

Abstract
Group A Streptococcus pyogenes (GAS) is a causative agent of pharyngeal and dermal infections in humans. A major virulence determinant of GAS is its dimeric signature fibrillar M-protein (M-Prt), which is evolutionarily designed in modules, ranging from a hypervariable extracellular N-terminal region to a progressively more highly conserved C-terminus that is covalently anchored to the cell wall. Of the >250 GAS isolates classified, only the subset of skin-trophic Pattern D strains expresses a specific serotype of M-Prt, PAM, that directly binds to host human plasminogen (hPg) via its extracellular NH2-terminal variable A-domain region. This interaction allows these GAS strains to accumulate components of the host fibrinolytic system on their surfaces to serve extracellular functions. While structure-function studies have been accomplished on M-Prts from Pattern A-C GAS isolates with different direct ligand binding properties from PAM, much less is known regarding the structure-function relationships of PAM-type M-Prts, particularly their dimerization determinants. To examine these questions, PAMs from seven GAS strains with sequence variations in the NH2-terminal ligand binding domains, as well as truncated versions of PAM, were designed and studied. The results from bioinformatic and biophysical analyses show that the different domains of PAM are disparately engaged in dimerization. From these data, we propose an experimentally-based model for PAM secondary and quaternary structures that is highly dependent on the conserved helical C-terminal C-D-domains. In addition, while the N-terminal regions of PAMs are variable in sequence, the binding properties of hPg and its activated product, plasmin, to the A-domain, remain intact.
Keywords: Protein domains, protein chemistry, protein-protein interaction, plasminogen, bacteria, receptor structure-function, recombinant protein expression
Graphical Abstract
1. Introduction
Group A Streptococcus pyogenes (GAS) is a spherical Gram-positive β-hemolytic pathogen that selectively infects humans, causing a range of infections, from currently antibiotic-sensitive pharyngitis and impetigo to serious invasive diseases, such as necrotizing fasciitis and streptococcal toxic shock, with a mortality >25%. In addition, immune sequela of these infections, such as acute rheumatic fever (ARF), are common causes of heart disease worldwide (Lee et al., 2009a). Thus, the morbidity and sometimes mortal GAS infections represent serious burdens to human health.
Numerous virulence factors are present in GAS, ranging from those present on the bacterial chromosomal to those expressed by incorporated bacteriophages (Bao et al., 2014). The GAS genome is very plastic with many integration sites, and horizontal transfer with recombination of genetic materials between GAS strains (Bao et al., 2014) and between GAS strains with other β-hemolytic streptococcal strains (McNeilly and McMillan, 2014) lead to new isolates with variable properties that result from selective pressures from the immune system of human hosts.
All known GAS isolates contain a signature high copy number M-Protein (M-Prt), encoded by the emm gene. This is the most widely recognized virulence determinant of GAS. Approximately 250 different antigenically distinct M-Prt-types of GAS isolates have been identified through sequencing ~90 nucleotides in the hypervariable region (HVR) of the emm gene (Beall et al., 1996; Bessen, 2016). M-Prts contain ~350–450 amino acids arranged in a modular fashion (Hollingshead et al., 1986). The N-terminus is HVR, which is sequentially followed by variable A- and B- domains and highly conserved C-, D-, and Pro/Gly-modules (Fischetti, 1989; Fischetti, 1991). The C-terminus of mature M-Prts includes a sortase A (LPXTG)-sensitive membrane spanning site, which, when fully processed, is covalently linked to cell wall peptidoglycans. Conformationally, the mature M-Prt is a fibrillar protein consisting primarily of α-helices interrupted by loops, with its N-terminal regions (HVR-A-B-C-D) protruding through the cell wall into extracellular solutions. This provides GAS with a hair-like surface and allows the binding of many different types of host proteins to variably interact with strain-specific M-Prts.
The emm gene is located within a virulence-determining region of the bacterial chromosome, viz., the multiple gene activator (Mga) regulon (Hondorp and McIver, 2007), which can contain up to five other sortase A-anchored proteins (Liang et al., 2013). This entire regulon is situated between the mga and laminin binding protein (lmb) genes in Pattern D strains. Thus, the environmentally-sensitive transcriptional factor, Mga, is an important regulator of M-Prt expression (McIver and Scott, 1997; McIver et al., 1995). The number and arrangement of the genes in the Mga regulon is used to type the pattern of this chromosomal region. Five patterns (A-E) have been identified with different gene arrangements in this regulon (Bessen et al., 1996). The characterization of this region of the chromosome is important for association of the subject GAS strain with the epidemiology of GAS infections, as well as their tissue selectivities and virulences.
In many pattern A-C strains, GAS utilizes M-Prts for adherence and colonization on oral and skin epithelial cells (Berge and Sjobring, 1993; Walker et al., 2014) to provide the inflammatory response accompanying infection. In addition to direct causation of the infection, GAS has evolved survival mechanisms in different host niches to assist the organism in evasion of the host innate immune response. For example, several Pattern A-C M-Prts interact with hemostasis system proteins, e.g., fibrinogen (Ghosh, 2011; Glinton et al., 2017), to provide fibrin that encapsulates the bacteria to allow their colonization. This serves as an initial protective antiphagocytosis mechanism. Some other M-Prts of the Pattern A-C class react with complement binding proteins, e.g., C4BP, and Factor H (FH), through the HVR- region, and inhibit C3b deposition on GAS cells (Berggård et al., 2001; Gustafsson et al., 2013).
Pattern D GAS strains are unique with regard to binding of host proteins and are the only GAS subtype that directly interacts with human plasma plasminogen (hPg), a key component of the fibrinolytic system, through their M-Prts (PAM). This specific and very tight interaction occurs via the variable A-domains of PAM (Ringdahl et al., 1998; Rios-Steiner et al., 2001; Wang et al., 2010b; Wistedt et al., 1995). The hPg-PAM interaction is central to the pathogenesis of this microorganism (Sanderson-Smith et al., 2006b). PAM-bound hPg is activated by a secreted subtype (2b) of streptokinase (SK2b), produced only by Pattern D strains (Zhang et al., 2012; Zhang et al., 2013). This process results in formation of a serine protease plasmin (hPm) (Lahteenmaki et al., 2001), that remains bound to GAS, thereby providing GAS with a surface-bound protease that is employed to solubilize host fibrin that encapsulates the GAS cells to permit ultimate dissemination. In addition, cell-bound hPm serves to degrade extracellular matrix (ECM) components and cellular tight junctions thereby facilitating the invasion of GAS into deep tissues of the host (Sumitomo et al., 2016; Wong et al., 1992). Thus, the exclusive PAM-hPg interaction generally favors virulence of GAS (Sanderson-Smith et al., 2008; Sun et al., 2004).
Isolated M-Prts are generally believed to exist as non-ideal coiled-coil dimers in solution (Ghosh, 2011; Phillips et al., 1981), but limited experimental biophysical evidence lies at the basis of this conclusion. Nonetheless, the relative population of dimeric M-Prts on the cell surface is not known but would be expected to be limited by spacing constraints of the covalently bound monomers. While the functions of M-Prts bound to GAS cells have received much attention, M-Prts are also released from the cell surface during invasive infections, principally by a bacterial secreted cysteine protease, SpeB (Berge and Björck, 1995), and by neutrophil proteases (Herwald et al., 2004). The soluble forms of these M-Prts retain much their functional N-termini and thus possess important functions as soluble proteins. Thus, the structure-function relationships of M-Prts in solution are equally important to a fuller understanding of these virulence factors. In this regard, Pattern D M-Prts are not nearly as well studied as Pattern A-C M-Prts (Cedervall et al., 1997; Ghosh, 2018), but we know that PAM dimers are present in solution and possess functional properties (Agrahari et al., 2013; Agrahari et al., 2016; Bhattacharya et al., 2014). However, some Pattern D GAS strains possess PAMs with significant sequence differences in their hPg-binding sites of the A-domain (Sanderson-Smith et al., 2007). It is of central importance to understand whether natural variations in these PAMs result in conformational differences that could affect their structure-function properties, since it is also not expected that the highly variable number of amino acids in the N-terminal domains of M-Prts (Cedervall et al., 1997; Fischetti, 1989; Fischetti et al., 1988; Ghosh, 2011; McNamara et al., 2008; Nilson et al., 1995; Stewart et al., 2016) are compatible with a generalization that these regions all exist as α-helical parallel coiled-coils of similar stability, even in solution. Thus, the determinants for secondary and quaternary structures of PAM, as well as functional interactions of PAMs, were investigated herein, using PAMs from different GAS isolates with variable N-terminal sequences.
2. Materials and methods
2.1. Bacterial strains and cultures
The GAS strains investigated are listed by their isolate names (and emm serotypes): AP53 (emm53), NS88.2 (emm98.1), NS223 (emm91), NS455 (emm52), SS1448 (emm86.2), SS1572 (emm223), SS1574 (emm224). The AP53 strain was provided by G. Lindahl (Lund, Sweden), strains NS88.2, NS223, and NS455 were gifts from M. J. Walker (Queensland, Australia) (Sanderson-Smith et al., 2006a), and strains SS1448, SS1572, and SS1574 were obtained from the Centers for Disease Control and Prevention. These GAS isolates were cultured at 37° C, 5% CO2 on sheep blood agar plates (Teknova, Hollister, CA) or in Todd-Hewitt broth (BD Bacto, Franklin Lakes, NJ) supplemented with 1% (w/v) yeast extract (BD Bacto) (THY).
