

Abstract
Among the repertoire of immunoregulatory proteins encoded by myxoma virus, M013 is a viral homologue of the viral pyrin domain-only protein (vPOP) family. In myeloid cells, M013 protein has been shown to inhibit both the inflammasome and NF-κB signaling pathways by direct binding to ASC1 and NF-κB1, respectively. In this study, a three-dimensional homology model of the M013 pyrin domain (PYD) was built based on similarities to known PYD structures. A distinctive feature of the deduced surface electrostatic map of the M013 PYD is the presence of a negatively region consisting of numerous aspartate and glutamate residues in close proximity. Single-site mutations of aspartate and glutamate residues reveal their role in interactions with ASC-1. The biological significance of charge complementarity in the M013–ASC-1 interaction was further confirmed by functional assays of caspase-1 activation and subsequent secretion of cytokines. M013 also has a unique 33-residue C-terminal tail that follows the N-terminal PYD, and it is enriched in positively charged residues. Deletion of the tail of M013 significantly inhibited the interactions between M013 and NF-κB1, thus compromising the ability of the viral protein to suppress the secretion of pro-inflammatory cytokines. These results demonstrate that vPOP M013 exploits distinct structural motifs to regulate both the inflammasome and NF-κB pathways.
Keywords: poxvirus, inflammasome, innate immunity, NF-kappa B (NF-KB), host-pathogen interaction, ASC-1, immune evasion, myxoma virus, PYRIN domain
Introduction
Molecular mimicry by pathogens, including viruses, can be exploited for the modulation of the host immune system in a fashion that favors the virus. In turn, host organisms have evolved their own immune strategies to overcome viral countermeasure mechanisms. Thus, characterization of viral anti-immune evasion mechanisms can provide insight into virus-host immunology (1). Poxviruses encode a diverse array of immunomodulatory proteins that is a reflection of their evolutionary history. These viral proteins target and subvert multiple elements of the innate immune machinery and play a fundamental role in virus replication and pathogenesis (2, 3). Members of the Poxviridae family include a diverse group of virus members, which infect a wide range of hosts. Among these, myxoma virus (MYXV)6 is an antecedent member of the genus Leporipoxvirus that also includes rabbit (Shope) fibroma virus, hare fibroma virus, and squirrel fibroma virus (4). MYXV causes a rapid systemic and lethal infection called myxomatosis in the European rabbit (Oryctolagus cuniculus) (4, 5). The manifestation of this vector-transmitted lethal disease in rabbits occurs in the form of extensive fulminating internal and external lesions and severe immunosuppression accompanied by supervening Gram-negative bacterial secondary infections of the respiratory tract. Among the considerable repertoire of immunomodulatory proteins encoded by MYXV, ORF M013L, known as M013, is a member of the PYD superfamily (6, 7) and is referred to as a viral pyrin-only protein (vPOP). It has been studied previously as an effector of immune evasion and inhibits the innate immune responses in rabbits (6). However, its immunomodulatory activities have been shown to be fully operative in both human and rabbit cells, suggesting strong conservation of the host cell innate immune targets of this vPOP (6, 8).
The 126-residue M013 protein is a strong antagonist of inflammasome activation via direct interactions with ASC-1 (apoptosis-associated speck-like protein containing caspase-recruitment domain 1) (6). The inflammasome, a multiprotein assembly, is a key component of innate immunity and is found principally in myeloid cells. Activated inflammasomes are composed of at least one pathogen sensor protein and a caspase, plus the adapter protein ASC-1. Inflammasomes are formed in response to pattern- and/or danger-associated molecular patterns and activate inflammatory caspases such as caspase-1 and/or -11 (9–11). Inflammasome-mediated activation of caspases leads to the proteolytic cleavage and secretion of specific cytokine precursors into mature pro-inflammatory cytokines, such as interleukin (IL)-1β and IL-18. Activation of inflammasomes is under the regulatory control of pyrin domains (PYD) (also known as PAAD/DAPIN) proteins, which adopt a death-domain fold. Human PYDs have a tertiary structure of six to seven tightly coiled α-helices arranged in a “Greek key” fold to allow homotypic and heterotypic protein interactions (12, 13). ASC-1 contains both a pyrin domain and a caspase-recruitment domain, whereas regulatory PYDs include the cellular pyrin-only proteins (cPOPs) (13, 14). cPOPs are endogenous inhibitors and regulatory proteins that possess single pyrin domains. Human cPOP1 and cPOP2 both have an inhibitory action on NF-κB signaling, and both inhibit the interaction between inflammasome ASC-1 and pathogen sensor molecules (13). The cPOP1 (also known as ASC-2), interacts with IκB kinase α and is postulated to prevent the downstream phosphorylation of IκBα (15). In addition to its interaction with ASC-1, cPOP2 also blocks NF-κB activation by interfering with nuclear import (16). M013 protein was shown to interact directly with ASC-1, although the mechanism by which M013 and cPOPs regulate signaling remains unknown.
In addition to the antagonism of the inflammasome, M013 protein also regulates NF-κB signaling via direct binding to NF-κB1 p105 (8). Like the inflammasome, the NF-κB pathway acts as a central regulator of cellular responses in normal physiology and disease. Activation of the NF-κB complex in response to signals detected by various sensors/receptors such as those triggered by cytokines (e.g. TNF receptor) or pattern recognition receptors (e.g. Toll-like receptors, nucleotide-binding oligomerization domain-like receptors, etc.) mediates the degradation of IκBα. This degradation leads to the nuclear translocation of p50, p65, and c-Rel–containing dimers that activate the transcription of downstream pro-inflammatory and anti-viral genes (17–19). NF-κB signaling not only operates as an effector of anti-pathogen responses, but this transcriptional regulator also plays a role in a wide variety of other conditions, including cancer (18, 20, 21). The functional importance of M013 was determined by experiments in which the M013 gene knocked out by insertional disruption, and the resulting virus (vMyxM013-KO) was unable to infect rabbit leukocytes productively (6). In myxoma virus, M013 is the only confirmed antagonist of NF-κB signaling identified thus far. It protects the inhibitor of NF-κB (IκBα) from degradation, disrupting the NF-κB signaling cascade by binding to and preventing degradation of NF-κB1 p105 (8).
