

Abstract
Background
Helitrons play an important role in shaping eukaryotic genomes due to their ability to transfer horizontally between distantly related species and capture gene fragments during the transposition. However, the mechanisms of horizontal transfer (HT) and the process of gene fragment capturing of Helitrons still remain to be further clarified.
Results
Here, we characterized a novel Helitron family discontinuously distributed in 27 out of 256 insect genomes. The most prominent characteristic of Hel1 family is its high sequence similarity among species of different insect orders. Related elements were also identified in two spiders, representing the first report of spider Helitrons. All these elements were classified into 2 families, 9 subfamilies and 35 exemplars based on our new classification criteria. Autonomous partners of Helitron were reconstructed in the genomes of three insects and one spider. Integration pattern analysis showed that majority of Hel1A elements in Papilio xuthus and Pieris rapae inserted into introns. Consistent with filler DNA model, stepwise sequence acquisition was observed in Sfru_Hel1Aa, Sfru_Hel1Ab and Sfru_Hel1Ac in Spodoptera frugiperda. Remarkably, the evidence that Prap_Hel1Aa in a Lepdidoptera insect, Pieris rapae, was derived from Cves_Hel1Aa in a parasitoid wasp, Cotesia vestalis, suggested the role of nonregular host-parasite interactions in HT of Helitrons.
Conclusions
We proposed a modified classification criteria of Helitrons based on the important role of the 5′-end of Helitrons in transposition, and provided evidence for stepwise sequence acquisition and recurrent HT of a novel Helitron family. Our findings of the nonregular host-parasite interactions may be more conducive to the HT of transposons.
Electronic supplementary material
The online version of this article (10.1186/s13100-019-0165-4) contains supplementary material, which is available to authorized users.
Keywords: Helitron, Transposable elements, Horizontal transfer, Insects, Genome evolution
Introduction
As the single largest component of the genetic material of most eukaryotic and proeukaryotic species, transposable elements (TEs) play key roles in the epigenetic regulation of the genome and generation of genomic novelty [1, 2]. Depending on the mode of transposition, TEs are traditionally categorized as class-I elements or retrotransposons and class-II elements or DNA transposons [1, 3]. Copy and paste retrotransposons replicate via reverse transcription of an RNA intermediate of a source element, and can be further divided into long terminal repeat (LTR) and non-LTR retrotransposons. DNA transposons move through a single or double-stranded DNA intermediate, and are classified into three major subclasses, including the classic “cut-and-paste” transposons, rolling-circle (RC) transposons called Helitrons, and self-synthesizing transposons called Mavericks/Polintons. Both retrotransposons and DNA transposons exist as self-mobilizing autonomous elements or non-autonomous elements relying on trans-mobilization by the enzymatic machinery of their autonomous counterparts [4].
Helitrons, a novel superfamily of transposons, were originally discovered by in silico genome-sequence analysis [5], and later identified in a wide range of organisms, from protists to mammals [6, 7]. Helitrons are fundamentally different from classical transposons in terms of enzymatic activity and structure. Helitrons encode a RepHel protein homologous to RCR prokaryotic transposases, which comprises the replication initiator (Rep) and helicase (Hel) domains and is predicted to have both HUH (His-hydrophobe-His) endonuclease activity and 5′ to 3′ helicase activity [8]. Helitrons do not create target site duplications or contain terminal inverted repeats, and recent studies show that they transpose via copy-and-paste rather than cut-and-paste mechanism [9]. The characteristic features of Helitrons include a ‘TC’ motif on the 5′-end and a ‘CTRR’ motif on the 3′-end, and a palindromic sequence of 16–20 bp near the 3′-end, which can form a hairpin structure. Because of the minimal sequence feature and high sequence heterogeneity among Helitron copies, a classification system for family and subfamily definition has been proposed based on genome-wide analysis of Helitrons in the maize, Zea mays [10].
Helitrons have attracted widespread attention because their remarkable ability to capture gene fragments at the DNA level makes them play an important role in the host genome evolution. This process appears to have been particularly remarkable in the maize genome, where it is estimated that at least 20,000 gene fragments have been picked up and shuffled by Helitrons [10–12]. High frequency of Helitron-mediated gene capture is also reported in bats [13]. A recent study revealed that Helitrons have captured 3724 fragments from 268 genes in the silkworm, Bombyx mori [14]. Several models have been proposed to explain the mechanism of gene capture at the DNA level including end bypass and filler DNA model [8, 15].
Horizontal transfer (HT) is the non-vertical exchange of genetic material between reproductively isolated species. The inherent mobility and replication abilities of TEs facilitate them to undergo vector-mediated HT between organisms to avoid co-evolved host suppression mechanisms leading to vertical inactivation [1, 16–18]. The first evidence for the repeated HT of four different families of Helitrons including Heligloria, Helisimi, Heliminu, and Helianu, was described in an unprecedented array of organisms, including mammals, reptiles, fish, invertebrates, and polydnaviruses [19]. Subsequent identification of horizontally transferred Helitrons, such as Hel-2 [20], Lep1 [21], suggesting that Helitrons rely heavily on HT for their propagation and maintenance throughout evolution [21]. However, the physiological or ecological factors favoring the high frequency of HT still remains elusive.
Here, we have conducted a thorough search for the distribution of a novel Helitron family by analyzing the sequenced genomes of 256 insects and 22 spiders. We found that Hel1 elements distributed in 27 investigated insect genomes as well as the genome of a distantly related spider, Nephila clavipes, which were classified into 9 subfamilies and 34 exemplars. A related Hel2 family was identified in the genome of a spider, Parasteatoda tepidariorum. Furthermore, we provided evidence for stepwise sequence acquisition and recurrent HT of this novel Helitron family. Our results provided new insights into the classification and evolution of Helitrons, and suggested that the Helitrons can undergo horizontal transfer by diverse means.
Results
Identification and distribution of a novel Helitron transposon
A novel Helitron element was occasionally found during the analysis of flanking sequence of one short interspersed nuclear element (SINE) in Chilo suppressalis (Additional file 1: Figure S1). Subsequent database search detected a total of 884 similar sequences in C. suppressalis genome (Additional file 2: Table S1). Sequence analysis showed that these sequences present the typical structural features of the Helitron transposons: almost all copies have characteristic 5′-TC and 3′-CTRY nucleotide termini. The integration occurs precisely between the host A and T nucleotides, without duplications or deletions of the target sites, consistent with the RC mechanism (Additional file 1: Figure S2a). Further analysis showed that these Helitron sequences could be divided into three exemplars of two subfamilies, Csup_Hel1Aa, Csup_Hel1Ab and Csup_Hel1Ea. Their consensus sequences are of 162, 257 and 195 bp long, respectively. The conserved 3′-stem-loops (hairpins) were also predicted upstream of the 3′-CTRR termini of Csup_Hel1A exemplar (Additional file 1: Figure S2b).
A broad homology-based search of contemporary whole genome shotgun (WGS) databases of 256 insects identified similar elements in 27 species, including 12 out of 42 Lepidoptera, 4 out of 110 Diptera, 4 out of 52 Hymenoptera, 1 out of 2 Orthoptera, 2 out of 2 Phasmatodea, 1 out of 12 Coleoptera, 2 out of 23 Hemiptera, 1 out of 3 Blattodea. Among 22 spider WGS genome database, similar elements were only found in P. tepidariorum and N. clavipes (Additional file 1: Figure S3 and Fig. 1). These elements vary in size from 162 bp to 8065 bp. The number of copies also varies from exemplar to exemplar. Cvir_Hel1Ea in Calephelis virginiensis showed the highest copy numbers of 5578, occupies 0.479% of genome, while only 4 copies were found in Aros_Hel1Aa in Athalia rosae. The average percentage divergence varied from 0.01 to 15.672, indicating different invasion time (Table 1).
Fig. 1.

The taxonomy tree of 29 species containing Csup_Hel1A-like Helitrons
Table 1.