2.1. DNA manipulations, cloning constructs, and protein purification
Genomic DNAs of the GAS isolates described above were extracted as previously reported (Ward and Leigh, 2002). To construct pam expression plasmids, the coding sequence of each PAM, excluding the N-terminal signal peptide and C-terminal LPXTG membrane insertion region, was amplified via the polymerase chain reaction (PCR). PCR was conducted using Phusion high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA). The forward primer was designed specifically for each PAM, which was necessitated by their 5’-variabilities, while the reverse primer was the same for each pam and contained a sequence that encoded a His6-tag for subsequent protein purification (Table S1). The PCR products as well as the plasmid, pET-28a (EMD4Biosciences, Darmstadt, Germany), were digested with Nco I and EcoR I (New England BioLabs) at 37° C. The digested PAM inserts and the vector were purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI). The digested PCR products and plasmid pET-28a were next ligated at 14° C overnight using T4 DNA ligase (New England BioLabs). Escherichia coli DH5α (Invitrogen, Carlsbad, CA) cells were transformed with ligated mixtures employing the MicroPulser Electroporation System (Bio-Rad, Hercules, CA). The transformed cells were then cultured on Luria Bertani (LB) agar plates supplemented with 40 μg/mL kanamycin. The plasmid DNAs of the surviving colonies were extracted for PCR screening, and sequence analysis using the EZNA plasmid DNA Midi Kit (Omega, Norcross, GA). The same cloning strategy was applied to truncated PAMAP53, viz., PAMAP53_short (residues 42–175), PAMAP53_medium (residues 42–207), and PAMAP53_long (residues 42–338) (Zhang et al., 2012).
2.2. Protein expression and purification
An engineered vector pET-15b (Novagen, Gibbstown, NJ) was employed to construct plasmids for expression of shortened peptides, including AGL55NS88.2 (residues 95–149 of PAMNS88.2), KTI55SS1448 (residues 85–139 of PAMss1448), and VEK75AP53 (residues 97–171 of PAMAP53). Standard digestions and ligations were carried out as above. Colony screening was conducted on LB agar plates containing 100 μg/mL ampicillin. The final expression plasmid sequentially contains an ATG initiation codon, a His6-tag, the GB1 subunit of the GABA receptor used for enhanced solubility, a 9-residue linker, a thrombin cleavage site (LVPR↓GS), the peptide coding sequence, and a translation stop codon. Consequently, all recombinant peptides contained residual Gly-Ser at their N-termini. Tyr present in the sequences was used for concentration determinations by A280nm measurements (Bhattacharya et al., 2014).
E. coli BL21/DE3 cells (New England BioLabs) were transformed with the expression plasmids. Protein expression and purification procedures for r-PAMs, unlabeled shortened peptides, and 13C/15N-labeled peptides have been described previously (Yuan et al., 2017).
2.3. Protein identification and concentration determinations
The integrity of all proteins and peptides was confirmed by MALDI-TOF mass spectrometry on an Autoflex III spectrometer (Bruker Daltonics) (Tables 1, 2). To obtain accurate extinction coefficients at 280 nm of all full-length PAMs, as well as PAMAP53_long, the A280nm refractive index increments at 670 nm were measured on an Optima XL-1 analytical ultracentrifuge (Beckman Coulter, Brea, CA) using a synthetic boundary centerpiece (Babul and Stellwagen, 1969). The cell was loaded in one chamber with 400 μL sodium phosphate, pH 7.4, and 190 μL and the protein sample was placed in the connecting chamber. The rotational speed at 20° C was increased to 3,000 rpm to allow a sharp boundary to be formed between solvent and protein and the refractive increment measured at 670 nm. All measurements were completed in ~10 min in order that diffusion of protein at the boundary was limited. The number of interference fringes at the boundary is proportional to the protein concentration (4.1 fringes = 1 mg/mL) (Babul and Stellwagen, 1969). The protein concentration was accordingly determined from fringe displacement data and this, along with the A280nm of the protein solution, provided accurate extinction coefficients (Table S2). This method has not been verified with shorter peptides. Thus, these extinction coefficients were calculated with ExPASY based upon Trp or Tyr residues in the sequences.
Table 1.
Molecular weights and α-helical contents of naturally occurring PAMs
| Protein | α-helical contenta | Mol. Wt. calcd. (MALDI-TOF)b | Mol. Wt. AUCc | |
|---|---|---|---|---|
| 18,000 rpm | 20,000 rpm | |||
| PAMNS88.2 | 33 % | 40,004 (40,020) | 81,900 ± 300 | 81,900 ± 1,200 |
| PAMNS223 | 61 % | 41,728 (41,760) | 83,700 ± 1,000 | 84,800 ± 1,000 |
| PAMNS455 | 72 % | 41,916 (42,055) | 83,400 ± 1,200 | 80,200 ± 1,700 |
| PAMSS1448 | 33 % | 38,544 (38,644) | 77,700 ± 400 | 76,200 ± 900 |
| PAMSS1572 | 59 % | 41,529 (41,635) | 83,200 ± 900 | 84,000 ± 500 |
| PAMSS1574 | 54 % | 37,266 (37,401) | 78,800 ± 1,200 | 72,300 ± 300 |
CD spectroscopy was conducted at 25° C, and the α-helix content was calculated for each protein employing the mean residue ellipticities at 222 nm.
Calculated molecular mass from the protein linear sequence calculated by ExPASy and the (experimentally determined molecular masses from MALDI-TOF).
Molecular weight (Mol. Wt.) of protein in solution from AUC at 25° C. In all cases, single molecular weight species was observed throughout the concentration gradient in the cell. Data from the top, top, and top channels were collected and presented as the mean ± S.D.
Table 2.
Molecular weights and α-helical contents of PAMAp53 and its derivative truncated peptides
| Protein | α-helical contenta | Mol. Wt. calcd. (MALDI-TOF)b | Mol. Wt. AUCc | ||
|---|---|---|---|---|---|
| PAMAP53_short | 39 % | 16,845 (16,917) | 28,000 rpm 15,200 ± 1,000 | 32,000 rpm 14,800 ± 2,000 | 36,000 rpm 14,400 ± 2,300 |
| PAMAP53_medium | 32 % | 20,498 (20,348) | 24,000 rpm 22,700 ± 500 | 28,000 rpm 21,500 ± 600 | 32.0 rpm 18.0 ± 200 |
| PAMAP53_long | 49 % | 35,232 (34,937) | 18,000 rpm 67,600 ± 2,500 | 21,000 rpm 67,600 ± 3,800 | 24,000 rpm 67,500 ± 4,500 |
| PAMAP53 | 27 % | 40,941 (41,083) | 81,507d | ||
| VEK75ap53 | 9,119 (9136) | 40,000 rpm 9,700 ± 300 | 46,000 8,800 ± 100 | ||
| AGL55NS88.2 | 6,726 (6,733) | 40,000 rpm 6,400 ± 200 | 46,000 rpm 6,700 ± 100 | ||
| KTI55SS1448 | 6,784 (6,791) | 40,000 6,900 ±120 | 46,000 6,400 ± 100 | ||
CD spectroscopy was conducted at 25° C, and α-helical content was calculated for each protein employing the mean residue ellipticities at 222 nm.
Calculated molecular mass from the protein linear sequence calculated by ExPASy and the (experimentally determined molecular masses from MALDI-TOF).
Molecular weight (Mol. Wt.) of the proteins in solution from AUC at 25° C. In all cases, single molecular weight species was observed throughout the concentration gradient in the cell. Data from the top, top, and top channels were collected and are presented as the mean ± S.D.
From (Bhattacharya et al., 2014).
2.4. Circular dichroism (CD)
Far-UV CD spectra were collected for r-PAMs for secondary structure analyses. Measurements were made on an Aviv model 202 SF Spectrometer (Aviv, Lakewood, NJ) in 10 mM sodium phosphate/50 mM NaCl, pH 7.4. CD spectral data were continually recorded from 195 to 250 nm in the wavelength scan mode. Ellipticities were also monitored at 222 nm in the temperature scan mode, and collected from 4° C to 90° C. The spectra represented the average of three scans in both applications. An average buffer reference scan was subtracted from that of each sample. The mean residue ellipticities ([θ]) of the protein/peptides were calculated using: [θ] = (θ × MRW)/(1 × c), where θ is the original signal in mdeg, MRW is the mean residue weight in g/mol, l is the path length in mm, and c is the protein concentration in mg/mL (Greenfield, 2006). The α-helical content was estimated from [θ]222 nm = − 30,300fH – 2,340, in which fH refers to the fraction of helices in the protein (Chen et al., 1972). The data were plotted using GraphPad Prism 7.
2.5. Analytical ultracentrifugation (AUC)
Sedimentation equilibrium analytical ultracentrifugation was performed at 25° C using an Optima XL-1 analytical ultracentrifuge (Beckman Coulter). The proteins/peptides were dissolved in 10 mM sodium phosphate/50 mM NaCl, pH 7.4, and diluted to A280nm ~ 0.1 prior to loading. Three channels of a six-channel centerpiece were loaded with the buffer as a reference solution and protein samples were added to the adjacent channels. Three cell housings, containing one centerpiece each, were assembled and placed into a Beckman An-60 Ti rotor, together with the counterbalance (Bhattacharya et al., 2014). Samples were rotated individually at the speeds of 18,000 and 20,000 rpm for all full-length PAMs; 28,000, 32,000 and 36,000 rpm for PAMAP53_short; 24,000, 28,000 and 32,000 rpm for PAMAP53_medium; 18,000, 21,000, and 24,000 rpm for PAMAP53_long. Scans were recorded hourly at 280 nm until three scans overlapped, at which point equilibrium was attained. The partial specific volume of each protein or peptide was calculated from the amino acid compositions using Sednterp (Table S2). The density of the buffer was 1 g/mL. Apparent weight average molecular weights for the whole sample were calculated from non-linear fitting of the data with Optima XL-A/XL-I data analysis software (Beckman Coulter), using concentration data from the meniscus (cm) to the cell bottom (cb) (Schuck et al., 2014). AUC sedimentation equilibrium assays were conducted in triplicate for each peptide or protein.