In contrast to parental MYXV, infection of human THP-1 monocytic cells with the vMyxM013-KO virus induced rapid and dramatic secretion of pro-inflammatory cytokines associated with NF-κB pathway, such as TNF, IL-6, and MCP-1. Infecting THP-1 cells with vMyxM013-KO virus led to the activation of the IκB kinase kinases and degradation of IκBα at an early phase of infection, resulting in the activation and nuclear translocation of NF-κB (8). In addition, the M013 protein was shown to interact directly with ASC-1, an integral component of the cellular inflammasome complex and inhibited caspase-1 activation and the processing of pro-inflammatory cytokines IL-1β and IL-18 (6). Secreted proteins to block the activity of these pro-inflammatory cytokines have not been identified in myxoma virus, despite their presence in other orthopoxviruses (22). After infection with myxoma virus, M013 binds host ASC-1 protein in both rabbit and human cells to inhibit the subsequent activation of the inflammasome complex (6). The current working model for this inhibition is that M013 protein becomes positioned as a competitive inhibitor in the complex somewhere between ASC-1 and pro-caspase-1, but there have been no structural studies of these larger complexes to date. Evidence for the importance of charged residues in PYRIN domain homotypic interactions has been demonstrated in ASC-1 but remains to be elucidated for vPOPs (23).
In this study, we report that site-directed mutants of the M013 vPOP protein can selectively inhibit one pathway over another by specifically interfering with interactions between ASC-1 and/or NF-κB1. We found that mutation in selected charged residues of M013 pyrin domain diminished the interaction between M013 and ASC-1. On the other hand, deletion of a C-terminal tail region selectively reduced the protein–protein interactions between M013 and NF-κB1. These selectively deficient mutants of M013 offer the prospect of uniquely inhibiting either the inflammasome and/or NF-κB signaling pathways in vitro or in vivo.
Results
M013 homology modeling and mutagenesis rationale
The amino acid sequence of M013_PYD was aligned to the known host cPOPs and selected other cellular pyrin domain–containing proteins using Clustal Omega (24). There is modest conservation of hydrophobic residues in the predicted six-helix bundle of M013, especially in α-helix 2 (Fig. 1A). Overall, M013_PYD exhibits very low sequence identities and similarities to cellular pyrins. To provide more insight into sequence-structure relationships, a three-dimensional model was built under the presumption that M013 adopted a conventional pyrin domain fold. The Consensus and Homology suites in the software MOE were used to align the M013 sequence with known structures (25). The pyrin domain of myeloid cell nuclear differentiation antigen (Protein Data Bank code 2DBG) was ultimately used to thread the sequence of M013 into the pyrin fold. An energy minimization was performed to relieve mild steric repulsions. An independent model of M013 using Robetta software (26) resulted in a similar overall three-dimensional model, with the core of the six α-helices relatively conserved and modest changes in the size of the connecting loops. More details are provided in Fig. S1.
Figure 1.

A, multiple sequence alignment of viral and cellular pyrin domains. Amino acids are colored by their aliphatic (gray) or hydrophilic properties (red, negative; blue, positive; magenta, polar), except where sequence conservation is observed (background shading). The relative extent of conservation is represented as a color gradient from cyan (low) to red (high). The PYD helices (cylinders) correspond to the structure of myeloid cell nuclear differentiation antigen (MNDA; Protein Data Bank code 2DBG). Black dots denote the key negatively charged residues (Glu-28, Asp-29, Asp-41, and Asp-57) that are discussed under “Results.” The positively charged tail of M013 and S013 is annotated along with the conserved Arg/Lys residues (blue +). B, homology modeling of the pyrin domain of M013. Left panel, ribbon model of the six α-helices (residues 1–93). Right panel, corresponding surface electrostatic representation of the same view, with red and blue indicating negative and positive charges, respectively. The view is looking direct at the type Ib face of the pyrin domain. Asp-41 resides at the end of α4 on the opposite face from the current view and is not visible on the electrostatic surface (denoted by the arrow).
Strikingly, the model of M013 reveals a negatively charged cluster with numerous aspartate and glutamate residues (Fig. 1B). This predicted feature is unique to M013 and lies on the type 1b face of the pyrin domain (27, 28). The predominance of negative charges confer an estimated pI of 4.6 for the pyrin domain of M013 (residues 1–93). The identical face of the pyrin domain in all other known structures lacks this feature (Fig. S2), suggesting that residues comprising this negatively charged face may be important for the distinct functions of M013. A second unique aspect of the viral M013 POP is an extended tail at the C terminus (residues 94–126). Based on these distinct sequence and structural considerations, a series of site-directed and truncation mutants were generated to understand the molecular basis for M013 modulatory functions. Selected aspartate and glutamate residues were mutated to asparagine and glutamine, respectively. In addition, a truncation mutant of M013 comprising the pyrin domain (residues 1–93; henceforth referred to as M013PYD) was also generated to understand the contribution of this novel domain to M013 antagonism of innate immunity.
M013 mutants interact with differential affinity to ASC-1 and NF-κB1
Monocytes and macrophages are the primary cells expressing the genes associated with active inflammasomes (29). To assess how WT M013 (WT-M013) or M013 mutants interact with the endogenous ASC-1 or NF-κB1 in human myeloid cells, we performed in vitro transfection and co-IP assays in THP-1 monocytic leukemia cells (30). The cells were transfected with WT-M013 or mutant plasmid DNA that allows expression of each protein in cells infected with MYXV: that is, the mutant protein expression is under the control of a poxvirus synthetic early/late promoter (pSE/L). 18 h after plasmid transfection, the cells were infected with MYXV from which the M013 gene had been disrupted (vMyx-M013KO), so that the only M013 protein expressed was from the transfected plasmid. To determine whether an early or late expression of M013 optimally modulates the interaction of M013 with ASC-1 or NF-κB1 and thereby the immune activation of these pathways, we chose to test at time points 6 and 24 h after virus infection (22). To determine the role of specific charged residues, we focused our attention on the selected single mutations (Table 1). Because the function of C-terminal region of M013 is unknown, we deleted the last 33 residues (residues 94–126) of M013 to generate the pyrin-only variant, M013PYD.
Table 1.