Characteristics of 35 Helitron exemplars from 29 species
| Group | Average divergencea | Length (bp)b | Copies (% Genome) | |
|---|---|---|---|---|
| Araneae (2/5) | ||||
| Parasteatoda tepidariorum | Ptep_Hel2Aa | 0.322 | 2903 (11) | 345 (0.069) |
| Nephila clavipes | Ncla_Hel1Aa | 0.532 | 293 (79) | 4611 (0.055) |
| Blattaria (1/3) | ||||
| Blattella germanica | Bger_Hel1Aa | ND | 664 (5) | 91 (0.003) |
| Orthoptera (1/2) | ||||
| Locusta migratoria | Lmig_Hel1Aa | 0.319 | 2080 (3) | 86 (0.003) |
| Hemiptera (2/23) | ||||
| Myzus persicae | Mper_Hel1Fa | 1.278 | 551 (30) | 420 (0.067) |
| Homalodisca vitripennis | Hvit_Hel1Ga | 1.024 | 930 (165) | 1914 (0.081) |
| Coleoptera (1/12) | ||||
| Anoplophora glabripennis | Agla_Hel1Aa | 0.740 | 195 (17) | 27 (0.001) |
| Lepidoptera (12/42) | ||||
| Bombyx mori | Bmor_Hel1 | 702 | ||
| Bmor_Hel1Aa | 15.672 | 249 (4) | 8 (0.000) | |
| Bmor_Hel1Ca | 3.004 | 217 (67) | 680 (0.037) | |
| Spodoptera frugiperda | Sfru_Hel1 | 3404 | ||
| Sfru_Hel1Aa | 0.852 | 196 (60) | 495 (0.022) | |
| Sfru_Hel1Ab | 0.164 | 262 (86) | 463 (0.028) | |
| Sfru_Hel1Ac | 0.016 | 378 (69) | 1113 (0.096) | |
| Mamestra configurata | Mcon_Hel1Aa | 0.663 | 223 (63) | 1814 (0.071) |
| Melitaea cinxia | Mcin_Hel1Ca | 0.132 | 356 (62) | 271 (0.025) |
| Pieris rapae | Prap_Hel1Aa | 0.054 | 447 (449) | 1520 (0.276) |
| Papilio glaucus | Pgla_Hel1Ga | 0.467 | 165 (35) | 461 (0.020) |
| Papilio xuthus | Pxut_Hel1 | 2326 | ||
| Pxut_Hel1Aa | 0.069 | 296 (56) | 796 (0.097) | |
| Pxut_Hel1Ab | 0.066 | 204 (153) | 722 (0.060) | |
| Papilio machaon | Pmac_Hel1Ha | 0.015 | 178 (20) | 39 (0.002) |
| Operophtera brumata | Obru_Hel1Ka | 12.134 | 193 (30) | 437 (0.013) |
| Chilo suppressalis | Csup_Hel1 | 1760 | ||
| Csup_Hel1Aa | 0.010 | 162 (95) | 103 (0.005) | |
| Csup_Hel1Ab | 0.018 | 257 (16) | 95 (0.008) | |
| Csup_Hel1Ea | 0.034 | 195 (86) | 752 (0.047) | |
| Calephelis nemesis | Cnem_Hel1Ea | 0.919 | 271 (62) | 5578 (0.481) |
| Calephelis virginiensis | Cvir_Hel1Ea | 2.171 | 269 (158) | 2703 (0.231) |
| Diptera (4/110) | ||||
| Bactrocera tryoni | Btry_Hel1Ba | 0.207 | 1291 (4) | 1325 (0.330) |
| Bactrocera oleae | Bole_Hel1Aa | 0.302 | 1206 (4) | 186 (0.056) |
| Phortica variegata | Pvar_Hel1Da | 0.205 | 343 (5) | 38 (0.008) |
| Calliphora vicina | Cvic_Hel1Ca | 0.436 | 264 (52) | 214 (0.012) |
| Hymenoptera (4/52) | ||||
| Pseudomyrmex gracilis | Pgra_Hel1Aa | ND | 748 (5) | 83 (0.022) |
| Athalia rosae | Aros_Hel1Aa | ND | 8065 (1) | 4 (0.020) |
| Cotesia vestalis | Cves_Hel1Aa | 0.372 | 278 (76) | 137 (0.020) |
| Polistes dominula | Pdom_Hel1Ia | 0.968 | 395 (3) | 165 (0.031) |
| Phasmatodea (2/2) | ||||
| Dryococelus australis | Daus_Hel1Aa | 0.077 | 667 (27) | 1326 (0.026) |
| Timema cristinae | Tcri_Hel1Aa | 0.099 | 3435 (5) | 105 (0.035) |
ND, not determined
aAverage divergence is calculated between copies within a species
bThe number in bracket is the number of copies used to reconstruct the consensus sequences
Multiple alignment of the consensus sequences showed that the 30 bp fragments at the 5′-end showed as high as 93.1% average identity, however, the 30 bp fragments at the 3′-end showed somewhat sequence divergence. Among 35 exemplars, 22 showed above 80% pairwise identities, the rest 13 exemplars showed less than 80% identities with above 22 exemplars (Fig. 2). The high sequence identity of 5′-end extended to 126 bp (89% identity) except Ptep_Hel2Ca (Additional file 2: Table S2 and Additional file 1: Figure S4). According to our novel classification method, these elements were divided into 2 families, Hel1 and Hel2, and 9 subfamilies, Hel1A-Hel1I. While the 30 bp fragment at the 3′-end of Ptep_Hel2Ca showed over 80% identity with Hel1C, as low as 73.3% identity was found between 30 bp fragment at the 5′-end of Ptep_Hel2Ca and Csup_Hel1A (Additional file 1: Figure S5). Different exemplars were found in the same species. For example, in the genome of C. suppressalis, three exemplars of two subfamilies, Csup_Hel1Aa, Csup_Hel1Ab, and Csup_Hel1Eb, were detected, which showed average percentage divergence of 0.01, 0.018 and 0.034, respectively (Table 1). The classification was supported by evolutionary analysis, which showed that these three exemplars are polyphyletic in origin and separated into three distinct clades. Similarly, two exemplars of Helitron subfamily, Pxut_Hel1Aa and Pxut_Hel1Ab were found in the genome of Papilio xuthus with the average percentage divergence of 0.064 and 0.066, respectively, and cluster into two distinct clades (Fig. 3).
Fig. 2.

Multiple alignment of 34 Hel1 exemplars. a Multiple alignment of 30 bp region at the 5′-ends from Hel1 families and 30-bp region at the 3′-ends from Hel1A subfamilies. The alignment was graphically edited using TeXshade package. b Multiple alignment of 30 bp region at the 3′-ends from Hel1B-Hel1I subfamilies. c Genetic distance analysis of Hel1 and Hel2 from all species, the average distance is 0.069
Fig. 3.

Phylogenetic analysis of all copies of Hel1 Helitrons in C. suppressalis (a) and P. xuthus (b). Copies of different exemplars are indicated by distinct colors. The phylogenetic tree was constructed by the neighbor-joining method using MEGA 7.0 software
Characterization of reconstructed potential autonomous DNA Helitrons
A total of 7 Helitrons with degenerated remnants of Helitron coding sequences were initially detected in the genome of N. clavipes, P. tepidariorum, Papilio machaon, Cotesia vestalis, Homalodisca vitripennis, A. rosae and Timema cristinae (Additional file 3: Data S1). The longest Helitron was found in A. rosa, with the length of 8065 bp, and at position 785–1860, there was an insertion of 1076 bp fragment putatively encoding C-terminal catalytic domain of Cre recombinase (INT_Cre_C). Due to the short sequencing length of WGS sequence, the characteristic 5′-TC nucleotide termini was not found in P. machaon, however, 2 copies of 247 bp tandem repetitive sequences (TRS) were detected at 5′-end of this contig (Additional file 1: Figure S6).
Further blast searches were executed and 4 potential autonomous Helitrons with an uninterrupted ORF coding for Rep/helicase of 1495, 1467, 1495 and 1496 amino acids were reconstructed in C. vestalis (Cves_Hel1), P. tepidariorum (Ptep_Hel2), P. machaon (Pmac_Hel1) and A. rosae (Aros_Hel1), respectively (Additional file 3: Data S2). Multiple alignment showed that the predicted Rep/helicase proteins are composed of a Rep domain containing “two-His” replication initiator motifs and two conserved tyrosine residues, and a helicase domain containing eight conserved motifs of the SF1 superfamily of DNA helicases [22] (Fig. 4). More than 86% amino acid identities were observed among these 4 Rep/helicase proteins. Specially, the amino acid identity of Helitrons in P. machcaon and A. rosae is more than 98% (Additional file 1: Figure S7).
Fig. 4.