2.6. Helical wheel modeling
Helical wheel diagrams (Schiffer and Edmundson, 1967) for each domain in different PAMs were constructed in the parallel mode through the online server DrawCoil 1.0.
2.7. Nuclear magnetic resonance (NMR)
NMR spectra were recorded at 25° C on a Bruker AVANCE 800 spectrometer, equipped with a 5 mm triple resonance (TCI, 1H, 13C, 15N) cryoprobe, for both resonance assignments and NOE identifications. All 13C/15N-labeled peptides, viz. AGL55NS88.2, KTI55ss1448, and VEK75Ap53, were dissolved in 20 mM BisTris-d19/2 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS)/0.1% NaN3, and 5% 2H2O/95% H2O, pH 6.7. The NMR data were processed with Bruker TopSpin 3.5 and analyzed with Sparky (Lee et al., 2009b). 1H chemical shifts were referenced to internal DSS, whereby C and N chemical shifts were indirectly referenced to DSS (Markley et al., 1998). For resonance assignments of the peptide backbone and side chains, the following pulse sequences were used: 15N-HSQC (Kay et al., 1992; Mori et al., 1995), HNCO/HN(CA)CO (Clubb et al., 1992; Kay et al., 2011), HNCA (Löhr and Rüterjans, 2002), HNCACB/CBCA(CO)NH (Wittekind and Mueller, 1993), (H)CC(CO)NH, and H(CC)(CO)NH (Löhr and Rüterjans, 2002). For intramolecular NOE distance restraints, both 15N-NOESY-HSQC (Talluri and Wagner, 1996) and 13C-NOESY-HSQC (Ikura et al., 1990) spectra were obtained.
2.8. NMR solution structures
The TALOS-N program was applied to predict the secondary structures of AGL55, KTI55, and VEK75 in their apo-forms based on assigned backbone chemical shifts (Shen and Bax, 2013). The CS-Rosetta program (Shen et al., 2009; Shen et al., 2008) was applied to generate the starting template for each peptide based on the chemical shifts. Structural calculations were then carried out with combined dihedral angle restraints (ϕ and ψ) from TALOS-N and the NOE restraints observed from 2D-NOESY and 3D-NOESY-HSQC experiments. Approximately 200 structures were generated through a simulated annealing protocol in the program Xplor-NIH 2.37 (Schwieters et al., 2006; Schwieters et al., 2018; Schwieters et al., 2003). Ten conformers with the lowest restraint energy values, and without distance and angle violations, were further refined with implicit water. The validity of the structures was analyzed PSVS version 1.5 with also integrates PROCHECK version 3.5.4 (Laskowski et al., 1993) in a single interface (Bhattacharya et al., 2007). Visualization of the structures was performed using PyMOL.
2.9. Surface plasmon resonance (SPR)
The binding kinetics of r-PAMs to hPg were measured via refractive index changes upon analyte/ligand binding in real-time using a BIAcore X100 Biosensor system (GE Healthcare). All binding experiments were conducted at 25° C. The running buffer was HBS-EP (10 mM Hepes, 0.15 M NaCl, 3 mM EDTA, 0.005% polysorbate 20, pH 7.4) flowed at 30 μL/min. hPg (30 μg/mL) in 10 mM sodium acetate, pH 4.5, was adsorbed onto flow cell-2 of a CM5 sensor chip (GE Healthcare), to a level of ~1,000 response units (RUs), using an amine coupling kit (GE Healthcare) (Bhattacharya et al., 2014; Chandrahas et al., 2015; Glinton et al., 2017). Non-bound sites on the gold surface of sensor chip were subsequently blocked by injection of 1 M ethanolamine, pH 8.5.
In the binding experiments, various concentrations of analytes in HBS-EP buffer were injected for a 120 sec association time and a 360 sec dissociation time with hPg-coupled CM5 (Yuan et al., 2017). The gold surface was regenerated between cycles using 10 mM glycine, pH 2. The responses in flow cell-2 were subtracted from the background response of the analyte in the reference cell (flow cell-1) which does not contain immobilized ligand. The sensorgrams were analyzed using BIA evaluation software 2.0.1 (GE Healthcare). Equilibrium dissociation constants (KD) were calculated from the ratio of the dissociation (koff) and association rates (kon). Nonlinear fitting of the association and dissociation curves with a 1:1 binding model was employed. Binding curves were presented in GraphPad Prism 7.
2.10. Flow cytometric analysis (FCA)
GAS cells were grown in THY media to an O.D.600nm of ~0.6 at 37° C, after which they were washed with phosphate-buffered-saline (PBS, pH 7.4) and then resuspended in PBS plus 1% ovalbumin for blocking. The cells were then incubated with hPg (ERL, South Bend, IN), after which the primary antibody, mouse-anti-hPg (ERL), was incubated with the cell suspensions. For detection of bound hPg, a secondary antibody, AlexaFluor 488-donkey-anti-mouse IgG (Invitrogen), was incubated with the cells. FCA was conducted using a FACSAria III (BD Biosciences) cytometer with the 488 nm laser at a flow rate of 10 μL/min, with 10,000 events recorded for analysis. Data were collected by gating on fluorescence (FITC-A) and on side-scatter.
The histograms obtained were then analyzed using FCS Express Version 4 software (De Novo Software, Los Angeles, CA). The median fluorescence intensity for each histogram was used to calculate the events accepted by the gating formula that fall within the specified marker. The percentage of anti-PAM or hPg-binding for each strain was normalized to strain, AP53, which was set at 100%. FCA of each GAS strain was conducted in triplicate, and all data were plotted using GraphPad Prism 7.
3. Results
3.1. Homology increases from N-termini to C-termini among different recombinant (r) PAMs
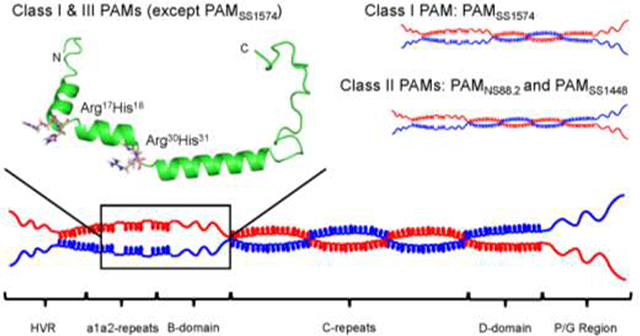
We selected seven recombinant (r)-PAMs from Pattern D GAS isolates to examine how the sequence variability affects the conformations, propensity to dimerize in solution, and the ligand binding properties of this class of M-Prt (Fig. 1A). PAM from the GAS AP53 (PAMAP53) isolate was chosen as the prototype PAM for this work since most of our structure-function studies on Pattern D M-Prts have been performed on PAMAP53 and PAMAP53 fragments (Cnudde et al., 2006; Ringdahl et al., 1998; Ringdahl et al., 1998; Wang et al., 2010a; Wang et al., 2010b), thus allowing facile comparisons with the present work. Amino acid sequence alignments show that these seven PAMs contain similar modular structures and sequence variabilities decrease from the extracellular N-terminus to the C-terminus, which is closer to the bacterial capsule (Fig. S1). The HVR and A-domains of PAMs, that direct many of their functional properties, are the most diverse regions in the entire protein in terms of their lengths and amino acid sequences. Importantly, because a1- and a2-repeats within the A-domain are the binding loci for hPg, the variabilities in this domain directly relate to the function of PAMs. On the basis of the differences within the A-domain, seven full-length PAMs were categorized into three structural classes in this study (Fig. 1A). Class I contains intact a1- and a2-repeats, each of which harbors one Arg-His dipeptide (simply referred to as RH1 in repeat a1 and RH2 in repeat a2). These RH regions have been shown to be essential residues for hPg binding (Fig. 1B) (Rios-Steiner et al., 2001; Sanderson-Smith et al., 2006b; Schenone et al., 2000; Wang et al., 2010a; Wang et al., 2010b). PAMAP53 and PAMSS1574, which are very similar in sequence to each other, were classified as Class I PAMs. Class II PAMs encompassed those lacking the a1-repeat and only containing the single RH2 binding locus in the a2-repeat. These PAMs included PAMNS88.2 and PAMSs1448. Class III PAMs, PAMNS223, PAMNS455, and PAMSS1572, also contained intact a1a2-repeats as shown in Class I, but were interrupted by V-H-D or D-H-D tripeptide insertions at the end of the a1-repeat.
Fig. 1.

Schematics of full-length r-PAMs and truncated r-PAMAP53 peptides. All recombinant proteins start with the first residue (#42) immediately after the signal peptide. (A) Within all naturally occurring PAMs, homology progressively increases from their N- to C-termini. Domains align in the following order from the N-terminus: hypervariable region (HVR), A-domai n containing a1a2- or a2-repeats, B-domain, C-domain containing c1-c3 repeats, D-domain, and the Pro/Gly (P/G)-rich region. (B) Three truncated PAMAP53 proteins were prepared that terminated at different locations in the PAMAP53 sequence. The a1a2-repeats are zoomed in to manifest the variability of this domain among different r-PAMs. Residues in red refer to the sequence of a1a2-repeats, and residues in blue indicate the Arg-His (RH) motifs that specifically binding to hPg (Rios-Steiner et al., 2001; Sanderson-Smith et al., 2006b; Schenone et al., 2000; Wang et al., 2010a; Wang et al., 2010b). Dashes are applied to increase the homology of alignment when necessary. The schematic is drawn to relative scale, and numbers indicate the first and last residue of each domain.