Mutational analyses of vPOP M013 protein from myxoma virus
The results from NF-κB activation and caspase-1 activation in THP-1 cells are summarized: +, weak binding; ++, moderate binding; +++, strong binding; −, no binding. Details of the assays are described under “Experimental procedures.”
| Mutation | Interaction with ASC-1 | Interaction with NF-κB | NF-κB activation | Caspase cleavage |
|---|---|---|---|---|
| D41A | +++ | + | ++ | − |
| E28Q | − | ++ | − | +++ |
| D29N | + | + | ++ | ++ |
| D57N | + | + | − | + |
| M013PYD (delete 94–126) | ++ | + | +++ | − |
Western blots were used to evaluate the level of M013 proteins expressed 6 h after virus infection (Fig. 2A). WT-M013 was expressed robustly, and the C-terminal deletion M013PYD and E28Q were readily detectable at this time point (Fig. 2A, lanes 2, 4, and 5), whereas weak or no protein levels were observed for mutants D29N, D57N, and D41A (Fig. 2A, lanes 6–8), suggesting that these mutant proteins did not accumulate at this early time point. On the other hand, 24 h after virus infection, WT-M013, and all of the mutant proteins were comparably expressed in THP-1 cells (Fig. 2B, lanes 2–8). To detect interaction between the expressed V5-tagged M013 proteins and endogenous ASC-1, co-immunoprecipitation of cell lysate was performed using anti-V5 antibody to pull down M013. Subsequently, the levels of bound ASC-1 (∼22 kDa) was assessed by anti-ASC-1 antibody by a Western blotting. The co-immunoprecipitation assay for the cell lysates at 6 h postinfection (h.p.i.), revealed the strong interaction between ASC-1 and WT-M013 and the mutants D41A and M013PYD (Fig. 2C, lanes 2, 6, and 7). In the cell lysates harvested at 24 h.p.i., WT-M013, M013PYD, and D41A interacted strongly with ASC-1 (lanes 2, 3, and 7), whereas little or no interaction occurred for the mutants E28Q, D29N, and D57N (22-kDa band in Fig. 2D, lanes 4–6 and 8).
Figure 2.

M013 mutants interact with the differential affinity with ASC-1 and NF-κB1. A and B, expression (input) of V5-tagged WT-M013 and M013 mutants in the THP-1 cells transfected and infected with vMyx-M013KO and harvested 6 h (A) and 24 h (B) postinfection. C and D, detection of WT-M013 and its mutant's interaction with endogenous ASC-1 in THP1 cells by co-immunoprecipitation. After transfection and infection, co-IP was performed using anti-V5 antibody, and the interacting endogenous ASC-1 in the complex was detected using anti-ASC-1 antibody by Western blotting analysis. The samples were prepared 6 h (C) and 24 h (D) after infection. E and F, detection of WT-M013 and its mutant's interaction with endogenous NF-κB1 (p105/p50) in THP1 cells by co-immunoprecipitation. After transfection and infection, co-IP was performed using anti-NF-κB1 antibody, and the interacting expressed V5-tagged M013 proteins in the complex were detected using anti-V5 antibody by Western blotting analysis. The samples were prepared 6 h (E) and 24 h (F) post infection. Lane M, protein marker; lane WT, WT M013; C1, cells infected with vMyx-M013KO; lane M013PYD, M013 with C terminus deletion; lanes E28Q, D29N, D57N, and D41A, M013 with mutations E28Q, D29N, D57N, and D41A, respectively; lane C2, cell lysates with no transfection or infection; *, a lane was deleted in the figure because of loading sample not described in the manuscript.
To detect interaction between M013 proteins and endogenous NF-κB1, antibody to human NF-κB1 was used for co-immunoprecipitation, and the presence of V5-tagged M013 proteins was detected in the immunoprecipitates as a 15-kDa band in Western blots using anti-V5 antibody. Co-immunoprecipitation of NF-κB1 revealed numerous nonspecific high-molecular-weight bands that were also visible in control cells infected only with vMyx-M013KO (C1) (Fig. 2E, lanes 1 and 2). However, at 6 h.p.i., WT-M013 (15-kDa band) but no other mutant proteins were detectable in the IP complex with NF-κB1 (Fig. 2E, lanes 3–7). Notably, at 24 h.p.i. M013PYD remained unable to associate with NF-κB1 (Fig. 2F, lane 3), whereas WT-M013 and all the mutants D41A, E28Q, D29N, and D57N could interact with NF-κB1 (15-kDa bands in Fig. 2F, lanes 2 and 4–7). Protein bands corresponding to ASC-1 (22 kDa) or M013 (15 kDa) were not detected in co-IP assay using cells infected with vMyx-M013KO alone (C1) or uninfected cells (C2) (Fig. 2, D and F, lanes 1 and 8).
In addition to human THP-1 cells, we also tested the interaction between M013 variants and NF-κB1 or ASC1 in HeLa cells, which lack endogenous inflammasome components but maintain NF-κB signaling capacity. A set of transfection and co-IP assays were performed using selected M013 variants. For detection of interactions between ASC and M013 mutants, HeLa cells were co-transfected with plasmids expressing ASC-1 and one of the V5-tagged M013 variants. The expression of the 15-kDa WT-M013 and mutant proteins are shown in the input samples (Fig. S3, A and B). Co-IP was performed using anti-V5 antibody to pull down M013 proteins, and the presence of ASC-1 in the complex was detected using anti–ASC-1 antibody (Fig. S3C). In a simultaneous experiment, co-IP was done using anti–NF-κB1 antibody to detect endogenous NF-κB1 (Fig. S4, A and B), and the presence of V5-tagged M013 in the complex was detected using anti-V5 antibody (∼15 kDa) by Western blotting analysis (Fig. S3D). No protein bands were observed in the controls; C1 (HeLa cells infected only with vMyx-M013KO) and C2 (HeLa cells without infection or transfection). Co-IP data suggested that M013 mutants E28Q, D29N, and D57N showed weak or no interaction with ASC-1, whereas WT-M013, D41A, and M013PYD were pulled down with ASC-1 in the IP complex (Fig. S3C). Endogenous NF-κB1 exhibited strongest interactions with WT-M013 and mutant D41A, consistent with findings from THP-1 cells. Weak interactions were observed between NF-κB1 and mutants E28Q and D29N and essentially no interactions with M013PYD or D57N. These results suggest that protein–protein interactions with some M013 mutants (such as D57N interactions with NF-κB1) differ in different cell lines. However, in general, the binding studies in different cell lines are consistent with structural determinants for ASC-1 binding in the pyrin domain and NF-κB binding in the C-terminal tail of M013.
