2 can modulate ERK1/2 signaling in macrophages via heteromer formation with C5a1 and β-arrestin recruitment. Immunol. Cell Biol. 92, 631–639 10.1038/icb.2014.32 [DOI] [PubMed] [Google Scholar]
- 44. Wood A. J. T., Vassallo A., Summers C., Chilvers E. R., and Conway-Morris A. (2018) C5a anaphylatoxin and its role in critical illness-induced organ dysfunction. Eur J. Clin. Invest. 48, e13028 10.1111/eci.13028 [DOI] [PubMed] [Google Scholar]
- 45. Dey M., Cao C., Dar A. C., Tamura T., Ozato K., Sicheri F., and Dever T. E. (2005) Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell 122, 901–913 10.1016/j.cell.2005.06.041 [DOI] [PubMed] [Google Scholar]
- 46. Sutterwala F. S., Haasken S., and Cassel S. L. (2014) Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 1319, 82–95 10.1111/nyas.12458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grailer J. J., Canning B. A., Kalbitz M., Haggadone M. D., Dhond R. M., Andjelkovic A. V., Zetoune F. S., and Ward P. A. (2014) Critical role for the NLRP3 inflammasome during acute lung injury. J. Immunol. 192, 5974–5983 10.4049/jimmunol.1400368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luo B., Huang F., Liu Y., Liang Y., Wei Z., Ke H., Zeng Z., Huang W., and He Y. (2017) NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front. Physiol. 8, 519 10.3389/fphys.2017.00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li S., Liang F., Kwan K., Tang Y., Wang X., Tang Y., Li J., Yang H., Chavan S. S., Wang H., Andersson U., Lu B., and Tracey K. J. (2018) Identification of ethyl pyruvate as a NLRP3 inflammasome inhibitor that preserves mitochondrial integrity. Mol. Med. 24, 8 10.1186/s10020-018-0006-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang T., Garstka M. A., and Li K. (2017) The controversial C5a receptor C5aR2: its role in health and disease. J. Immunol. Res. 2017, 8193932 10.1155/2017/8193932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. An L.-L., Mehta P., Xu L., Turman S., Reimer T., Naiman B., Connor J., Sanjuan M., Kolbeck R., and Fung M. (2014) Complement C5a potentiates uric acid crystal-induced IL-1β production. Eur. J. Immunol. 44, 3669–3679 10.1002/eji.201444560 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.