Progressively more limited variability has been observed throughout the B-, C-, D- and Pro/Gly-domains of these PAMs (Fig. S1). It is noteworthy that PAMSS1574 is devoid of the third C-repeat, resulting in a significantly smaller protein compared to other PAMs (Fig. S1). Because PAMSs1574 lacks one C-domain, it is possible to generate a fourth class of Pattern D PAMS when considering this aspect of the structure. Our three classifications were based strictly on the a1a2 domains.
3.2. The α-helix is the predominant secondary structure of PAMs
All full-length proteins and truncated peptides were readily purified by affinity chromatography employing the His6 adduct, as described earlier (Bhattacharya et al., 2014; Chandrahas et al., 2015), and were of high purity as shown by MALDI-TOF, which displayed the values nearly identical to the expected molecular weights (Tables 1, 2).
Despite extensive variations in the N-terminal sequences of PAMs, all full-length proteins displayed overall high α-helix contents, ranging from 30%−70% at 25° C, as determined by Circular Dichroism (CD) measurements. Specifically, α-helices were comprised of ~30% of overall secondary structures in PAMNS88.2, PAMSS1448, and PAMAp53, while PAMSS1574 demonstrated a more moderate α-helical content. More strikingly, PAMNS223, PAMNS455, and PAMSS1572 contained an α-helical fractional amount of >60% (Fig. 2A, Table 1). Since sequence variabilities progressively decrease from the N- to the C-terminus and the C-D-Pro/Gly domains of PAMs are the most conserved domains, we designed truncated versions at the N-terminus for the prototype PAMAP53 to assess the helical contents of the individual regions. The truncated PAMs were constructed from Asn42, the first residue downstream of the signal peptide. The polypeptides, PAMAP53_short, PAMAP53_medium, and PAMAP53_long, extend to Lys175 (the beginning of C1-domain), Ala207 (the end of the C1-domain), and Lys (the middle of D-domain), respectively (Fig. 1B).
Fig. 2.

CD spectra of naturally occurring r-PAMs and truncated r-PAMAp53 peptides. Mean residue ellipticities (MRE) of each protein and peptide were calculated from their CD signals. The MRE is plotted as a function of wavelength from 195 nm to 250 nm at 25° C (A and B), and as a function of temperature from 4° C to 90° C at 222 nm (C and D). The results are shown as data points that represent the average of triplicate scans at a given wavelength or temperature.
CD spectra of the truncated PAMAP53 polypeptides were first obtained using the wavelength scan mode. At 25° C, both PAMAP53 and PAMAP53_long showed troughs at 208 nm and 222 nm, a strong signature of α-helix motifs (Fig. 2B). The highest relative percentage of α-helices, at ~50%, was observed from PAMAP53_long, rather than full-length PAMAP53, which has the Pro/Gly helical destabilizing domain at C-terminus (Table 2). Under the same conditions, PAMAP53_short and PAMAP53_medium displayed a spectral pattern with a single trough at 208 nm, demonstrating a low propensity of α-helices (Fig. 2B).
The temperature scan mode was then employed to evaluate changes of α-helical content with temperature. For all full-length (Fig. 2C) and truncated (Fig. 2D) r-PAMs, temperature scanning revealed that the mean residue ellipticity (MRE) at 222 nm increased at increasing temperatures, corresponding to a decreased α-helical content. Of note, the MRE values of all the full-length PAMs, together with PAMAP53_long, showed sigmoidal temperature-dependent curves, while the MRE of PAMAP53_short and PAMAP53_medium increased hyperbolically with temperature changes (Fig. 2C, D).
3.3. The C-repeats mediate the dimerization of PAM
Previous data have shown that PAMAP53 exists as a dimer at room temperature (Bhattacharya et al., 2014). Similarly, at 25° C, as shown by molecular weight determinations via analytical ultracentrifugation (AUC) (Table 1), all full-length r-PAMs were monodisperse throughout the concentration gradient established at two different rotor speeds and were found to be dimeric (Table 1). On the other hand, the AUC results of truncated r-PAMs demonstrated that PAMAP53_short, with only the HVR, A(a1a2)-, and B-domains, and PAMAp53_medium, with the HVR-A(a1a2)-B-, and one of three c-repeats (c1) in the C-domain, were also monodisperse and existed as monomers. PAMAP53_long, extended to contain all three c-repeats in the C-domain and part of the D-domain, dimerized similarly to the full-length r-PAMs (Table 2).
To further reveal the domains essential for dimerization, heptad registers of the amino acid sequences within these seven different PAMs were analyzed and organized according to the classical helical wheel representation (Fig. S2). The Pro/Gly region was not included in these analyses since Pro and Gly would not be expected to exist in helical conformations. Because there are only scattered amino acid substitutions in the three c-repeats and in the D-domain among various naturally occurring PAMs, PAMAP53 was chosen as a representative to illustrate the arrangement of amino acids in the helical wheel model of all PAMs (Fig. 3), and to correlate with function-studies accomplished on this PAMAP53. The requisite for such an alignment is a dominance of α-helices and the presence of hydrophobic residues throughout the sequences represented. Optimally, heptad positions a and d are mainly occupied by Leu and Val residues, and face the inner surfaces in a coiled-coil dimer, while positions b, c, e, f, and g are exposed to the outer surfaces. The three c-repeats (c1-c3) of all PAMs, except for PAMSS1574, contain a total of 119 residues, rendering the C-domain as the longest of all the PAM domains (Fig. S1). Considering their amino acid compositions, Leu and Val account for over 50% of positions a and d in the C-domain, thereby providing the opportunity to form hydrophobic clusters in the inner surface of a helical coiled-coil. Additionally, Leu residues were found at positions a and d in regions of HVRs, as well as A-, B-, and D-domains (Fig. S2), but these domains are shorter than the C-domain. Notably, the typical seven-residue hydrophobic periodicities were interrupted by sequences in a1a2 containing hPg-binding motifs, viz., the Arg-His binding site dipeptides (RH1 and RH2), thus generating flexibility in the α-helices, perhaps providing spacing between the chains for binding of hPg.
Fig. 3.

Helical wheel representations of the highly conserved C- and D-domains of PAMAp53. Construction of helical wheels for (A) c1-c3 repeats of the C-domain and (B) the D-domain was performed on the server, DrawCoil 1.0 (http://www.grigoryanlab.org/drawcoil/). The position of each residue is identical to that from the heptad register alignment (Fig. S2). Residues are categorized and highlighted in colored circles: grey, apolar residues; blue, positively charged residues; red, negatively charged residues; orange, polar residues. The blue and red dash lines indicate potential electrostatic attractions and repulsions, respectively, between residues at positions e and g (Mason et al., 2008).
Alignments, using heptad registers, were then applied to other domains of PAMs to investigate how the non-polar residues existing in each region form dimers. While highly diverse among PAMs, the HVR also contained some nonpolar residues, e.g., Leu and Val. These residues occupied nearly one-third of positions a and d in this domain, making it possible to create a portion of a hydrophobic coiled-coil interface (Fig. 4). In this regard, it has been shown that while HVRs of M-Prts from Pattern E strains, viz., M22, M28, and M49, contained even fewer apolar residues at positions a and d than PAMs, they nonetheless formed canonical dimeric coiled-coils at 4° C (Buffalo et al., 2016), a temperature that stabilized coiled-coil formation. All PAMs, except PAMSS1448, contain a negatively charged cluster in the HVRs at position c and/or g in the helical wheel. Accordingly, PAMSS1448 has a significant higher isoelectric point (pi ~9.3) than the other PAMs (pi ~7.5).
Fig. 4.

Helical wheel representations of the hypervariable region (HVR) in different r-PAMs. Construction of helical wheels for the HVR in (A) PAMAP53, (B) PAMNS88.2, (C) PAMNS223, (D) PAMNS455, (E) PAMSS1448, (F) PAMSS1572, and (G) PAMSS1574 was performed using DrawCoil 1.0 (http://www.grigoryanlab.org/drawcoil/). The position of each residue is identical to that from the heptad register alignments (Fig. 3). The residues are categorized and highlighted in colored circles: grey, apolar residues; blue, positively charged residues; red, negatively charged residues; orange, polar residues. The blue and red dash lines indicate potential electrostatic attraction and repulsion, respectively, between residues at positions e and g, respectively.
Thus, from the heptad register analyses and the AUC results presented above, we propose that the complete C-domain is essential for PAMs to form stable dimers, confirming an assumption solely based on high sequence conservation for several M-Prts (not of the Pattern D type) (Cedervall et al., 1997; Nilson et al., 1995). Further, hydrophobic residues in other domains can also contribute, albeit to a lesser degree, to the energetics of dimerization. However, it was not known prior to this study whether other highly variable domains could destabilize the C-domain helices. This does not appear to be the case.
3.4. The hPg binding motifs in PAMs readily form mobile loops rather than rigid helices
The homologous a1a2-repeats in PAMs are responsible for binding hPg with very high affinity, and residues in such tandem repeats are not strictly aligned in heptad registers (Fig. S2). Therefore, NMR experiments were performed for three peptides harboring hPg-binding modules (Fig. 5A). Specifically, AGL55NS88.2 and KTI55SS1448 both originate from the N-terminal region of the HVR of the PAMNS88.2 and PAMSS1448 (Ala95 and Lys85, respectively) to the middle of the B-domain (Y149 and Y139, respectively). These two peptides contain a single Arg-His (RH2) motif in the a2 region, due to the natural loss of the a1-repeat in corresponding full-length PAMs. VEK75AP53 begins with V97 in the HVR region of the N-terminus of PAMAP53 and extends to the last residue of B-domain, Q171. This latter peptide contains two Arg-His motifs (RH1 and RH2) present in the a1 and a2 regions, respectively. The MALDI-derived molecular weights of these peptides (Table 2) attest to their high purity. Further, the sedimentation equilibrium data (Table 2) clearly shows that these three peptides exist as monomers in solution and were monodisperse at all points in the concentration gradient at two different rotor speeds. We employed these peptides to determine their 3D structures by NMR in solution to determine how these functional regions are initially conformationally presented to hPg.