Differential interaction of M013 mutants with ASC-1 and NF-κB1 alter the activation of inflammasome and NF-κB signaling cascades
Interaction of WT-M013 with NF-κB1 appears to regulate the processing of NF-κB1/p105 and thereby the accumulation of p50 in THP-1 cells (8). To evaluate the impact of M013 mutations on the processing of NF-κB1/p105, Western blotting analysis was performed. THP1 cells were transfected with WT-M013 or M013 mutant expression plasmids for 18 h and then infected with vMyx-M013KO virus, and the cells were then harvested at 6 and 24 h.p.i. for the detection of processed NF-κB1. Specific bands of NF-κB1/p105 and p50 were detected at 6 and 24 h.p.i. along with some nonspecific bands that were also seen in the nontransfected controls. The presence of p105 and not p50 for the WT-M013 lane at the 6-h time point suggested that processing of p105 was inhibited in the presence of WT-M013 (Fig. 3A, lane 1). Very little processed p50 could be detected in samples from control C1 (cells infected with vMyx-M013KO alone) and the mutants E28Q and D41A (Fig. 3A, lanes 2, 3, and 6). In contrast, slightly higher levels of processed p50 could be detected from samples transfected with the plasmids expressing M013 mutants D29N and D57N and M013PYD (Fig. 3A, lanes 4 and 5). Subsequently, with the accumulation of expressed M013 protein and its mutants after 24 h.p.i., an increase in the level of the processed p50 form was observed in samples from C1 (vMyx-M013KO alone), M013PYD, E28Q, D29N, and D57N (Fig. 3B, lanes 2–6). As a control, samples from WT-M013, cells without infection or transfection (C2) and cells transfected with the empty plasmid (P) showed little or no processed p50 (Fig. 3B, lanes 1, 8, and 9). In all these functional assays, for the positive control, THP-1 cells were first stimulated with phorbol 12-myristate 13-acetate (PMA) for 3 h and then with LPS for 5 or 23 h followed by the addition of 5 mm ATP 1 h prior to harvesting the cells (31). LPS stimulation of THP-1 cells resulted in the efficient activation of NF-κB signaling pathway and processing of NF-κB1/p105 at the 6- and 24-h time points (Fig. 3A, lane 8, and B, lane 10).
Figure 3.

Differential activation of NF-κB1 p50 and caspase-1 with WT-M013 and mutants. A and B, levels of processed NF-κB1/p50 in the cell lysates (input) transfected with V5-tagged WT-M013 and mutant plasmids and infected with vMyx-M013KO for 6 h (A) and 24 h (B). C and D, detection of activated caspase-1 p20 in the lysates from THP-1 cells transfected with V5-tagged WT-M013 and mutant plasmids and infected with vMyx-M013KO for 6 h (C) and 24 h (D). E and F, β-tubulin (50 kDa) showing equal protein loading for the levels of NF-KB1 and caspase-1 in the THP-1 cells transfected with V5 tagged-M013 and mutants and infected with vMyx-M013KO for 6 h (E) and 24 h (F). Lane WT, WT M013; lane C1, cells infected with vMyx-M013KO; lanes E28Q, D29N, D57N, and D41A, M013 with mutations E28Q, D29N, D57N, and D41A, respectively; M013PYD, M013 with C terminus deletion; lane LPS, positive control for the activation of NF-κB1 and caspase-1; lane M, protein marker; lane C2, cell lysates with no transfection or infection.
To test whether the variable levels of interactions among M013 mutants with ASC-1 may influence the activation of the inflammasome in response to viral stimulation, we examined the activation of caspase-1 (p20) by Western blotting analysis. Binding of WT-M013 with ASC-1 inhibited the cleavage of caspase-1 resulting in little or no expression of activated caspase-1 p20 at 6 and 24 h.p.i. in the THP-1 cell lysate (Fig. 3, C, lane 2, and D, lane 1). An increase in the level of activated caspase-1, p20 form was observed for C1 (vMyx-M013KO alone) and the M013 mutants including E28Q, D29N, and D57N at 6 and 24 h.p.i. (Fig. 3, C, lanes 3 and 5–7, and D, lanes 2 and 4–6). Although M013 mutants M013PYD and D41A showed strong interaction with ASC-1 similarly to WT-M013, they were not able to inhibit activation of caspase-1 at 6 or 24 h.p.i. (Fig. 3, C, lanes 4 and 8, and D, lanes 3 and 7). In the same assay, no or very little activated caspase-1 p20 was detectable from the negative controls, C2 and P (Fig. 3, C, lane 9, and D, lane 9). As a positive control, THP-1 stimulated with LPS along with PMA and ATP revealed activated caspase-1 at the 24-h time point (Fig. 3D, lane 8). Antibody against tubulin was used to show equal total protein loading in all the tested samples for NF-κB (Fig. 3, E and F).
Activation of caspase-1 was also measured in THP1 cells using a bioluminescence assay (Promega Caspase-Glo® 1 inflammasome assay). At 6 h.p.i., the level of activated caspase-1 was significantly reduced for the cells transfected with WT-M013 compared with C1 (i.e. cells infected with vMyx-M013KO virus alone), LPS treatment as a positive control, or cells transfected with plasmids expressing mutants M013PYD, D29N, D57N, and D41A (Fig. 4A). This pattern of caspase-1 activation was also observed at 24 h.p.i., when WT-M013 continuously blocked caspase-1 activation more than M013PYD, E28Q, D29N, and D57N (Fig. 4B). Interestingly, the level of activated caspase-1 remained consistent at both time points for mutant D41A, which showed strong interaction with ASC-1. These results suggest that the increase in caspase-1 activation was time-dependent, and some mutations in M013 seemed to weaken the interaction of M013 with ASC-1 and thus alter the activation of caspase-1.
Figure 4.