Gene structure (a), predicted Rep motif (b) and helicase domain (c) of reconstructed putative autonomous Hel1 and Hel2 Helitrons from C. vestalis, P. machaon, A. rosae and P. tepidariorum. In the alignment of the Rep motif, representative structure was from Myotis lucifugus (HeliBat1), Arabidopsis thaliana (AtHEL2P), Anopheles gambiae (Helitron_Ag1), Streptomyces cyaneus plasmid (Rep_SC) and Bacillus borstelensis plasmid (Re_BB). In the alignment of helicase domain, representative structure was from Myotis lucifugus (HeliBat1), Arabidopsis thaliana (AtHEL2P), Anopheles gambiae (Helitron_Ag1), Danio rerio (Dr_Helentron1) and Saccharomyces cerevisiae (PIF1_YEAST)
Contribution of Hel1 to gene and genome evolution
We further analyzed the integration pattern relative to the annotated genes in two representative genomes, P. xuthus and Pieris rapae. Out of the 796, 722 and 1520 copies of Pxut_Hel1Aa, Pxut_Hel1Ab and Prap_Hel1Aa, 463 (58%) of Pxut_Hel1Aa Helitrons, 413 (57%) of Pxut_Hel1Ab Helitrons, and 740 (52%) of Prap_Hel1Aa Helitrons were found in introns. Only 4, 7 and 3 copies of Pxut_Hel1Aa, Pxut_Hel1Ab and Prap_Hel1Aa were found to insert into exons, respectively (Fig. 5a). Further analysis revealed the insertion of multiple copies of Hel1 into introns of the same gene. For example, as many as 4 copies of Prap_Hel1Aa inserted into introns of LOC110995424 gene, and the fifth copy inserted into 3′-end of coding sequence (CDS) (Additional file 1: Figure S8). However, in most cases, only one copy was detected in intron regions of a specific gene. Notably, a 127 bp copy of Pxut_Hel1A (NW_013531711.1: 4069476–4,069,349) inserted into CDS of a gene encoding an unclassified protein (Fig. 5b). Thus, the P. xuthus and P. rapae Hel1 Helitrons mainly contribute to structural variation in introns, which might influence the regulation of gene expression.
Fig. 5.

Gene association of Hel1A Helitrons in P. xuthus and P. rapae. a Overall proportions of Pxut_Hel1Aa, Pxut_Hel1Ab and Prap_Hel1Aa in the respective genome are represented as pie charts. b Integration of a Pxut_Hel1A element within the coding sequence (CDS) of a gene encoding an unclassified protein in P. rapae
Sequence acquisition and new Hel1 creation
Among all Helitrons identified in this study, Hel1 in Spodoptera frugiperda attracted our attention. In addition to Sfru_Hel1Aa、Sfru_Hel1Ab and Sfru_Hel1Ac, a 161 bp copy (FJUZ01003913.1: 464817–464,657) was detected (Fig. 6). Sequence analysis showed that the consensus sequences of these three exemplars shared almost 99% identity with this short sequence excluding insertions, thus this short copy was designated as core sequence (Sfcore) (Fig. 6). Further analysis showed that compared with the core sequence, a 35 bp fragment named “A” region inserted into core sequence 130 bp downstream of the 5′-end in Sfru_Hel1Aa, and a 66 bp fragment named “B” region inserted into “A” region in Sfru_Hel1Ab, while a 108 bp fragment named “C” region inserted into “B” region in Sfru_Hel1Ac (Fig. 6a). Alignment of the consensus sequences showed that the insertion sites were consistent with the overlapping region (Fig. 6b). The search of putative source loci of these insertions revealed that “A” region consisted of “A1” and “A2” regions, among which “A1” region was derived from sequence NJHR01000652 (137291–137,357) and “A2” region from NJHR01000244 (961324–961,247) (Additional file 1: Figure S9). While sequence NJHR01000585 (153969–153,870) showed high identity with “B” region, we did not find source locus of “C” region, putative due to the incomplete genome sequencing. Additionally, 2-13 bp end junctions were identified in each source locus, supporting the filler DNA model [23] (Additional file 1: Figure S9). Furthermore, the average percentage divergence was 0.852, 0.164 and 0.016, respectively, indicating a clear evolutionary order (Table 1).
Fig. 6.
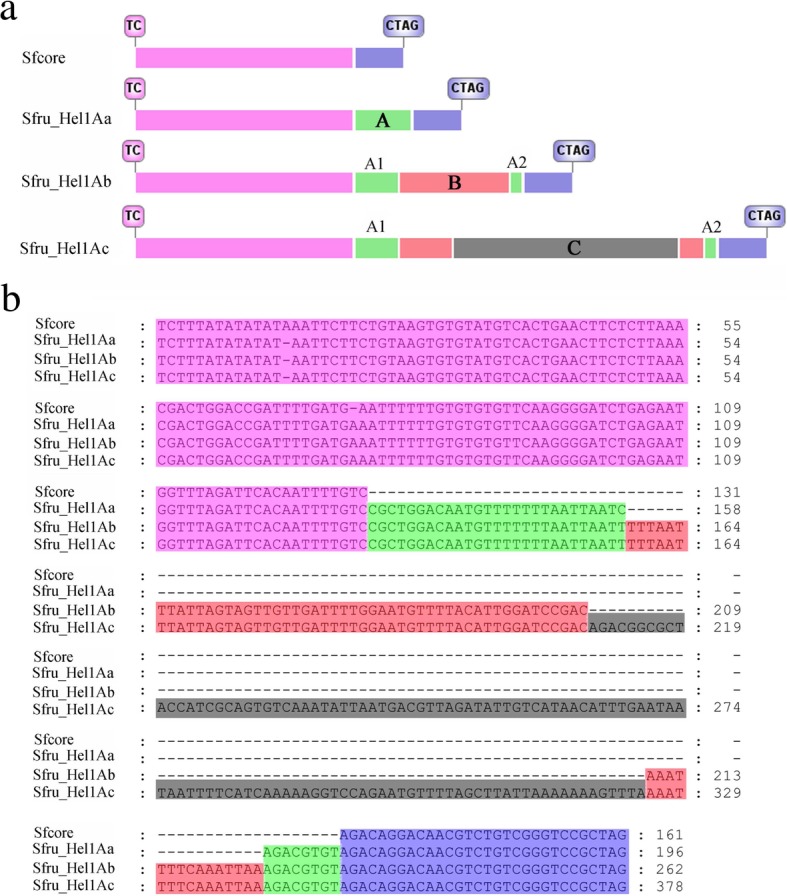
Stepwise transposition of Sfru_Hel1A in S. frugiperda. a Schematic structures of core sequence and exemplars in S. frugiperda. The 131-bp 5′-end and 30-bp 3′-end sequences are colored in pink and blue, respectively. The A, B and C regions are in green, red and black, respectively. Typical structural features of Helitron elements including characteristic 5′-TC and 3′-CTRY nucleotide termini were boxed. b Multiple alignment of core sequence and the consensus sequences of Hel1 exemplars in S. frugiperda
The sequence acquisition of P. xuthus Hel1 is different from that of S. frugiperda. Two exemplars of Hel1A subfamily, Pxut_Hel1Aa and Pxut_Hel1Ab, were found in P. xuthus, with the length of 296 bp and 204 bp, respectively, and only 172 bp region was shared by these two exemplars (Additional file 1: Figure S10a). The average percentage divergence of Pxut_Hel1Aa and Pxut_Hel1Ab was 0.069 and 0.066, respectively (Table 1). It seems unlikely that Pxut_Hel1Ab was formed by the sequence acquisition of Pxut_Hel1Aa. Furthermore, we also found a core sequence (BBJE01004687.1: 58267–58,430) highly similar to that of S. frugiperda (Additional file 1: Figure S10b). We speculated that Pxut_Hel1Aa and Pxut_Hel1Ab were independently derived from the core sequence by sequence acquisition during transposition.
In case of C. suppressalis, the 162 bp consensus sequence of Csup_Hel1Aa was over 96% identical to the above core sequences of P. xuthus and S. frugiperda (Additional file 1: Figure S10b). Compared with Csup_Hel1Ab, a 7 bp fragment (AGACGTG) was unique to Csup_Hel1Aa (Additional file 1: Figure S10b). Given similar average percentage divergence in these two exemplars, it seems that Csup_Hel1Ab was not derived from Csup_Hel1Aa. On the other hand, 5 core sequences with high similarity to Csup_Hel1Ea were also found in the genome of C. suppressalis (Additional file 1: Figure S10c). Considering that the average percentage divergence of Csup_Hel1Ea was 0.034, we inferred that Csup_Hel1Ea was evolutionarily earlier than Csup_Hel1A, and had different origin with Csup_Hel1Aa.
Evolution and horizontal transfer of Hel1
Using HeligloriaAi_DW1 and HeligloriaAi_Rp1 as out group [19], the phylogenetic tree of the 35 Helitron consensus sequences showed that Ptep_Hel2Ca was evolutionarily different from other Hel1 elements, and insects of the same order were not clustered together. The incongruence of Hel1 elements and host phylogeny as well as the patchy distribution and high sequence similarity of Hel1 elements among distantly related lineages suggest the recurrence of HT and that multiple mechanisms may underlie the horizontal spread of Hel1. Notably, Lepidopteran Prap_Hel1Aa and Hymenopteran Cves_Hel1Aa, Dipteran Cvic_Hel1Ca and Lepidopteran Bmor_Hel1Ca, Hemipteran Hvit_Hel1Ga and Lepidopteran Pgla_Hel1Ga were clustered into distinct clades, which diverged 325, 272 and 358 million years ago, respectively (http://www.timetree.org/) [24] (Fig. 7). Furthermore, several paralogous and orthologous empty sites were also detected in these insect genomes (Additional file 1: Figure S11). It is also noteworthy that the genetic distance between species of the same cluster was less than 0.1, indicating that these elements have spread horizontally among these species within a relatively narrow timeframe.