Fig. 5.

NMR solution structures of AGL55NS88.2, KTI55SS1448, and VEK75AP53. (A) Coding sequences of three peptides were cloned from their corresponding naturally occurring PAMs, viz., PAMNS88.2, PAMSS1448, and PAMAP53. PAM-specific hPg-binding a1a2-repeats or the a2-repeat of the A-domain are shown in red. The Arg-His hPg-binding residues are shown in blue. The ribbon diagram of the lowest energy structure is presented for (B) AGL55NS88.2, (C) KTI55SS1448, and (D) VEK75AP53. The Arg-His motifs are shown as sticks, and the N- and C-termini of the peptides are labeled.
Initial NOESY experiments for these peptides show that residues in the N- and C-terminal regions only have sequential connectivities, suggesting a lack of defined secondary structural elements at the termini. Additionally, the VEK75AP53 structure presented a long unstructured tail from residue D49 to Q75 at the C-terminus, corresponding to the majority of the B-domain (E133 to Q171) in PAMAP53. Nonetheless, a significant fraction of α-helical conformations is predicted in the central regions of the peptides based on chemical shifts using TALOS-N and CS-Rosetta (Shen et al., 2010), and confirmed by non-sequential NOEs observed from 2D-NOESY and 3D-NOESY-HSQC experiments. Using the predicated structure from CS-Rosetta as the template, the XPLOR-NIH program was performed to generate structure clusters (Fig. S3) with the lowest energies and without violations in dihedral angle and distance restraints. The structural statistics for each peptide are provided in Table S3. Small average pairwise root mean square derivatives (RMSD) were observed for structured residues indicating that these truncated peptides exist as an ensemble consisting of defined secondary structures. Accordingly, from the ensembles of ten solution structures for AGL55, KTI55, and VEK75, overlaid according to the longest helical structure in each (Fig. S3), the lowest energy structure of each of the three uncomplexed peptides is illustrated in Fig. 5B–D.
In AGL55NS88.2, two α-helical segments, from R8 to E24, and from D27 to D40, are connected with a mobile loop consisting of the R25H26 dipeptide (Fig. 5B). Similarly, both RH1 and RH2 dipeptides (R17H18 and R30H31) of VEK75 are also located in mobile loops. Thus, the structures of AGL55NS88.2 and VEK75AP53 demonstrated flexible hPg-binding modules flanked by rigid helices (Fig. 5B, D). It is noted that the N-terminus of KTI55SS1448 contains two short helical segments, and the RH2 dipeptide of KTI55SS1448 is situated in a loop region consisting of eight residues (K23-D30) (Fig. 5C). The larger portion of loops upstream of the hPg-binding motif made KTI55SS1448 a less compact conformation than the other single-Rî2-containing peptide, AGL55NS88.2. On the basis of solution structures obtained here, we conclude that the RH1 and RH2 hPg-binding motifs readily breach rigid helical structures and become flexible, which likely aids in the binding of hPg. Also, helices are not readily observed in the B-domain of VEK75AP53, thereby rendering this region as a mobile structural element (Rios-Steiner et al., 2001; Wang et al., 2010b).
It was also found that in the AGL55NS88.2 sequence, oppositely charged residues are staggered every three positions, i.e., Glu7-Arg10-Glu13-Lys16-Glu19-Arg22. This staggered alignment can stabilize the α-helix through side-chain electrostatic attractions, but such a feature nonetheless is not present in KTI55SS1448, rendering this region as less structured than AGL55NS88.2.
3.5. r-PAMs demonstrate tight binding to hPg
SPR experiments were next performed to evaluate hPg-binding affinities of the r-PAMs. Class I and III PAMs were similar in their tight binding to hPg, with relatively slow off-rates and resultant KD values of ~10 nM (Fig. 6 A–E, Table 3). However, two members of C l ass II PAMs somewhat diverged regarding their hPg-binding patterns. Binding of PAMNS88.2 to hPg was as strong as Class I and Class III PAMs (Fig. 6F), but PAMSS1448 exhibited a KD of ~30 nM, due to its relatively fast dissociation rate (Fig. 6G, Table 3). But, on the whole, in spite of the variability in a1a2-repeats among different r-PAMs, these dimeric full-length PAMs all tightly associated with hPg with KD values at the low nM scale. Three truncated variants of Class I PAM, i.e., PAMAP53_short, PAMAP53_medium, and PAMAP53_long, also show tight binding to hPg at the nM scale, suggesting that for PAMAP53, dimerization is not a determining factor for its binding to hPg, which is consistent with the previous report on this subject (Bhattacharya et al., 2014).
Fig. 6.

Binding assays of full-length r-PAMs to hPg. All binding experiments were conducted by SPR at 25° C. Kinetic analyses were performed for (A) PAMAP53, (B) PAMSS1574, (C) PAMNS223, (D) PAMNS455, (E) PAMss1572, (F) PAMns88.2, (G) PAMss1448. The data were fit using a 1:1 Langmuir binding model. The corresponding binding constants (KD) for these curves were calculated from the average values of koff/kon, are provided in Table 3.
Table 3.
Binding kinetics of naturally occurring PAMs and truncated PAMAP53 to hPg at 25° C
| Protein | kon (×104 Ms−1) | koff (×10−4 s−1) | KD (nM)a |
|---|---|---|---|
| Class 1 PAMs | |||
| PAMapsb | 34 ± 15 | 5.6 ± 1.1 | 1.6 ± 0.8 |
| PAMss1574 | 10 ± 3 | 1.1 ± 0.2 | 1.1 ± 0.4 |
| Class II PAMs | |||
| PAMNS88.2 | 20 ± 4 | 2.3 ± 0.3 | 1.2 ± 0.3 |
| PAMSS1448 | 3.6 ± 0.7 | 10 ± 1 | 28 ± 6 |
| Class III PAMs | |||
| PAMNS223 | 24 ± 9 | 3.3 ± 0.4 | 1.4 ± 0.5 |
| PAMNS455 | 54 ± 12 | 1.7 ± 0.2 | 0.3 ± 0.07 |
| PAMSS1572 | 12 ± 6 | 5.9 ± 0.1 | 4.9 ± 2.4 |
| Truncated PAMAP53 | |||
| PAMAP53_short | 4.0 ± 0.7 | 7.7 ± 1.6 | 1.9 ± 0.5 |
| PAMAP53_medium | 2.3 ± 0.1 | 5.4 ± 0.9 | 2.3 ± 0.4 |
| PAMAP53_long | 0.9 ± 0.4 | 1.3 ± 0.1 | 1.4 ± 0.6 |
KD values were calculated from the average values of koff/kon in the Table.
3.6. hPg binding to PAM-containing GAS strains is a measure of surface exposure of PAM
After assuring that all r-PAMS fully bound to hPg, we examined the binding of to the seven PAM-positive GAS cells by flow cytometry in order to assess the PAM protein expression levels on these cell lines. In this assay, the negative control was AP53Δpam. In contrast to the relatively very similar Kd values for hPg to the purified r-PAMs, these GAS strains displayed diversity in their abilities to capture hPg (Fig. 7). SS1574, one of Class I PAM expressing strains, bound the highest amount of hPg, at <2-fold than that of the AP53. Both strains harboring Class II PAMs, viz., NS88.2 and SS1448, associated with hPg at a level comparable to that of AP53. Nevertheless, in Class III PAM-expressing strains, SS1572 only presented a very low constitutive association with hPg, Collectively, we have found that PAM-positive GAS strains diverged in PAM expression, as evaluated by hPg binding.
Fig. 7.

Whole cell PAM expression. After incubation of hPg with GAS, the cells were sequentially incubated with mouse anti-hPg (primary antibody) and Alexa Fluor 488-donkey anti-mouse IgG (secondary antibody). The cells were washed with PBS, pH 7.4, between incubation steps. The median fluorescence intensity at 488 nm of original histograms was used to determine the amount of cell-bound hPg. The capacity of binding to hPg for each strain was normalized to AP53, which was set at 100%. AP53/Δpam was used as the negative control.