WT-M013 and mutants modulate the caspase-1 activation and cytokine response in THP-1 cells. A and B, luminescence assay result to quantitate the caspase-1 activation in THP-1 cell supernatants transfected with V5-tagged WT-M013 and mutant plasmids and infected with vMyx-M013KO for 6 h (A) and 24 h (B) (as described under “Results”). C–E represent the ELISA results for TNF, IL-6, and IL-18 in the supernatants from THP-1 cells transfected with V5-tagged WT-M013 and mutants and infected with vMyx-M013KO for 6 h. F–H, ELISA results for TNF, IL-6, and IL-18 in the supernatants from THP-1 cells transfected with V5-tagged WT-M013 and mutants and infected with vMyx-M013KO 24 h. WT, WT M013; C1, cells infected with vMyx-M013KO; M013PYD, M013 with C terminus deletion; E28Q, D29N, D57N, and D41A, M013 with mutations E28Q, D29N, D57N, and D41A, respectively; C2, cells with no transfection or infection (negative control); LPS, positive control; P, cells transfected with an empty plasmid. Significance was tested using one-way analysis of variance, with multiple comparison correction. A value of p < 0.05 was considered significant.
Because activation of the inflammasome would be predicted to lead to the elevation of numerous pro-inflammatory cytokines, either directly or indirectly, we checked whether the differential interactions of M013 mutants with ASC-1 and/or NF-kB1 would alter the subsequent release of downstream cytokines. To test the levels of secreted cytokines, supernatants from the THP-1 cells that were transfected with different M013 expression plasmids and infected with vMyx-M013KO virus were collected at 6 and 24 h.p.i. for ELISA assays. ELISA showed that the levels of secreted TNF were significantly higher for the mutants M013PYD, E28Q, D29N, and D57N compared with WT-M013 at 6 h.p.i. (Fig. 4C). No significant differences were observed for WT-M013 compared with mutant D41A (Fig. 4C). IL-6 was found to be considerably higher in the cell supernatants from mutants M013PYD, D29N, D57N, and D41A compared with WT-M013 (Fig. 4D). However, levels of IL-6 were not statistically significant between WT-M013, E28Q, and the control C2 (no transfection or infection) (Fig. 4D). We then tested the accumulation of IL-18, a signature cytokine marker of NLRP3 inflammasome activation and a critical member of the IL-1β family. Significantly higher levels of IL-18 were found for C1 and mutants E28Q, D29N, and D57N. However, D41A levels were comparable with WT-M013 at 6 h.p.i. (Fig. 4E). This suggests that despite the low expression (Fig. 2A), M013 mutants were able to induce the secretion of signature cytokines of both the NLRP3 inflammasome and NF-κB1 pathways at 6 h.p.i. A robust increase was observed for most of the cytokines in transfected THP-1 cell supernatants at, 24 h.p.i. In contrast to D41A and C2, levels of IL-6 were substantially higher for control C1, mutants M013PYD, E28Q, D29N, and D57N (Fig. 4G). At 24 h.p.i, the experimental control C1 (vMyx-M013KO alone) and the mutants E28Q, D29N, and D57N revealed significantly higher levels of IL-18 compared with the WT-M013 (Fig. 4H). These findings appear consistent with these mutants showing little or no binding with ASC-1. In contrast, no statistically significant differences in IL-18 levels were observed for the M013PYD and D41A mutants, both of which retain affinity for ASC-1 (Fig. 2D) when compared with the WT-M013. These results suggest that weakened binding of M013 mutants to ASC-1 correlates with the increased activation of caspase-1 and the production of specific indicator cytokines.
Discussion
The role of M013 in immunosuppression and the pathogenesis of MYXV in rabbits is well-known from previous studies (6). However, its deletion does not alter the replication of MYXV in cultured cells (6). The vPOP M013 is known to co-regulate both the PYRIN-containing inflammasomes (e.g. NLRP3) and NF-κB pathways (8). However, its mechanism of action by interacting with different cellular protein components of the two pathways ASC-1 and NF-κB1 are unknown. Here, we show for the first time that M013 interacts with both endogenous ASC-1 and NF-κB1 in either human myeloid cells that are competent for both pathways (THP1 cells) or human nonmyeloid cells that are competent for only NF-κB signaling (HeLa cells). This allowed us to conduct mutational studies on M013 that have the potential to map the interactions of M013 with ASC-1 or NF-κB1 and study the consequences on the independent activation of NF-κB and inflammasome pathways.
Using homology modeling, we identified distinguishing features of M013 that may encode the specificity for interactions with ASC-1 or NF-κB1. Strikingly, single-site mutations that reduced the negative charge on a distinct region of M013 (E28Q, D29N, and D57N) resulted in weak or no interaction of M013 with ASC-1 (Table 1). These observations suggest that even modest changes in the primary sequence of M013 result in severe effects on its functions. The role of negatively charged residues is site-specific, as evidenced by the D41A mutation, which resides on the opposite face of the pyrin domain (Fig. 1B). This mutation does not affect ASC-1 interactions and has mild effects on the antagonism of inflammatory signaling, relative to WT-M013 (Fig. 4). Overall, the binding and functional data are consistent with a model in which a negatively charged M013 region constitutes a key epitope for binding to the pyrin domain of ASC-1. The role of both charged and hydrophobic residues in PYD–PYD interaction have been described by Moriya et al. (23), and structure determination of the M013 complex with ASC-1 will be required to fully understand the molecular basis for specificity.
Viral and cellular PYDs are generally 90 residues in length. The vPOPs of M013 and S013 have an extended tail beyond the predicted PYD that contains a highly conserved region of positive charges at the C terminus (Fig. 1A). The S013L protein from Shope fibroma virus has 60% sequence identity to M013 and has been shown to be recruited into ASC-1 specks (32). The tail deletion mutant M013PYD (residues 1–93) retained the interaction with ASC-1, as shown by a co-IP assay, suggesting that the PYD of M013 is indeed the key determinant for PYD–PYD interactions. Given the related sequences of the C-terminal tails of M013/S013, we investigated whether this region is important for antagonism of NF-κB pathways, which is a conserved functional property of these two vPOPs. Results from our co-IP assay for NF-κB1 and M013 demonstrate that M013 mutants E28Q, D29N, and D57N still interact with NF-κB1, suggesting that charged residues in the PYD domain of M013 do not strongly impact this interaction with NF-κB1. However, deletion of C-terminal tail (residues 94–126) resulted in a complete loss of interaction between NF-κB1 and M013.