Fig. 7.

Phylogenetic relationships among 35 exemplars from 29 species. Taxa showing Hel1 and Hel2 are colored taxonomically, with Lepidopteran insects in red, Diptera insects in purple, Hymenoptera wasps in yellow, Phasmatodea species in blue, Araneae species in green. The opaque projection indicates the HT between the two species
The clustering of Prap_Hel1Aa from P. rapae (Lepidoptera: Pieridae) and Cves_Hel1Aa from C. vestalis (Hymenoptera: Braconidae) into the same clade is of particular interest. While the calculated genetic distances of orthologous genes calreticulin, Hsc70 and opsin between P. rapae and C. vestalis were 0.325, 0.229 and 0.312, respectively (Additional file 1: Figure S12a, b, c), sequence comparison showed that the consensus sequences of Prap_Hel1Aa and Cves_Hel1Aa shared over 98% identity excluding a 169 bp insertion in Prap_Hel1Aa (Additional file 1: Figure S12d). Considering the average percentage divergence of Prap_Hel1Aa and Cves_Hel1Aa were 0.054 and 0.372, respectively, we speculated that Prap_Hel1Aa was derived from C. vestalis through HT, followed by the capture of 169 bp fragment and a rapid burst in transposition. This hypothesis was partly supported by the reconstructed phylogeny in which the Cves_Hel1Aa copies are generally nested within clades made of Prap_Hel1Aa copies, and the closely related Csup_Hel1Aa copies from C. suppressalis were phylogenetically separated from both Prap_Hel1Aa and Cves_Hel1Aa (Additional file 1: Figure S13). Additionally, the 169 bp insertion fragment was almost entirely absent in a short copy of Prap_Hel1Aa (Prap0202, LWME01000202.1: 138955–138,682), which was over 94% identical to eight copies of Cves_Hel1Aa, and specifically over 97% identity was observed at the 30 bp 3′-ends of Prap0202 and these Cves_Hel1Aa copies (Additional file 1: Figure S12e). Interestingly, PCR amplification and sequencing revealed orthologous empty site of Prap0202 in a local population of P. rapae, suggesting Prap_Hel1Aa elements mobilized recently (Additional file 1: Figure S14). Furthermore, as many as 11 elements in P. rapae genome were found to be completely same as the consensus sequence of Prap_Hel1Aa, indicating recent invasion of the P. rapae genome by Prap_Hel1Aa elements (Additional file 1: Figure S15).
There are few reports on the occurrence of HT between Lepidoptera and Diptera [19]. In this study, we found that Cvic_Hel1Ca and Bmor_Hel1Ca, Pgla_Hel1Ga and Hvit_Hel1Ga were clustered into same clade, respectively, and the corresponding consensus sequences were highly similar (Additional file 1: Figure S16a and Figure S17), suggesting the occurrence of HT between these insects. Furthermore, orthologous empty sites were detected in both Lepidoptera and Diptera insects (Additional file 1: Figure S11). Notably, a putative Hel1 sequence (GEND01024785.1: 446–129) was found in the transcriptome shotgun assembly (TSA) database of Entomophthora muscae, which shared 90% identity with two copies of Cvic_Hel1Ca and 74% identity with Bmor_Hel1Ca (Additional file 1: Figure S16b), suggesting a possible role of E. muscae in the HT between Lepidoptera and Diptera insects.
Discussion
The classification of Helitron has always been ambiguous. The classical classification system was proposed based on genome-wide analysis of maize Helitrons, in which the sequences with the most similar 3′-ends (30 bp with at least 80% identity) were classified as members of the same family and sequences with the most similar 5′-ends (30 bp with at least 80% identity) were classified as members of the same subfamily [25]. This criteria has been followed by several other studies [19]. In addition, the unique internal sequence that was > 20% different at the nucleotide level from any other Helitron internal regions was defined “exemplars” [10]. However, based on genome-wide analysis of silkworm Helitrons, Han et al. (2013) suggested that sequences with identities > 80% in the 30 bp of both their 5′- and 3′-ends were classified as members of the same family, and full-length sequences with identity > 80% were classified in the same subfamily. Due to the lack of knowledge regarding Helitron cis- or trans- activation of Helitron, these classification criteria are exploratory. According to the end bypass model, which was proposed to explain the mechanism of gene capture of Helitron, transposition initiates at the 5′-end and gene capture occurs if the 3′-end signal is missed. A random cryptic sequence located downstream would then act as the termination signal and all intervening sequences would be captured [8, 26]. This model was supported by the fact that the xanA gene fragment was captured by a Helitron in Aspergilus nidulans genome [27]. Recent study showed that the modification or deletion of the hairpin loop or palindrome sequence had little effect on the transposon colony-forming activity of the reconstructed active bat Helitron, Helraiser. However, the deletion of 5′-end of Heliraiser resulted in complete loss of activity [9]. Given the importance role of the 5′-end of Helitrons in transposition, we think it seems more reasonable to classify the family with 5′-end of Helitron. Thus, we proposed a new classification standard, as described in methods. This new criteria was supported by our phylogenetic analysis of P. xuthis and C. suppressalis Helitrons, in which the copies of different subfamilies or exemplars of Helitron are well separated phylogenetically (Fig. 3).
The distinct copy and paste transposition process of Helitrons ensures them the capability of reaching high genomic copy numbers. For example, maize and silkworm Helitrons constitute 6.6% and 4.23% of the genome, respectively [11, 14]. In this study, as many as 5578 copies of Cvir_Hel1Ea were found in C. virginiensis, which account for 0.479% of genome. Besides their direct effect on genome size, evidence has accumulated in recent years that Helitrons can also impact the gene structure and expression as well as genome organization [28, 29]. For example, the insertion of two non-autonomous Helitron elements, AtREP3 and AtREP1, into upstream of ETT and ARF4 genes in tebichi (teb) mutant Arabidopsis thaliana resulted in the upregulation of these two genes [30]. In the tetraploid sour cherry, Prunus cerasus, the insertion of a small non-autonomous Helitron element into 38 bp downstream of the stop codon of SFB gene is proposed to interfere with the polyadenylation process, resulting in a loss of function of the SFB gene involved in gametophytic self-incompatibility [31]. In this study, we found that, similar to silkworm Helitrons [14], majority copies of Pxut_Hel1A and Prap_Hel1Aa insert into introns of host genome, suggesting that Hel1 duplication and transposition led to structural variation in introns, which might influence the regulation of gene expression. Notably, a copy with 3′-end deletion of Pxut_Hel1A inserted into coding region of an unclassified gene (Gene accession: LOC110995424) in P. xuthus genome, while the impact of the insertion on gene function is unknown at present.
A predominant characteristic of Helitrons is their ability to capture and amplify host genome sequences. Among 1649 Helitron-like transposons identified in genome of maize inbred line B73, over 90% of maize Helitrons have captured gene fragments [32]. While end bypass and filler DNA models [8, 15] have been proposed to explain Helitron gene capture and transposition, the exact mechanisms is far from clear. It has been proposed that gene capture during Helitron transposition occurs in a stepwise or sequential way [33]. In this study, three exemplars of Helitron, Sfru_Hel1Aa, Sfru_Hel1Ab and Sfru_Hel1Ac, were identified in S. frugiperda together with a shorter core sequence sharing high identity with these three exemplars. Multiple sequence alignment showed that these three exemplar Helitrons have high sequence identity in shared sequences, but differ due to additional captured regions internal to the elements. The gene fragment trapped within Helitrons excluded the end bypass model. Alternatively, filler DNA model suggests that Helitrons acquire DNA from the host during the repair of double-strand breaks (DSBs) internal to the element, and predicts that short regions flanking the DSB in the acceptor transposon should be homologous to DNA sequences flanking the original host sequence captured by the transposon [6]. The identification of end junctions in the putative source loci suggested that Hel1 Helitrons acquire DNA from the host putatively by filler DNA insertion during the repair of DSBs. Notably, the average percentage divergence of these three exemplars were 0.852, 0.164 and 0.016, respectively, strongly supporting the occurrence of stepwise transposition and amplification putatively using the core sequence as the source element. However, while shorter core sequences were also identified in respective host genomes, exemplars of the Pxut_Hel1A and Csup_Hel1 seems to capture host gene fragment during independent transposition events.