4. Discussion
The GAS surface-protruding rod-like M-Prt, composed entirely of secondary structure elements, is considered as an essential virulence factor in the pathogenesis of this microbe (Fischetti, 1989; Ghosh, 2011; Smeesters et al., 2010). Recruitment of host proteins to suppress innate immune pathways is a major virulence mechanism of M-Prt and other M-like proteins. Thus, these surface proteins interact with several components of the human host, e.g., hemostasis factors (Loof et al., 2014; Oehmcke et al., 2012), complement inhibitors (Agrahari et al., 2013; Agrahari et al., 2016), and epithelial cell receptors (Frick et al., 2003; Siemens et al., 2011; Tamura and Nittayajarn, 2000), and the great variability of M-Prts in different GAS serotypes allows GAS to display different abilities to colonize and/or invade. Previous research has unveiled structure-function relationships of M-Prts with virulence properties in Pattern A-C and Pattern E strains (Buffalo et al., 2016; Cedervall et al., 1997; Fischetti, 1989; Ghosh, 2011; McNamara et al., 2008; Nilson et al., 1995; Smeesters et al., 2010; Stewart et al., 2016), but more limited research has been accomplished on M-Prts expressed by Pattern D strains. Among the major properties of Pattern D M-Prts (PAMs) is that they serve as the major direct receptors for tight binding to host hPg, a property not shared by other M-Prt pattern types. Genetic linkage of PAM with a specific subtype of SK produced only by Pattern D strains, SK2b, that selectively activates hPg bound to PAM has been established (Zhang et al., 2012). The net result of these interactions is to provide a surface bound protease (hPm) that GAS can employ for release from initially protective fibrin nets and ultimate dissemination via degradation of innate immune barriers. Based on studies with a large peptide region of M1, a M-Prt of the Pattern A-C group which does not directly bind hPg, it is believed that M-Prts exist as parallel kinked coiled-coils with significant exposure of its N-terminal domains on the GAS surface (Buffalo et al., 2016; Ghosh, 2011; McNamara et al., 2008). But, PAMs possess large differences in their hPg binding N-termini (the A-domain, composed of homologous a1a2 repeats) as compared to M1, and do not directly bind to fibrinogen, and secondary structural predictions of the N-termini of PAM do not suggest the presence of stable helical structures. However, studies with isolated r-PAMAP53 show that it does, in fact, dimerize (Bhattacharya et al., 2014). Thus, we have addressed in this report the extent to which the important protein binding N-terminus of PAM stabilizes dimerization of PAM and dictates the tight binding of PAM to hPg.
4.1. Different domains are disparately engaged in PAM dimerization
Three truncated PAMAP53 proteins, PAMAP53_short, PAMAP53_medium, and PAMAP53_long, were constructed and expressed (Fig. 1B) to evaluate the contributions to the dimerization of PAM from each domain. The results demonstrated that PAMAP53_long, as a result of containing the three c-repeats in the C-domain, displayed a higher fractional α-helix (49%) than the other two shortened PAMAP53 peptides, PAMAP53_short (39%) and PAMAP53_medium (32%), indicating that c-repeats stabilized the α-helical content. PAMAP53 (29%), which extended the C-terminus further than PAMAP53_long also contains the Pro/Gly-region, which not unexpectedly disrupted helical structures. PAMAP53_short and PAMAP53_medium demonstrated a transition from partly folded to well-folded peptides at low temperatures (Fig. 2D). These three truncated PAMs demonstrated a transition from partly folded α-helical segments to well-folded dimeric proteins (Fig. 2D). At lower temperatures (4° C), PAMAP53_short and PAMAP53_medium also showed dimerization (C. Q., unpublished data), suggesting that Leu residues in domains upstream of the C-repeats had a propensity to form a hydrophobic core in dimers. However, limited numbers of such residues in a single c-repeat could not facilitate PAMAP53_medium to dimerize at 25° C, whereas PAMAP53_long, incorporating three intact C-repeats, exhibited complete and stable dimerization at 25° C. This implies that the length of C-repeats dominates the balance between monomer and dimer.
In this investigation, it is shown that naturally occurring pAMs from seven different GAS strains are dimeric without exception, as shown by AUC experiments at 25° C (Table 2). Related conclusions can be made from CD temperature scan spectra. The MREs of all the full-length pAMs, as well as pAMAp53_long, increased sigmoidally as the temperatures were increased, reminiscent of cooperative transitions of DNA melting curves with disruption of helical stacking occurred followed by disruption of the dimeric structures. Thus, these CD results suggested that coiled-coil dimerization is a key feature of those pAMs that harbor intact c-repeats. In contrast, PAMAP53_short and PAMAP53_medium displayed hyperbolic shapes in their melting curves, indicating that helical disruption, but not dissociation, occurred as a function of temperature, which probably preceded dissociation. These hyperbolic melting patterns support the AUC data showing that these latter two peptides existed as monomers at 25° C.
Previous work has demonstrated that the B-domain of M1, from Pattern A-C strains, contain residues that disrupted segments of the coiled-coil helices, allowing kinks in the helices that are important in the binding of M1 to fibrinogen (McNamara et al., 2008). Further, the B-domain of Pattern A-C M6 protein show interruptions in the seven-residue periodicity, thus making this region more flexible (Fischetti et al., 1988). Whether PAMs form kinked dimers to allow hPg binding is not known. However, the NMR studies indicated that an extensive mobile loop in the B-domain has been observed from solution structure of VEK75AP53, suggesting that this region is predominantly unstructured in the unbound form. If rigid helical structures predominated in the B-domain, recruitment of the large hPg, via a1a2-repeats, would require conformational changes, possibly destabilizing the coiled-coil dimer at the adjacent C-repeats. In this sense, the B-domain can serve as a cushion to release potential torsional forces generated from association of PAMs with hPg.
It is clear that the c-repeats are the major contributors to the coiled-coil dimerization of PAM. In this region, Leu and Val residues from two helical chains generate a hydrophobic core to stabilize the dimeric conformation. On the other hand, Arg and Lys residues are scattered along C-domain, which would destabilize dimerization through electrostatic repulsions. However, it has been demonstrated that the alkyl group of Arg39 in the protein, C4BPa1, contributed to a hydrophobic nook and clustered with apolar side chains in the HVRs of M2, M22, M28, and M49 proteins (Buffalo et al., 2016). While, in the case of PAMs, the HVR does not bind to C4BPa1 (Agrahari et al., 2013), we can nonetheless infer that in PAM c-repeats, the Lys and Arg residues at positions a and d in the heptads have the potential to produce an internal hydrophobic niche through their alkyl groups. Once the inner hydrophobic core forms, most of the charged residues align at the solvent surface (i.e., positions b, c, e, f, and g in the heptad). Of importance, positions a and e of the heptads are mainly occupied by positively charged residues, while predominant amino acids of position g are negatively charged Asp or Glu, which are positioned to provide charge attractions.
4.2. A model of PAM structures
We herein propose a secondary structural model to describe the dimerization pattern for PAMs (Fig. 8). Based upon AUC results and the helical wheel modeling, the C-domain has been shown to be the most important region for dimer formation, and hence proposed as the hinge part of the model. To a lesser extent, the D-domain and HVR contribute to dimerization. Contrarily, the α-helices of the a1a2-repeats of the A-domain in different PAMs are disrupted by the hPg binding Arg-His motifs, upstream of the B-domain, which mainly consists of mobile loops. Accordingly, these two coiled-coil destabilizing domains constitute separated concave rings in the illustrated model.
Fig. 8.

The structural model of r-PAMs. Models were drawn to scale in ChemDraw Professional 16.0. According to the results from CD, AUC, and heptad register alignments, the c-repeats of the C-domain, together with partial HVR and D-domain regions, form helical coiled-coil dimers in a rod-like conformation in solution. But a1a2-repeats and most of the B-domain manifest coiled-coil disrupting properties. (A) Class I and III PAMs, except PAMSS1574, encompass three c-repeats and intact a1a2-repeats. (B) PAMSS1574 is the shortest among all naturally occurring PAMs due to the loss of one c-repeat, despite the complete a1a2-repeats as other Class I and III PAMs. (C) Class II PAMs, viz. PAMNS88.2 and PAMSS1448, lack the whole a1-repeat but express three c-repeats. In the C-domain.
It has been shown that coiled-coil oligomers exist in proteins in both eukaryotes and prokaryotes, e.g., tropomyosin (Sodek et al., 1972) and tropocollagen (Bianchi et al., 1966). It has also been reported that the minimum length of a chemically synthesized polyheptapeptide, Ac-(Lys-Leu-Glu-Ala-Leu-Glu-Gly)n-Lys-amide, required for the coiled-coil dimerization, was 29 residues (Lau et al., 1984). It therefore seems plausible for bacteria to synthesize such polyheptapeptides only up to 30–40 residues when they express dimeric surface virulence factors, such as PAMs. Thus, the polar or charged residues, which are substituted for Leu, Ile, or Val at positions a and d in heptad registers, interrupt the coiled-coil oligomer. Therefore, to accomplish the formation of functional PAM dimers in solution, GAS strains synthesize significantly more amino acids in each chain at the expense of energy. This strategy encompasses the long dimeric C-domain, together with the D-domain, to maintain a1a2-repeats distant from the cell surface, which would otherwise interfere with binding of PAMs to hPg and other proteins.
Next, residues at position a and d in heptad registers, other than Leu or Val, may function to create a hydrophilic niche, especially at higher temperatures. In this case, the aliphatic side chains of Leu or Val residues are exposed to an aqueous environment, and the potential of a rod-like conformation would be high. It is thus suggested that to achieve the most energetically favorable format, the hydrophilic heads of Lys, Arg, and Ser of the C-domain become coiled-coil destabilizing residues and interact with the aqueous environment to retain the apolar environment in the interior.
4.3. Regulation of PAM expression in different GAS strains
Although it has been reported that hPg binds to streptococcal enolase and GAPDH, metabolically necessary enzymes present in all GAS strains (Pancholi and Fischetti, 1998; Winram and Lottenberg, 1996), the binding affinities of hPg to these receptors are much weaker than that of PAM. Because all naturally occurring PAMs bind to hPg with similar affinities at 25° C, divergence in whole-cell binding among pam-positive GAS strains will depend on the PAM expression levels on the cell surface. Thus, hPg binding to Pattern D GAS cells is a determinant for the integrity of PAM on these same cells.