We examined whether these differences in the interaction of M013 protein and its mutants with ASC-1 or NF-κB1 would compromise the ability to modulate signaling pathways. Data clearly show that mutation of negatively charged residues compromised the ability of M013 to inhibit cleavage of pro-caspase to active caspase-1, resulting in increased levels of IL-18 secretion. In contrast to these binding/signaling correlations with the ASC-1 interface, the M013 variants M013PYD and D41A maintain direct protein interactions with ASC-1, yet are unable to block activation of caspase-1 and release of IL-18 at both the early and later time points of infection (Fig. 4). These results suggest that interactions with ASC-1 alone are insufficient to inhibit caspase-1 activation. D41A is situated on the opposite face relative to the negatively charged ASC-1–binding region (Fig. 1B), and it is possible that this region mediates oligomerization of M013–ASC-1 complexes that contributes to antagonism of caspase activation. Similarly, the tail region of M013 (residues 94–126) may also contribute to protein–protein interactions that antagonize the inflammasome. A model for the mechanism of binding and antagonism is shown in Fig. 5.
Figure 5.
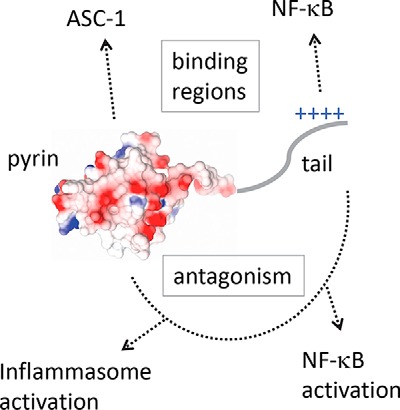
Model for antagonism by M013 via two structural domains. The N-terminal pyrin domain M013 is represented as an electrostatic surface, followed by the tail enriched in positively charged residues. The binding of M013 to its cellular targets is imparted by the two domains indicated by the boxed text above the model. However, antagonism of downstream signaling requires the interplay between both domains, as indicated by the dotted line connecting the two regions below the model.
Functional analyses of M013PYD revealed that the tail region of M013 is required for the inhibition of NF-κB1/p105 processing to p50 and subsequent release of NF-κB regulated pro-inflammatory cytokines TNF and IL-6. Interestingly, single-site mutants E28Q and D29N also compromised NF-κB antagonism despite maintaining interactions with NF-κB1. Overall, these data are consistent with a model suggesting that interactions with NF-κB1 and ASC-1 are necessary but not sufficient to enable inhibition of downstream signaling pathways. Although distinct structural motifs enable direct recognition of ASC-1 and NF-κB, downstream regulatory effects require a cooperation between the two segments of M013. Thus, vPOP M013 protein has evolved dual regulatory functions through C-terminal extension of its PYD, and future structural and functional studies will be necessary to enable an understanding of how these regions work together to suppress innate immunity. M013 is currently being studied as an anti-inflammatory gene therapy for ocular inflammation in mouse models of uveitis (33), but it is currently unknown whether this M013-mediated inhibition is via inflammasome inhibition, the blockade of NF-κB signaling, or both. A better understanding of the interactions between MYXV and cancerous tissues within human hosts may aid in the development and translation of modified MYXV as an oncolytic therapy. Finally, anti-inflammatory mimics of the viral protein, such as the short anti-NF-κB tail region that is unique to M013 and S013, could be developed as future anti-inflammatory peptides in clinical or research settings.
Experimental procedures
Gene cassette preparation
Primers with complementarity to the nucleotides coding for the N and C termini of the M013 protein were utilized in a modified overlap-extension PCR technique to flank the gene with the vaccinia virus synthetic early/late promoter at the upstream end and the coding sequence for the V5 epitope at the downstream end. These primers were used to amplify the gene from a viral genomic DNA preparation of myxoma virus strain Lausanne. The stop codon of M013 was omitted to allow for expression of an M013-V5 tag fusion protein. Secondary PCR was performed to amplify this fragment with primers designed to incorporate 5′ overhangs including the attB1 and attB2 sequences for Gateway BP recombination (Thermo Fisher Scientific). Accuprime Pfx DNA polymerase (Life Technologies) was used for all PCR.
Mutagenic primer design and polymerase chain reaction
For each single point mutation, two complementary oligonucleotides were designed to include 15–30 nucleotides complementary to M013 on each side of the substituted codon. Each oligonucleotide was interrogated with Primer3 software to ensure the absence of predicted hairpins, primer-dimers, etc. All oligonucleotides were purchased from IDT with standard desalting.
The attB-flanked pSE/L-M013-V5 cassette was amplified by PCR in two separate reactions with AccuPrime Pfx: the first with the attB1 forward primer in combination with the minus strand mutagenic primer and the second with the attB2 reverse primer in combination with the plus strand mutagenic primer. The unpurified product of each reaction was diluted 1:50 in a second step reaction with attB1 forward and attB2 reverse primers to ligate the two fragments together to form a complete cassette once again.
Plasmid preparation and bacterial transformation
Gene cassettes were cloned into pDONR221 with BP Clonase II (Thermo Fisher Scientific) following the manufacturer's protocol. The plasmids were transformed into calcium chloride-competent Escherichia coli strain DH5α and selected by kanamycin resistance. Following confirmation of coding sequences, specific mutant M013 constructs were also cloned into cytomegalovirus promoter-driven plasmids. To generate these vectors, the M013 gene was amplified from the pSE/L-M013-V5 plasmid using primers specific to the beginning and end of the M013 sequence, omitting the stop codon. These primers were designed with 5′ overhangs containing the attB1 and attB2 sequences. These fragments were also cloned into pDONR221 using BP Clonase II. Finally, the pDONR221 vector containing the mutant M013 gene was recombined with pcDNA-DEST40 by LR Clonase II (Thermo Fisher Scientific), following the manufacturer's protocol. The resulting vector expressed M013 fused to V5+His6 tag driven by the constitutively active cytomegalovirus promoter. M013L ORF lacking the C-terminal tail (residues 94–126) was constructed by PCR amplification using appropriate primers and cloned in the entry vector pDONR221.