No less than 2836 horizontal transposon transfer (HTT) events have been recorded so far in multicellular eukaryotes [34], however, the mechanisms underlying HTT remain largely mysterious. The role of a host-parasite relationship has been proposed recently as a major mechanism of horizontal DNA transfer [21, 35, 36]. In this study, we provide evidence that Prap_Hel1Aa might derive from Cves_Hel1Aa. While C. vestali is larval parasitoid of the diamondback moth, Plutella xylostella (Lepidoptera: Plutellidae), we did not find any Cves_Hel1-like sequences in the genome database of P. xylostella (http://iae.fafu.edu.cn/DBM/), putatively due to the evolutionary dead-end of parasitized caterprillars. On the other hand, parasitoids are likely to oviposit within marginal (or even completely unsuitable) hosts in the laboratory or field, even if suitable hosts are present [37], and C. vestalis has been reared from several species belonging to different Lepidopteran families [38], thus we propose that C. vestalis might be a nonregular parasite of P. rapae, and this nonregular host-parasite interactions contribute to the HT of Hel1 between these two species. The origin of Cves_Hel1Aa in C. vestalis seems to be a mystery. A number of core sequences were found in Lepidoptera genome including S. frugiperda, P. xuthus and C. suppressalis, thus as a vector for HT in Lepidoptera insects, C. vestalis is more likely to acquire and transfer Cves_Hel1Aa to P. rape from other Lepidoptera insects.
While our results indicate the role of nonregular host-parasite interactions in HT of Prap_Hel1Aa and Cves_Hel1Aa, the evidence of 2 additional cases of HTT (Cvic_Hel1Ca and Bmor_Hel1Ca, Pgla_Hel1Ga and Hvit_Hel1Ga) based on their patchy distribution and incongruence of Hel1 and host phylogeny is somewhat intriguing due to the absence of host-parasite relationship among these species. It has been proposed that mechanisms of HT include insect-associated facultative symbionts [39–45]. In addition, the Lep1-like elements identified in the genome of Nosema bombycis suggested that the intracellular microsporidia parasite is also a potential vector for HT [21]. Recent studies have also suggested that both baculovirus and polydnaviruses might be important vectors of HTT [46–48]. While Lep1-like and Hel-2 Helitrons had been identified in C. vestalis and Cotesia sesamiae bracovirus and AcNPV, respectively [20, 21], we did not find Hel1 in the genomes of bracovirus and NPV. However, the discovery of Hel1-like sequence in TSA database of E. muscae suggests that pathogen may also serve as a vector mediating HT of insect TEs. More widespread sequencing would be required to find exact vectors that would facilitate the HT of Hel1 Helitrons in these species.
Conclusion
In the current report, we conducted a thorough search for a novel Helitron family by analyzing the sequenced genomes of 256 insects and 22 spiders. We modified the classical classification system for family and subfamily definition of Helitrons, and classified Hel1 family into 9 subfamilies and 34 exemplars, among which three exemplars in S. frugiperda exhibited stepwise sequence acquisition, supporting the filler DNA model. We proposed that nonregular host-parasite interactions plays an important role in HT of Helitrons. Our data may have implications for understanding the evolution and HT mechanisms of Helitrons.
Materials and methods
Data resources
The publicly available 256 Insecta and 22 Arachnida WGS from National Center for Biotechnology Information (NCBI) (last accessed September 30, 2017) were used in this study. P. rapae and P. xuthus WGS were downloaded from NCBI. A list of the analyzed species and corresponding amount of sequence data is provided in Additional file 4: Data S3 online. As corresponding gene annotation files, we used the GFF files GCF_001856805.1 for P. rapae and GCF_000836235.1 for P. xuthus, respectively.
Database searches and copy number estimation of Helitrons
Database searches were performed and comprise three steps. Firstly, the novel Helitron sequence located downstream of a SINE in C. suppressalis was used as a query in BLASTN searches against the NCBI C. suppressalis WGS database. Sequences of high homology as well as 200 bp upstream and downstream flanking regions were extracted and analyzed for hallmarks of Helitrons such as characteristic 5′-TC and 3′-CTRY nucleotide termini, and the consensus sequences of three Helitron exemplars of Hel1 families in C. suppressalis, Csup_Hel1Aa, Csup_Hel1Ab and Csup_Hel1Ea, were determined. Secondly, a total of 255 insect WGS collections were searched using 161 bp common sequence of Csup_Hel1Aa and Csup_Hel1Ab (Additional file 1: Figure S18) as query to detect sequences with high identity with Csup_Hel1Aa and Csup_Hel1Ab in other insect species, and 9 subfamilies, Hel1A-Hel1I were identified. Finally, WGS collections of other invertebrates were searched using 161 bp common sequence of Csup_Hel1Aa and Csup_Hel1Ab as query to detect Hel1-like sequences in other species, and the second family Hel2 was identified. In total, 2 families, 9 subfamilies, and 35 Helitron exemplars were identified, and consensus sequences for each Helitron exemplars were reconstructed based on a multiple alignment of at least 10 individual copies [36]. Specially, the copies of Aros_Hel1Aa and Bmor_Hel1Aa were less than 10, thus all copies were used for multiple alignment to determine consensus sequence. All consensus sequences are provided in Additional file 5: Data S4.
To estimate copy number and average percentage divergence of Helitrons, we used respective consensus sequences to search against related genomes where these Helitron elements were found using BLASTN. All contiguous fragments with at least 80% identity at the nucleotide level to the consensus over 100 bp were used to estimate copy number in all species [36, 49]. Given that 3′-ends deletion occurred in several copies of different subfamilies/exemplas in the same organism species, all those undistinguishable copies were counted as members of families. For example, two Helitron exemplars in P. xuthus, Pxut_Hel1Aa and Pxut_Hel1Ab shared high identity of 128 bp sequence at 5′-ends, thus all copies aligned only with part or full of this 128 bp region in the consensus sequence were estimate as members of family (Additional file 1: Figure S19). Furthermore, all fragments sharing at least 80% identity over at least 80% of the length of the consensus sequence were aligned and used for average percentage divergence calculation with Kimura-2 parameter model [50] in all species except A. rosae, Blattella germanica, Locusta migratoria, P. tepidariorum, Pseudomyrmex gracilis, T. cristinae and Phortica variegata, in which a high level of fragmentation was observed in multiple Helitron copies.
Reconstruction of potential autonomous Helitron
The reconstruction of autonomous Helitron comprise three steps. Firstly, large DNA fragments ranging from 1000 bp to 10 kb that shared similar terminal sequences to the above families were retrieved from WGS databases, and their potential transposase were predicted using getorf in EMBOSS-6.3.1 package [51]. Secondly, these candidates with degenerated remnants of Helitron coding sequences were used as queries in BLAST searches against both WGS databases, TSA and non-redundant protein databases. Finally, the query sequence and hit sequences were aligned to reconstruct the uninterrupted coding sequences with complete Rep/helicase gene ORF of Helitron by removing frameshifts and insertions.
Nomenclature
To distinguish Helitron elements from 29 species, we assume a set of concept names that consist of short Latin of single species, the type of TEs, the family, subfamily and exemplars of Helitron, just like Csup_Hel1Aa. Given that the 3′-end sequences of Helitrons were more variable than the 5′-end sequences [12], and the 5′-end sequence was strictly necessary for Helitron transposition [9], we modified Yang and Bennetzen′s method to reclassify Helitron TEs [25]. Generally, the sequences with the most similar 5′-ends (30 bp with at least 80% identity) were classified as members of the same family and sequences with the most similar 3′-ends (30 bp with at least 80% identity) were classified as members of the same subfamily. Due to the internal sequence divergence of copies in the same Helitron subfamily, the unique internal sequences with more than 80% identity were classified as members of exemplars.
Gene association and genomic show cases
The site of the Helitron integration relative to annotated genes was analyzed with a custom Perl script [29]. All copies of Prap_Hel1Aa, Pxut_Hel1Aa and Pxut_Hel1Ab, were determined for their positions in the genome through BLAST analysis with respective genome database and the GFF annotation files. The Helitrons in coding and untranslated gene regions as well as the distances of intergenic copies to the closest neighboring gene were determined and the numbers were counted [29]. All the genic and genomic loci harboring Helitrons were refined and visualized with the respective annotations using Perl script. All the figures used CorelDRAW to beatify the fine tune.
Sequence analysis and phylogeny
RNAstructure (http://rna.urmc.rochester.edu/RNAstructureWeb) was used to predict and analyze DNA secondary structure [52]. Multiple alignment of Helitrons were created by MUSCLE [53], and subsequently visualized with GENEDOC (www.psc.edu/biomed/genedoc) and TeXshade [54].
The phylogeny of Helitron elements was built using MrBayes 3.2 [55] after removing ambiguously aligned regions using BMGE [56] (Additional file 5: Data S5). Nucleotide substitution models were chosen using the AIC criterion in Modeltest [57] (HKY + G). The robustness of the nodes was evaluated for all phylogenies by performing a bootstrap analysis involving 1000 pseudo replicates of the original matrix [36].