5. Conclusions
The significance of this study is the proposal of an experimentally-based model that depicts the dimerization of PAMs. Biophysical methods were employed to reveal the manner in which dimerization of PAMs has occurred, mainly stabilized by the C-domain with three c-repeats. The HVR and D-domain are also engaged to a lesser extent in the stabilization of PAM dimers. Coiled-coil disrupting regions, including the a1a2 repeats and the B-domain, are relevant to allow high affinity binding of PAMs to hPg.
From the evolutionary perspective, it is necessary for GAS to develop a strategy that permits synthesis of a long rod-like region of PAM, which maintains the functionally-relevant N-terminus distant from the cell surface. On the other hand, charged or polar residues at positions a and d in the C-repeats, which seemingly destabilize coiled-coil dimerization, provide PAMs and GAS cells with the ability to remain well-tuned to the higher temperatures in which they operate.
Supplementary Material
Highlights.
Group A S. pyogenes (GAS) is a causative agent in pharyngeal and dermal infections.
A major virulence determinant of GAS is the signature dimeric modular M-protein.
Host human plasminogen binds to the A-domain of all Pattern D M-proteins and governs virulence.
The determinants of Pattern D M-protein dimerization reside in its helical C-domain.
The modular nature of M-proteins are evolutionarily designed for bacterial survival in hostile host niches.
Acknowledgments
Funding
This work was supported by NIH Grant HL013423.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interests with this work.
Data deposition
Backbone chemical shift assignments and corresponding experimental restraints used in the structure calculations for AGL55NS88.2, KTI55SS1448, and VEK75AP53 have been deposited in the BioMagResBank with the accession numbers, 30389, 30390, and 30391, respectively. The coordinates of calculated structure ensembles have been deposited in the Protein Data Bank with accession codes 6BZJ (AGL55NS88.2), 6BJK (KTI55SS1448), and 6BZL (VEK75AP53).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrahari G, Liang Z, Mayfield JA, Balsara RD, Ploplis VA, Castellino FJ, 2013. Complement-mediated opsonization of invasive Group A Streptococcus pyogenes strain AP53 is regulated by the bacterial two-component cluster of virulence responder/sensor (CovRS) system. J. Biol. Chem 288, 27494–27504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrahari G, Liang Z, Glinton K, Lee SW, Ploplis VA, Castellino FJ, 2016. Streptococcus pyogenes employs strain-dependent mechanisms of C3b inactivation to inhibit phagocytosis and killing of bacteria. J. Biol. Chem 291, 9181–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babul J, Stellwagen E, 1969. Measurement of protein concentration with interferences optics. Anal. Biochem 28, 216–221. [DOI] [PubMed] [Google Scholar]
- Bao Y, Liang Z, Booyjzsen C, Mayfield JA, Li Y, Lee SW, Ploplis VA, Song H, Castellino FJ, 2014. Unique genomic arrangements in an invasive serotype M23 strain of Streptococcus pyogenes identify genes that induce hypervirulence. J. Bacteriol 196, 4089–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall B, Facklam R, Thompson T, 1996. Sequencing emm-specific PCR products for routine and accurate typing of group A streptococci. J. Clin. Microbiol 34, 953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge A, Sjobring U, 1993. PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem 268, 25417–25424. [PubMed] [Google Scholar]
- Berge A, Bjorck L, 1995. Streptococcal cysteine proteinase releases biologically active fragments of streptococcal surface proteins. J. Biol. Chem 270, 9862–9870. [DOI] [PubMed] [Google Scholar]
- Berggard K, Johnsson E, Morfeldt E, Persson J, Stalhammar-Carlemalm M, Lindahl G, 2001. Binding of human C4BP to the hypervariable region of M protein: a molecular mechanism of phagocytosis resistance in Streptococcus pyogenes. Mol. Microbiol 42, 539–551. [DOI] [PubMed] [Google Scholar]
- Bessen DE, 2016. Molecular basis of serotyping and the underlying genetic organization of Streptococcus pyogenes. In: Streptococcus pyogenes: Basic Biology to Clinical Manifestations (Ferretti JJ, Stevens DL, Fischetti VA), editors. [PubMed] [Google Scholar]
- Bessen DE, Sotir CM, Readdy TL, Hollingshead SK, 1996. Genetic correlates of throat and skin isolates of group A streptococci. J. Infect. Dis 173, 896–900. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Tejero R, Montelione GT, 2007. Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Liang Z, Quek AJ, Ploplis VA, Law R, Castellino F,J, 2014. Dimerization is not a determining factor for functional high affinity human plasminogen binding by the group A streptococcal virulence factor PAM and is mediated by specific residues within the PAM a1a2 domain. J. Biol. Chem 289, 21684–21693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi E, Conio G, Ciferri A, 1966. The helix-coil transformation for tropocollagen solutions and its relationship to transformations involving the crystalline form of the protein. Biopolymers 4, 957–970. [DOI] [PubMed] [Google Scholar]
- Buffalo C, Bahn-Suh AJ, Hirakis SP, Biswas T, Amaro RE, Nizet V, Ghosh P, 2016. Conserved patterns hidden within group A Streptococcus M protein hypervariability recognize human C4b-binding protein. Nat. Microbiol 1, 16155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedervall T, Johansson MU, Akerstrom B, 1997. Coiled-coil structure of group A streptococcal M proteins. Different temperature stability of class A and C proteins by hydrophobic-nonhydrophobic amino acid substitutions at heptad positions a and d. Biochemistry 36, 4987–4994. [DOI] [PubMed] [Google Scholar]
- Chandrahas V, Glinton K, Liang Z, Donahue DL, Ploplis VA, Castellino FJ, 2015. Direct host plasminogen binding to bacterial surface M-protein in Pattern D strains of Streptococcus pyogenes Is required for activation by Its natural coinherited SK2b protein. J. Biol. Chem 290, 18833–18842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Yang JT, Martinez HM, 1972. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 11, 4120–4131. [DOI] [PubMed] [Google Scholar]
- Clubb RT, Thanabal V, Wagner G, 1992. A constant-time 3-dimensional triple-resonance pulse scheme to correlate intraresidue H-1(N), N-15, and C-13(‘) chemical-shifts in N-15-C-13-labeled proteins. J. Magn. Reson. 97, 213–217. [Google Scholar]
- Cnudde SE, Prorok M, Castellino FJ, Geiger JH, 2006. X-ray crystallographic structure of the angiogenesis inhibitor, angiostatin, bound to a peptide from the group A streptococcal surface protein PAM. Biochemistry 45, 11052–11060. [DOI] [PubMed] [Google Scholar]
- Fischetti VA, 1989. Streptococcal M protein: molecular design and biological behavior. Clin. Microbiol. Rev 2, 285–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischetti VA, 1991. Streptococcal M protein. Sci. Am 264, 58–65. [DOI] [PubMed] [Google Scholar]
- Fischetti VA, Parry DA, Trus BL, Hollingshead SK, Scott JR, Manjula BN, 1988. Conformational characteristics of the complete sequence of group A streptococcal M6 protein. Proteins 3, 60–69. [DOI] [PubMed] [Google Scholar]
- Frick IM, Schmidtchen A, Sjobring U, 2003. Interactions between M proteins of Streptococcus pyogenes and glycosaminoglycans promote bacterial adhesion to host cells. Eur. J. Biochem 270, 2303–2311. [DOI] [PubMed] [Google Scholar]
- Ghosh P, 2011. The nonideal coiled coil of M protein and its multifarious functions in pathogenesis. Adv. Exp. Med. Biol 715, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, 2018. Variation, indispensability, and masking in the M protein. Trends Microbiol. 26, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinton K, Beck J, Liang Z, Qiu C, Lee SW, Ploplis VA, Castellino FJ, 2017. Variable region in streptococcal M-proteins provides stable binding with host fibrinogen for plasminogen-mediated bacterial invasion. J. Biol. Chem 292, 6775–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield NJ, 2006. Analysis of the kinetics of folding of proteins and peptides using circular dichroism. Nat. Protoc 1, 2891–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MCU, Lannergård J, Nilsson OR, Kristensen BM, Olsen JE, Harris CL, Ufret-Vincenty RL, Stålhammar-Carlemalm M, Lindahl G, 2013. Factor H binds to the hypervariable region of many streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog. 9, e1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwald H, Cramer H, Morgelin M, Russell W, Sollenberg U, Norrby-Teglund A, Flodgaard H, Lindbom L, Bjorck L, 2004. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell 116, 367–379. [DOI] [PubMed] [Google Scholar]
- Hollingshead SK, Fischetti VA, Scott JR, 1986. Complete nucleotide sequence of type 6 M protein of the group A Streptococcus. Repetitive structure and membrane anchor. J. Biol. Chem 261, 1677–1686. [PubMed] [Google Scholar]
- Hondorp ER, McIver KS, 2007. The Mga virulence regulon: infection where the grass is greener. Mol. Microbiol 66, 1056–1065. [DOI] [PubMed] [Google Scholar]
- Ikura M, Kay LE, Tschudin R, Bax A, 1990. 3-Dimensional NOESY-HMQC spectroscopy of a C-13-labeled protein. J. Magn. Reson 86, 204–209. [Google Scholar]
- Kay L, Keifer P, Saarinen T, 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc 114, 10663–10665. [Google Scholar]
- Kay LE, Ikura M, Tschudin R, Bax A, 2011. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. 1990. J. Magn. Reson 213, 423–441. [DOI] [PubMed] [Google Scholar]
- Lahteenmaki K, Kuusela P, Korhonen TK, 2001. Bacterial plasminogen activators and receptors. FEMS Microbiol. Rev 25, 531–552. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM, 1993. PROCHEK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291. [Google Scholar]
- Lau SY, Taneja AK, Hodges RS, 1984. Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded alpha-helical coiled-coils. J. Biol. Chem 259, 13253–13261. [PubMed] [Google Scholar]
- Lee JL, Naguwa SM, Cheema GS, Gershwin ME, 2009a. Acute rheumatic fever and its consequences: a persistent threat to developing nations in the 21st century. Autoimmun. Rev 9, 117–123. [DOI] [PubMed] [Google Scholar]