Sequencing
Sequencing reactions were performed by the University of Florida ICBR Sanger Sequencing core using BigDye chemistry (Applied Biosystems). The chromatograph was generated using an Applied Biosystems DNA Analyzer capillary electrophoresis instrument. The bases were called to create the sequence, using 4Peaks software (Nucleobytes).
Reagents and antibodies
Rabbit polyclonal antibodies for NFκB1 (p105/p50) (catalog no. 3035S, lot no. 5) and caspase-1 (catalog no. 2225S, lot no. 3) were purchased from Cell Signaling Technology. Anti-V5 mAb (catalog no. R960-25, lot no. 1900119) was purchased from Invitrogen. ASC-1 (catalog no. AB3607, lot no. 2801940) and c-Myc (catalog no. Sc-40, lot no. B0116) antibodies were purchased from EMD Millipore and Santa Cruz Biotechnology. PMA and LPS (lipopolysaccharide) were purchased from Sigma.
Cell lines and cell culture
HeLa and THP1 cells were cultured in Dulbecco's modified Eagle's medium and RPMI medium, respectively, supplemented with 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 100 units/ml penicillin, and 100 mg/ml streptomycin. THP1 cell line was cultured in RPMI 1640 medium (Lonza) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 mg/ml streptomycin. For the control assay, THP1 cells were stimulated for 100 ng/ml PMA (for 3 h) and again later with LPS for a total of 5 or 23 h followed by the final activation with 5 mm ATP 1 h prior to the harvesting of the cells.
Viral preparation
Construction of a WT myxoma virus (vMyx-GFP) that expresses a GFP cassette under the control of a poxvirus synthetic early/late promoter was described previously (34). Construction of the vMyxM013-KO virus was previously described (6). Viruses were purified by centrifugation through a sucrose cushion and successive sucrose gradient sedimentations as described previously (6).
Transfections assays
THP1 cells were seeded at 80% confluence onto 6-well plates, the day before being transfected. The cells were transfected according to the manufacturer's protocol using Effectene transfection reagent (Qiagen) and 0.5 μg of DNA, in serum-free RPMI medium per each well.
Western blotting
For detection of protein, cells were harvested at different time points, washed with PBS, and stored at −80 °C or processed immediately with RIPA lysis buffer (50 mm Tris, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Roche). Protein samples were separated on SDS-PAGE gels and transferred to polyvinylidene difluoride membrane (GE Healthcare) using a wet transfer apparatus (Bio-Rad). Membranes were blocked in 5 ml of LI-COR odyssey blocking buffer for 1 h at room temperature and then incubated overnight with primary antibody at 4 °C. The membranes were washed three times for 15 min each with TBST and incubated with HRP-conjugated goat anti-mouse (1:5000) or goat anti-rabbit (1:5000) secondary antibody in 5 ml of LI-COR odyssey blocking buffer for 1 h at room temperature with gentle agitation. The membranes were washed three times for 15 min each with TBST. The blots were scanned for the signal using LI-COR Odyssey® CLx scanner.
Quantification of cytokine secretion by ELISA
THP1 cells were transfected with the plasmids expressing M013L WT and the mutants and subsequently infected with vMyx-M013KO for 6 and 24 h. At the indicated time points after virus infection, the cells were separated from supernatants by low speed centrifugation, and the supernatants were used for ELISA. The level of secreted cytokines TNF, IL-6 (eBiosciences), IL-1β (BD Biosciences), and IL-18 (RayBiotech) were determined in the supernatants using ELISA assay kits following manufacturer protocol.
Caspase-1 luminescence assay
Caspase-Glo® 1 inflammasome assay kit (Promega) was used to measure the caspase-1 activation in THP1 cells transfected with plasmids expressing WT M013L and the mutants and subsequently infected with vMyx-M013KO. Caspase-1 and caspase-1 inhibitor reagents were prepared and added to the transfected cells directly as described by the manufacturer.
Author contributions
R. R. G., C. J., M. M. R., A. R. K., A. S. L., and G. M. conceptualization; R. R. G., C. J., M. M. R., and A. R. K. formal analysis; R. R. G., C. J., and M. M. R. validation; R. R. G., C. J., M. M. R., A. R. K., and G. M. investigation; R. R. G., C. J., M. M. R., and A. R. K. methodology; R. R. G., C. J., M. M. R., A. R. K., A. S. L., and G. M. writing-original draft; R. R. G., C. J., M. M. R., A. R. K., A. S. L., and G. M. writing-review and editing; M. M. R., A. S. L., and G. M. supervision; A. R. K., A. S. L., and G. M. funding acquisition; G. M. resources.
Supplementary Material
This work was supported by Science Foundation Ireland Grant SFI-12/IA/1239 (to A. R. K.), National Institutes of Health Grant R01 AI100987 (to G. M. and M. M. R.), start-up funding from Arizona State University (to G. M.), and the Shaler Richardson Professorship Endowment (to A. S. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- MYXV
- myxoma virus
- PYD
- pyrin domain
- vPOP
- viral pyrin-only protein
- cPOP
- cellular pyrin-only protein
- IL
- interleukin
- TNF
- tumor necrosis factor
- IP
- immunoprecipitation
- h.p.i.
- h postinfection
- PMA
- phorbol 12-myristate 13-acetate
- LPS
- lipopolysaccharide.