Specifically, in the evolutionary analysis of subfamilies from P. xuthus and C. suppressalis, we conducted local BLAST analysis and got a CSV file based on location information to obtain all sequences that are larger than 80% coverage of and 80% identity to the consensus sequences. Finally, we extracted these sequences from each genome using TBtools [58]. A Neighbor-Joining (NJ) phylogenetic tree of these sequences in C. suppressalis and P. xuthus were constructed using MEGA 7.0.
Detection of insertion polymorphism of Prap_Hel1Aa
In P. rapae, using one pair of primers flanking the insertion site (Forward primer: 5′-ACGAGAGATGGCTACAACAG-3′; Reverse primer: 5′- AACACACCCACACCCTAAAC -3′), the insertion polymorphism of one short copy of Prap_Hel1Aa (Prap0202, LWME01000202.1: 138955–138,682) was assessed by performing a PCR survey. The PCR products were cloned into the pMD18-T vector (TaKaRa, Dalian, China) and sequenced.
Additional files
Figure S1. Insertion of a novel Helitron element into downstream region of a SINE element in C. suppressalis genome. The SINE and Helitron sequences are highlighted in blue and pink, respectively. The nucleotides highlighted in purple are the target site duplication (TSD) of SINE. Figure S2. Characteristic of Csup_Hel1A. (a), Multiple alignment of 30-bp end sequences as well as the flanking host nucleotides at the 5′- and 3′-end from Csup_Hel1A elements. The alignment was graphically edited using TeXshade package. (b), Predicted secondary structure of 30-bp region at the 3′-end of Csup_Hel1A consensus sequence. Figure S3. The distribution of Csup_Hel1A-like Helitrons in insect and spider genomes. Taxa showing Hel1A-like Helitrons are colored taxonomically, with Lepidopteran insects in red, Diptera insects in purple, Hymenoptera wasps in yellow, Araneae species in green. Figure S4. Multiple alignment of 126 bp region at the 5′-end consensus sequence of Hel1 Helitrons. Figure S5. Multiple alignment (a) and genetic distance analysis (b) of 30-bp region at the 5′-end of consensus sequences of Ptep_Hel2Ca and Csup_Hel1Ab. Figure S6. Structural analysis of degenerated remnants potential autonomous Helitrons found in N. clavipes, P. tepidariorus, P. machaon, C. vestalis, H. vitripennis, A. rosae and T. cristinae. Figure S7. Multiple alignment (a) and genetic distance analysis (b) of Rep/helicase protein sequences of reconstructed potential autonomous Helitrons. Figure S8. The typical integration pattern of Hel1A within genomes of P. xuthus and P. rapae. (a), A copy of Pxut_Hel1Aa inserted into the coding sequence (CDS) of a gene. (b), A copy of Pxut_Hel1Ab inserted into intron. (c), Several copies of Prap_Hel1Aa inserted into introns and exons of the same gene. Figure S9. Identification of source loci and end junctions of insertions in Sfru_Hel1A. The end junction sequences are shaded. Figure S10. Multiple alignment of Pxut_Hel1Ab consensus sequence and core sequence in P. xuthus (a), Csup_Hel1Aa and Csup_Hel1Ab consensus sequences in C. suppressalis as well as core sequence in genome of P. xuthus (BBJE) and S. frugiperda (FJUZ) (b) and Csup_Hel1Ea consensus sequence and four short core sequences in C. suppressalis (c). Figure S11. Paralogous or orthologous empty sites of Prap_Hel1Aa in P. rapae, Cves_Hel1Aa in C. vestalis, Csup_Hel1Aa in C. suppressalis, and Mcin_Hel1Ca in Melitaea cinxia. The 4-letter project ID of WGS accession number and corresponding species are listed as following: LWME for P. rapae, JZSA for C. vestalis, ANCD for C. suppressalis, APLT for Melitaea cinxia, JXPT for Bactrocera oleae and AZMT for Microplitis demolitor. Figure S12. Multiple alignment of orthologous gene of calreticulin from P. rapae (EU826537.1) and C. vestalis (KX384605.1) (a), heat shock protein 70 from P. rapae (KJ573767.1) and C. vestalis (JX088378.1) (b), opsin from P. rapae (AB177984.1) and C. vestalis (KY368220.1) (c), consensus sequences of Prap_Hel1Aa and Cves_Hel1Aa as well as a short copy of Prap_Hel1Aa (Prap0202, LWME01000202.1: 138955–138,682) (d) and Prap0202 and eight individual sequences of Cves_Hel1Aa including Cves_Hel1Aa.1(JZSA01006637.1: 22335–22,612), Cves_Hel1Aa.2 (JZSA01007293.1: 718–441), Cves_Hel1Aa.3 (JZSA01002845.1: 21212–21,486), Cves_Hel1Aa.4 (JZSA01005118.1: 18777–18,500), Cves_Hel1Aa.5 (JZSA01002791.1: 17198–17,475), Cves_Hel1Aa.6 (JZSA01001525.1: 718–441), Cves_Hel1Aa.7 (JZSA01000595.1: 12281–12,558) and Cves_Hel1Aa.8 (JZSA01006408.1: 3165–2888) (e). Figure S13. Phylogenetic analysis of all copies of Hel1 Helitrons in C. vestalis, P. rapae and C. suppressalis. The phylogenetic tree was constructed by the neighbor-joining method using MEGA 7.0 software. Figure S14. Detection of orthologous empty site of a short copy of Prap_Hel1Aa (Prap0202, LWME01000202.1: 138955–138,682) in P. rapae larvae collected from Yangzhou, China. Figure S15. Multiple alignment of sequences completely same as the consensus sequence of Prap_Hel1Aa in P. rapae. Figure S16. Multiple alignment of Emus_Hel1Ca and the consensus sequences of Bmor_Hel1Ca, Cvic_Hel1Ca (a) as well as Emus_Hel1Ca and the individual sequences of Bmor_Hel1Ca (Bmor_Hel1Ca.1, AADK01000158.1: 43487–43,268) and Cvic_Hel1Ca (Cvic_Hel1Ca.1, JXOT01107662.1: 1253–1516; Cvic_Hel1Ca.2, JXOT01181287.1: 382–645) (b). Figure S17. Multiple alignment (a) and genetic distance ananlysis (b) of Pgla_Hel1Ga and Hvit_Hel1Ga. Figure S18. Multiple alignment of Csup_Hel1Aa and Csup_Hel1Ab. Figure S19. Multiple alignment of Pxut_Hel1Aa and Pxut_Hel1Ab from P. xuthus. (PDF 3085 kb)
Table S1. List of copies of the Helitron based on searches of WGS database in C. suppressalis. Table S2. Estimates of evolutionary divergence between Hel1 transposon of 28 speices . The number of base differences per site from between sequences are shown. The analysis involved 34 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All ambiguous positions were removed for each sequence pair. There were a total of 131 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.0. (XLSX 66 kb)
Data S1. The Hel1 elements with degenerated remnants of Helitron coding sequences identified in insect and spider genome databases. Data S2. The reconstructed potential autonomous Hel1 Helitrons. (PDF 67 kb)
Data S3. The list of the analyzed species and corresponding amount of sequence data. (XLSX 2613 kb)
Data S4. The consensus sequences of 35 Helitron exemplars. Data S5. The sequences used for evolutionary analysis. (PDF 101 kb)
Acknowledgements
The authors would like to thank anonymous referees for their helpful comments on the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 30871642 and 31701792), National Rice Industry Technology System Project (Grant No. Cars-001-25), Jiangsu Agricultural Science and Technology Innovation Fund (Grant No. ZX (17)2002) and Jiangsu Science Project of China (Grant No. BK20181215).
Availability of data and materials
All the data supporting the findings are included in this published article and its supplementary information files.
Authors′ contributions
GH did most of the experimental work and wrote the manuscript; NZ analysed the genome database; JX designed the experiments; HJ reconstructed the autonomous Helitrons; CJ performed genomic DNA extraction and PCR; ZZ analysed the data and revised the manuscript; QS and DS revised the manuscript; JF designed the experiments and wrote the manuscript; JW designed the experiments, supervised all of the experimental work and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Guangjie Han, Email: hanguangjie177@163.com.
Nan Zhang, Email: 857512580@qq.com.
Jian Xu, Email: bio-xj@163.com.
Heng Jiang, Email: 1041010655@qq.com.
Caihong Ji, Email: 181068913@qq.com.
Ze Zhang, Email: zezhang@cqu.edu.cn.
Qisheng Song, Email: songq@missouri.edu.
David Stanley, Email: stanleyd@missouri.edu.
Jichao Fang, Phone: +86-25-84390395, Email: fangjc@jaas.ac.cn.