- Lee W, Westler WM, Bahrami A, Eghbalnia HR, Markley JL, 2009b. PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics 25, 2085–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Zhang Y, Agrahari G, Chandrahas V, Glinton K, Donahue DL, Balsara RD, Ploplis VA, Castellino FJ, 2013. A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D Streptococcal pyogenes strain influences virulence-associated genes. J. Biol. Chem 288, 6561–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr F, Ruterjans H, 2002. Correlation of backbone amide and side-chain 13C resonances in perdeuterated proteins. J. Magn. Reson 156, 10–18. [DOI] [PubMed] [Google Scholar]
- Loof TG, Deicke C, Medina E, 2014. The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Frontiers in cellular and infection microbiology 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wuthrich K, 1998. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J. Biomol. NMR 12, 1–23. [DOI] [PubMed] [Google Scholar]
- Mason JM, K MM, Arndt KM, 2008. iPEP: peptides designed and selected for interfering with protein interaction and function. Biochem. Soc. Trans 36, 1442–1447. [DOI] [PubMed] [Google Scholar]
- McIver KS, Scott JR, 1997. Role of mga in growth phase regulation of virulence genes of the group A streptococcus. J. Bacteriol 179, 5178–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIver KS, Heath AS, Green BD, Scott JR, 1995. Specific binding of the activator Mga to promoter sequences of the emm and scpA genes in the group A streptococcus. J. Bacteriol 177, 661906624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara C, Zinkernagel AS, Macheboeuf P, Cunningham MW, Nizet V, Ghosh P, 2008. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 319, 1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeilly CL, McMillan DJ, 2014. Horizontal gene transfer and recombination in Streptococcus dysgalactiae subsp. equisimilis. Front. Microbiol. 5, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Abeygunawardana C, Johnson MO, van Zijl PC, 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108, 94–98. [DOI] [PubMed] [Google Scholar]
- Nilson BH, Frick IM, Akesson P, Forsen S, Bjorck L, Akerstrom B, Wikstrom M, 1995. Structure and stability of protein H and the Ml protein from Streptococcus pyogenes. Implications for other surface proteins of gram-positive bacteria. Biochemistry 34, 13688–13698. [DOI] [PubMed] [Google Scholar]
- Oehmcke S, Morgelin M, Malmstrom J, Linder A, Chew M, Thorlacius H, Herwald H, 2012. Stimulation of blood mononuclear cells with bacterial virulence factors leads to the release of pro-coagulant and pro-inflammatory microparticles. Cell. Microbiol 14, 107–119. [DOI] [PubMed] [Google Scholar]
- Pancholi V, Fischetti VA, 1998. alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J. Biol. Chem 273, 14503–14515. [DOI] [PubMed] [Google Scholar]
- Phillips GN, Flicker PF, Cohen C, Manjula BN, Fischetti VA, 1981. Streptococcal M protein: alpha-helical coiled-coil structure and arrangement on the cell surface. Proc. Natl. Acad. Sci. USA 78, 4689–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringdahl U, Svensson M, Wistedt AC, Renn T, Kellner R, Muller-Esterl W, Sjobring U, 1998. Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J. Biol. Chem 272, 6424–6230. [DOI] [PubMed] [Google Scholar]
- Rios-Steiner JL, Schenone M, Mochalkin I, Tulinsky A, Castellino FJ, 2001. Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J. Mol. Biol 308, 705–719. [DOI] [PubMed] [Google Scholar]
- Sanderson-Smith M, Batzloff M, Sriprakash KS, Dowton M, Ranson M, Walker MJ, 2006a. Divergence in the plasminogen-binding group a streptococcal M protein family: functional conservation of binding site and potential role for immune selection of variants. J. Biol. Chem 281, 3217–3226. [DOI] [PubMed] [Google Scholar]
- Sanderson-Smith ML, Walker MJ, Ranson M, 2006b. The maintenance of high affinity plasminogen binding by group A streptococcal plasminogen-binding M-like protein is mediated by arginine and histidine residues within the a1 and a2 repeat domains. J. Biol. Chem 281, 25965–25971. [DOI] [PubMed] [Google Scholar]
- Sanderson-Smith ML, Dowton M, Ranson M, Walker MJ, 2007. The plasminogen-binding group A streptococcal M protein-related protein Prp binds plasminogen via arginine and histidine residues. J. Bacteriol 189, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson-Smith ML, Dinkla K, Cole JN, Cork AJ, Maamary PG, McArthur JD, Chhatwal GS, Walker MJ, 2008. M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22, 2715–2722. [DOI] [PubMed] [Google Scholar]
- Schenone MM, Warder SE, Martin JA, Prorok M, Castellino FJ, 2000. An internal histidine residue from the bacterial surface protein, PAM, mediates its binding to the kringle-2 domain of human plasminogen. J. Pept. Res 56, 438–445. [DOI] [PubMed] [Google Scholar]
- Schiffer M, Edmundson AB, 1967. Use of helical wheels to represent the structures of proteins and to identify segments with helical potential. Biophys. J 7, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P, Gillis RB, Besong TM, Almutairi F, Adams GG, Rowe AJ, Harding SE, 2014. SEDFIT-MSTAR: molecular weight and molecular weight distribution analysis of polymers by sedimentation equilibrium in the ultracentrifuge. Analyst 139, 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Clore GM, 2006. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Mag. Res. Sp 48, 47–62. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Bermejo GA, Clore GM, 2018. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 27, 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM, 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Shen Y, Bax A, 2013. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A, 2009. TALOS plus : a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Bryan PN, He Y, Orban J, Baker D, Bax A, 2010. De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds. Protein Sci. 19, 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker DM, Bax A, 2008. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 105, 4685–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens N, Patenge N, Otto J, Fiedler T, Kreikemeyer B, 2011. Streptococcus pyogenes M49 plasminogen/plasmin binding facilitates keratinocyte invasion via integrin-integrin linked kinase (ILK) pathways and protects from macrophage killing. J. Biol. Chem 286, 21612–21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeesters PR, McMillan DJ, Sriprakash KS, 2010. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 18, 275–282. [DOI] [PubMed] [Google Scholar]
- Sodek J, Hodges RS, Smillie LB, Jurasek L, 1972. Amino-acid sequence of rabbit skeletal tropomyosin and its coiled-coil structure. Proc. Natl. Acad. Sci. USA 69, 3800–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CM, Buffalo CZ, Valderrama JA, Henningham A, Cole JN, Nizet V, Ghosh P, 2016. Coiled-coil destabilizing residues in the group A Streptococcus M1 protein are required for functional interaction. Proc. Natl. Acad. Sci. USA 113, 9515–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo T, Nakata M, Higashino M, Yamaguchi M, Kawabata S, 2016. Group A Streptococcus exploits human plasminogen for bacterial translocation across epithelial barrier via tricellular tight junctions. Sci Rep 7, 20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Ringdahl U, Homeister JW, Fay WP, Engelberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D, 2004. Plasminogen is a critical host pathogenicity factor for Group A streptococcal infection. Science 305, 1283–1286. [DOI] [PubMed] [Google Scholar]
- Talluri S, Wagner G, 1996. An optimized 3D NOESY-HSQC. J. Magn. Reson 112, 200–205. [DOI] [PubMed] [Google Scholar]
- Tamura GS, Nittayajarn A, 2000. Group B streptococci and other gram-positive cocci bind to cytokeratin 8. Infect. Immun. 68, 2129–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V, 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin. Microbiol. Rev 27, 264–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Prorok M, Castellino FJ, 2010a. NMR backbone dynamics of VEK-30 bound to the human plasminogen kringle 2 domain. Biophys. J 99, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zajicek J, Geiger JH, Prorok M, Castellino FJ, 2010b. Solution structure of the complex of VEK-30 and plasminogen kringle 2. J. Struct. Biol 169, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PN, Leigh JA, 2002. Characterization of PauB, a novel broad-spectrum plasminogen activator from Streptococcus uberis. J. Bacteriol. 184, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winram SB, Lottenberg R, 1996. The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 142, 2311–2320. [DOI] [PubMed] [Google Scholar]
- Wistedt AC, Ringdahl U, Mulleresterl W, Sjobring U, 1995. Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol. Microbiol 18, 569–578. [DOI] [PubMed] [Google Scholar]
- Wittekind M, Mueller L, 1993. HNCACB, A high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha-carbon and beta-carbon resonances in proteins. J. Magn. Reson 101, 201–205. [Google Scholar]
- Wong AP, Cortez SL, Baricos WH, 1992. Role of plasmin and gelatinase in extracellular matrix degradation by cultured rat mesangial cells. Am. J. Physiol 236, F1112–F1118. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Zajicek J, Qiu C, Chandrahas V, Lee SW, Ploplis VA, Castellino FJ, 2017. Conformationally organized lysine isosteres in Streptococcus pyogenes M protein mediate direct high-affinity binding to human plasminogen. J. Biol. Chem 292, 15016–15027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liang Z, Hsueh HT, Ploplis VA, Castellino FJ, 2012. Characterization of streptokinases from Group A streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M-Protein and cluster 2b streptokinase. J. Biol. Chem 287, 42093–42103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liang Z, Glinton K, Ploplis VA, Castellino FJ, 2013. Functional differences between Streptococcus pyogenes cluster 1 and cluster 2b streptokinases are determined by their β-domains. FEBS Lett. 587, 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.