References
- 1. Alcami A. (2003) Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 3, 36–50 10.1038/nri980 [DOI] [PubMed] [Google Scholar]
- 2. Smith G. L., Benfield C. T., Maluquer de Motes C., Mazzon M., Ember S. W., Ferguson B. J., and Sumner R. P. (2013) Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J. Gen. Virol. 94, 2367–2392 10.1099/vir.0.055921-0 [DOI] [PubMed] [Google Scholar]
- 3. Seet B. T., Johnston J. B., Brunetti C. R., Barrett J. W., Everett H., Cameron C., Sypula J., Nazarian S. H., Lucas A., and McFadden G. (2003) Poxviruses and immune evasion. Annu. Rev. Immunol. 21, 377–423 10.1146/annurev.immunol.21.120601.141049 [DOI] [PubMed] [Google Scholar]
- 4. Kerr P. J., Liu J., Cattadori I., Ghedin E., Read A. F., and Holmes E. C. (2015) Myxoma virus and the Leporipoxviruses: an evolutionary paradigm. Viruses 7, 1020–1061 10.3390/v7031020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ross J., and Sanders M. F. (1977) Innate resistance to myxomatosis in wild rabbits in England. J. Hyg. (Lond.) 79, 411–415 10.1017/S0022172400053262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnston J. B., Barrett J. W., Nazarian S. H., Goodwin M., Ricciuto D., Wang G., and McFadden G. (2005) A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 23, 587–598 10.1016/j.immuni.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 7. Cameron C., Hota-Mitchell S., Chen L., Barrett J., Cao J. X., Macaulay C., Willer D., Evans D., and McFadden G. (1999) The complete DNA sequence of myxoma virus. Virology 264, 298–318 10.1006/viro.1999.0001 [DOI] [PubMed] [Google Scholar]
- 8. Rahman M. M., Mohamed M. R., Kim M., Smallwood S., and McFadden G. (2009) Co-regulation of NF-κB and inflammasome-mediated inflammatory responses by myxoma virus pyrin domain-containing protein M013. PLoS Pathogens 5, e1000635 10.1371/journal.ppat.1000635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Franchi L., Muñoz-Planillo R., and Núñez G. (2012) Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 10.1038/ni.2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lamkanfi M., and Dixit V. M. (2012) Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 10.1146/annurev-cellbio-101011-155745 [DOI] [PubMed] [Google Scholar]
- 11. Rathinam V. A., Vanaja S. K., and Fitzgerald K. A. (2012) Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342 10.1038/ni.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park H. H., Lo Y. C., Lin S. C., Wang L., Yang J. K., and Wu H. (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 25, 561–586 10.1146/annurev.immunol.25.022106.141656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Indramohan M., Stehlik C., and Dorfleutner A. (2018) COPs and POPs patrol inflammasome activation. J. Mol. Biol. 430, 153–173 10.1016/j.jmb.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mariathasan S., and Monack D. M. (2007) Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 7, 31–40 10.1038/nri1997 [DOI] [PubMed] [Google Scholar]
- 15. Stehlik C., Krajewska M., Welsh K., Krajewski S., Godzik A., and Reed J. C. (2003) The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor–κB and pro–caspase-1 regulation. Biochem. J. 373, 101–113 10.1042/bj20030304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bedoya F., Sandler L. L., and Harton J. A. (2007) Pyrin-only protein 2 modulates NF-κB and disrupts ASC:CLR interactions. J. Immunol. 178, 3837–3845 10.4049/jimmunol.178.6.3837 [DOI] [PubMed] [Google Scholar]
- 17. Wullaert A., Bonnet M. C., and Pasparakis M. (2011) NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 21, 146–158 10.1038/cr.2010.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pasparakis M. (2009) Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat. Rev. Immunol. 9, 778–788 10.1038/nri2655 [DOI] [PubMed] [Google Scholar]
- 19. Beinke S., and Ley S. C. (2004) Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 382, 393–409 10.1042/BJ20040544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayden M. S., and Ghosh S. (2004) Signaling to NF-κB. Genes Dev. 18, 2195–2224 10.1101/gad.1228704 [DOI] [PubMed] [Google Scholar]
- 21. Rahman M. M., and McFadden G. (2011) Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 9, 291–306 10.1038/nrmicro2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rahman M. M., and McFadden G. (2011) Myxoma virus lacking the pyrin-like protein M013 is sensed in human myeloid cells by both NLRP3 and multiple Toll-like receptors, which independently activate the inflammasome and NF-κB innate response pathways. J. Virol. 85, 12505–12517 10.1128/JVI.00410-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moriya M., Taniguchi S., Wu P., Liepinsh E., Otting G., and Sagara J. (2005) Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry 44, 575–583 10.1021/bi048374i [DOI] [PubMed] [Google Scholar]
- 24. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chemical Computing Group (2018) Molecular Operating Environment, Chemical Computing Group, Montreal, Canada [Google Scholar]
- 26. Kim D. E., Chivian D., and Baker D. (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 10.1093/nar/gkh468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferrao R., and Wu H. (2012) Helical assembly in the death domain (DD) superfamily. Curr. Opin. Struct. Biol. 22, 241–247 10.1016/j.sbi.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liepinsh E., Barbals R., Dahl E., Sharipo A., Staub E., and Otting G. (2003) The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J. Mol. Biol. 332, 1155–1163 10.1016/j.jmb.2003.07.007 [DOI] [PubMed] [Google Scholar]
- 29. Awad F., Assrawi E., Jumeau C., Georgin-Lavialle S., Cobret L., Duquesnoy P., Piterboth W., Thomas L., Stankovic-Stojanovic K., Louvrier C., Giurgea I., Grateau G., Amselem S., and Karabina S. A. (2017) Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS One 12, e0175336 10.1371/journal.pone.0175336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsuchiya S., Yamabe M., Yamaguchi Y., Kobayashi Y., Konno T., and Tada K. (1980) Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26, 171–176 10.1002/ijc.2910260208 [DOI] [PubMed] [Google Scholar]
- 31. Cordle S. R., Donald R., Read M. A., and Hawiger J. (1993) Lipopolysaccharide induces phosphorylation of MAD3 and activation of c-Rel and related NF-κB proteins in human monocytic THP-1 cells. J. Biol. Chem. 268, 11803–11810 [PubMed] [Google Scholar]
- 32. Dorfleutner A., Talbott S. J., Bryan N. B., Funya K. N., Rellick S. L., Reed J. C., Shi X., Rojanasakul Y., Flynn D. C., and Stehlik C. (2007) A Shope fibroma virus PYRIN-only protein modulates the host immune response. Virus Genes 35, 685–694 10.1007/s11262-007-0141-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ildefonso C. J., Jaime H., Rahman M. M., Li Q., Boye S. E., Hauswirth W. W., Lucas A. R., McFadden G., and Lewin A. S. (2015) Gene delivery of a viral anti-inflammatory protein to combat ocular inflammation. Hum. Gene Ther. 26, 59–68 10.1089/hum.2014.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnston J. B., Barrett J. W., Chang W., Chung C. S., Zeng W., Masters J., Mann M., Wang F., Cao J., and McFadden G. (2003) Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J. Virol. 77, 5877–5888 10.1128/JVI.77.10.5877-5888.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.