Jianjun Wang, Phone: +86-514-87977284, Email: wangjj@yzu.edu.cn.
References
- 1.Barron MG, Fiston-Lavier AS, Petrov DA, Gonzalez J. Population genomics of transposable elements in Drosophila. Annu Rev Genet. 2014;48:561–581. doi: 10.1146/annurev-genet-120213-092359. [DOI] [PubMed] [Google Scholar]
- 2.Jangam D, Feschotte C, Betrán E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017;33(11):817–831. doi: 10.1016/j.tig.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O. A unified classification system for eukaryotic transposbale elments. Nat Rev Genet. 2007;8(12):973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 4.Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8(4):272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 5.Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A. 2001;98(15):8714–8719. doi: 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapitonov VV, Jurka J. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007;23(10):521–529. doi: 10.1016/j.tig.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Rossato DO, Ludwig A, Depra M, Loreto EL, Ruiz A, Valente VL. BuT2 is a member of the third major group of hAT transposons and is involved in horizontal transfer events in the genus Drosophila. Genome Biol Evol. 2014;6(2):352–365. doi: 10.1093/gbe/evu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas J, Pritham EJ. Helitrons, the eukaryotic rolling-circle transposable elements. Microbiology Spectrum. 2015;3(4):MDNA3–0049-2014. doi: 10.1128/microbiolspec.MDNA3-0049-2014. [DOI] [PubMed] [Google Scholar]
- 9.Grabundzija I, Messing SA, Thomas J, Cosby RL, Bilic I, Miskey C, et al. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat Commun. 2016;7:10716. doi: 10.1038/ncomms10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Bennetzen JL. Distribution, diversity, evolution, and survival of Helitrons in the maize genome. Proc Natl Acad Sci U S A. 2009;106(47):19922–19927. doi: 10.1073/pnas.0908008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong W, He L, Lai J, Dooner HK, Du C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc Natl Acad Sci U S A. 2014;111(28):10263–10268. doi: 10.1073/pnas.1410068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du C, Fefelova N, Caronna J, He L, Dooner HK. The polychromatic Helitron landscape of the maize genome. Proc Natl Acad Sci U S A. 2009;106(47):19916. doi: 10.1073/pnas.0904742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas J, Phillips CD, Baker RJ, Pritham EJ. Rolling-circle transposons catalyze genomic innovation in a mammalian lineage. Genome Biol Evol. 2014;6(10):2595–2610. doi: 10.1093/gbe/evu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han MJ, Shen YH, Xu MS, Liang HY, Zhang HH, Zhang Z. Identification and evolution of the silkworm Helitrons and their contribution to transcripts. DNA Res. 2013;20(5):471–484. doi: 10.1093/dnares/dst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41(41):331. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartl DL, Lohe AR, Lozovskaya ER. Modern thoughts on an ancyent marinere: function, evolution, regulation. Annu Rev Genet. 1997;31(31):337–358. doi: 10.1146/annurev.genet.31.1.337. [DOI] [PubMed] [Google Scholar]
- 17.Peccoud J, Loiseau V, Cordaux R, Gilbert C. Massive horizontal transfer of transposable elements in insects. Proc Natl Acad Sci U S A. 2017;114(18):4721–4726. doi: 10.1073/pnas.1621178114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sormacheva I, Smyshlyaev G, Mayorov V, Blinov A, Novikov A, Novikova O. Vertical evolution and horizontal transfer of CR1 non-LTR retrotransposons and Tc1/mariner DNA transposons in Lepidoptera species. Mol Biol Evol. 2012;29(12):3685–3702. doi: 10.1093/molbev/mss181. [DOI] [PubMed] [Google Scholar]
- 19.Thomas J, Schaack S, Pritham EJ. Pervasive horizontal transfer of rolling-circle transposons among animals. Genome Biol Evol. 2010;2:656–664. doi: 10.1093/gbe/evq050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coates BS. Horizontal transfer of a non-autonomous Helitron among insect and viral genomes. BMC Genomics. 2015;16:137. doi: 10.1186/s12864-015-1318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo X, Gao J, Li F, Wang J. Evidence of horizontal transfer of non-autonomous Lep1 Helitrons facilitated by host-parasite interactions. Sci Rep. 2014;4:5119. doi: 10.1038/srep05119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pritham EJ, Feschotte C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc Natl Acad Sci U S A. 2007;104(6):1895–1900. doi: 10.1073/pnas.0609601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorbunova V, Levy AA. Non-homologous DNA end joining in plant cells is associated with deletions and filler DNA insertions. Nucleic Acids Res. 1997;25(22):4650. doi: 10.1093/nar/25.22.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22(23):2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Bennetzen JL. Structure-based discovery and description of plant and animal Helitrons. Proc Natl Acad Sci U S A. 2009;106(31):12832–12837. doi: 10.1073/pnas.0905563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendiola MV, Bernales I, Cruz FDL. Differential roles of the transposon termini in IS91 transposition. Proc Natl Acad Sci U S A. 1994;91(5):1922–1926. doi: 10.1073/pnas.91.5.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cultrone A, Domínguez YR, Drevet C, Scazzocchio C, Fernández-Martín R. The tightly regulated promoter of the xanA gene of Aspergillus nidulans is included in a helitron. Mol Microbiol. 2007;63(6):1577–1587. doi: 10.1111/j.1365-2958.2007.05609.x. [DOI] [PubMed] [Google Scholar]
- 28.Stuart T, Eichten SR, Cahn J, Karpievitch YV, Borevitz JO, Lister R. Population scale mapping of transposable element diversity reveals links to gene regulation and epigenomic variation. Elife. 2016;5:e20777. doi: 10.7554/eLife.20777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seibt KM, Wenke T, Muders K, Truberg B, Schmidt T. Short interspersed nuclear elements (SINEs) are abundant in Solanaceae and have a family-specific impact on gene structure and genome organization. Plant J. 2016;86(3):268–285. doi: 10.1111/tpj.13170. [DOI] [PubMed] [Google Scholar]
- 30.Soichi I, Kenzo N, Atsushi M. A link among DNA replication, recombination, and gene expression revealed by genetic and genomic analysis of TEBICHI gene of Arabidopsis thaliana. PLoS Genet. 2009;5(8):e1000613. doi: 10.1371/journal.pgen.1000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsukamoto T, Hauck NR, Tao R, Ning J, Iezzoni AF. Molecular and genetic analyses of four nonfunctional S haplotype variants derived from a common ancestral S haplotype identified in sour cherry (Prunus cerasus L.) Genetics. 2010;184(2):411. doi: 10.1534/genetics.109.109728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong Y, Lu X, Song W, Shi L, Zhang M, Zhao H, et al. Structural characterization of helitrons and their stepwise capturing of gene fragments in the maize genome. BMC Genomics. 2011;12:609. doi: 10.1186/1471-2164-12-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lal SK, Hannah LC. Plant genomes: massive changes of the maize genome are caused by Helitrons. Heredity. 2005;95(6):421–422. doi: 10.1038/sj.hdy.6800764. [DOI] [PubMed] [Google Scholar]
- 34.Dotto BR, Carvalho EL, da Silva AF, Dezordi FZ, Pinto PM, TdL C, et al. HTT-DB: new features and updates. Database. 2018;2018. [DOI] [PMC free article] [PubMed]
- 35.Schaack S, Gilbert C, Feschotte C. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 2010;25(9):537–546. doi: 10.1016/j.tree.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilbert C, Schaack S, Pace JK, II, Brindley PJ, Feschotte C. A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature. 2010;464(7293):1347–1350. doi: 10.1038/nature08939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heimpel GE, Neuhauser C, Hoogendoorn M. Effects of parasitoid fecundity and host resistance on indirect interactions among hosts sharing a parasitoid. Ecol Lett. 2003;6(6):556–566. [Google Scholar]
- 38.Cameron PJ, Walker GP. Host specificity of Cotesia rubecula and Cotesia plutellae, parasitoids of white butterfly and diamondback moth. New Zealand Plant Protection. 1997;50(50):236–241. [Google Scholar]
- 39.Oliver K, Degnan P, Burke G, Moran N. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol. 2010;55(55):247–266. doi: 10.1146/annurev-ento-112408-085305. [DOI] [PubMed] [Google Scholar]
- 40.Husnik F, Nikoh N, Koga R, Ross L, Duncan RP, Fujie M, et al. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell. 2013;153(7):1567–1578. doi: 10.1016/j.cell.2013.05.040. [DOI] [PubMed] [Google Scholar]
- 41.Sloan DB, Nakabachi A, Richards S, Qu J, Murali SC, Gibbs RA, et al. Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol Biol Evol. 2014;31(4):857–871. doi: 10.1093/molbev/msu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunning Hotopp JC, Clark ME, Oliveira DC, Foster JM, Fischer P, Muñoz Torres MC, et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317(5845):1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- 43.Venner S, Miele V, Terzian C, Biemont C, Daubin V, Feschotte C, et al. Ecological networks to unravel the routes to horizontal transposon transfers. PLoS Biol. 2017;15(2):e2001536. doi: 10.1371/journal.pbio.2001536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Werren JH. Biology of Wolbachia. Annu Rev Entomol. 1997;42(1):587. doi: 10.1146/annurev.ento.42.1.587. [DOI] [PubMed] [Google Scholar]
- 45.Nikoh N, Tanaka K, Shibata F, Kondo N, Hizume M, Shimada M, et al. Wolbachia genome integrated in an insect chromosome: evolution and fate of laterally transferred endosymbiont genes. Genome Res. 2008;18(2):272–280. doi: 10.1101/gr.7144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilbert C, Feschotte C. Horizontal acquisition of transposable elements and viral sequences: patterns and consequences. Curr Opin Genet Dev. 2018;49:15–24. doi: 10.1016/j.gde.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilbert C, Chateigner A, Ernenwein L, Barbe V, Bezier A, Herniou EA, et al. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat Commun. 2014;5:3348. doi: 10.1038/ncomms4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herniou EA, Olszewski JA, O'Reilly DR, Cory JS. Ancient coevolution of baculoviruses and their insect hosts. J Virol. 2004;78(7):3244–3251. doi: 10.1128/JVI.78.7.3244-3251.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang HH, Xu HE, Shen YH, Han MJ, Zhang Z. The origin and evolution of six miniature inverted-repeat transposable elements in Bombyx mori and Rhodnius prolixus. Genome Biol Evol. 2013;5(11):2020. doi: 10.1093/gbe/evt153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lerat E, Rizzon C, Biémont C. Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Res. 2003;13(8):1889–1896. doi: 10.1101/gr.827603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends in Genetics Tig. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 52.Mathews JSR, David H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11(1):129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beitz E. TEXshade: shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics. 2000;16(2):135–139. doi: 10.1093/bioinformatics/16.2.135. [DOI] [PubMed] [Google Scholar]
- 55.Ronquist F, Teslenko M, DMP V, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Criscuolo A, Gribaldo S. BMGE (block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10(1):210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 58.Chen C, Xia R, Chen H, He Y. TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. BioRxiv. 2018:289660.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Insertion of a novel Helitron element into downstream region of a SINE element in C. suppressalis genome. The SINE and Helitron sequences are highlighted in blue and pink, respectively. The nucleotides highlighted in purple are the target site duplication (TSD) of SINE. Figure S2. Characteristic of Csup_Hel1A. (a), Multiple alignment of 30-bp end sequences as well as the flanking host nucleotides at the 5′- and 3′-end from Csup_Hel1A elements. The alignment was graphically edited using TeXshade package. (b), Predicted secondary structure of 30-bp region at the 3′-end of Csup_Hel1A consensus sequence. Figure S3. The distribution of Csup_Hel1A-like Helitrons in insect and spider genomes. Taxa showing Hel1A-like Helitrons are colored taxonomically, with Lepidopteran insects in red, Diptera insects in purple, Hymenoptera wasps in yellow, Araneae species in green. Figure S4. Multiple alignment of 126 bp region at the 5′-end consensus sequence of Hel1 Helitrons. Figure S5. Multiple alignment (a) and genetic distance analysis (b) of 30-bp region at the 5′-end of consensus sequences of Ptep_Hel2Ca and Csup_Hel1Ab. Figure S6. Structural analysis of degenerated remnants potential autonomous Helitrons found in N. clavipes, P. tepidariorus, P. machaon, C. vestalis, H. vitripennis, A. rosae and T. cristinae. Figure S7. Multiple alignment (a) and genetic distance analysis (b) of Rep/helicase protein sequences of reconstructed potential autonomous Helitrons. Figure S8. The typical integration pattern of Hel1A within genomes of P. xuthus and P. rapae. (a), A copy of Pxut_Hel1Aa inserted into the coding sequence (CDS) of a gene. (b), A copy of Pxut_Hel1Ab inserted into intron. (c), Several copies of Prap_Hel1Aa inserted into introns and exons of the same gene. Figure S9. Identification of source loci and end junctions of insertions in Sfru_Hel1A. The end junction sequences are shaded. Figure S10. Multiple alignment of Pxut_Hel1Ab consensus sequence and core sequence in P. xuthus (a), Csup_Hel1Aa and Csup_Hel1Ab consensus sequences in C. suppressalis as well as core sequence in genome of P. xuthus (BBJE) and S. frugiperda (FJUZ) (b) and Csup_Hel1Ea consensus sequence and four short core sequences in C. suppressalis (c). Figure S11. Paralogous or orthologous empty sites of Prap_Hel1Aa in P. rapae, Cves_Hel1Aa in C. vestalis, Csup_Hel1Aa in C. suppressalis, and Mcin_Hel1Ca in Melitaea cinxia. The 4-letter project ID of WGS accession number and corresponding species are listed as following: LWME for P. rapae, JZSA for C. vestalis, ANCD for C. suppressalis, APLT for Melitaea cinxia, JXPT for Bactrocera oleae and AZMT for Microplitis demolitor. Figure S12. Multiple alignment of orthologous gene of calreticulin from P. rapae (EU826537.1) and C. vestalis (KX384605.1) (a), heat shock protein 70 from P. rapae (KJ573767.1) and C. vestalis (JX088378.1) (b), opsin from P. rapae (AB177984.1) and C. vestalis (KY368220.1) (c), consensus sequences of Prap_Hel1Aa and Cves_Hel1Aa as well as a short copy of Prap_Hel1Aa (Prap0202, LWME01000202.1: 138955–138,682) (d) and Prap0202 and eight individual sequences of Cves_Hel1Aa including Cves_Hel1Aa.1(JZSA01006637.1: 22335–22,612), Cves_Hel1Aa.2 (JZSA01007293.1: 718–441), Cves_Hel1Aa.3 (JZSA01002845.1: 21212–21,486), Cves_Hel1Aa.4 (JZSA01005118.1: 18777–18,500), Cves_Hel1Aa.5 (JZSA01002791.1: 17198–17,475), Cves_Hel1Aa.6 (JZSA01001525.1: 718–441), Cves_Hel1Aa.7 (JZSA01000595.1: 12281–12,558) and Cves_Hel1Aa.8 (JZSA01006408.1: 3165–2888) (e). Figure S13. Phylogenetic analysis of all copies of Hel1 Helitrons in C. vestalis, P. rapae and C. suppressalis. The phylogenetic tree was constructed by the neighbor-joining method using MEGA 7.0 software. Figure S14. Detection of orthologous empty site of a short copy of Prap_Hel1Aa (Prap0202, LWME01000202.1: 138955–138,682) in P. rapae larvae collected from Yangzhou, China. Figure S15. Multiple alignment of sequences completely same as the consensus sequence of Prap_Hel1Aa in P. rapae. Figure S16. Multiple alignment of Emus_Hel1Ca and the consensus sequences of Bmor_Hel1Ca, Cvic_Hel1Ca (a) as well as Emus_Hel1Ca and the individual sequences of Bmor_Hel1Ca (Bmor_Hel1Ca.1, AADK01000158.1: 43487–43,268) and Cvic_Hel1Ca (Cvic_Hel1Ca.1, JXOT01107662.1: 1253–1516; Cvic_Hel1Ca.2, JXOT01181287.1: 382–645) (b). Figure S17. Multiple alignment (a) and genetic distance ananlysis (b) of Pgla_Hel1Ga and Hvit_Hel1Ga. Figure S18. Multiple alignment of Csup_Hel1Aa and Csup_Hel1Ab. Figure S19. Multiple alignment of Pxut_Hel1Aa and Pxut_Hel1Ab from P. xuthus. (PDF 3085 kb)
Table S1. List of copies of the Helitron based on searches of WGS database in C. suppressalis. Table S2. Estimates of evolutionary divergence between Hel1 transposon of 28 speices . The number of base differences per site from between sequences are shown. The analysis involved 34 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All ambiguous positions were removed for each sequence pair. There were a total of 131 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.0. (XLSX 66 kb)
Data S1. The Hel1 elements with degenerated remnants of Helitron coding sequences identified in insect and spider genome databases. Data S2. The reconstructed potential autonomous Hel1 Helitrons. (PDF 67 kb)
Data S3. The list of the analyzed species and corresponding amount of sequence data. (XLSX 2613 kb)
Data S4. The consensus sequences of 35 Helitron exemplars. Data S5. The sequences used for evolutionary analysis. (PDF 101 kb)
Data Availability Statement
All the data supporting the findings are included in this published article and its supplementary information files.